
Atomistic simulation of volumetric properties of epoxy networks: effect of monomer length
 a
and
Cameron F.
Abrams
*a
a
and
Cameron F.
Abrams
*a
Abstract
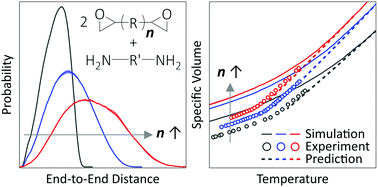
Properties of epoxy thermosets can be varied broadly to suit design requirements by altering the chemistry of the component agents. Atomistically-detailed molecular dynamics simulations are well-suited for molecular insight into the structure–property relationship for a rational tailoring of the chemistry. Since the macroscopic properties of interest for applications emerge hierarchically from molecular-scale chemical interactions, seamless integration of experiment, computation, and theory is of great interest. Recently, a Specific Volume–Cooling Rate analysis protocol was successfully developed to quantitatively compare the volumetric properties of an epoxy network model with experimental results in the literature, in spite of the nine orders of magnitude mismatch in the accessible time-scales. Here, we extend the application of the method for two epoxy networks in the same class of chemistry but whose monomers have a higher number of repeating units compared to the previous one for validating the generality of our approach. We observed that atomistic simulations are able to predict the experimental temperature trend of the specific volume within 0.4% for both these networks. Using the William–Landel–Ferry equation to account for rate differences, we also see good agreement between the computational and experimental values of the glass transition temperature.
 Please wait while we load your content...
Please wait while we load your content...

