
Ultrafast unidirectional chiral rotation in the Z–E photoisomerization of two azoheteroarene photoswitches†

Abstract
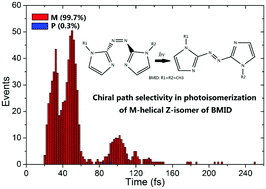
Unidirectional rotation represents a very important functional feature in photochemistry, such as in the design of light-driven molecular rotary motors. Great attention has recently been devoted to the unidirectional preference of the torsional motion of azobenzene and other molecules. Azoheteroarenes offer functional advantages over their more conventional azobenzene counterparts due to the introduction of heteroaromatic rings. In this paper, the Z–E photoisomerization dynamics of two azoheteroarenes, 1,2-bis(1-methyl-1H-imidazol-2-yl)diazene and 1,2-bis(1H-imidazol-2-yl)diazene, are investigated with trajectory surface-hopping molecular dynamics at the semi-empirical OM2/MRCI level. Starting from the S1 excited state of the M-helical Z-isomer of both azoheteroarenes, more than 99% of the trajectories decay to their ground states through the M-helical conical intersections by twisting about the central N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond. This chiral path preference can be well understood by the energy profiles generated by the linear interpolation between the Franck–Condon geometry of the M-helical Z-isomer and the relevant S1/S0 conical intersections. The Z–E photoisomerization mechanisms of these two azoheteroarenes display a higher preference for unidirectional rotary dynamics under a chiral path than their counterpart azobenzene.
N double bond. This chiral path preference can be well understood by the energy profiles generated by the linear interpolation between the Franck–Condon geometry of the M-helical Z-isomer and the relevant S1/S0 conical intersections. The Z–E photoisomerization mechanisms of these two azoheteroarenes display a higher preference for unidirectional rotary dynamics under a chiral path than their counterpart azobenzene.
 Please wait while we load your content...
Please wait while we load your content...

