
Unveiling CO adsorption on Cu surfaces: new insights from molecular orbital principles†

Abstract
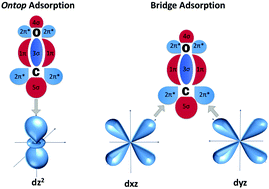
CO adsorption on Cu(100), (110), and (111) surfaces has been extensively studied using Kohn–Sham density functional theory calculations. A holistic analysis of adsorption energies, charge transfer, and structural changes has been employed to highlight the variations in adsorption mechanisms upon changing the surface type and the adsorption site. Each surface, with its unique arrangement of atoms, resulted in a varying adsorbate behavior, although the same adsorption site is considered. This directly reflects the influence of the atomic arrangement on the substrate–adsorbate interactions. Site-interactions are rigorously investigated using molecular-orbital and charge transfer principles taking into account the fundamental interaction of frontier (5σ and 2π*) orbitals. Considering the effects of the surface atomic arrangement and density of metal interacting orbitals, along with the relative d-5σ and d-2π* energy spacings, the calculated adsorption preference to higher coordination sites is explained, which also revealed valuable interpretations to the well-known DFT CO adsorption puzzle. In addition, we studied the perturbations occurring upon adsorption to the 3σ and 1π orbitals, which hold the internal C–O bond. Studying 3σ and 1π orbital perturbations provided a wealth of theoretical interpretations for the varying behavior of the adsorbate molecule when similar adsorption sites are compared at different facets.
 Please wait while we load your content...
Please wait while we load your content...

