
Computational investigations of Dienes defect- and vacancy-induced changes in the electronic and vibrational properties of carbon fiber structural units†‡

Abstract
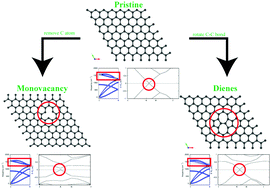
Carbon fiber (CF) is a promising lightweight alternative to steel and is of significant interest for energy applications. As CF continues to find new uses and is exposed to new external conditions, a noninvasive method of monitoring its structural integrity is critical. Raman spectroscopy is a commonly used method for this monitoring; however, it is highly inferential, and the interpretation of the data is not always straightforward. In this work, we perform density functional theory (DFT) calculations to investigate changes in the vibrational properties of CF structural units (i.e., graphene and graphite) caused by monovacancy and Dienes defects as a foundation for modeling more complex defects that move our model toward that of realistic CF. Using large computational supercells, we can understand how these defects change the electronic structure and vibrational properties of graphene and graphite for interdefect distances near those of the lower experimental limit. The monovacancy opens an electronic bandgap at the K point. Although no such electronic gap is opened by the Dienes defect, both defects introduce flat defect bands near the Fermi energy. The Dienes defect creates long-range deviations of the phonons, leading to substantial broadening of the highest frequency optical modes in the band structure compared to that of the pristine material. In contrast, the phonon changes caused by the monovacancy are short range, and only minor changes in the band structure or phonon density of states were observed. These findings can assist in the interpretation of experimental results by providing atomic-scale insight into key electronic and vibrational features.
 Please wait while we load your content...
Please wait while we load your content...

