Baker’s yeast mediated enantioselective synthesis of the bisabolene sesquiterpenes (+)-epijuvabione and (−)-juvabione
Claudio
Fuganti
and
Stefano
Serra
*
Dipartimento di Chimica del Politecnico, Centro CNR per lo Studio delle Sostanze Organiche Naturali, Via Mancinelli, 7, I-20131, Milano, Italy
First published on 24th December 1999
Abstract
Fermenting baker’s yeast converts the unsaturated aldehydes 7 and 11 into the saturated alcohols 8 and 12, respectively. The microbial saturation of the double bond proceeds in high chemical yields and the stereoselectivity of the reduction is strongly influenced by the E∶Z ratio of the substrate. Enantiopure 8 and 12 are chiral building blocks for the synthesis of bisabolene sesquiterpenes and their usefulness is shown in the preparation of (+)-epijuvabione 1 and (−)-juvabione 3.
Introduction
The sesquiterpenes of the bisabolene family widely occur in nature and are constituents of a large number of essential oils. Among these compounds, the most challenging ones are those which contain two contiguous secondary stereogenic centres: one in the cyclohexene ring and the other in the side chain. Since many natural products of this kind show biological activity, which is strictly related to their absolute configuration, several stereoselective![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 1 approaches have been reported for the construction of the bisabolene skeleton. In order to develop a diastereoselective path to alicyclic bisabolene sesquiterpenes, we report now a study on the baker’s yeast reduction of (R)- and (S)-3-(4-methylcyclohex-3-enyl)but-2-enal. We find that the enzymic reduction of the latter aldehydes affords the saturated alcohols 8 and 12 with high chemical yield. The stereochemical course of the reduction is not affected by the pre-existing stereocentres, but by the E∶Z ratio of the conjugate double bond. The enantiopure alcohols 8 and 12 are useful building blocks for the synthesis of the bisabolene sesquiterpenes. In this context, we use the latter compounds for preparing two isomeric forms of juvabione, a well known sesquiterpenes with juvenile hormone activity (Fig. 1). Many different syntheses2 of enantiopure juvabiones have been developed in the past, but only few enzymic approaches to this kind of compound have been reported. The first enzyme-mediated preparation described in the literature3 was based essentially on the baker’s yeast reductive enantiodifferentiation of a bicyclic diketone followed by multistep chemical transformations of the obtained enantiopure ketol in the bisabolene skeleton. Moreover a different synthesis of juvabione4 also utilised baker’s yeast in the preparation of a chiral sulfoxide followed by stereocontrolled conjugate addition of its lithium salt to cyclohexenone. Recently, an alternative construction of 1 and 2 from racemic norcamphor has been developed5 by employing lipase mediated kinetic resolution. Up to now, to our knowledge, no approach to juvabiones is based on the direct stereocontrolled enzyme mediated saturation of a prochiral double bond. Herein we report the enatioselective preparation of the natural (+)-epijuvabione 1 and (−)-juvabione 3 which were synthesised in few chemical steps, starting from alcohols 8 and 12 respectively.
1 approaches have been reported for the construction of the bisabolene skeleton. In order to develop a diastereoselective path to alicyclic bisabolene sesquiterpenes, we report now a study on the baker’s yeast reduction of (R)- and (S)-3-(4-methylcyclohex-3-enyl)but-2-enal. We find that the enzymic reduction of the latter aldehydes affords the saturated alcohols 8 and 12 with high chemical yield. The stereochemical course of the reduction is not affected by the pre-existing stereocentres, but by the E∶Z ratio of the conjugate double bond. The enantiopure alcohols 8 and 12 are useful building blocks for the synthesis of the bisabolene sesquiterpenes. In this context, we use the latter compounds for preparing two isomeric forms of juvabione, a well known sesquiterpenes with juvenile hormone activity (Fig. 1). Many different syntheses2 of enantiopure juvabiones have been developed in the past, but only few enzymic approaches to this kind of compound have been reported. The first enzyme-mediated preparation described in the literature3 was based essentially on the baker’s yeast reductive enantiodifferentiation of a bicyclic diketone followed by multistep chemical transformations of the obtained enantiopure ketol in the bisabolene skeleton. Moreover a different synthesis of juvabione4 also utilised baker’s yeast in the preparation of a chiral sulfoxide followed by stereocontrolled conjugate addition of its lithium salt to cyclohexenone. Recently, an alternative construction of 1 and 2 from racemic norcamphor has been developed5 by employing lipase mediated kinetic resolution. Up to now, to our knowledge, no approach to juvabiones is based on the direct stereocontrolled enzyme mediated saturation of a prochiral double bond. Herein we report the enatioselective preparation of the natural (+)-epijuvabione 1 and (−)-juvabione 3 which were synthesised in few chemical steps, starting from alcohols 8 and 12 respectively.
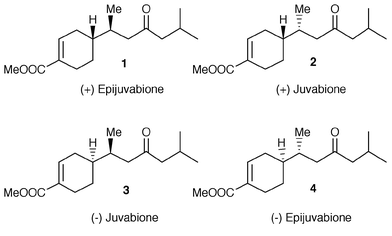 | ||
| Fig. 1 | ||
Results and discussion
It is known that the baker’s yeast reduction of non-chiral activated trisubstituted olefins proceeds with high enantioselectivity.6 Preliminary studies,7 aimed to the synthesis of the bisabolane sesquiterpenes, have shown that (E)-3-(p-tolyl)but-2-en-1-ol is reduced by baker’s yeast to enantiopure (S)-(+)-3-(p-tolyl)butan-1-ol, confirming the general trend of enantioselectivity for this kind of substrate. In considering a similar approach for the synthesis of bisabolene sesquiterpenes, we looked for a method to prepare an analogous C11 diastereomerically and enantiomerically pure building block. In this case a problem of double selectivity arose. Since it seemed suitable to investigate whether the microbial saturation can be influenced by the presence of a contiguous stereocentre, we selected as substrates the aldehydes 7 and 11 (Scheme 1) which bear a stereogenic centre in the γ position. These latter were obtained in their enantiopure form starting from the commercially available (+)-(R)-limonene and (−)-(S)-limonene, respectively. The selective metalation at C-10 of limonene8 was performed using n-butyllithium–TMEDA complex to produce the allyllithium species 5 and 9. These intermediates were converted to the β,γ-unsaturated aldehydes 6 and 10 by reaction with dry DMF at low temperature (−78 °C) followed by acidic work-up. The crude products were not isolated, but isomerized directly with activity grade I alumina to afford the desired aldehydes 7 and 11 as a mixture of E∶Z isomers in 85∶15 ratio. These two geometrical isomers can be separated by chromatography and we prepared various samples with different E∶Z ratios, in order to study their influence on the stereochemical course of the microbial reduction. The experimental conditions were the same for each substrate (see Experimental section) and we decided to perform the baker’s yeast reduction in the presence of a non-polar resin. This technique7 allowed the large-scale preparation of 8 and 12, since high concentration of substrate (6 g l−1) was achieved and the work-up procedure was simplified.
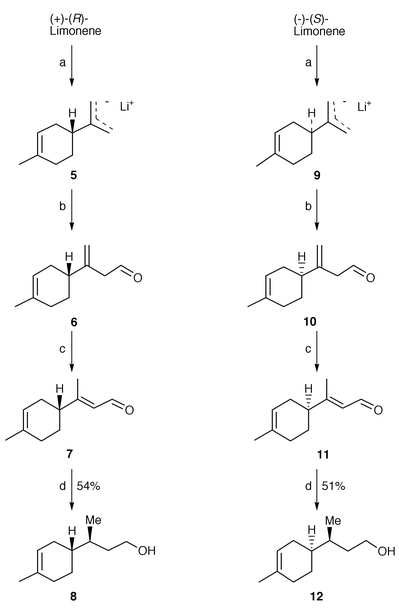 | ||
| Scheme 1 Reagents and conditions: a) n-BuLi–TMEDA; b) DMF–hexane; c) Al2O3 activity grade 1–Et2O; d) baker’s yeast, 4 d, 51–54%. | ||
The results of the biotransformations (Table 1), seen together, suggest some interesting considerations. The reduction of the conjugated double bond by baker’s yeast gives high yields of saturated alcohols (51–54%) and the diastereoisomeric ratio in the products is independent of the enantiomeric form of the substrates. The new chiral centre is formed with satisfactory preference for the (S) absolute configuration independently from the stereogenic centre in γ position. These results (high chemical yield, de <90%) are very different from those obtained in the reduction of the aromatic analogous 3-(p-tolyl)but-2-en-1-ol,7 which was reduced by baker’s yeast in lower chemical yields (20% after 5 days of fermentation) and in high enantioselectivity (ee >95%). Moreover the de of the saturated alcohols 8 and 12 are influenced by the E∶Z ratio of the substrates. The analytical data for E∶Z 95∶5 (entry 1 in Table 1) and 5∶95 (entry 4 in Table 1) show that both aldehydes were reduced. Moreover the reduction of the E∶Z mixture, obtained by thermodynamic equilibration, gave an alcohol with a strictly analogous diastereoisomeric ratio (entry 2 in Table 1). These results can be interpreted assuming that a slow isomerization of the aldehydes occurs whilst the saturation of the double bond proceeds at a slower rate for the Z than for the E isomer.
Thus alcohols 8 and 12 were obtained in diastereomerically enriched form but their purity was unsatisfactory for use as chiral building blocks in sesquiterpene synthesis. For this reason, we performed the reduction on a multigram scale of the easily available mixture of aldehydes obtained by alumina isomerization and then we purified the resulting alcohols via fractional crystallisation of their corresponding 3,5-dinitrobenzoates (Scheme 2). A few crystallisations from methanol gave enantiomerically and diastereomerically pure esters 13 and 14 (99 and 97% de respectively) which were converted into pure alcohols 8 and 12 by saponification with methanolic KOH.
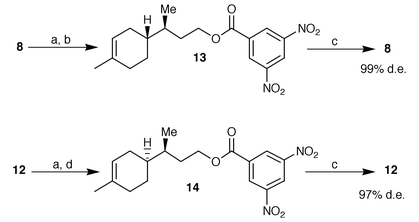 | ||
| Scheme 2 Reagents and conditions: a) 3,5-dinitrobenzoyl chloride–Py, 96%; b) three crystallisations from methanol, 67%; c) KOH–MeOH then H3O+; 95%; d) four crystallizations from methanol, 47%. | ||
The transformation of these latter alcohols into the title compounds was achieved through a common pathway (Scheme 3). Oxidation with pyridinium chlorochromate afforded the aldehydes 15 and 18 which, after treatment with isobutyl magnesium bromide, gave the alcohols 16 and 19, respectively, as a mixture of diastereoisomers. The epoxidation of the cyclohexenic double bond by m-chloroperbenzoic acid, followed by treatment with LDA at reflux,9 afforded regioselectively the allylic alcohols 17 and 20. These latter were submitted to a known oxidative isomerization protocol. Indeed, the reaction of the above mentioned diols with pyridinium chlorochromate in the presence of toluene-p-sulfonic acid,10 followed by oxidation of the resulting isomerized aldehydes with silver oxide,11 gave (+)-epitodomatuic acid and (−)-todomatuic acid, which were not isolated, but esterified directly by diazomethane treatment. An accurate chromatographic purification of the obtained crude esters afforded enantiomerically and diastereomerically pure (+)-epijuvabione and (−)-juvabione, whose analytical data agreed with those reported in the literature.4,5,11
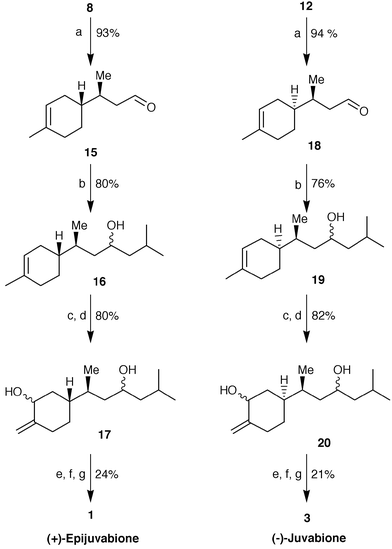 | ||
| Scheme 3
Reagents and conditions: a) PCC–CH2Cl2; b) iBuMgBr–THF; c) m-chloroperbenzoic acid; d) LDA–THF, reflux; e) PCC–CH2Cl2, PTSA cat.; f) Ag2O–MeOH; g) CH2N2–Et2O.
| ||
Experimental
Optical rotations were measured on a Propol automatic digital polarimeter, and are given in 10−1 deg cm2 g−1. IR spectra were recorded on a Perkin-Elmer 2000 FTIR spectrometer. 1H NMR spectra were recorded in CDCl3 solutions at room temperature unless otherwise stated, on a Bruker AC-250 spectrometer (250 MHz 1H). The chemical-shift scale is based on internal tetramethylsilane. J-Values are given in Hz. Mass spectra were measured on a FINNIGAN-MAT TSQ 70 spectrometer. TLC analyses were performed on Merck Kieselgel 60 F254 plates. All the chromatographic separations were carried out on silica gel columns.3-(4-Methylcyclohex-3-enyl)but-2-enal 7 and 11
To a stirred solution of 1.7 M n-butyllithium in hexane (175 ml, 298 mmol) under nitrogen was added dropwise N,N,N′,N′-tetramethylethylenediamine (35 g, 300 mmol). To the resulting yellow solution, (+)-limonene (110 ml, 678 mmol) was added slowly and the mixture was stirred overnight at room temperature. The obtained dark red solution of metalated limonene was added to a cooled (−78 °C) and vigorously stirred mixture of dry DMF (100 ml) in hexane (150 ml). When the exothermic reaction had ceased, the mixture was allowed to warm to 0 °C and then quenched with diluted HCl (5%). The layers were separated, and the aqueous solution was extracted with ether. The combined organic solution was washed with brine, dried (Na2SO4) and evaporated in vacuo. The residue was diluted with dry ether (250 ml) and was stirred with activity grade I alumina (50 g) for 2 h. The alumina was separated by filtration and washed with diethyl ether. The combined filtrate was concentrated in vacuo and distillation of the residue gave (+)-limonene (60 ml, 370 mmol) and pure (3R)-3-(4-methylcyclohex-3-enyl)but-2-enal (20.9 g, 127 mmol, 43% based on n-butyllithium) as a mixture of E∶Z isomers (85∶15). These can be separated by chromatography using hexane–ethyl acetate (95∶5) as eluent to give pure 7-(E), [α]20D
= +115.4 (c 1.1, CHCl3) (Found: C, 80.65; H, 9.90; C11H16O requires C, 80.44; H, 9.82%); νmax(film)/cm−1 2925, 1673, 1438, 1379, 1156; δH 1.55–2.35 (7H, m, cyclohexenic ring), 1.67 (3H, s, CyMe), 2.19 (3H, s, C![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) CMe), 5.41 (1H, br s, CCH), 5.91 (1H, dt, J 7.9, 0.9, MeCCH), 10.04 (1H, d, J 7.9, CHO); m/z (EI) 164 (17%), 149 (12), 131 (23), 121 (17), 107 (28), 95 (51), 79 (53), 68 (94), 67 (100), 53 (48), 39 (78); and 7-(Z), [α]20D
= −8.4 (c 2.1, CHCl3) (Found: C, 80.72; H, 9.85; C11H16O requires C, 80.44; H, 9.82%); νmax(film)/cm−1 2929, 1671, 1439, 1378, 1189, 1114; δH 1.55–2.35 (7H, m, cyclohexenic ring), 1.69 (3H, s, CyMe), 1.92 (3H, s, CCMe), 5.43 (1H, br s, CCH), 5.85 (1H, d, J 8.3, MeCCH), 10.05 (1H, d, J 8.3, CHO); m/z (EI) 164 (10%), 149 (43), 131 (69), 121 (30), 107 (32), 95 (48), 93 (53), 91 (59), 79 (64), 67 (88), 53 (50), 39 (100).
CMe), 5.41 (1H, br s, CCH), 5.91 (1H, dt, J 7.9, 0.9, MeCCH), 10.04 (1H, d, J 7.9, CHO); m/z (EI) 164 (17%), 149 (12), 131 (23), 121 (17), 107 (28), 95 (51), 79 (53), 68 (94), 67 (100), 53 (48), 39 (78); and 7-(Z), [α]20D
= −8.4 (c 2.1, CHCl3) (Found: C, 80.72; H, 9.85; C11H16O requires C, 80.44; H, 9.82%); νmax(film)/cm−1 2929, 1671, 1439, 1378, 1189, 1114; δH 1.55–2.35 (7H, m, cyclohexenic ring), 1.69 (3H, s, CyMe), 1.92 (3H, s, CCMe), 5.43 (1H, br s, CCH), 5.85 (1H, d, J 8.3, MeCCH), 10.05 (1H, d, J 8.3, CHO); m/z (EI) 164 (10%), 149 (43), 131 (69), 121 (30), 107 (32), 95 (48), 93 (53), 91 (59), 79 (64), 67 (88), 53 (50), 39 (100).
Compounds 11-(E) and 11-(Z)were obtained starting from commercially (−)-limonene using the above described procedure. Their analytical data are identical with those described for 7-(E) and 7-(Z) except for the optical rotation; 11-(E), [α]20D
= −105.9 (c 2.5, CHCl3); 11-(ZZZ), [α]20D
= +7.4 (c 1.8, CHCl3).
Baker’s yeast reduction of aldehydes 7 and 11
A 10 l open cylindrical glass vessel equipped with a mechanical stirrer was charged with tap water (5 l) and glucose (350 g). Fresh baker’s yeast (1.5 kg) was added in small pieces to the stirred mixture and the fermentation allowed to proceed for 2 h. The aldehyde 7 (35 g, 213 mmol), adsorbed on the resin XAD 1180 (150 g), was added in one portion. The vigorous stirring was continued for 4 days at room temperature. During this time further baker’s yeast (250 g) and glucose (100 g) were added after 24 h, and again after 48 h since the fermentation started. The resin was then separated by filtration on a sintered glass funnel (porosity 0, >160 μm) and the water phase extracted again with further resin (50 g). The combined resin crops were extracted with ethyl acetate (4 × 150 ml) and the acetate solution was washed with brine. The dried organic phase (Na2SO4) was concentrated under reduced pressure to give an oil (39 g). The latter was dissolved in CHCl3 and treated with MnO2 (80 g) stirring at reflux the mixture for 5 h. The residue obtained upon filtration and evaporation of the CHCl3 phase was purified by column chromatography using hexane–ethyl acetate (9∶1 to 3∶1) as eluent to give pure 8 (19.5 g, 116 mmol, 54%) as a pale yellow oil. The diastereoisomeric ratio was 82∶18 (determined by NMR analysis).The same procedure performed on (S)-(4-methylcyclohex-3-enyl)but-2-enal 11 gave the saturated alcohol 12 in 51% yield and with similar diastereoselectivity (see Table 1).
Purification of alcohol 8 and 12
Three crystallisations of 13 from methanol afforded pure (99% de) 3-(4-methylcyclohex-3-enyl)butanol dinitrobenzoate ester 13 (20.5 g, 57 mmol, 67%), [α]20D
= +41 (c 1.4, CHCl3); mp 83–84 °C (Found: C, 59.48; H, 6.05; N, 7.85; C18H22N2O6 requires C, 59.66; H, 6.12; N, 7.73%); νmax(film)/cm−1 3109, 2913, 1722, 1634, 1548, 1463, 1346, 1296, 1179, 1075, 918, 721; δH 0.97 (3H, d, J 6.4, CHMe), 1.20–2.10 (10H, m), 1.65 (3H, s, cyMe), 4.40–4.60 (2H, m, CH2CH2OCOAr), 5.38 (1H, s, CCH), 9.16 (2H, d, J 1.9, ArH), 9.23 (1H, m).
Four crystallisations of 14 from methanol afforded pure (97% de) 3-(4-methylcyclohex-3-enyl)butanol dinitrobenzoate ester 14 (14.4 g, 40 mmol, 47%), [α]20D
= −50.4 (c 1.1, CHCl3); mp 66–68 °C (Found: C, 59.35; H, 6.08; N, 7.66; C18H22N2O6 requires C, 59.66; H, 6.12; N, 7.73%); νmax(film)/cm−1 3096, 2913, 1729, 1627, 1545, 1456, 1347, 1278, 1168, 1078, 956, 721; δH 0.98 (3H, d, J 6.4, CHMe), 1.20–2.10 (10H, m), 1.65 (3H, s, cyMe), 4.40–4.60 (2H, m, CH2CH2OCOAr), 5.38 (1H, s, CCH), 9.16 (2H, d, J 1.9, ArH), 9.23 (1H, m).
CH); m/z (EI) 168 (M+, 32%), 135 (10), 124 (21), 121 (25), 107 (11), 95 (16), 81 (19), 79 (37), 69 (33), 68 (88), 67 (100), 55 (24); and pure 3-(4-methylcyclohex-3-enyl)butanol 12, [α]20D
= −105 (c 1.8, CHCl3) (Found: C, 78.58; H, 12.05; C11H20O requires C, 78.51; H, 11.98%); νmax(film)/cm−1 3332, 2920, 1436, 1378, 1056, 912, 798; δH 0.88 (3H, d, J 6.6, CHMe), 1.20–2.10 (11H, m), 1.64 (3H, s, CyMe), 3.56–3.80 (2H, m, CH2CH2OH), 5.37 (1H, br s, CCH); m/z (EI) 168 (M+, 7%), 135 (51), 124 (44), 121 (24), 107 (23), 95 (98), 81 (43), 79 (78), 68 (50), 67 (100), 55 (47).
3-(4-Methylcyclohex-3-enyl)butanal 15 and 18
Alcohol 8 or 12 (1.5 g, 9 mmol) in CH2Cl2 (10 ml) was added in one portion to a suspension of pyridinium chlorochromate (3 g, 14 mmol) in CH2Cl2 (50 ml) and stirred at room temperature until no more starting alcohol was detected by TLC analysis (1 h). The mixture was then diluted with diethyl ether (120 ml) and filtered on a layer of Celite. The organic phase was washed with water, dried (Na2SO4) and concentrated under reduced pressure. Bulb to bulb distillation of the residue (oven temperature 85 °C/0.2 Torr) gave pure 15 (1.4 g, 8.4 mmol, 93%), [α]20D = +127.8 (c 1, CHCl3) (Found: C, 79.60; H, 11.00; C11H18O requires C, 79.46; H, 10.91%); νmax(film)/cm−1 2962, 2916, 2715, 1726, 1452, 1378, 1019, 912, 798; δH 0.94 (3H, d, J 6.7, CHMe), 1.20–2.10 (8H, m), 1.64 (3H, CyMe), 2.15–2.26 (1H, m, CHHCHO), 2.46 and 2.51 (1H, 2 dd, J 4.5, 1.9, CHHCHO), 5.36 (1H, br s, CCH), 9.77 (1H, dd, J 2.9, 1.9, CHO); m/z (EI) 166 (M+, 7%), 164 (10), 149 (10), 148 (M+ − H2O, 12), 123 (19), 122 (42), 107 (36), 93 (64), 81 (39), 79 (61), 67 (100), 55 (62); or 18 (1.42 g, 8.4 mmol, 94%), [α]20D
= −77.7 (c 2.2, CHCl3) (Found: C, 79.65; H, 10.96; C11H18O requires C, 79.46; H, 10.91%); νmax(film)/cm−1 2962, 2915, 2835, 1728, 1451, 1378, 1018, 913, 798; δH 0.95 (3H, d, J 7.0, CHMe), 1.18–2.10 (8H, m), 1.64 (3H, s, CyMe), 2.16–2.30 (1H, 2 dd, J 8.7, 3.1, 2.6, CHHCHO), 2.46 and 2.52 (1H, 2 dd, J 4.8, 1.7, CHHCHO), 5.36 (1H, br s, CCH), 9.76 (1H, dd, J 2.6, 1.7, CHO); m/z (EI) 166 (M+, 4%), 148 (39), 133 (30), 121 (17), 107 (31), 106 (33), 93 (90), 79 (57), 67 (100), 53 (43).
2-(4-Methylcyclohex-3-enyl)-6-methylheptan-4-ol 16 and 19
The aldehyde 15 or 18 (2 g, 12 mmol) in dry THF (60 ml) was cooled to 0 °C and treated under nitrogen with isobutylmagnesium chloride (5 ml of 3 M solution in THF, 15 mmol). The mixture was stirred at this temperature for 1 h, then quenched with saturated NH4Cl aq. (60 ml) and finally it was extracted with ethyl acetate. The organic phase was dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by chromatography column using hexane–ethyl acetate (9∶1) as eluent to give pure alcohol 16 (2.15 g, 9.6 mol, 80%) (Found: C, 80.52; H, 12.45; C15H28O requires C, 80.29; H, 12.58%); νmax(film)/cm−1 3346, 2957, 2924, 1467, 1378, 1150, 1022, 800; δH 0.84–0.95 (9H, m, MeCHMe + CHMe), 1.16–2.10 (14H, m), 1.64 (3H, s, CCMe), 3.68–3.82 (1H, m, CHOH), 5.38 (1H, br s, CCH); m/z (EI) 224 (M+, 30%), 206 (M+ − H2O, 44), 191 (27), 177 (18), 149 (30), 127 (22), 121 (43), 107 (16), 95 (36), 94 (100), 79 (54), 67 (42); or alcohol 19 (2.05 g, 76%) (Found: C, 80.60; H, 12.50; C15H28O requires C, 80.29; H, 12.58%); δH 0.85–0.97 (9H, m, MeCHMe + CHMe), 1.16–2.10 (14H, m), 1.64 (3H, s, CCMe), 3.68–3.82 (1H, m, CHOH), 5.38 (1H, br s, CCH); IR and mass spectrum are identical with those reported for compound 16.
5-(3-Hydroxy-1,5-dimethylhexyl)-2-methylenecyclohexanol 17 and 20
m-Chloroperbenzoic acid (1.25 g of 70% wet acid, 5 mmol) was added to a cooled solution (0 °C) of alcohol 16 or 19 (0.9 g, 4 mmol) in CH2Cl2. The mixture was stirred for 1 h and then the m-chlorobenzoic acid formed was filtered off on a Celite pad. The obtained clear solution was washed with a 5% Na2SO3 aq. solution, with a saturated NaHCO3 aq. solution and then dried (Na2SO4). The solvent was evaporated and the residue, dissolved in dry THF (60 ml), was treated at reflux with lithium diisopropylamide (6 ml of 2 M solution in THF, 12 mmol) under a static atmosphere of nitrogen. After 2 h the mixture was cooled, poured into ice and extracted three times with ethyl acetate. The organic phase was dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by column chromatography eluting with hexane–ethyl acetate (2∶1) to afford pure 17 (780 mg, 3.2 mmol, 80%) (Found: C, 75.19; H, 11.80; C15H28O2 requires C, 74.95; H, 11.74%); νmax(film)/cm−1 3365, 2930, 1654, 1467, 1368, 1217, 1063, 900, 758; δH 0.84–0.96 (9H, m, CHMe + CHMeMe), 1.1–2.26 (14H, m), 2.43 (1H, m), 3.75 (1H, m, CH2CHOHCH2), 4.00–4.15 and 4.35 (1H, 2 m, CH2CCHOH), 4.72–4.79 (1H, m, CCH2), 4.83 and 4.92 (1H, 2 br s, CCH2); m/z (EI) 240 (M+, 3%), 223 (M+ − OH, 16), 222 (M+ − H2O, 23), 205 (72), 165 (27), 149 (32), 138 (31), 111 (57), 109 (99), 93 (95), 81 (76), 69 (85), 43 (100); or pure 20 (800 mg, 3.3 mmol, 82%) (Found: C, 75.05; H, 11.76; C15H28O2 requires C, 74.95; H, 11.74%); IR H1 NMR and mass spectrum are identical to those reported for compound 17.
(+)-Epijuvabione 1 and (−)-juvabione 3
The diol 17 or 20 (0.7 g, 2.9 mmol) in CH2Cl2 (10 ml) was added in one portion to a stirred suspension of pyridinium chlorochromate (2.6 g, 12 mmol) and PTSA (0.6 g, 3 mol) in CH2Cl2 (50 ml). The reaction was stirred until no more starting diol was detected by TLC analysis (3 h) and then was diluted with diethyl ether (100 ml). The mixture was filtered through a layer of Celite and the organic phase was washed with water, dried (Na2SO4) and concentrated under reduced pressure. The residue was dissolved in a solution of AgNO3 (0.85 g, 5 mmol) in methanol (60 ml) and NaOH (0.2 g, 5 mmol) in water (20 ml) was added dropwise. The resulting black mixture was heated at reflux for 8 h and then filtered. The filtrate was treated with 5% HCl aq. and extracted three times with ethyl acetate. The organic phase was washed with brine, was dried (Na2SO4) and concentrated under reduced pressure. The residue, dissolved in dry diethyl ether, was treated with an excess of a 5% solution of diazomethane in diethyl ether. Evaporation of the solvent and chromatographic purification of the residue afforded pure 1 as a colourless oil (175 mg, 0.7 mmol, 24% overall); [α]20D = +94.3 (c 0.65, benzene) {lit.,5 [α]20D = +95.8 (c 0.5, benzene) and lit.,11 [α]20D = −94.14 (c 0.64, benzene) for the (−)-epijuvabione} (Found: C, 72.35; H, 9.85; C16H26O3 requires C, 72.14; H, 9.84%); νmax(film)/cm−1 2956, 1716, 1653, 1436, 1367, 1255, 1085, 1035; δH 0.88 (3H, d, J 6.6, CHMe), 0.91 (3H, d, J 6.4 MeCHMe), 0.92 (3H, d, J, 6.4, MeCHMe), 1.25 (1H, m), 1.45 (1H, m), 1.77 (1H, dm, J 12), 1.85–2.30 (8H, m), 2.45 (1H, dd, J 4.5, 16), 2.47 (1H, dm, J 18), 3.73 (3H, s, COOMe), 6.96 (1H, br s, CCH); m/z (EI) 266 (M+, 1%), 234 (M+ − MeOH, 12), 207 (6), 206 (5), 177 (7), 167 (23), 166 (22), 134 (100), 127 (14), 107 (36), 91 (12), 85 (19), 79 (29), 57 (42), 41 (34); or pure 3 (160 mg, 21% overall); [α]20D
= −64.3 (c 0.5, benzene) {lit.,11 [α]20D
= −64.45 (c 0.568, benzene)} (Found: C, 72.40; H, 9.90; C16H26O3 requires C, 72.14; H, 9.84%); νmax(film)/cm−1 2958, 1718, 1710, 1651, 1435, 1365, 1251, 1081; δH 0.88 (3H, d, J 6.8, CHMe), 0.91 (3H, d, J 6.5 MeCHMe), 0.92 (3H, d, J, 6.5, MeCHMe), 1.22 (1H, m), 1.46 (1H, m), 1.8 (1H, dm, J 12), 1.90–2.30 (8H, m), 2.44 (1H, dd, J 4.7, 16), 2.47 (1H, dm, J 16), 3.72 (3H, s, COOMe), 6.95 (1H, br s, CCH); m/z (EI) 266 (M+, 1%), 248 (1), 235 (3), 234 (M+ − MeOH, 12), 207 (2), 206 (5), 177 (7), 167 (21), 166 (22), 134 (100), 127 (10), 107 (32), 91 (13), 85 (16), 79 (33), 57 (44), 41 (40).
Acknowledgements
We thank MURST for the partial financial support.References
- J. ApSimon , in The Total Synthesis of Natural Products, vol. 5, Wiley and Sons, New York, 1983, p. 55. Search PubMed.
- B. M. Fraga, Nat. Prod. Rep., 1995, 12, 303 RSC.
- E. Nagano and K. Mori, Biosci. Biotechnol. Biochem., 1992, 56, 1589 Search PubMed.
- H. Watanabe, H. Shimizu and K. Mori, Synthesis, 1994, 1249 CrossRef CAS.
- H. Nagata, T. Taniguchi, M. Kawamura and K. Ogasawara, Tetrahedron Lett., 1999, 40, 4207 CrossRef CAS.
- C. Fuganti and P. Grasselli , in Enzymes in Organic Synthesis, CIBA Foundation Symposium, ed. R. Porter and S. Clark, Pitman, London, 1985, p. 112; Search PubMed; S. Servi Synthesis 1990 1 Search PubMed.
- C. Fuganti, S. Serra and A. Dulio, J. Chem. Soc., Perkin Trans. 1, 1999, 279 RSC and references cited therein..
- R. J. Crawford, W. F. Erman and C. D. Broaddus, J. Am. Chem. Soc., 1972, 94, 4298 CrossRef CAS.
- G. Guella, M. Cavazza, A. Guerriero and F. Pietra, Helv. Chim. Acta, 1984, 67, 1248 CrossRef CAS.
- G. Cainelli and G. Cardillo , in Chromium Oxidations in Organic Chemistry, Springer Verlag, New York, 1984. Search PubMed.
- B. A. Pawson, H. C. Cheung, S. Gurbaxani and G. Saucy, J. Am. Chem. Soc., 1970, 92, 336 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2000 |