
Repulsion–dispersion parameters for the modelling of organic molecular crystals containing N, O, S and Cl†
 a
and
a
and
Abstract
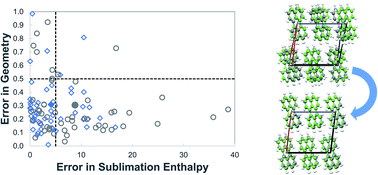
In lattice energy models that combine ab initio and empirical components, it is important to ensure consistency between these components so that meaningful quantitative results are obtained. A method for deriving parameters of atom–atom repulsion dispersion potentials for crystals, tailored to different ab initio models, is presented. It is based on minimization of the sum of squared deviations between experimental and calculated structures and energies. The solution algorithm is designed to avoid convergence to local minima in the parameter space by combining a deterministic low-discrepancy sequence for the generation of multiple initial parameter guesses with an efficient local minimization algorithm. The proposed approach is applied to derive transferable exp-6 potential parameters suitable for use in conjunction with a distributed multipole electrostatics model derived from isolated molecule charge densities calculated at the M06/6-31G(d,p) level of theory. Data for hydrocarbons, azahydrocarbons, oxohydrocarbons, organosulphur compounds and chlorohydrocarbons are used for the estimation. A good fit is achieved for the new set of parameters with a mean absolute error in sublimation enthalpies of 4.1 kJ mol−1 and an average rmsd15 of 0.31 Å. The parameters are found to perform well on a separate cross-validation set of 39 compounds.
- This article is part of the themed collection: Methods and applications of crystal structure prediction
 Please wait while we load your content...
Please wait while we load your content...

