
First-principles stability ranking of molecular crystal polymorphs with the DFT+MBD approach†
 a
and
Alexandre
Tkatchenko
*a
a
and
Alexandre
Tkatchenko
*a
Abstract
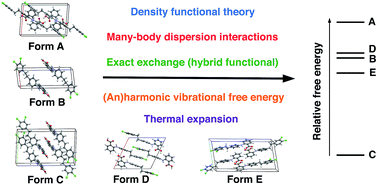
The ability to accurately calculate the relative stabilities of numerous polymorphs of a given molecular crystal is crucial for the success of any molecular crystal structure prediction (CSP) approach. We have recently presented a hierarchical CSP procedure based on van-der-Waals-inclusive density functional theory [Hoja et al., 2018, arXiv:1803.07503], which yields excellent stability rankings for molecular crystals involving rigid molecules, salts, co-crystals, and highly polymorphic drug-like molecules. This approach includes many-body dispersion effects, exact exchange, as well as vibrational free energies. Here, we discuss in detail the impact of these effects on the obtained stability rankings. In addition, we assess the impact of the approximations used in our hierarchical procedure. We show that our procedure is generally robust to 1–2 kJ mol−1 for the systems in the latest CSP blind test but vibrational free energies for crystals involving flexible molecules would benefit from directly including many-body dispersion interactions. In addition, we also discuss the effect of temperature on the structure of molecular crystals and a simple but effective method for estimating anharmonic effects.
- This article is part of the themed collection: Methods and applications of crystal structure prediction
 Please wait while we load your content...
Please wait while we load your content...

