
Thermodynamic stability and transport properties of tavorite LiFeSO4F as a cathode material for lithium-ion batteries†
Abstract
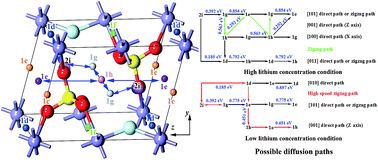
First principles computation methods play an important role in developing and optimizing new energy storage and conversion materials. The thermodynamic stability, transport properties of the carriers and the lithium diffusion mechanisms of LiFeSO4F compounds are calculated by using the first principles computation. According to the calculated formation enthalpies and electronic properties, it can be concluded that both LiFeSO4F and delithiated FeSO4F have high thermodynamic stability during cycling. Band structure analysis reveals that the conduction and valence bands that are mainly composed of Fe3d states are rather localized, leading to large band gaps and effective masses of the carriers. LiFeSO4F and FeSO4F thus exhibit poor electronic conductivities. To improve the electronic conductance of the materials, introduction of delocalized states in the band gap region via doping or nano-crystallization of the electrode material is still necessary. Further investigations on lithium diffusion dynamics suggest that suitable amounts of lithium vacancies at the 2i sites are particularly crucial. Under high lithium concentration conditions, these vacancies are very helpful to initiate the transfer of lithium into the empty positions by eliminating the Li–Li repulsions and then activate the diffusion of lithium through the channels. While under low lithium concentration conditions, they can act as intermediate sites effectively for several high-speed diffusion channels. As the calculated activation energies for the possible diffusion paths (0.185–0.563 eV) are very small, LiFeSO4F and FeSO4F thus show excellent ionic conductance.
 Please wait while we load your content...
Please wait while we load your content...

