
Mechanisms and energetics of free radical initiated disulfide bond cleavage in model peptides and insulin by mass spectrometry†
Abstract
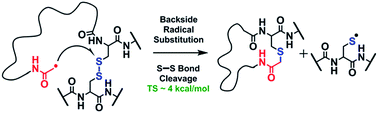
We investigate the mechanism of disulfide bond cleavage in gaseous peptide and protein ions initiated by a covalently-attached regiospecific acetyl radical using mass spectrometry (MS). Highly selective S–S bond cleavages with some minor C–S bond cleavages are observed by a single step of collisional activation. We show that even multiple disulfide bonds in intact bovine insulin are fragmented in the MS2 stage, releasing the A- and B-chains with a high yield, which has been challenging to achieve by other ion activation methods. Yet, regardless of the previous reaction mechanism studies, it has remained unclear why (1) disulfide bond cleavage is preferred to peptide backbone fragmentation, and why (2) the S–S bond that requires the higher activation energy conjectured in previously suggested mechanisms is more prone to be cleaved than the C–S bond by hydrogen-deficient radicals. To probe the mechanism of these processes, model peptides possessing deuterated β-carbon(s) at the disulfide bond are employed. It is suggested that the favored pathway of S–S bond cleavage is triggered by direct acetyl radical attack at sulfur with concomitant cleavage of the S–S bond (SH2). The activation energy for this process is substantially lower by ∼9–10 kcal mol−1 than those of peptide backbone cleavage processes determined by density functional quantum chemical calculations. Minor reaction pathways are initiated by hydrogen abstraction from the α-carbon or the β-carbon of a disulfide, followed by β-cleavages yielding C–S or S–S bond scissions. The current mechanistic findings should be generally applicable to other radical-driven disulfide bond cleavages with different radical species such as the benzyl and methyl pyridyl radicals.
 Please wait while we load your content...
Please wait while we load your content...

