Selenium speciation in enriched and natural samples by HPLC-ICP-MS and HPLC-ESI-MS with perfluorinated carboxylic acid ion-pairing agents†
Mihály Kotrebaia, Marc Birringerb, Julian F. Tysona, Eric Blockb and Peter C. Uden*a
aDepartment of Chemistry, University of Massachusetts, Box 35410, Amherst, MA 01003-4510, USA
bDepartment of Chemistry, SUNY–Albany, Albany, NY 12222, USA
First published on UnassignedUnassigned24th December 1999
Abstract
Selenium-enriched plants, such as hyperaccumulative phytoremediation plants (Astragalus praleongus, 517 μg g−1 Se, and Brassica juncea, 138 μg g−1 Se in dry sample), yeast (1200, 1922 and 2100, μg g−1 Se in dry sample), ramp (Allium tricoccum, 48, 77, 230, 252, 405 and 524 μg g−1 Se in dry sample), onion (Allium cepa, 96 and 140 μg g−1 Se in dry sample) and garlic (Allium sativum, 68, 112, 135, 296, 1355 μg g−1 Se in dry sample) were analyzed by HPLC-ICP-MS for their selenium content and speciation after hot water and enzymatic extractions. Reference samples with natural selenium levels, such as onion and garlic controls, cooking garlic powder, baking yeast powder and a commercial garlic supplement were also analyzed. Selected samples were also examined by HPLC-electrospray ionization (ESI)-MS. HPLC was mostly carried out with 0.1% heptafluorobutanoic acid (HFBA) as ion-pairing agent in 1 + 99 v/v methanol–water solution, but 0.1% trifluoroacetic acid (TFA) in 1 + 99 v/v methanol–water solution was also utilized to permit chromatography for compounds that did not elute with HFBA. More than 75% of the total eluting selenium compounds, based upon element specific detection, were identified from retention time data and standard spiking experiments, and between 60 and 85% of compounds were identified by MS, with up to 25% of the total eluting molecular selenium species being unidentified as yet. Limits of quantification (LOQ, defined as the concentration giving an S/N of 10) for HPLC-ICP-MS were in the range 2–50 ng mL−1 Se in the injected extracts for the selenium-enriched samples and 2–10 ng mL−1 Se for the natural selenium level samples. LOQ values for HPLC-ESI-MS were ca. 100 times higher than those measured by HPLC-ICP-MS.
Introduction
The importance of selenium as a trace element in the human diet has long been established. A major clinical development was the finding by Clark et al.l that human dietary supplementation with selenium-enriched yeast decreased cancer incidence and mortality rate by nearly 50%. Increase in selenium intake to the 200 μg d−1 level, used in the Clark study and suggested by the United States National Research Council, is not feasible by consuming a diet with natural selenium concentrations owing to the low abundance of selenium in common foods.2 Hence a knowledge of the selenium content of selenium-enriched supplements or proposed supplements is important. The cancer preventive effect of selenium has been tentatively attributed to the biological functions of selenoamino acids.3,4 Our earlier publications5–8 and other review and summary papers9–16 give a good coverage of the research in this field.Ion-pair chromatography, often used for the separation of amino acids,17,18 is increasingly popular for the speciation of selenoamino acids owing to its superior performance.6,7,19,20 The ion-pairing role of trifluoroacetic acid (TFA) and other perfluorinated carboxylic acids for the separation of common amino acids has been evaluated.21–23
Casiot et al.20 identified Se-adenosylselenohomocysteine in a yeast extract with off-line ESI-MS detection. We independently reported this finding and the identification of selenomethionine in yeast by on-line HPLC-ESI-MS.19 Identification of γ-glutamyl-Se-methylselenocysteine and possibly γ-glutamyl-Se-selenomethionine in garlic were also made in the same publication. Diverse sulfur compounds, including γ-glutamyl derivatives of S-alk(en)ylcysteines and methionine, have already been identified in Allium species by MS and other methods.24–26
Many different samples were analyzed by various methods for their selenium content with some success. Zheng et al.27 and Uden et al.7 analyzed commercially available selenium supplements. Stemming from the Clark study, an important area of research has been the determination of selenium species in yeast.6,20,28–31 Some other frequently analyzed samples are Allium plants,25,31 marine algae,32–35 soybean,36 urine37 and fish.38
As part of an ongoing study of the cancer chemopreventive activity of selenium, ion-pairing reversed-phase separation methods developed earlier39 were utilized to analyze various selenium-enriched samples. These samples were ramp (Allium tricoccum), onion (Allium cepa), garlic (Allium sativum), yeast and phytoremediation plants with several different selenium concentrations for each sample type. Selenium compounds were identified (a) by retention time matching with peaks in the chromatogram of a standard mixture containing over 20 different selenium species and (b) by spiking experiments with 0.1% heptafluorobutanoic acid (HFBA) or 0.1% TFA containing mobile phase using HPLC-ICP-MS. Compounds at sufficiently high concentrations were also identified by HPLC-ESI-MS. The percentage of the total selenium present in the various species in each sample was calculated.
Experimental
Instrumentation
Elan 5000 (for selenium-enriched samples) and Elan 5000a (for samples with natural selenium levels) inductively coupled plasma mass spectrometers (Perkin-Elmer SCIEX, Thornhill, Ontario, Canada) were used as HPLC detectors. Samples were introduced by a Meinhard nebulizer with an in-house fabricated spray chamber containing an impact bead.8 The spray chamber had a path length of 8.4 cm and a volume of 14 mL. Instrumental conditions were as follows: rf forward power, 1100 W; plasma flow rate, 15.0 L min−1; auxiliary flow rate, 0.8 L min−1; nebulizer flow rate, 0.80–0.95 L min−1; resolution, normal; scanning mode, peak hop; dwell time, 500 or 1000 ms; and isotope monitored, mass 82.The chromatographic system consisted of a liquid chromatographic pump (SP8810, Spectra-Physics, San Jose, CA, USA) and a 5 μm Symmetry Shield RP8 (15 cm × 3.9 mm id) column (Waters, Milford, MA, USA), which has a polar modifier group between the C8 group and the silica base.40,41 The column was connected to the nebulizer with PEEK tubing (30 cm × 0.25 mm id). The mobile phase compositions were as follows: 99 + 1 v/v water–methanol was used in each case, containing (a) 0.1% TFA or (b) 0.1% HFBA.
A Bruker-Hewlett-Packard Esquire-LC mass spectrometer (Bruker-Franzen Analytik, Bremen, Germany) was used for the molecular mass spectral studies. For HPLC-ES-MS analysis the 1 mL min−1 column eluate was split 1∶5 with a T splitter. The T splitter was connected to the ESI source with PEEK tubing (8 cm × 0.25 mm id). Mass calibration and optimization of the operating parameters were performed daily and generally followed the manufacturer’s guidelines.
Instrumental conditions were as follows: positive ion, standard scan range, normal scan resolution; ESI source, capillary −3500 V, capillary exit 65 V, end plate −3000 V; nebulizer (N2) pressure, 20 lb in−2; drying gas (N2) flow rate, 12 L min−1; drying gas temperature, 350 °C; lens pass voltages, skimmer 1 15 V, skimmer 2 5 V; ion charge control, off; accumulation time, 0.5 ms; cut-off, m/z 45; scan, m/z 50–800; averages; 15; rolling average, no.
Peak integration and other chromatographic calculations were performed using PeakFit software.
Chemicals
Barnstead (Boston, MA, USA) E-pure 18 MΩ water , nitric acid, hydrochloric acid (purified by sub-boiling), TFA, HFBA (Aldrich, Milwaukee, WI, USA), and methanol (HPLC grade) were used.The selenium compounds in the standard mixture are listed in Table 1. Compounds 1, 2, 3, 6, 7, 11, 18 and protease XIV were obtained from Sigma (St. Louis, MO, USA) and 4, 9, 17, and 14 from Professor H. Ganther (University of Wisconsin, Madison, WI, USA); 5, 8, 10, 13, 15, 16, 19, 21 and 22 were synthesized in-house. Plasma selenium standard solution (1000 μg mL−1) was obtained from Spex Industries (Edison, NJ, USA).
| 1 | Selenic acid, selenate, SeO42− (Na2SeO4) |
| 2 | Selenous acid, selenite, SeO32− (Na2SeO3) |
| 3 | Selenocyanate, SeCN− (KSeCN) |
| 4 | Methaneseleninic acid, CH3Se(O)OH |
| 5 | Se-Lanthionine, NH2CH(COOH)CH2SeCH2CH(COOH) NH2 |
| 6 | Trimethylselenonium, (CH3)3Se+ [(CH3)3SeI] |
| 7 | Selenocystine, NH2CH(COOH)CH2SeSeCH2CH(COOH)NH2 |
| 8 | Se-Cystathionine, NH2CH(COOH)CH2SeCH2CH2CH(COOH) NH2 |
| 9 | Se-MethyIseIenocysteine, CH3SeCH2CH(COOH)NH2 |
| 10 | Se-2-Propynylselenocysteine,
HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CCH2SeCH2CH(COOH)NH2 CCH2SeCH2CH(COOH)NH2 |
| 11 | Selenomethionine, CH3SeCH2CH2CH(COOH)NH2 |
| 12 | Degradation product of Se-2-methyl-2-propenylselenocysteine |
| 13 | γ-Glutamyl-Se-methylselenocysteine, CH3SeCH2CH(COOH)NHC(O)CH2CH2CH (COOH)NH2 |
| 14 | Se-Allylselenocysteine,
CH2![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) CHCH2SeCH2CH(COOH)NH2 CHCH2SeCH2CH(COOH)NH2
|
| 15 | cis-Se-1-Propenylselenocysteine,
CH3CHCHSeCH2CH(COOH)NH2 |
| 16 | trans-Se-1-Propenylselenocysteine,
CH3CHCHSeCH2CH(COOH)NH2 |
| 17 | Se-1-Propylselenocysteine, CH3CH2CH2SeCH2CH(COOH)NH2 |
| 18 | Selenoethionine, CH3CH2SeCH2CH2CH(COOH)NH2 |
| 19 | Selenohomocystine, NH2CH(COOH)CH2CH2SeSeCH2CH 2CH(COOH)NH2 |
| 20 | Degradation product of Se-1-methyl-2-propenylselenocysteine |
| 21 | Se-2-Methyl-2-propenylselenocysteine,
CH2C(CH3)CH2SeCH2CH(CO
OH)NH2 |
| 22 | Se-1-Methyl-2-propenylselenocysteine,
CH2CHCH(CH3)SeCH2CH(COOH)NH2
|
| 23 | Se-Adenosylselenohomocysteine, NH2CH(COOH)CH2CH2SeCH2C4 H5O3C5N4NH2 |
Selenium-enriched yeast (1922 μg g−1 Se in dry sample) was obtained from Nutrition 21 (San Diego, CA, USA) and from a commercial source; 1200 and 2100 μg g−1 Se in dry sample). Selenium-enriched ramp (48, 77, 230, 252, 405 and 524 μg g−1 Se in dry sample) was provided by Professor P. Whanger (Oregon State University, Corvallis, OR, USA). Selenium-enriched garlic (0.2, 68, 112, 235, 296 and 1355 μg g−1 Se in dry sample) and onion (0.2, 96 and 140 μg g−1 Se in dry sample) samples were obtained from Professor D. Lisk (Cornell University, Ithaca, NY, USA). Astragalus praleongus (517 μg g−1 Se in dry sample) and Brassica juncea (138 μg g−1 Se in dry sample) were obtained from Dr. G. S. Bañuelos (USDA, Fresno, CA, USA). The total selenium concentrations were as supplied with the samples. Other non-selenium-enriched samples, garlic, baking yeast, cooking garlic powder and commercial garlic supplement were purchased locally. Stock standard solutions of selenoamino acids were prepared in 0.2 mol L−1 HCl. A stock standard solution of selenate was prepared in 2% v/v HNO3. All solutions were stored in the dark at 0–4 °C.
Sample preparation
Enzymatic and hot water extractions followed the procedures reported earlier,7 and are summarized here. For the hot water extraction, 5 mL of distilled, de-ionized water were added to a 0.2 g sample in a 15 mL centrifuge tube and the tube was placed in a double boiling-water bath for 1 h. The mixture was shaken well every 15 min. For the enzymatic extraction, 5 mL of distilled, de-ionized water were added to 0.2 g of sample and 0.02 g of protease XIV enzyme in a 15 mL centrifuge tube, then the mixture was shaken for 24 h at room temperature. After the extraction, the samples were centrifuged and filtered. Samples were acidified before injection into the HPLC system (60 μL of 0.5 mol L−l HCl to 300 μL of sample extract).39Procedures
Selenium-enriched plants, such as hyperaccumulative phytoremediation plants (Astragalus praleongus, 517 μg g−1 Se, and Brassica juncea, 138 μg g−1 Se in dry sample), yeast (1200, 1922 and 2100 μg g−1 Se in dry sample), ramp (48, 77, 230, 252, 405 and 524 μg g−1 Se in dry sample), onion (96 and 140 μg g−1 Se in dry sample) and garlic (68, 112, 135, 296 and 1355 μg g−1 Se in dry sample) and reference samples with natural selenium levels, such as onion and garlic controls, cooking garlic powder, baking yeast powder and a commercial garlic supplement, were analyzed by HPLC-ICP-MS for their selenium content and speciation after hot water and enzymatic extractions. Selected samples were also examined by HPLC-ESI-MS.Measurements of the selenium in natural level samples were made in two laboratories with different instrumentation with replicate sample extracts from different days and indicated good reproducibility. Selenium-enriched samples were not prepared or measured on the same days as the natural samples to avoid the risk of cross-contamination.
Results and discussion
HPLC-ICP-MS analysis of selenium standards
Fig. 1 shows the HPLC-ICP-MS chromatogram of the compounds listed in Table 1. HFBA was used as ion-pairing agent at 0.1% concentration. The compound identification numbers in Table 1 are given on the peaks. The peak intensities were normalized to the highest peak. The concentrations of the standards were in the 0.2–2 μg ml−1 range, with increasing concentrations for the later eluting compounds. | ||
| Fig. 1 HPLC-ICP-MS chromatograms of selenium standard mixture (peak labels correspond to compound numbers listed in Table 1) using HFBA with 16 and 70 min (inset) time-scales. | ||
The ICP-MS detection system is not able to tolerate more than a few per cent of organic content in the mobile phase without serious hardware modifications, which represented a limitation to the possible use of ion-pairing agents. This led to the choice of HFBA and 1% methanol, for which the retention times typically increased by a factor of five compared with TFA, to over 1 h in the case of the last standard peak.39 Selenite and methaneseleninate (MeSeO2) also could be resolved by the use of PeakFit chromatographic software. In individual standard injections they were easily distinguishable based on retention times. Selenocyanate was not included in the standard mixture as it decomposes to elemental selenium under the acidic conditions employed.
HPLC-ICP-MS analysis of selenium-enriched samples
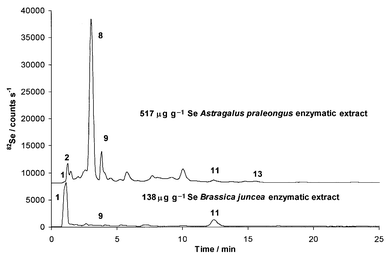 | ||
| Fig. 2 HPLC-ICP-MS chromatograms of enzymatic extracts of phytoremediation samples: 515 μg g−1 Se Astralagus praleongus (upper) and 138 μg g−1 Se Brassica juncea (lower) using HFBA. | ||
Fig. 3(a) shows a comparison of the water (lower chromatogram and left intensity scale) and the enzymatic (upper chromatogram and right intensity scale) extracts of the 1922 μg g−1 Se yeast on a 50 min time-scale. The water extract contains, on the basis of integration, 10% of the selenium found in the enzymatic extract. Earlier studies6,7 also showed that the enzymatic extraction efficiency for yeast was around 95%, indicating almost complete extraction, while the efficiency of water extraction was approximately 10%. These results suggest that only 10% of the total selenium species are in free, non-protein bound form, while the rest, mainly in the form of selenomethionine (11), is part of different proteins.42Se-Adenosylselenohomocysteine (23) was only identified when TFA was used [Fig. 3(c), upper and middle] and not when HFBA was used. The two NH2 groups of this molecule are distant enough from each other to be able to engage fully in double ion-pairing action. Hence the size and polarity of the doubly ion-paired 23 precludes elution from the column at a 1% methanol concentration. Compounds 11 and 23 were also identified by ESI-MS when TFA was used, as reported earlier.19 As shown in Fig. 3(b), minor components identified by retention time matching followed by spiking experiments were selenite (2), Se-lanthionine (5), selenocystine (7), Se-cystathionine (8), Se-methylselenocysteine (9) and y-glutamyl-Se-methylselenocysteine (13). The only difference between the comparison of the TFA and HFBA chromatograms was the detection of compound 23.
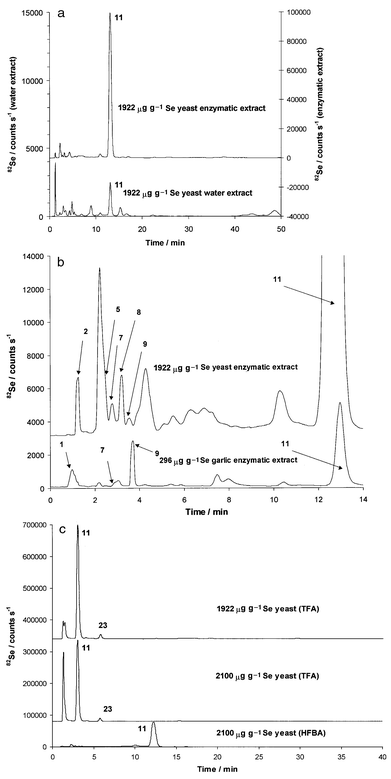 | ||
| Fig. 3 (a) HPLC-ICP-MS chromatograms of enzymatic (upper) and water (lower) extracts of 1922 μg g−1 Se yeast sample using HFBA. (b) First 15 min of HPLC-ICP-MS chromatograms of enzymatic extract of 1922 μg g−1 Se yeast (upper) and 296 μg g−1 Se garlic (lower) samples using HFBA . (c) HPLC-ICP-MS chromatograms of enzymatic extracts of 1922 μg g−1 Se yeast using TFA (upper) and 2100 μg g−1 Se yeast using TFA (middle) and HFBA (lower). | ||
Comparison of the TFA (middle) and HFBA (lower) chromatograms of the enzymatic extract of 2100 μg g−1 Se yeast sample in Fig. 3(c) reveals major differences. The TFA chromatogram (middle) indicates the presence of an unretained compound (or compounds) and selenomethionine (11), both as major constituents. The HFBA chromatogram (lower) shows, within the parallel elution window, only 11 as the major peak plus a very poorly defined broad peak in the 10 min elution region, which, on the basis of area comparison, could account for the area of the unretained peak seen under TFA conditions. ESI-MS identification of this unknown peak has so far been unsuccessful. The 1200 μg g−1 Se yeast sample contained about 15% selenite, which indicates a poorer degree of conversion from the inorganic selenium growth medium to the organic selenium forms. Selenium can be accumulated in the human body in the form of proteins as a result of supplementation. This is undesirable since selenium compounds replace their sulfur counterparts causing gene damage. Supplementation with different selenium species will result in different degrees of accumulation. The accumulation from inorganic selenium is much higher than from 11. Hence the goal in selenium-enriched yeast cultivation is to limit the inorganic selenium content as much as possible, increasing the conversion rate to organic forms, particularly selenomethionine. The 1200 μg g−1 Se yeast clearly proved less than optimal in this regard, while the 1922 μg g−1 Se yeast has a much more desirable distribution. The 2100 μg g−1 Se yeast, while converting the inorganic selenium with the same efficiency as the 1922 μg g−1 Se yeast (residual selenite was 1% for each sample), produced an unknown selenium compound in the major concentration range. The same observation was made on a set of yeast samples from a different source containing 1250 μg g−1 Se. These findings indicate that the total selenium concentration measurement is not a good indication of selenium distribution. Furthermore, fluctuations in selenium distribution among different selenium-enriched yeast samples show the need to be cautious with selenium supplementation regimens.
(a) Garlic.. Fig. 4 shows the full time-scale chromatograms of enzymatic extracts of 68, 112, 235, 296, 1355 μg g−1 Se-containing garlic samples with percentage distributions as follows: 4% (1), 1.5% (8), 60%* (9), 13%* (11), 8%* (13), 87% (sum) for the 1355 μg g−1 Se garlic, 2% (1), 0.5% (7), 0.5% (8), 3% (9), 13%* (11), 73%* (13), 96% (sum) for the 296 μg g−1 Se garlic, 1.5% (1), 0.5% (7), 0.5% (8), 3% (9), 17%* (11), 70%* (13), 95% (sum) for the 235 μg g−1 Se garlic, 0.1% (1), 1% (7), 1.5% (8), 3% (9), 28%* (11), 52%* (13), 93% (sum) for the 112 μg g−1 Se garlic and 1% (1), 0.5% (7), 0.5% (8), 2.5% (9), 18%* (11), 68%* (13), 93% (sum) for the 296 μg g−1 Se garlic. Compounds with distribution percentage values marked with an asterisk were confirmed by HPLC-ESI-MS identification.
 | ||
| Fig. 4 HPLC-ICP-MS chromatograms of enzymatic extracts of 68, 112, 235, 296 and 1355 μg g−1 Se garlic samples using HFBA (the 1355 μg g−1 Se garlic extract was diluted 10-fold before injection). | ||
The lower chromatogram in Fig. 3(b) shows the first 14 min for the 296 μg g−1 Se garlic enzymatic extract. Several important observations may be made upon examining the chromatograms. The selenium distributions of garlic samples are very different from those of yeast. Although selenomethionine (11) is clearly present in each sample, it is never the dominating selenium species. In previous work5–7,19 selenomethionine was not identified in garlic samples, because only their water extracts were examined. As for the yeast samples, the selenomethionine could not be efficiently extracted by hot water and enzymatic extraction was needed to release 11 from the proteins. Careful comparison of the TFA and HFBA chromatograms did not indicate that any of the compounds eluting with the former ion-pair agent would not also elute with the latter. The major components in the low and middle range selenium-enriched garlic (68, 112, 235, 296 μg g−1 Se) were γ-glutamyl-Se-methylselenocysteine (13) and selenomethionine (11). Selenate (1), selenocystine (7), Se-cystathionine (8) and Se-methylselenocysteine (9) were also identified. Upon increase in the total selenium concentration to 1355 μg g−1 Se, the distribution of the selenium species changed, and 9 became the principal selenium species, while 13 and 11 were present at 13 and 8%, respectively.
The relative percentage concentrations of γ-glutamyl-Se-methylselenocysteine (13) and selenomethionine (11) were essentially constant up to the 296 μg g−1 Se total selenium level, hence their absolute concentrations had increased with increasing total selenium concentration. The concentration of selenium present in the form of 13 (simply calculated by multiplying the percentage levels by the total selenium concentration) was 46, 58, 164, 216 and 108 μg g−1 and as selenomethionine was 12, 31, 40, 38 and 176 μg g−1 as the total selenium concentration in the samples increased. These numbers show that the concentration of 13 decreased not only relatively, but also in absolute terms, between concentrations where the distribution conversion took place (296 and 1355 μg g−1 total selenium). The concentration of 11, with only a small discrepancy, increased as the total selenium concentration increased. Summation of the absolute concentrations of 11 and 13 gives values of 58, 89, 204, 254 and 284 μg g−1 Se. The 11 and 13 content of the 68 μg g−1 Se containing garlic was taken as the base value. The total selenium values were used to calculate the increase factors in total selenium concentration (112/68 = 1.65, 235/68 = 3.46, 296/68 = 4.35), then the ‘ideal’ sums were calculated by multiplying 58 (the sum in the lowest selenium containing sample) by the factors. Upon comparing the calculated values (95, 200 and 252) and the actual values (89, 204 and 254) with the total selenium values, a linear relationship is revealed between the total selenium concentration and the sum of the concentrations of 11 and 13 in the lower to middle total selenium concentration range. Meanwhile, the proportion of the Se-methylselenocysteine (9) remained at 3%. On passing from the 296 to the 1355 μg g−1 total selenium concentration samples, the proportion of 9 had increased to 60% and the sum of concentrations of 11 and 13, to 284 μg g−1 Se. This indicated that the above-mentioned linear relationship no longer applied and there was a plateau for the sum with the excess selenium being left in the form of Se-methylselenocysteine. We could also calculate a presumptive theoretical total selenium limit from the 284 μg g−1 Se sum by doing the above calculation in reverse [(284/58) × 68 = 333]. It may be concluded that at concentrations below 333 μg g−1 the selenium is incorporated mainly into γ-glutamyl-Se-methylselenocysteine and selenomethionine, and above 333 μg g−1Se-methylselenocysteine is the major product of the selenium conversion processes.
(b) Ramp.. Selenium-enriched ramp with six different total selenium concentrations (48, 77, 230, 252, 405 and 524 μg g−1 Se) were analyzed. Chromatograms of the enzymatic extracts of 48, 77, 230 and 524 μg g−1 Se ramp are presented in Fig. 5, and the selenium distributions for all of the samples are as follows: 22% (1), 1.5% (8), 44% (9), 5% (11), 1.5% (13), 74% (sum) for the 524 μg g−1 Se ramp, 25% (1), 0.5% (8), 44% (9), 5% (11), 1.5% (13), 76% (sum) for the 405 μg g−1 Se ramp, 15% (1), 2% (8), 50% (9), 8% (11), 1% (13), 76% (sum) for the 203 μg g−1 Se ramp, 25% (1), 3% (8), 35% (9), 10% (11), 73% (sum) for the 48 μg g−1 Se ramp, 1% (1), 1% (8), 34% (9), 21% (11), 3% (13), 60% (sum) for the 77 μg g−1 Se ramp and 42% (1), 5% (8), 35% (9), 3% (11), 2% (13), 85% (sum) for the 252 μg g−1 Se ramp.
 | ||
| Fig. 5 HPLC-ICP-MS chromatograms of enzymatic extracts of 48, 77, 230 and 524 μg g−1 Se ramp samples using HFBA. | ||
The 48, 230, and 524 μg g−1 samples were grown as part of a pilot study with various concentrations of selenium added to the growth mixture. The 77 μg g−1 ramp grew in the same growth bed as used in the previous year in the pilot study. These ramps were overlooked when the 48 μg g−1 Se ramps were harvested, and thus they are called ‘second year’ ones, their only selenium exposure being that which remained in the growth bed used in the previous spring. The 252 μg g−1 Se ramp grew in a large bed of growth mixture that was situated in the open; substantial rain resulted in water on the growth bed, which presented less than optimal growing conditions.
The chromatographic profiles of the ramp extracts are very similar. The 252 μg g−1 Se ramp (not shown in Fig. 5) was different, presenting a lower inorganic to organic conversion rate than the other samples. This could be explained by the less than optimal growing conditions. In these samples, inorganic selenate ion (1) and Se-methylselenocysteine (9) were identified as the major forms of selenium, while Se-cystathionine (8) and γ-glutamyl-Se-methylselenocysteine (13) were minor forms with selenomethionine (11) at an intermediate level. The 77 μg g−1 Se ‘second year’ ramp had much higher inorganic to organic conversion with only 1% selenate remaining, presumably owing to the extra year that it had for the conversion and to the depleted inorganic selenium supply in the growth bed. Compound 11 was present only in the enzymatic extracts of the different samples and not in the water extracts. Comparison of TFA and HFBA chromatograms did not indicate any discrepancies.
In contrast to the behavior observed with garlic, there was no selenium distribution change in selenium-enriched ramp samples over a wide range of total selenium concentration. The distribution in the ramp was similar to that of high selenium garlic even at the lowest level of 48 μg g−1.
(c) Onion.. At this stage, onion samples with a wide range of selenium concentrations were not available, hence conclusions regarding the change in selenium distribution upon increase in total selenium content could not be made. Fig. 6 shows the HFBA chromatogram of 96 (lower) and 140 (upper) μg g−1 onion samples with selenium distributions as follows: 10% (1), 1% (7), 1% (9), 5% (11), 63%* (13), 96% (sum) for the 140 μg g−1 Se onion and 33% (1), 4% (8), 5% (9), 10% (11), 35%* (13), 92% (sum) for the 96 μg g−1 Se onion. Compounds with distribution percentage values marked with an asterisk were confirmed by HPLC-ESI-MS identification.
 | ||
| Fig. 6 HPLC-ICP-MS chromatograms of enzymatic extracts of 96 and 140 μg g−1 Se onion samples using HFBA. | ||
The samples contained selenate (1) and γ-glutamyl-Se-methylselenocysteine (13) as major components, selenocystine (7) (140 μg g−1) and Se-cystathionine (8) (96 μg g−1) as minor components with Se-methylselenocysteine (9) and selenomethionine (11) at intermediate levels. The selenium distribution in onion is similar to that of the low- and mid-level garlic, except that the higher inorganic concentration indicates a lower inorganic to organic conversion like the ramp, despite the presence of 13.
HPLC-ICP-MS analysis of non-selenium-enriched samples
Fig. 7 shows the chromatograms of the enzymatic extracts of samples with natural selenium levels. The total selenium concentrations were estimated to be less than 0.5 μg g−1 Se, which is below the detection limit of our method used for total selenium determination.7 The samples were onion and garlic control samples harvested along with the selenium-enriched counterparts, and yeast, garlic powder and garlic supplement from the authors’ households. The selenium distribution of these samples are as follows: 100% (1), 100% (sum) for onion control, 10% (1), 90% (11), 100% (sum) for commercial yeast, 13% (9), 34% (11), 53% (13), 100% (sum) for commercial garlic powder, 4% (1), 12% (9), 53% (11), 31% (13), 100% (sum) for garlic control and 10% (1), 14% (9), 28% (11), 48% (13), 100% (sum) for commercial garlic supplement.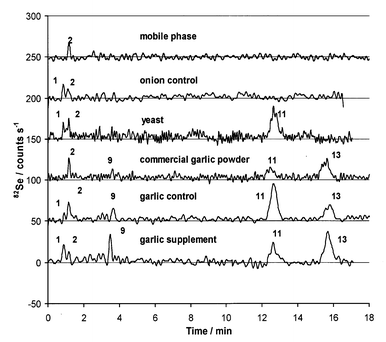 | ||
| Fig. 7 HPLC-ICP-MS chromatograms of enzymatic extracts of samples containing natural level selenium: onion, yeast, garlic from different sources and mobile phase using HFBA. | ||
The upper chromatogram in Fig. 7 shows a blank injection of the mobile phase (0.1% HFBA in 1% methanol) indicating a slight (20 counts s−1) selenite contamination, which remained in the blank even after repeated attempts to clean the chromatographic system. The S/N of these chromatograms were brought to this acceptable level by Fourier transformation followed by deleting the high frequency content of the signal using PeakFit software. The peaks were identified by retention time matching and then by spiking of the samples with the appropriate standards. The selenium species identified in these samples were mostly the same as the species in the enriched samples. The compounds present above the detection limit were selenate (1), Se-methylselenocysteine (9), selenomethionine (11) and y-glutamyl-Se-methylselenocysteine (13) in the garlic samples, inorganic selenium and selenomethionine (11) in the yeast and inorganic selenium in the natural onion.
HPLC-ESI-MS analysis of selenium-enriched samples
We earlier reported on the identification of principal selenium compounds in 1922 μg g−1 Se yeast and in 296 μg g−1 Se garlic using on-line HPLC-ESI-MS.19 In those experiments TFA was used as ion-pairing agent, because the compounds identified [selenomethionine (11), γ-glutamyl-Se-methylselenocysteine (13) and Se-adenosylselenohomocysteine (23)] eluted in the later parts of the chromatograms where interference from coeluting compounds were less severe and there was no need for the separation power of HFBA. Also, by not using HFBA with the resulting longer retention times, the peak dispersion was kept at a relatively lower level, thus maximizing peak intensities and providing a better signal to noise correlation.The upper trace of Fig. 8 shows the overlaid selected ion chromatograms (SIC) of m/z 167, 198 and 313 and the background corrected ion sum of the mass spectra under each SIC peak, from the enzymatic extract of the 1355 μg g−1 Se garlic sample using HFBA. The retention times and the mass spectra indicate the presence of Se-methylselenocysteine (9), 11 and 13 as the HPLC-ICP-MS measurements had indicated. The use of HFBA was necessary in this case because 9 was only slightly retained when TFA was used, and mass spectra could not be recorded despite its very high concentration.
 | ||
| Fig. 8 HPLC-ESI-MS SIC (upper trace) and mass spectra (lower traces) of enzymatic extract of 1355 μg g−1 Se garlic sample using HFBA. | ||
Conclusion
The selenium distribution of 23 different selenium-enriched or non-enriched natural sample extracts were determined after HPLC separation with ICP-MS or ESI-MS detection. The samples analyzed fell into three distinct categories. In the first category, selenomethionine was the principal component of the different yeast samples. The second category includes the Allium samples (ramp, garlic, onion) containing mainly Se-methylselenocysteine or related compounds. Phytoremediation samples form category three, with very poor inorganic to organic conversion in Brassica juncea and with the formation of Se-cystathionine in Astragalus praleongus.The experiments showed that the total selenium content affects the selenium distribution in samples of the same type with different selenium levels, while having no effect on others. This was even true among samples that belonged to the same family (onion and ramp). On the other hand, samples with similar total selenium contents can have different distributions and inorganic to organic conversion rates, depending on the environment during selenium accumulation. The analysis of non-selenium-enriched natural samples showed the presence of the same compounds observed as the principal components of the selenium-enriched samples. The on-line HPLC-ESI-MS analysis of the high selenium garlic provided a further example wherein the identity of the principal components were confirmed by molecular mass spectrometry in addition to retention time identification.
Acknowledgements
This work was supported in part by the NIH (CA45164) and the NRI Competitive Grants Program/USDA (Award No. 96-355003351). We thank the Schering Plough Research Institute for a fellowship (M.K.). The provision of the Elan 5000 plasma source mass spectrometer by the Perkin-Elmer Company and of the Symmetry Shield RP8 columns by Waters Chromatography Corporation are gratefully acknowledged. Eric Denoyer (Perkin-Elmer) and Ray Crowley (Waters Chromatography) are thanked for their interest and assistance. The authors also thank John Gray (ETP) for providing the detector upgrade for the ICP mass spectrometer and Professor R. M. Barnes for the use of the Elan 5000a ICP-MS.Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the specific granting agency.
References
- L. C. Clark, B. W. Turnball, E. H. Slate, D. K. Chalker, J. Chow, L. S. Davis, R. A. Glover, D. K. Graham, E. G. Gross, A. Krongrad, J. L. Lesher, H. K. Park, B. B. Sanders, C. L. Smith, J. R. Taylor, D. S. Alberts, R. J. Allison, J. C. Bradshaw, D. Curtus, D. R. Deal, M. Dellasega, J. D. Hendrix, J. H. Herlong, L. J. Hixon, F. Knight, J. Moore, J. S. Rice, A. I. Rogers, B. Schuman, E. H. Smith and J. C. Woodard, J. Am. Med. Assoc., 1996, 276, 1957 Search PubMed.
- V. C. Morris and O. A. Levander, J. Nutr., 1970, 100, 1383 Search PubMed.
- J. Ip and H. Ganther, in Cancer Chemoprevention, ed. L. Wattenberg, M. Lipkin, C. W. Boone and G. J. Kelloff, CRC Press, Boca Raton, FL, 1992, p. 479. Search PubMed.
- C. Ip, K. Elbayoumy, P. Upadhyaya, H. Ganther, S. Vadhanavikit and H. Thompson, Carcinogenesis, 1994, 15, 187 CAS.
- S. M. Bird, H. H. Ge, P. C. Uden, J. F. Tyson, E. Block and E. Denoyer, J. Chromatogr. A, 1997, 789, 349 CrossRef CAS.
- S. M. Bird, P. C. Uden, J. F. Tyson, E. Block and E. Denoyer, J. Anal. At. Spectrom., 1997, 12, 785 RSC.
- P. C. Uden, S. M. Bird, M. Kotrebai, P. Nolibos, J. F. Tyson, E. Block and E. Denoyer, Fresenius’ J. Anal. Chem., 1998, 362, 447 CrossRef CAS.
- M. Kotrebai, S. M. Bird, J. F. Tyson, E. Block and P. C. Uden, Spectrochim. Acta, Part B, 1999, 54, 1573 CrossRef.
- K. Pyrzynska, Analyst, 1996, 121, 77R RSC.
- G. Zoorob, M. Tomlinson, J. Wang and J. Caruso, J. Anal. At. Spectrom., 1995, 10, 853 RSC.
- C. D. Thomson, Analyst, 1998, 123, 827 RSC.
- R. Lobinski, Appl. Spectrosc., 1997, 51, 260 Search PubMed.
- A. D’Ulivo, Analyst, 1997, 122, 117R RSC.
- X. Dauchy, M. Potingautier, A. Astruc and M. Astruc, Fresenius’ J. Anal. Chem., 1994, 348, 792 CAS.
- H. M. Crews, Spectrochim. Acta, Part B, 1998, 53, 213 CrossRef.
- D. Behne, C. Hammel, H. Pfeifer, D. Rothlein, H. Gessner and A. Kyriakopoulos, Analyst, 1998, 123, 871 RSC.
- S. M. Furness-Green, T. R. Inskeep, J. J. Starke, L. Ping, H. R. Greenleaf-Schumann and T. E. Goyne, J. Chromatogr. A, 1998, 36, 227 CAS.
- L. Campanella, G. Crescentini and P. Avino, J. Chromatogr. A, 1999, 833, 137 CrossRef CAS.
- M. Kotrebai, M. Birringer, J. F. Tyson, E. Block and P. C. Uden, Anal. Commun., 1999, 36, 249 RSC.
- C. Casiot, V. Vacchina, H. Chassaigne, J. Szpunar, M. Potin-Gautier and R. Lobinski, Anal. Commun., 1999, 36, 77 RSC.
- M. Patthy, J. Chromatogr. A, 1994, 660, 17 CrossRef CAS.
- K. N. Petritis, P. Chaimbault, C. Elfakir and M. Dreux, J. Chromatogr. A, 1999, 833, 147 CrossRef CAS.
- G. Inchauspe, P. Delrieu, P. Dupin, M. Laurent and D. Samain, J. Chromatogr. A, 1987, 404, 53 CrossRef CAS.
- E. Block, Angew. Chem., Int. Ed. Engl., 1992, 31, 1135 CrossRef.
- L. D. Lawson, in Garlic: the Science and Therapeutic Application of Allium Sativum L. and Related Species, ed. H. P. Koch and L. D. Lawson, Wilkins and Wilkins, Baltimore, 1996, p. 37. Search PubMed.
- S. G. Edwards, D. Musker, H. A. Collin and G. Britton, Phytochem. Anal., 1994, 5, 4 CAS.
- J. Zheng, W. Goessler and W. Kosmus, Trace Elem. Electr., 1998, 15, 70 Search PubMed.
- R. M. Olivas, O. F. X. Donard, N. Gilon and M. Potin-Gautier, J. Anal. At. Spectrom., 1996, 11, 1171 RSC.
- N. Gilon, A. Astruc, M. Astruc and M. Potingautier, Appl. Organomet. Chem., 1995, 9, 623 CrossRef CAS.
- F. T. Awadeh, M. M. Abdelrahman, R. L. Kincaid and J. W. Finley, J. Dairy Sci., 1998, 81, 1089 Search PubMed.
- X. J. Cai, E. Block, P. C. Uden, B. D. Quimby and J. Sullivan, J. Agric. Food Chem., 1995, 43, 1754 CrossRef CAS.
- Y. Koike, Y. Nakaguchi and K. Hiraki, Bunseki Kagaku, 1993, 42, 285 Search PubMed.
- W. Maher, S. Baldwin, M. Deaker and M. Irving, Appl. Organomet. Chem., 1992, 6, 103 CrossRef CAS.
- N. R. Bottino, C. H. Banks, K. J. Irgolic, P. Micks, A. E. Wheeler and R. A. Zingaro, Phytochem., 1984, 23, 2445 Search PubMed.
- A. E. Wheeler, R. A. Zingaro, K. Irgolic and N. R. Bottino, J. Exp. Mar. Biol. Ecol., 1982, 57, 181 CrossRef CAS.
- K. Yasumoto, T. Suzuki and M. Yoshida, J. Agric. Food Chem., 1988, 36, 463 CrossRef CAS.
- M. M. Gomez, T. Gasparic, M. A. Palacios and C. Camara, Anal. Chim. Acta, 1998, 374, 241 CrossRef CAS.
- H. M. Crews, P. A. Clarke, D. J. Lewis, L. M. Owen, P. R. Strutt and A. Izquierdo, J. Anal. At. Spectrom., 1996, 11, 1177 RSC.
- M. Kotrebai, J. F. Tyson, E. Block and P. C. Uden, J. Chromatogr. A, in the press. Search PubMed.
- J. W. Dolan, LC-GC-Mag Sep. Sci., 1998, 16, 350 Search PubMed.
- J. E. O’Gara, B. A. Alden, T. H. Walter, J. S. Petersen, C. L. Niedarlander and U. D. Neue, Anal. Chem., 1995, 67, 3809 CrossRef CAS.
- C. Casiot, J. Szpunar, R. Lobinski and M. Potin-Gautier, J. Anal. At. Spectrom., 1999, 14, 645 RSC.
Footnote |
| † Presented at SAC 99, Dublin, Ireland, July 25–30, 1999. |
| This journal is © The Royal Society of Chemistry 2000 |