
Crystal structure prediction of flexible pharmaceutical-like molecules: density functional tight-binding as an intermediate optimisation method and for free energy estimation†
 a
and
Jan Gerit
Brandenburg
‡*a
a
and
Jan Gerit
Brandenburg
‡*a
Abstract
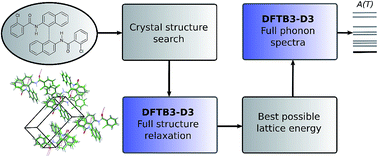
Successful methodologies for theoretical crystal structure prediction (CSP) on flexible pharmaceutical-like organic molecules explore the lattice energy surface to find a set of plausible crystal structures. The initial search stages of CSP studies use relatively simple lattice energy approximations as hundreds of thousands of minima have to be considered. These generated crystal structures often have poor molecular geometries, as well as inaccurate lattice energy rankings, and performing reasonably accurate but computationally affordable optimisations of the crystal structures generated in a search would be highly desirable. Here, we seek to explore whether semi-empirical quantum-mechanical methods can perform this task. We employed the dispersion-corrected tight-binding Hamiltonian (DFTB3-D3) to relax all the inter- and intra-molecular degrees of freedom of several thousands of generated crystal structures of five pharmaceutical-like molecules, saving a large amount of computational effort compared to earlier studies. The computational cost scales better with molecular size and flexibility than other CSP methods, suggesting that it could be extended to even larger and more flexible molecules. On average, this optimisation improved the average reproduction of the eight experimental crystal structures (RMSD15) and experimental conformers (RMSD1) by 4% and 23%, respectively. The intermolecular interactions were then further optimised using distributed multipoles, derived from the molecular wave-functions, to accurately describe the electrostatic components of the intermolecular energies. In all cases, the experimental crystal structures are close to the top of the lattice energy ranking. Phonon calculations on some of the lowest energy structures were also performed with DFTB3-D3 methods to calculate the vibrational component of the Helmholtz free energy, providing further insights into the solid-state behaviour of the target molecules. We conclude that DFTB3-D3 is a cost-effective method for optimising flexible molecules, bridging the gap between the approximate methods used in CSP searches for generating crystal structures and more accurate methods required in the final energy ranking.
- This article is part of the themed collection: Methods and applications of crystal structure prediction
 Please wait while we load your content...
Please wait while we load your content...

