
Assessing the capability of in silico mutation protocols for predicting the finite temperature conformation of amino acids†

Abstract
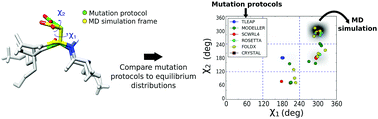
Mutation protocols are a key tool in computational biophysics for modelling unknown side chain conformations. In particular, these protocols are used to generate the starting structures for molecular dynamics simulations. The accuracy of the initial side chain and backbone placement is crucial to obtain a stable and quickly converging simulation. In this work, we assessed the performance of several mutation protocols in predicting the most probable conformer observed in finite temperature molecular dynamics simulations for a set of protein–peptide crystals differing only by single-point mutations in the peptide sequence. Our results show that several programs which predict well the crystal conformations fail to predict the most probable finite temperature configuration. Methods relying on backbone-dependent rotamer libraries have, in general, a better performance, but even the best protocol fails in predicting approximately 30% of the mutations.
 Please wait while we load your content...
Please wait while we load your content...

