Synthesis and reactions of α-fluorovinylphosphonium salts
Takeshi Hanamoto*, Keiko Shindo, Miki Matsuoka, Yasuhide Kiguchi and Michio Kondo
Department of Chemistry, Saga University, Honjyo-machi, Saga, 840-8502, Japan
First published on UnassignedUnassigned5th January 2000
Abstract
The α-fluorovinyltriphenylphosphonium triflate 4 is prepared in high yields by the diphenylphosphinylation of 1,1-difluoroethylene 1 and subsequent quaternization of the phosphine 2 with diphenyliodonium triflate in the presence of CuCl. The salt 4 then undergoes Michael addition followed by Wittig olefination to give the corresponding monofluoroethylene compounds in good yields. The reaction of 4 with the caesium salts of salicylaldehyde derivatives in DMF at 130 °C affords the corresponding monofluorinated chromenes in a one-pot synthesis. The hydrolysis of 4 in the presence of sodium hydroxide gives triphenylphosphine oxide in quantitative yield.
Introduction
Much effort has been made to introduce fluorine into organic compounds due to its dramatic effects on their structure, stability, reactivity and biological activity.1 Among them, the preparation of monofluorinated compounds can be divided into two main methods: one is the direct exchange of hydrogen by fluorine,2 and the other is the use of monofluorinated building blocks. As the former requires special techniques and equipment, the latter has recently received much attention and has become a significant procedure in this field.3 Vinylphosphonium salts are useful building blocks for the construction of two-carbon-extended olefinic compounds.4 Although some substituted vinylphosphonium salts have been synthesized and their reactivities examined,5 the syntheses of fluorine-substituted vinylphosphonium salts have been rarely reported.6 We have considered the α-fluorovinyltriphenylphosphonium triflate 4 as a promising candidate for the monofluorinated building block which would be stable and easy to handle. We have reported our preliminary findings![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 7 and now provide a full account of the work.
7 and now provide a full account of the work.Results and discussion
Previously used methods for the preparation of the vinyltriphenylphosphonium salts generally include elimination reactions of the corresponding alkyltriphenylphosphonium salts. Recently, Stang and co-workers have introduced a new method using the palladium-catalyzed vinylation of triphenylphosphine with vinyl triflates.8 However, these convenient methods appeared to be not applicable to the salt 4, which required the development of a new method from us. Our synthetic route for the preparation of the α-fluorovinylphosphonium salts is depicted in Scheme 1. We initially attempted to examine the convenient preparation of α-fluorovinyldiphenylphosphine 2 as the precursor for the salt. Since it is well documented that nucleophilic addition followed by elimination of the fluoride for producing 1,1-difluoroethylene derivatives is easy,9 it was therefore anticipated that the reaction of lithium diphenylphosphide10 and 1,1-difluoroethylene 1 should afford a convenient preparation of 2. Thus, to a THF solution of lithium diphenylphosphide prepared from lithium and chlorodiphenylphosphine was bubbled 1,1-difluoroethylene at −78 °C and the resulting mixture was stirred for 16 h at the same temperature. After the usual work-up, we obtained 2 in modest yield. In order to improve the yield of 2, we conducted the reaction at elevated reaction temperatures (−78–0 °C); however, the undesirable 1,1-bis(diphenylphosphinyl)ethylene was produced as a by-product. We next carefully investigated the reaction conditions and found that when the reaction was conducted over the temperature range of −60 to −40 °C in THF–toluene under a 1,1-difluoroethylene atmosphere, the yield was improved up to 86%. The added co-solvent, toluene, facilitated stirring of the reaction mixture at these low temperatures. The obtained 2 was generally oxidized in air to give the corresponding phosphine oxide.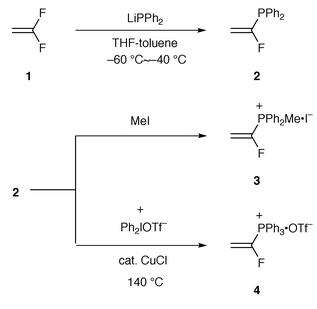 | ||
| Scheme 1 | ||
Quaternization of 2 with iodomethane smoothly proceeded to give the α-fluorovinyl(methyl)diphenylphosphonium iodide 3 in 91% yield. However, the alkyldiphenylphosphonium salts exhibited less performance compared with the corresponding triphenylphosphonium salts;11 therefore, we examined the quaternization of 2 with aryl compounds.12 The reaction of 2 with aryl halides or aryl triflates did not take place under certain conditions. Kitamura et al. have described the direct S-arylation of benzo[b]thiophenes to produce the 1-arylbenzo[b]thiophenium salt.13 This prompted us to use the diaryliodonium salts for the P-arylation of 2 instead of the S-arylation of the sulfides. We attempted to use the method of Kitamura.13 Compound 2 was treated with diphenyliodonium triflate in the presence of a catalytic amount of Cu(OAc)2 at 140 °C for 30 min under an Ar atmosphere. The reaction mixture turned into a brown-black oil. It was difficult to solidify the residual oil after the addition of an excess of diethyl ether to the oil. However, based on the NMR spectra, we confirmed the formation of 4. After many attempts, we optimized the best reaction conditions. Thus, the α-fluorovinyltriphenylphosphonium triflate 4 was obtained by the reaction of diphenyliodonium triflate in the presence of CuCl in 1,1,2,2-tetrachloroethane at 140 °C in 82% yield. We believe that this P-arylation method provides a new route for the synthesis of aryl phosphonium salts.
For comparison of the reactivities of the vinylphosphonium bromide (Schweizer’s reagent), the reaction of 4 with sodium ethoxide and benzaldehyde was carried out (Scheme 2). The β-ethoxy-α-fluoroethyltriphenylphosphonium ylide, generated via the addition of an ethoxide anion to 4 in ethanol at room temperature, reacted with benzaldehyde to provide 3-ethoxy-2-fluoro-1-phenylpropene 5a as a 43∶57 mixture of E and Z isomers in 75% yield (Table 1, entry 4). The E/Z ratio was determined by capillary GC-MS analysis. The geometrical isomers were separated from each other on a silica gel column and characterized based on their NMR spectra. The coupling constants with fluorine showed a value of 20 Hz for the E isomer and 39 Hz for the Z isomer. As expected, the reactivity of 4 was much higher than that of 3 as compared with the yields (entries 1 vs. 2 and 3 vs. 4). Although aromatic aldehydes and an α,β-unsaturated aldehyde gave good results, the aliphatic aldehyde gave poor results. A similar reaction using sodium piperidide as the nucleophile instead of sodium ethoxide in THF at 50 °C gave the corresponding 2-fluoro-1-phenyl-3-piperidinopropene 6 as a mixture of diastereomers in 44% total yield (Scheme 3). Furthermore, no reaction occurred with ketones (cyclohexanone and acetophenone) due to possible steric hindrance in the Wittig reaction.
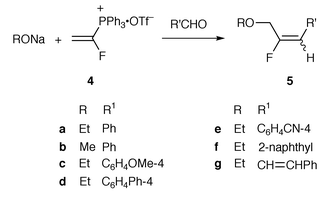 | ||
| Scheme 2 | ||
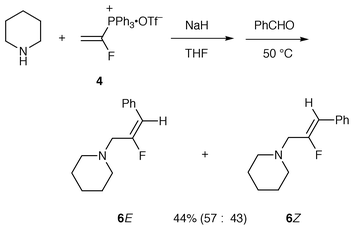 | ||
| Scheme 3 | ||
These results prompted us to utilize 4 for the synthesis of a variety of monofluorinated heterocyclic ring systems, since the synthesis of fluorinated heterocyclic compounds has also become an important area of agrochemical and pharmaceutical studies due to their unique biological properties.14 Among them, the syntheses of fluorinated chromenes are quite limited,15 although the chromene nucleus is also encountered in insecticides and natural products.16
We initially examined the synthesis of 3-fluoro-2H-chromene 8 as a model compound based on Schweizer’s protocol (Table 2).17 Contrary to our expectation, the reaction of the sodium salt of salicylaldehyde 7 with 4 in acetonitrile at 70 °C afforded no cyclized product (run 1), presumably due to the influence of the fluorine atom at the α-position of the salt. A further attempt to obtain the desired cyclized product in the above reaction was made by examining the effect of the solvent. However, the lack of satisfactory yield caused us to abandon the use of the sodium salt of the salicylaldehyde. Next, we focused on the effects of varying the cation on the salicylaldehyde and investigated three different bases. Unfortunately, the use of the potassium, magnesium, and lithium salts of the salicylaldehyde resulted in low yields or no reaction (runs 6, 7, and 8, respectively).
| ||||||
|---|---|---|---|---|---|---|
| Run | M | Base (mol. equiv.) | Solventa | T/°C | t/h | Yield (%) |
| a DEM: Diethoxymethane. | ||||||
| 1 | Na | None | Acetonitrile | 70 | 88 | 0 |
| 2 | Na | None | tert-BuOH | 80 | 40 | 27 |
| 3 | Na | None | DMSO | 120 | 16 | 32 |
| 4 | Na | None | DMF | 130 | 40 | 20 |
| 5 | Na | None | DEM–DMF 4∶1 | 80 | 40 | 35 |
| 6 | H | TMSOK (1.0) | THF | 50 | 16 | 0 |
| 7 | H | EtMgBr (1.0) | DEM–DMF 4∶1 | 80 | 16 | 0 |
| 8 | H | BuLi (1.0) | DEM–DMF 4∶1 | 80 | 16 | 7 |
| 9 | H | CsF (1.7)/Si(OEt)4 (1.3) | DEM–DMF 4∶1 | 80 | 40 | 8 |
| 10 | H | CsF (1.7)/Si(OEt)4 (1.3) | DMF | 130 | 40 | 34 |
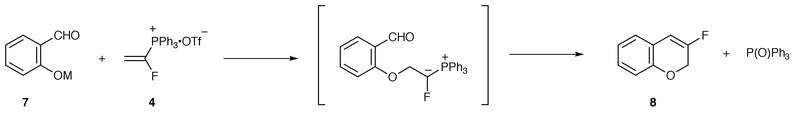
We next turned our attention to the potential of the CsF–Si(OEt)4 system.18 Although we examined the above reaction conditions for the cyclization of the salicylaldehyde, no improved yields were obtained. We also investigated the scope and limitations of the in situ preparation and reaction of the caesium salts of other salicylaldehyde derivatives. The cyclization of 3-methoxysalicylaldehyde and 2-hydroxy-1-naphthaldehyde proceeded to give the corresponding monofluorinated chromene derivatives 9, 10 in 47 and 31% yield, respectively (Chart 1). On the other hand, the cyclization of 5-nitrosalicylaldehyde resulted in a low yield (14%) of the product 11. It is noteworthy that the CsF–Si(OEt)4 system was effective for the above cyclization, because we confirmed that no cyclization of the sodium salts of 2-hydroxy-1-naphthaldehyde and 5-nitrosalicylaldehyde occurred under the conditions employed for the salicylaldehyde (entry 5 in Table 2). The reaction of the caesium salt of o-hydroxyacetophenone with the α-fluorovinyltriphenylphosphonium triflate did not give the anticipated product.
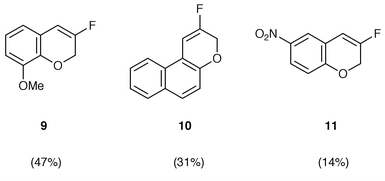 | ||
| Chart 1 Monofluorinated chromene derivatives. All reactions were conducted in DMF for 40 h at 130 °C. | ||
Furthermore, we examined the hydrogenation of the obtained monofluorinated chromenes to produce the corresponding monofluorinated chromanes.19 We employed three types of catalysts for the hydrogenation of 8.20 All the reactions were conducted under hydrogen at atmospheric pressure and room temperature. These results are shown in Scheme 4. The Rh/Al2O3 catalyst was the most effective for the transformation without any loss of the fluorine atom in the molecule. Thus, 3-fluorochromane 12 was obtained in 73% yield. In the case of 9, PtO2-catalyzed hydrogenation gave the corresponding 8-methoxychromane 13 with loss of the fluorine atom in the molecule in 63% yield.
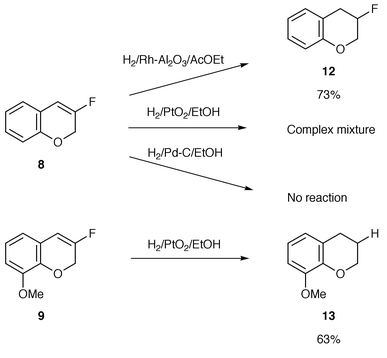 | ||
| Scheme 4 | ||
Finally, other reactivities of 4 were examined (Scheme 5). The hydrolysis of the salt was carried out in aq. alcohols or aq. THF containing excess of sodium hydroxide.21 Neither reaction produced the corresponding β-ethoxy-α-fluoroethyldiphenylphosphine oxide or the corresponding α-fluorovinyldiphenylphosphine oxide, but they did quantitatively produce triphenylphosphine oxide 14. On the other hand, the oxidation of 2 conducted in acetone containing hydrogen peroxide gave the corresponding α-fluorovinyldiphenylphosphine oxide 15 in 84% yield.
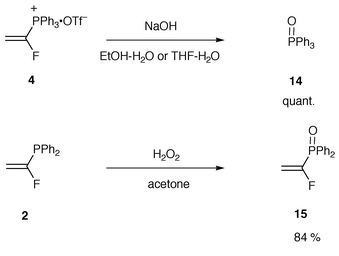 | ||
| Scheme 5 | ||
In conclusion, the α-fluorovinyltriphenylphosphonium triflate 4 can be a useful reagent for the synthesis of compounds containing monofluorinated olefinic moieties in one pot. Such systems are not readily available using general methods.
Experimental
Mps were measured with a Yanagimoto micro melting point apparatus MP-S3 and are uncorrected. IR spectra were recorded on a Perkin-Elmer Spectrum 2000. 1H NMR spectra were measured on a JEOL JNM-GX270. Chemical shifts are given by δ relative to that of internal Me4Si (TMS). Mass spectra were obtained with a Shimadzu GCMS QP-5000. Fast-atom-bombardment mass spectra (FABMS) were obtained with a JEOL JMS-HX110A. Elemental analyses were performed on a Yanagimoto MT3 CHN corder or were accomplished at the service center of the elementary analysis of organic compounds, Kyushu University. Analytical TLC was performed on silica gel plates (Merck, Kieselgel 60 F254, 20 × 20 cm, 0.25 mm). All solvents were purified before use: THF and diethoxymethane (DEM) were dried over sodium benzophenone ketyl radical; 1,1,2,2-tetrachloroethane, DMF, DMSO, MeCN and EtOH were distilled from calcium hydride. The procedures for the preparation of 2, 4 and 5 have already been reported.7α-Fluorovinyl(methyl)diphenylphosphonium iodide 3
A mixture of α-fluorovinyldiphenylphosphine 2 (1.50 g, 6.52 mmol) and iodomethane (1.2 mL, 19.3 mmol) in toluene (2.0 mL) was stirred at room temperature in the dark for 16 h. After the addition of diethyl ether (≈25 mL) to the mixture, the resulting precipitates were collected by filtration. Recrystallization from hexane–CH2Cl2 gave the desired product (2.20 g, 91%), mp 126.4–128.3 °C; ν(KBr)/cm−1 3103, 3062, 3007, 2959, 2884, 2801, 1636, 1588, 1482, 1435, 1407, 1370, 1322, 1298, 1274, 1186, 1162, 1114, 1070, 1036, 992, 900, 859, 791, 750, 685 and 644; δH(CDCl3) 3.27 (3H, d, J 13.7 Hz), 5.79 (1H, ddd, J 6.4, 7.3, 49.3 Hz), 6.49 (1H, ddd, J 6.4, 21.5, 29.8 Hz), 7.7–8.0 (m, 10 H); FABMS: m/z 245 (M+ − I) (Calc. for C15H15FIP: C, 48.41; H, 4.06. Found: C, 48.19; H, 3.91%).(E![[hair space]](https://www.rsc.org/images/entities/h3_char_200a.gif) )- and (Z)-2-Fluoro-3-methoxy-1-phenylpropene 5b
)- and (Z)-2-Fluoro-3-methoxy-1-phenylpropene 5b
Total yield 63%. (E)-isomer: ν(neat)/cm−1 3055, 3027, 2986, 2925, 2815, 1680, 1598, 1492, 1400, 1288, 1257, 1217, 1189, 1142, 1097, 954, 917, 750, 699 and 631; δH(CDCl3) 3.42 (3H, s), 4.14 (2H, d, J 23.0 Hz), 6.48 (1H, d, J 19.5 Hz), 7.24–7.38 (5H, m); MS m/z 166 (50%, M+), 151 (42), 135 (41), 133 (62), 115 (82), 109 (100) (Calc. for C10H11FO: C, 72.27; H, 6.67. Found: C, 72.10; H, 6.71%).(Z)-isomer: ν(neat)/cm−1 3055, 3027, 2986, 2993, 2822, 1693, 1600, 1530, 1495, 1451, 1383, 1349, 1220, 1189, 1165, 1155, 1101, 1091, 958, 917, 879, 753 and 694; δH(CDCl3) 3.44 (3H, s), 4.08 (2H, d, J 15.6 Hz), 5.76 (1H, d, J 38.6 Hz), 7.25–7.55 (5H, m); m/z 166 (75%, M+), 151 (47), 135 (59), 133 (69), 115 (95), 109 (100) (Found: C, 72.56; H, 6.75%).
(E)- and (Z)-3-Ethoxy-2-fluoro-1-(4′-methoxyphenyl)propene 5c
Total yield 80%. (E)-isomer: ν(neat)/cm−1 2978, 2932, 2897, 2870, 1683, 1609, 1574, 1515, 1464, 1383, 1295, 1252, 1180, 1142, 1096, 1035, 886, 862, 831, 770 and 715; δH(CDCl3) 1.25 (3H, t, J 6.8 Hz), 3.58 (2H, q, J 6.8 Hz), 3.81 (3H, s), 4.17 (2H, d, J 23.4 Hz), 6.40 (1H, d, J 20.5 Hz), 6.88 (2H, d, J 8.8 Hz), 7.20 (2H, d, J 8.8 Hz); m/z 210 (51%, M+), 166 (31), 165 (100), 139 (68), 133 (35), 121 (32), 115 (37), 91 (30) (Calc. for C12H15FO2: C, 68.55; H, 7.19. Found: C, 68.73; H, 7.37%).(Z)-isomer: ν(neat)/cm−1 2978, 2932, 2897, 2836, 1693, 1609, 1574, 1515, 1506, 1346, 1295, 1252, 1179, 1162, 1097, 1036, 879, 859, 821 and 770; δH(CDCl3) 1.26 (3H, t, J 6.8 Hz), 3.59 (2H, q, J 6.8 Hz), 3.81 (3H, s), 4.11 (2H, d, J 16.1 Hz), 5.70 (1H, d, J 38.6 Hz), 6.87 (2H, d, J 8.8 Hz), 7.46 (2H, d, J 8.8 Hz); m/z 210 (52%, M+), 166 (32), 165 (100), 139 (60), 133 (31), 115 (33), 91 (29) (Found: C, 68.65; H, 7.40%).
(E)- and (Z)-1-(4′-Biphenylyl)-3-ethoxy-2-fluoropropene 5d
Total yield 70%. (E)-isomer: ν(neat)/cm−1 3055, 3027, 2978, 2856, 1683, 1600, 1486, 1445, 1380, 1274, 1257, 1217, 1145, 1096, 1005, 886, 764 and 695; δH(CDCl3) 1.27 (3H, t, J 6.8 Hz), 3.60 (2H, q, J 6.8 Hz), 4.23 (2H, d, J 23.4 Hz), 6.48 (1H, d, J 20.0 Hz), 7.33–7.62 (9H, m); m/z 256 (68%, M+), 196 (43), 191 (53), 185 (100), 179 (38), 178 (56) (Calc. for C17H17FO: C, 79.66; H, 6.68. Found: C, 79.40; H, 6.80%).(Z)-isomer: mp 40.0–42.0 °C; ν(KBr)/cm−1 2979, 2897, 2869, 1693, 1679, 1594, 1506, 1482, 1444, 1403, 1349, 1328, 1257, 1162, 1083, 1018, 903, 869, 852, 818, 753 and 698; δH(CDCl3) 1.28 (3H, t, J 6.8 Hz), 3.62 (2H, q, J 6.8 Hz), 4.15 (2H, d, J 15.1 Hz), 5.81 (1H, d, J 38.6 Hz), 7.25–7.63 (9H, m); m/z 256 (76%, M+), 211 (33), 196 (46), 191 (57), 185 (100), 179 (36), 178 (55) (Found: C, 79.42; H, 6.70%).
(E)- and (Z)-1-(4′-Cyanophenyl)-3-ethoxy-2-fluoropropene 5e
Total yield 69%. (E)-isomer: ν(neat)/cm−1 2964, 2904, 2229, 1684, 1559, 1411, 1259, 1097, 1018, 869, 819 and 703; δH(CDCl3) 1.26 (3H, t, J 6.8 Hz), 3.59 (2H, q, J 6.8 Hz), 4.15 (2H, d, J 23.4 Hz), 6.44 (1H, d, J 19.1 Hz), 7.39 (2H, d, J 8.3 Hz), 7.64 (2H, d, J 8.3 Hz); m/z 205 (28%, M+), 177 (43), 160 (56), 158 (46), 156 (32), 148 (33), 140 (100), 134 (99), 133 (43), 130 (37), 129 (99), 128 (77), 116 (99), 101 (39), 75 (34) (Calc. for C12H12FNO: C, 70.23; H, 5.89. Found: C, 70.31; H, 5.99%).(Z)-isomer: ν(neat)/cm−1 2964, 2904, 2226, 1559, 1414, 1259, 1098, 1018, 865, 820 and 702; δH(CDCl3) 1.28 (3H, t, J 6.8 Hz), 3.62 (2H, q, J 6.8 Hz), 4.14 (2H, d, J 12.7 Hz), 5.82 (1H, d, J 37.6 Hz), 7.58 (2H, d, J 8.8 Hz), 7.62 (2H, d, J 8.8 Hz); m/z 205 (37%, M+), 177 (44), 161 (26), 160 (64), 158 (45), 148 (31), 140 (100), 134 (93), 133 (40), 130 (35), 129 (89), 128 (66), 116 (91), 101 (35), 75 (31) (Found: C, 69.87; H, 5.99%).
(E)- and (Z)-3-Ethoxy-2-fluoro-1-(2′-naphthyl)propene 5f
Total yield 77%. (E)-isomer: ν(neat)/cm−1 3055, 2977, 2871, 1681, 1598, 1557, 1507, 1380, 1273, 1182, 1138, 1097, 1018, 900, 868, 818 and 743; δH(CDCl3) 1.25 (3H, t, J 6.8 Hz), 3.60 (2H, q, J 6.8 Hz), 4.25 (2H, d, J 23.4 Hz), 6.60 (1H, d, J 20.0 Hz), 7.36–7.84 (7H, m); m/z 230 (93%, M+), 201 (38), 186 (39), 185 (59), 184 (31), 183 (95), 170 (38), 165 (100), 159 (86), 153 (93) (Calc. for C15H15FO: C, 78.24; H, 6.57. Found: C, 78.31; H, 6.68%).(Z)-isomer: mp 39.0–41.0 °C; ν(KBr)/cm−1 3068, 3034, 2977, 2863, 1699, 1676, 1605, 1564, 1489, 1445, 1404, 1377, 1349, 1288, 1261, 1213, 1162, 1099, 1008, 866, 832, 764, 729 and 697; δH(CDCl3) 1.29 (3H, t, J 6.8 Hz), 3.64 (2H, q, J 6.8 Hz), 4.18 (2H, d, J 15.1 Hz), 5.93 (1H, d, J 38.6 Hz), 7.40–7.95 (7H, m); m/z 230 (98, M+), 201 (34), 186 (30), 185 (72), 183 (81), 170 (37), 166 (30), 165 (100), 159 (87), 153 (78) (Found: C, 78.14; H, 6.63%).
(E)- and (Z)-(E)-5-Ethoxy-4-fluoro-1-phenylpenta-1,3-diene 5g
Total yield 82%. These compounds could not be separated. ν(neat)/cm−1 3021, 2977, 2870, 1673, 1492, 1448, 1346, 1138, 1097, 968, 886, 750 and 692; δH(CDCl3) 1.25 (3H, t, J 6.8 Hz), 1.26 (3H, t, J 6.8 Hz), 3.57 (2H, q, J 6.8 Hz), 3.59 (2H, q, J 6.8 Hz), 4.07 (2H, d, J 15.6 Hz), 4.29 (2H, d, J 20.5 Hz), 5.69 (1H, dd, J 10.7, 34.2 Hz), 6.13 (1H, dd, J 11.2, 18.1 Hz), 6.55 (2H, d, J 16.1 Hz), 6.80 (1H, ddd, J 1.0, 11.2, 16.1 Hz), 7.09 (1H, dd, J 10.7, 16.1 Hz), 7.20–7.45 (10H, m); (E)-isomer: m/z 206 (36%, M+), 159 (35), 147 (49), 146 (75), 142 (38), 141 (40), 129 (100), 128 (55), 115 (77), 91 (53); (Z)-isomer: m/z 206 (41%, M+), 159 (33), 147 (34), 146 (63), 142 (34), 141 (42), 129 (100), 128 (50), 115 (70), 91 (41) [Calc. for C13H15FO (M): 206.1096. Found: M+, 206.1109].(E)- and (Z)-2-Fluoro-1-phenyl-3-piperidinopropene 6
Total yield 44%. (E)-isomer: ν(neat)/cm−1 2936, 2853, 2777, 1676, 1453, 1302, 1282, 1223, 1133, 1098, 1065, 1039, 997, 882, 860, 787, 750 and 700; δH(CDCl3) 1.35–1.50 (2H, m), 1.55–1.70 (4H, m), 2.35–2.55 (4H, m), 3.28 (2H, d, J 22.5 Hz), 6.40 (1H, d, J 21.5 Hz), 7.22–7.36 (5H, m); m/z 219 (82%, M+), 218 (48), 136 (52), 135 (100), 133 (72), 128 (30), 116 (30), 115 (58), 109 (36), 98 (85), 97 (36), 84 (94), 83 (61) [Calc. for C14H19FN (M + H): 220.1501. Found: m/z, 220.1469].(Z)-isomer: ν(neat)/cm−1 2936, 2855, 2809, 2770, 1689, 1496, 1451, 1398, 1372, 1345, 1301, 1275, 1251, 1111, 1040, 996, 885, 862, 785, 754 and 694; δH(CDCl3) 1.35–1.50 (2H, m), 1.55–1.70 (4H, m), 2.40–2.60 (4H, m), 3.18 (2H, d, J 18.1 Hz), 5.65 (1H, d, J 38.6 Hz), 7.15–7.55 (5H, m); m/z 219 (100%, M+), 218 (51), 136 (22), 135 (82), 133 (33), 115 (73), 98 (42), 84 (86), 83 (27) [Calc. for C14H19FN (M + H): 220.1501. Found: m/z, 220.1461].
3-Fluoro-2H-chromene 8
To a suspension of CsF (65.5 mg, 0.43 mmol), Si(OEt)4 (72.0 μL, 0.32 mmol), and salicylaldehyde (26.0 μL, 0.25 mmol) in DMF at room temperature under argon atmosphere was added α-fluorovinyltriphenylphosphonium triflate 4 (114 mg, 0.25 mmol) in one portion. After being heated to 130 °C the reaction mixture was stirred for 40 h at that temperature. After usual work-up, the residual oil was chromatographed on silica gel (hexane as eluent) to give the desired product (12.9 mg, 34%) as a colorless oil, which generally turned yellow on storage under an argon atmosphere: ν(neat)/cm−1 3068, 2959, 2849, 1695, 1578, 1489, 1455, 1390, 1274, 1189, 1105, 1047, 1022, 879, 838, 752 and 658; δH(CDCl3) 4.81 (1H, d, J 1.0 Hz), 4.82 (1H, d, J 1.5 Hz), 6.04 (1H, dt, J 1.0, 12.2 Hz), 6.81 (1H, dd, J 2.0, 7.8 Hz), 6.87 (1H, dd, J 2.0, 7.8 Hz), 6.94 (1H, dt, J 2.0, 7.8 Hz), 7.07 (1H, dt, J 2.0, 7.8 Hz); m/z 150 (100%, M+), 131 (26), 121 (28), 101 (38), 96 (33), 75 (34), 74 (24), 63 (21) (Found: M+, 150.0467. Calc. for C9H7FO: M, 150.0481).3-Fluoro-8-methoxy-2H-chromene 9
47% Yield. ν(neat)/cm−1 2963, 2836, 1694, 1580, 1483, 1329, 1278, 1204, 1140, 1102, 1078, 1035, 971, 849, 730 and 641; δH(CDCl3) 3.87 (3H, s), 4.87 (1H, d, J 1.0 Hz), 4.88 (1H, d, J 1.5 Hz), 6.04 (1H, dt, J 1.0, 10.3 Hz), 6.61 (1H, dd, J 1.5, 7.3 Hz), 6.76 (1H, dd, J 1.5, 8.3 Hz), 6.86 (1H, ddd, J 1.0, 7.3, 8.3 Hz); m/z 180 (100%, M+), 179 (78), 149 (22), 137 (75), 109 (52), 101 (21), 89 (21), 83 (71), 75 (21), 63 (46), 62 (21) (Found: M+, 180.0587. Calc. for C10H9FO: M, 180.0617).2-Fluoro-3H-naphtho[2,1-b]pyran 10
31% Yield. Mp 38.1–39.5 °C; ν(neat)/cm−1 3034, 2849, 1683, 1631, 1506, 1230, 1185, 1083, 1032, 889, 814 and 749; δH(CDCl3) 4.91 (1H, d, J 2.0 Hz), 4.92 (1H, d, J 1.5 Hz), 6.72 (1H, d, J 12.0 Hz), 7.09 (1H, d, J 9.3 Hz), 7.36 (1H, ddd, J 1.0, 6.8, 9.3 Hz), 7.48 (1H, ddd, J 1.0, 6.8, 9.3 Hz), 7.62 (1H, d, J 9.3 Hz), 7.76 (1H, d, J 9.3 Hz), 7.80 (1H, d, J 9.3 Hz); m/z 200 (100%, M+), 199 (98), 171 (34), 170 (54), 153 (39), 152 (27) (Found: M+, 200.0638. Calc. for C13H9FO: M, 200.0637).3-Fluoro-6-nitro-2H-chromene 11
14% Yield. Mp 129.2–130.5 °C; ν(KBr)/cm−1 2917, 1699, 1615, 1583, 1505, 1404, 1338, 1261, 1208, 1154, 1093, 1027, 906, 849, 829, 748, 725, 699 and 625; δH(CDCl3) 4.98 (2H, d, J 1.5 Hz), 6.12 (1H, d, J 11.1 Hz), 6.87 (1H, d, J 8.8 Hz), 7.86 (1H, d, J 2.4 Hz), 7.99 (1H, dd, J 2.4, 8.8 Hz); m/z 195 (94%, M+), 194 (100), 149 (11), 148 (34), 120 (14), 101 (45), 83 (11), 75 (33), 74 (13) [Found: (M+ + H), 196.0423. Calc. for C9H7FNO3: m/z, 196.0410].3-Fluorochromane 12
To a solution of 23.9 mg of 3-fluoro-2H-chromene 8 (0.159 mmol) in 5 mL of ethyl acetate was added a catalytic amount of 5% Rh–Al2O3. Hydrogenation at atmospheric pressure was conducted for 16 h at room temperature. After usual work-up, the residual oil was chromatographed on silica gel (hexane–diethyl ether 100∶1 as eluent) gave the desired product (17.6 mg, 73%), ν(neat)/cm−1 2964, 1585, 1491, 1463, 1346, 1302, 1265, 1233, 1183, 1117, 1094, 1063, 1026, 1008, 948, 828, 785 and 756; δH(CDCl3) 2.95–3.25 (2H, m), 4.0–4.25 (1H, m), 4.25–4.40 (1H, m), 5.12 (1H, ddq, J 2.0, 47.4, 4.4 Hz), 6.86 (1H, dd, J 1.0, 7.3 Hz), 6.92 (1H, dt, J 1.0, 7.3 Hz), 7.07 (1H, dd, J 1.0, 7.3 Hz), 7.13 (1H, dt, J 1.0, 7.3 Hz); m/z 152 (100%, M+), 131 (31), 106 (44), 78 (56), 77 (22) (Found: M+, 152.0623. Calc. for C9H9FO: M, 152.0638).8-Methoxychromane 13
To a solution of 15.9 mg of 3-fluoro-8-methoxy-2H-chromene 9 (0.088 mmol) in 2 mL of EtOH was added a catalytic amount of PtO2. Hydrogenation at atmospheric pressure was conducted for 16 h at room temperature. After usual work-up, the residual oil was chromatographed on silica gel (hexane–diethyl ether 10∶1 as eluent) gave the desired product (9.2 mg, 63%), ν(neat)/cm−1 2964, 1261, 1094, 1019, 866 and 800; δH(CDCl3) 1.97–2.06 (2H, m), 2.79 (2H, t, J 6.3 Hz), 3.86 (3H, s), 4.27 (2H, t, J 5.4 Hz), 6.65–6.82 (3H, m); m/z 164 (100%, M+), 149 (55), 136 (40), 135 (39), 121 (41), 107 (36), 103 (41), 93 (37), 91 (55), 78 (34), 77 (59), 65 (49) [lit.,22m/z 164 (100%, M+), 150 (12), 149 (23), 136 (16), 135 (12), 133 (5), 122 (4), 121 (25), 108 (5), 107 (10), 105 (7), 91 (9), 79 (5), 78 (6.5), 77 (16), 65 (16), 51 (9)].α-Fluorovinyldiphenylphosphine oxide 15
To a solution of α-fluorovinyldiphenylphosphine 2 (51.8 mg, 0.22 mmol) in acetone (1 mL) was added 30% aq. hydrogen peroxide (0.1 mL) at 0 °C. After the mixture had been stirred for 30 min, the reaction was quenched with aq. NaHSO3. The mixture was extracted with diethyl ether, then the ethereal solution was dried over MgSO4. After removal of the solvent, the remaining oil was separated by column chromatography (silica gel; hexane–EtOAc 1∶1) to give the desired product (46.4 mg, 84%), mp 83.5–84.5 °C; ν(KBr)/cm−1 3096, 3055, 3021, 2959, 1632, 1588, 1489, 1435, 1336, 1319, 1203, 1179, 1121, 1097, 998, 913, 801, 757, 729, 712, 689 and 658; δH(CDCl3) 5.72 (1H, ddd, J 3.9, 22.0, 23.0 Hz), 5.82 (1H, ddd, J 3.9, 6.4, 51.3 Hz), 7.3–7.9 (m, 10 H); m/z 246 (14%, M+), 201 (43), 186 (38), 185 (100), 184 (21), 183 (88), 152 (35), 107 (21), 78 (21), 77 (64) (Calc. for C14H12FOP: C, 68.29; H, 4.91. Found: C, 68.30; H, 4.91%).Acknowledgements
This work was supported by the Mitsubishiyuka Foundation, the Yamanouchi Foundation, and a Grant-in-Aid for Scientific Research (10650836) from the Ministry of Education, Science, Sports, and Culture. We thank Central Glass Co., Ltd. for a gift of TfOH, which was used for the preparation of diphenyliodonium triflate. We also thank Chemetall Japan Co., Ltd. for a gift of CsF.References
- B. E. Smart, Chem Rev., 1996, 96, 1555 CrossRef CAS; G. Resnati and V. A. Soloshnok, Tetrahedron, 1996, 52, 1 CrossRef CAS.
- S. T. Purrington, B. S. Kagan and T. B. Patrick, Chem. Rev., 1986, 86, 997 CrossRef CAS.
- J. R. McCarthy, E. W. Huber, T.-B. Le, F. M. Laskovics and D. P. Matthews, Tetrahedron, 1996, 52, 45 CrossRef CAS; T. Ernet and G. Haufe, Tetrahedron Lett., 1996, 37, 7251 CrossRef CAS; S. A. Fontana, C. R. Davis, Y.-B. He and D. J. Burton, Tetrahedron, 1996, 52, 37 CrossRef CAS; P. J. Crowley, J. M. Percy and K. Stansfield, Tetrahedron Lett., 1996, 37, 8233 CrossRef CAS and references cited therein..
- E. E. Schweizer, A. T. Wehman and D. M. Nycz, J. Org. Chem., 1973, 38, 1583 CrossRef CAS; E. E. Schweizer, L. D. Smucker and R. J. Votral, J. Org. Chem., 1966, 31, 467 CAS; T. Minami and J. Motoyoshiya, Synthesis, 1992, 333 CrossRef CAS and references cited therein..
- A. G. Cameron and A. T. Hewson, J. Chem. Soc., Perkin Trans. 1, 1983, 2979 RSC; A. T. Hewson, Tetrahedron Lett., 1978, 3267 CrossRef CAS; J. M. McIntosh and R. S. Steevensz, Can. J. Chem., 1977, 55, 2442 CAS; J. M. McIntosh and H. B. Goodbrand, Synthesis, 1974, 862 CrossRef CAS.
- P. L. Heinze, T. D. Spawn, D. J. Burton and S. Shin-Ya, J. Fluorine Chem., 1988, 38, 131 CrossRef CAS.
- T. Hanamoto, Y. Kiguchi, K. Shindo, M. Matsuoka and M. Kondo, Chem. Commun., 1999, 151 RSC.
- R. J. Hinkle, P. J. Stang and M. H. Kowalski, J. Org. Chem., 1990, 55, 5033 CrossRef CAS.
- H. F. Koch and A. J. Kielbania, Jr, J. Am. Chem. Soc., 1970, 92, 729 CrossRef CAS.
- E. J. Corey and M. A. Tius, Tetrahedron Lett., 1980, 21, 3535 CrossRef CAS.
- J. R. Shutt and S. Trippett, J. Chem. Soc. C, 1969, 2038 RSC.
- L. Cassar and M. Foá, J. Organomet. Chem., 1974, 74, 75 CrossRef CAS; G. Wittig and H. Matzura, Angew. Chem., Int. Ed. Engl., 1964, 3, 231 CrossRef; D. Seyferth and J. M. Burlitch, J. Org. Chem., 1963, 28, 2463 CAS.
- T. Kitamura, M. Yamane, B.-X. Zhang and Y. Fujiwara, Bull. Chem. Soc. Jpn., 1998, 71, 1215 CAS; T. Kitamura, M. Yamane, R. Furuki, H. Taniguchi and M. Shiro, Chem. Lett., 1993, 1703 CAS.
- J. Ichikawa, Y. Wada, T. Okauchi and T. Minami, Chem. Commun., 1997, 1537 RSC; J. Ichikawa, M. Kobayashi, Y. Noda, N. Yokota, K. Amano and T. Minami, J. Org. Chem., 1996, 61, 2763 CrossRef CAS; L. Strekowski, A. S. Kiselyov and M. Hojjat, J. Org. Chem., 1994, 59, 5886 CrossRef CAS; K. Burger and B. Helmreich, J. Chem. Soc., Chem. Commun., 1992, 348 RSC; H. L. Sham and D. A. Betebenner, J. Chem. Soc., Chem. Commun., 1991, 1134 RSC and references cited therein..
- F. Camps, J. Coll, A. Messeguer and M. A. Pericás, J. Heterocycl. Chem., 1980, 17, 1377; Tetrahedron Lett., 1980, 21, 2361; J. Heterocycl. Chem., 1980, 17, 207 Search PubMed.
- F. M. Dean , in The Total Synthesis of Natural Products, ed. J. ApSimon, Wiley, New York, 1973, vol. 1, p. 467. Search PubMed.
- A general preparation of substituted 2H-chromenes from vinyltriphenylphosphonium bromide and salicylaldehyde derivatives has been studied by Schweizer et al. E. E. Schweizer, J. Am. Chem. Soc., 1964, 86, 2744 CrossRef CAS; E. E. Schweizer, J. Liter and D. J. Monaco, J. Org. Chem., 1968, 33, 2416 CrossRef.
- K. H. Ahn and S. J. Lee, Tetrahedron Lett., 1994, 35, 1875 CrossRef CAS.
- T. Yasunaga, R. Naito, T. Kontani, S. Tsukamoto, T. Nomura, T. Yamaguchi and T. Mase, J. Med. Chem., 1997, 40, 1252 CrossRef CAS.
- T. Allmendinger, C. Dandois and B. Walliser, Tetrahedron Lett., 1991, 32, 2735 CrossRef CAS.
- T. Minami, H. Sako, T. Ikehira, T. Hanamoto and I. Hirao, J. Org. Chem., 1983, 48, 2569 CrossRef CAS.
- B. Willhalm, A. F. Thomas and F. Gautschi, Tetrahedron, 1964, 20, 1185 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2000 |