
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRational design of a peptide capture agent for CXCL8 based on a model of the CXCL8:CXCR1 complex†
Dorothea
Helmer
ab,
Ina
Rink
a,
James A. R.
Dalton
c,
Kevin
Brahm
a,
Marina
Jöst
a,
Tobias M.
Nargang
b,
Witali
Blum
a,
Parvesh
Wadhwani
d,
Gerald
Brenner-Weiss
e,
Bastian E.
Rapp
b,
Jesús
Giraldo
c and
Katja
Schmitz
 *a
*a
aTechnische Universität Darmstadt, Clemens-Schöpf-Institut, Alarich-Weiss-Straße 4, 64287 Darmstadt, Germany. E-mail: schmitz@biochemie.tu-darmstadt.de; Fax: +49 6151 16 72058; Tel: +49 6151 16 6964
bKarlsruhe Institute of Technology, Institute of Microstructure Technology, Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
cLaboratory of Molecular Neuropharmacology and Bioinformatics, Institut de Neurociències and Unitat de Bioestadística, Universitat Autònoma de Barcelona, 08193 Bellaterra, Spain
dKarlsruhe Institute of Technology, Institute of Biological Interfaces (IBG-2), P.O. Box 3640, 76021 Karlsruhe, Germany
eKarlsruhe Institute of Technology, Institute of Functional Interfaces, Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
First published on 4th March 2015
Abstract
Protein-capture agents are widely used for the detection, immobilization and isolation of proteins and are the foundation for the development of in vitro diagnostic chips. The chemokine CXCL8 is an interesting protein target due to its involvement in the human inflammatory response. We constructed a novel structural model of CXCL8 interaction with its G-protein coupled receptor CXCR1, taking into account previously reported experimental data. From this CXCL8:CXCR1 model complex, the interaction of CXCL8 with residues near the extracellular domains 3 and 4 of CXCR1 were used as a scaffold for the rational design of a peptide capture agent called ‘IL8RPLoops’. A molecular dynamics simulation of IL8RPLoops indicates a stable helical conformation consistent with the CXCR1 structure from which it was derived. CXCL8 capture in fluorescence-based assays on beads and on glass demonstrates that IL8RPLoops is an effective capture agent for CXCL8. Additionally, we found IL8RPLoops to be a potent inhibitor of CXCL8-induced neutrophil migration and CXCL8:CXCR1 association. A theoretical binding model for IL8RPLoops:CXCL8 is proposed, which shows the peptide predominantly interacting with CXCL8 via electrostatic contacts with the ELR motif at the CXCL8 N-terminus.
Introduction
Protein-capture agents are widely used in the detection, immobilization and isolation of proteins and can facilitate the development of in vitro diagnostic chip technologies. Peptide capture agents can be displayed on phages1 or beads2 and are typically discovered by library screening methods. Effective protein capture on surfaces can also be achieved by co-immobilization of known low-affinity protein–ligands.3 A well-established method for the identification of protein capture agents consists of on-bead binding assays with fluorescently labelled proteins.4–6 TentaGel resin is typically chosen for fluorescence-based on-bead binding assays due to its excellent swelling ability in water and its low level of unspecific interactions.7,8 The detection of proteins by various capture agents, including peptides, can be exploited for protein diagnostic chips, where peptides or proteins are immobilized on surfaces such as glass slides.Chemokines are small signaling proteins that play a key role in the human immune response by activating leukocyte chemotaxis towards sites of inflammation or to lymph organs. CXCL8 (interleukin-8) was first described in 1987 as a tissue-derived neutrophil-activating protein.9 The 72 amino acid isoform is the most potent neutrophil attractant in vivo.10 Due to its involvement in several pulmonary diseases, rheumatism and cancer, CXCL8 can be used as a diagnostic marker. It is also a promising target for drug development.11–13 Only a few CXCL8-binding peptides have been previously characterized: the peptide Ac–RRWWCR–NH2 was reported to inhibit CXCL8-binding to human neutrophils and neutrophil chemotaxis,14 while the peptide VTTFFDYDYGAPC was reported as a low-affinity ligand for CXCL8.15 In addition, peptides derived from two short sequence motifs of the CXCR1 N-terminus connected by a linker with the general sequence Ac–MWDFDD–linker–MPPADEDYSP–NH2 possess Ki values in the low micromolar range.16,17 An NMR-based structural model (PDB id: 1ILQ) of the structure of one of these peptides, CXCR1–p1 (Ki = 7 μM)17 with a 6-aminohexanoic acid linker, in complex with CXCL8 has been described.18 CXCL8-binding peptides derived from the N-terminus of rabbit CXCR1 have also been reported.19 However, no peptides have been shown to capture CXCL8 on bead or on glass surface so far.
CXCL8 binds the G-protein coupled receptors (GPCR) CXCR1 and CXCR2 with an affinity of 2 nM.20 Detailed knowledge of the molecular interactions between CXCL8 and its GPCRs CXCR1 and CXCR2 can potentially enable the rational design of alternative binding peptides that can function as capture agents. The interaction between CXCL8 and CXCR1 has been studied experimentally by CXCR1/CXCL8 mutation studies and NMR. Two NMR experiments with 15N-labeled CXCL8 and CXCR1 N-terminal peptides CXCR1(1–38) and CXCR1(1–40) respectively, identified the CXCL8 N-loop, 310 helix, 40S loop and β3 strand as the major interaction sites of CXCL8 with the N-terminus of CXCR1.21,22 These interactions were largely retained in a complex of CXCL8 with CXCR1–p1, a peptide linking CXCR1(9–14) to CXCR1(20–29),17 of which a model is available in the protein database (PDB id: 1ILQ).18 Further studies suggested that the N-terminus of CXCL8 (residues 1–9) is essential for receptor activation, in particular, the glutamic acid–leucine–arginine motif (ELR motif) at the CXCL8 N-terminus (residues 4–6) was found necessary for biological activity.23,24 Residues R199 (extracellular domain 3, ECD3), R203 (transmembrane helix 5, TMH5) and D265 (TMH6) of the CXCR1 sequence were shown to be essential for receptor function and discussed as a possible interaction site for the ELR motif.18,25,26 Following the experimental data, a two-site mechanism has been proposed for the binding of CXCL8 to CXCR1 in which the CXCL8 N-loop first interacts with the CXCR1 N-terminus (site I) and the CXCL8 N-terminus containing the ELR motif then interacts with residues near ECD3 and 4 of the receptor (site II).27–29 Although NMR studies of CXCL8 with truncated constructs of CXCR1 in a lipid bilayer failed to provide proof of the interaction between the ELR motif and extracellular regions of CXCR1,22 binding studies showed that the region of ECD4, i.e. CXCR1(265–290), enhances binding affinity of monomeric CXCL8 for the CXCR1 N-terminus CXCR1(1–40) when juxtaposed onto a soluble scaffold (chemokine receptor elements on a soluble scaffold, CROSS).30 In addition to interactions with its GPCRs, CXCL8 also binds to glycosaminoglycans (GAG) on the surface of endothelial cells,31–34 thereby forming stable gradients of immobilized chemokine. The residues involved in GAG interactions are located at the C-terminal α-helix of CXCL8, mainly at residues K64 and K68.33–35 The GAG interaction at the chemokine C-terminus leaves the N-terminus accessible for GPCR interaction and activation. The crystal structure of CXCL8 (PDB id: 3IL8),36,37 as well as an NMR-based structure of CXCR1 in a lipid bilayer are available.38 However, the crystal structure of the CXCL8:CXCR1 complex has not been described so far. Liou et al. recently proposed a non-flexible docking of CXCL8 binding to CXCR1 supplemented with a homology modelled N-terminus based on the structure of bovine rhodopsin.39 This model presumably does not allow for CXCL8 interaction with endothelial GAG since K64 and K68 are involved in the CXCL8:CXCR1 interaction, suggesting the C-terminal α-helix as a major interaction site between CXCR1 and CXCL8. Furthermore, since the N-termini (residues 1–29) of CXCR1 and rhodopsin share only 23% sequence identity, rhodopsin is not an ideal template for chemokine receptor homology modelling. Thus there is scope for the development of a new structural binding model of CXCL8 with CXCR1 that is also consistent with experimental information. Such a model could be used for structure-based design of novel peptide capture agents.
The rational design of agonist-binding peptides or receptor mimics based on the sequence and structure of GPCR extracellular and intracellular loops has previously been shown to be a fruitful technique.40–42 These designed peptides can act as new leads for the discovery of novel high-affinity protein-capture agents as well as potential GPCR–agonist inhibitors. However, rational design approaches ideally require detailed structural knowledge of GPCR:agonist interactions. In accordance with this concept and based on experimentally confirmed CXCL8:CXCR1 interactions, we have constructed a novel structural binding model of CXCL8 to its GPCR CXCR1 through flexible protein–protein docking. In this model, the N-loop of CXCL8 interacts with the N-terminus of CXCR1 and the ELR-motif interacts with residues near ECD3 and ECD4 of CXCR1. Based on the proposed ELR-interaction sites near ECD3 and ECD4 of CXCR1, with a particular focus on the experimentally confirmed essential residues R199 (ECD3), R203 (TMH5) and D265 (TMH6),18,25 we have designed a peptide linking parts of ECD3 and ECD4 of CXCR1. We show that this peptide ‘IL8RPLoops’ is an effective CXCL8-capture agent in vitro on bead and on glass surfaces with patterns of immobilized protein. Additionally, we find IL8RPLoops to be an effective inhibitor of CXCL8-induced neutrophil migration. Based on further flexible protein–peptide docking, we also propose a binding model of the CXCL8:IL8RPLoops complex.
Results and discussion
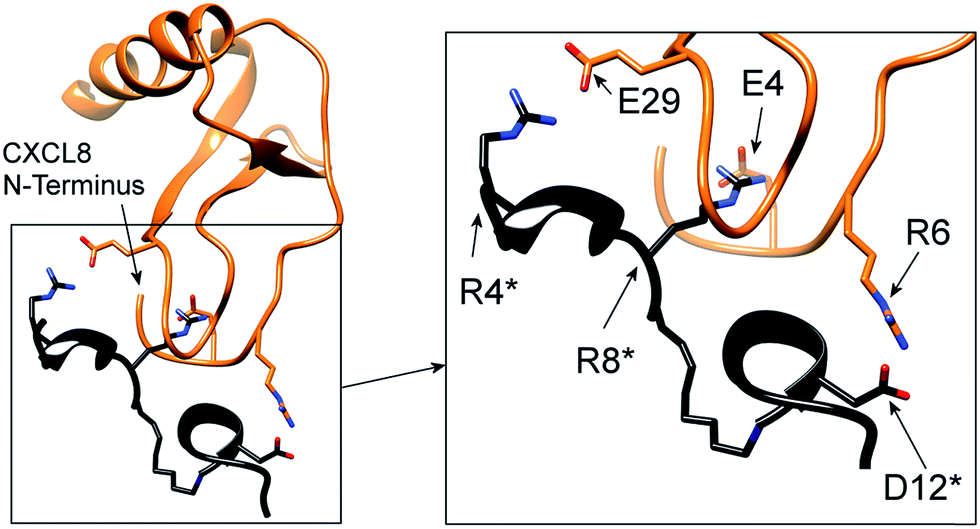
The ELR-motif in CXCL8 is crucial for receptor activation23,24 and is conceived to interact with R199 (ECD3), R203 (TMH5) and D265 (TMH6) of CXCR1.25 To confirm this hypothetical interaction, we performed a flexible protein–protein docking of CXCL8 to CXCR1 using ROSETTA,43 which has been shown to reliably predict native-like GPCR conformations.44 The published NMR structure of CXCR1 in the Protein Databank45 (PDB id: 2LNL) lacks its N-terminal residues 1–28 and shows the receptor in an inactive apo-state.38 Therefore, necessary conformational changes of the CXCR1 structure were induced upon docking of its agonist CXCL8. First, the missing N-terminal amino acid sequence of CXCR1, crucial for CXCL8:CXCR1 interaction, was homology-modelled using MODELLER based on the NMR-derived structure of CXCL8 in complex with the peptide CXCR1–p1 which was previously designed from the CXCR1 N-terminus by Attwood et al. by linking CXCR1(9–14) and CXCR1(20–29) by 6-aminohexanoic acid (PDB id: 1ILQ18).17 In the CXCL8:CXCR1–p1 complex, CXCR1–p1 interacts with CXCL8 N-loop, 310 helix, 40S loop and β3 strand in a very similar fashion to the interactions of CXCL8 with the complete CXCR1 N-terminus, i.e. CXCR1(1–40) or CXCR1(1–38), as shown by NMR.21,22 Thus the CXCL8:CXCR1–p1 complex is a suitable template for homology modelling of the CXCR1 N-terminus. Second, the CXCL8:CXCR1–p1 complex dictated the initial pre-docked orientation of CXCL8 (PDB id: 3IL8)36,37 relative to CXCR1, enabling experimentally established interactions to guide subsequent docking.18,21,38 Third, docking was performed with ROSETTA, allowing for full flexibility in CXCL8 and CXCR1 structures, thus permitting potential conformational changes in CXCL8 and CXCR1. Upon docking of CXCL8 and subsequent relaxation, CXCR1 underwent conformational changes involving a tilting of TMH7 in a mode reminiscent to the activation of other class A GPCRs, in particular adenosine A2A receptor.46,47 The tilting of TMH7 leads to the formation of two strong hydrogen bonds between E275 on TMH7 and T34 at the N-terminus (see ESI Fig. S1†). This is particularly interesting since Hébert et al. reported that a CXCR1 mutant in which E275 was changed to alanine lost all affinity for CXCL8.48 Since the two-site mechanism for chemokine receptor activation involves the interaction of several residues at two different receptor binding sites, this drastic effect seen by the change of one residue at one binding site is surprising. The model explains this finding by indicating that E275 stabilizes the active conformation of CXCR1 that is, as opposed to the inactive apo-form, capable of binding CXCL8.Important interactions in the modelled CXCL8:CXCR1 complex were characterized by measuring hydrogen-bonds distances. Distances between donor atoms (D) and acceptor atoms (A) should not exceed 3.9 Å and ideally lie between 2.7 Å and 2.9 Å.49–51Table 1 lists all intermolecular D–A distances in the proposed model (using 2.9 Å as a cut-off distance value for the formation of strong hydrogen bonds) and compares them to previously reported experimentally proven interactions. The model not only preserves the interactions between CXCL8 N-loop and β3 strand with the N-terminus of CXCR1 as given by the CXCR1:CXCR1–p1 template,18 it also predicts experimentally proven interactions between CXCL8 N-terminus and CXCR1 extracellular domains. A detailed comparison of our model with previously published data shows that the majority of interactions are in accordance with reported experimental data, suggesting good modelling reliability. The model confirms that the major interacting domains of CXCL8 comprise the N-loop, 310 helix, 40S loop and β3 strand, as well as supporting the proposed interaction between the CXCL8 N-terminus and residues near ECD3 and ECD4 of CXCR1 (see Fig. 1). Recently, after the construction of our model, a novel crystal structure of chemokine receptor CXCR4 in complex with a viral chemokine vMIP-II was released.52 This structure is remarkably similar to the herein proposed structure of the CXCR1:CXCL8 complex with the N-terminus of the chemokine reaching deep into the transmembrane helix bundle and the N-Loops and adjacent structures interacting with extracellular loops of CXCR4. We believe this further emphasizes the relevance of our model.
| CXCL8 domains (residue numbers) | CXCL8 residues that interact with CXCR1 (distance < 2.9 Å) in the proposed CXCL8:CXCR1 complex (Fig. 1) | CXCR1 residues that interact (distance < 2.9 Å) with the respective CXCL8 domains in the proposed CXCL8:CXCR1 complex (Fig. 1) | Specific CXCL8:CXCR1 interactions previously reported |
|---|---|---|---|
| N-terminus (1–9) | S1, A2, K3, E4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 23,24,53L5,23,24,53R6,23,24,53 C7, Q818,53 23,24,53L5,23,24,53R6,23,24,53 C7, Q818,53 |
P29,18 K117, H179, P180, N181, Ser184, V186, C187, Y188, E189, R203,23,25 L262, D265,23,25 T266, R269, T270, Q271, I273, R280,48 I283, G284, L287 | Q8:P291818 |
| N-loop (10–18) and 310-helix (19–21) |
I10,18,24K11,18T12,18,21,22Y13,18,54,55K15,18,21,54 P16, F17,18,21,22,56F2118,21,56,57 |
D11,26 F17, T18, G19, M20, P21,18D26,18Y27,18 P29, L32, E33 | (I10, K11, Y13):Y27,18 K11:(D24, E25, D26),18 (Y13, F17, F21):(P21, P22),18 CXCR1-N-terminus 1–40,21 CXCR1 N-terminus 1–39,22 H18, F21 CXCR1 N-terminus58 |
| β1-Strand (22–28) | E24, L25, R26, V27, I28 | F105, G106, T107, A177, Y178, H179, P180, Y188, L191 | |
| 30S-loop (29–37) | E29, G31, P32, H33,53 C34 | Y188, E189, L191, G192, R199,25 T266, L267, R269 | |
| β2-Strand (38–43) |
I40,18L4318,56 |
P21,18 H179 | I40:P29,18 L43:(P21, P22)18 |
| 40S-loop (44–47) | S44,18,21,22 D45, R4718 | T5, D6, P7, M9, F12, P22,18D24,18E2518 |
R47:(P21, P22),18 R47:(D24, E25, D26),18 CXCR1 N-terminus 1–38,22 CXCR1-N-terminus 1–4021 |
| β3-Strand (48–51) |
E48,18,21,22L49,18,21,22,55C5021,22 |
P22,18 E25, Y27,18 S28, W103, F105 | L49:(P21, P22),18 L49:Y27,18 CXCR1-N-terminus 1–40,21 CXCR1 N-terminus 1–3822 |
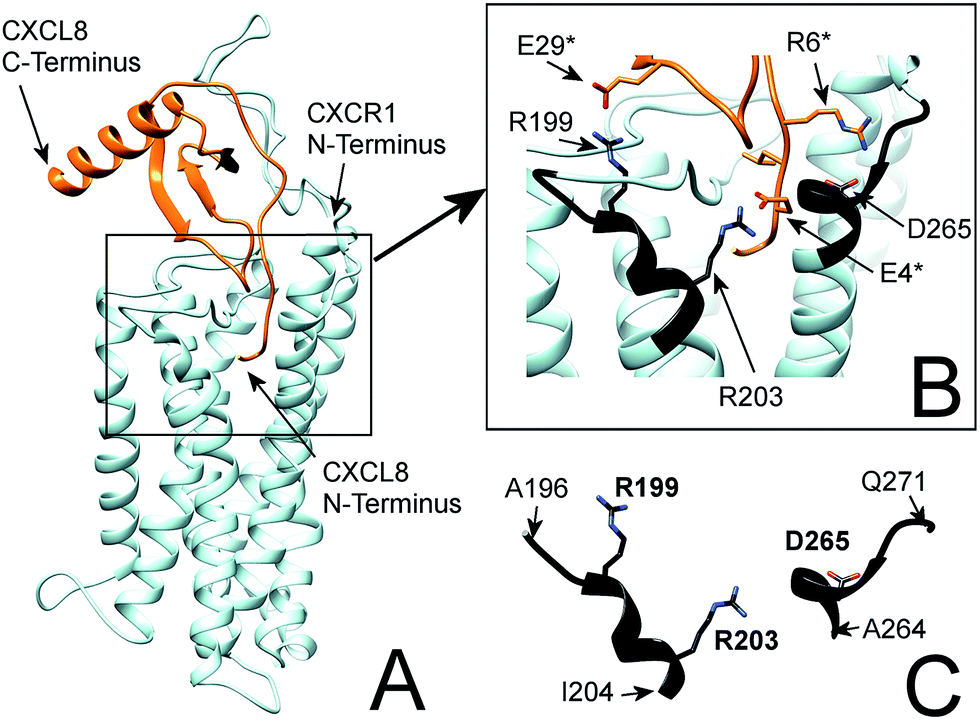 | ||
| Fig. 1 (A) Flexible protein–protein docking of CXCL8 to CXCR1 with ROSETTA, based on the previously reported interactions of the CXCL8 N-loop with the CXCR1 N-terminus18 and the proposed interaction of the CXCL8 N-terminus (ELR motif) with R199, R203 and D265 of CXCR1. (B) Focus on the interacting residues of CXCL8 and CXCR1; CXCL8 residues are marked by an asterisk. CXCR1 regions containing functionally important residues are highlighted in black. (C) Isolated CXCR1 residues near extracellular domains (ECD) 3 and 4: A196 to I204 (AKWRMVLRI) and A264 to Q271 (ADTLMRTQ) that contain residues R199 (ECD3), R203 (TMH5) and D265 (TMH6) that are essential for the interaction with CXCL8. In this model, the ELR motif of CXCL8 interacts with R203 (TMH5) and D265 (TMH6). Additionally, R199 (ECD3) interacts with E29 of the CXCL8 30S-loop. | ||
In particular, the proposed model supports the involvement of R199, R203 and D265 of CXCR1 in the CXCR1:CXCL8 interaction: charge–charge interactions between E4 of the CXCL8-ELR motif and R203 and R6 of the ELR motif and D265 as well as between E29 of the CXCL8 30S loop and R199 were found (see Fig. 1B). The two helical sequence motifs of the regions of ECD3/TMH5 and TMH6/ECD4 (see Fig. 1C) therefore constitute a candidate scaffold for the design of a peptide capable of binding CXCL8. We chose A196 to I204 and A264 to Q271 as appropriate parts of ECD3/TMH5 and TMH6/ECD4 because they collectively contain R199, R203 and D265 and both incorporate at least one full helix turn of TMH5 and TMH6, respectively, that could potentially be preserved in a peptide. We linked the two domains to bridge the distance between them in the full receptor structure (see Fig. 1C). We chose 6-aminohexanoic acid (Ahx) as a linker because it poses a good compromise between hydrophobicity, length and flexibility and has been employed successfully in the design of a different CXCL8-binding peptide, CXCR1–p1, based on the N-terminus of CXCR1.17 The resulting peptide AKWRMVLRI–Ahx–ADTLMRTQ was named ‘IL8RPLoops’. After a molecular dynamics simulation of 100 ns, the IL8RPLoops peptide retained a similar helical structure and spatial orientation of side chains R199, R203, D265 to that of the corresponding residues of CXCR1 (see Fig. 2). The helical structure of IL8RPLoops was later confirmed by circular dichroism spectroscopy (see Fig. 3).
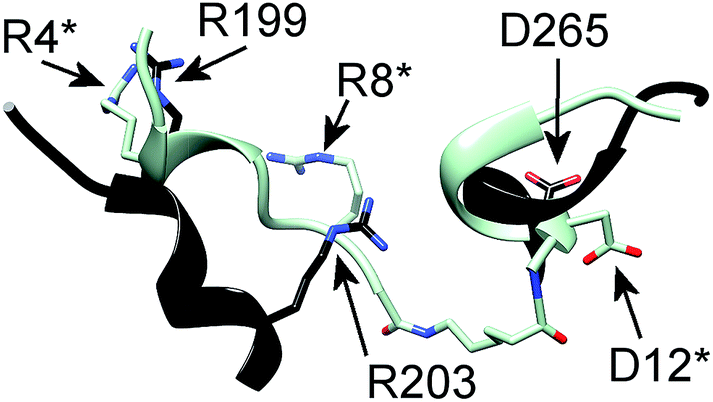 | ||
| Fig. 2 Molecular dynamics simulation of a rationally designed peptide corresponding to the two short sequence motifs shown in Fig. 1C. The structural motifs from Fig. 1C (in black) are superimposed to the designed peptide AKWRMVLRI–Ahx–ADTLMRTQ (in light blue), called ‘IL8RPLoops’ in which the two CXCR1 domains are linked by 6-aminohexanoic acid (Ahx). After 100 ns of molecular dynamics simulation the peptide assumes a partially helical secondary structure with a similar conformation to the original CXCR1 domains. The corresponding residues of IL8RPLoops were renumbered and marked by an asterisk. | ||
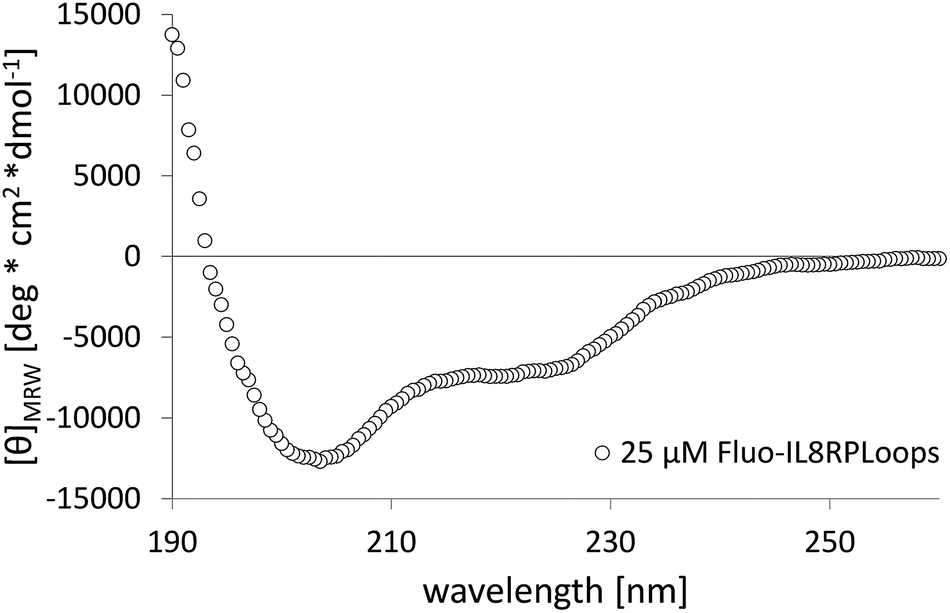 | ||
| Fig. 3 Circular dichroism spectrum of 25 μM Fluo-IL8RPLoops in low-salt PBS (ls-PBS, 40 mM Na2HPO4, 35 mM NaCl at pH 7.41). The shoulder at 222 nm together with the positive values at 190 nm is characteristic for helical structure and the minimum at 201 nm is consistent with the presence of unstructured parts in the peptide. | ||
IL8RPLoops was synthesized via solid phase peptide synthesis (SPPS) using standard Fmoc/tBu strategy.59 The circular dichroism spectrum of purified Fluo-IL8RPLoops confirmed a mixture of α-helix and unordered structure as predicted by the peptide model (Fig. 3). CXCL8(1–72) was expressed as previously described.60 The dissociation constant of IL8RPLoops and CXCL8 was determined by fluorescence anisotropy measurements.61 Four individual measurements gave an average Kd of 0.5 ± 0.3 μM (Fig. 4), which compares favorably with the Kd values of other previously reported capture agents.3,4
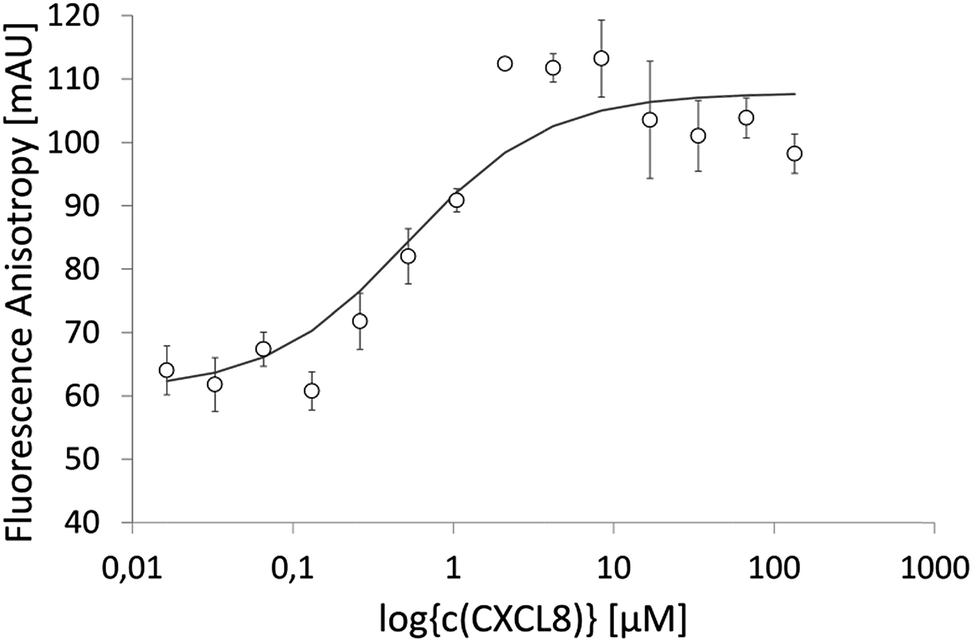 | ||
| Fig. 4 Fluorescence anisotropy data of 200 nM Fluo-IL8RPLoops binding to CXCL8 in ls-PBS. One-site saturation f = Bmin + Bmax × abs(x)/(Kd + abs(x)), regression was done with SigmaPlot. Four individual measurements were conducted, giving an average Kd of 0.5 ± 0.3 μM. The graph shows a single experiment with STD (for this experiment alone, a Kd of 0.5 ± 0.2 μM was calculated). | ||
To test the peptide's specificity, a randomized peptide with the sequence (Fluo)-LWMIVRKAR–Ahx–RAMQLTDT was synthesized. In circular dichroism measurements the randomized peptide showed no tendency for helical folding (see ESI Fig. S2†). It also showed no affinity for CXCL8 under 100 μM (see ESI Fig. S3†). Therefore the interaction between CXCL8 and IL8RPLoops is not merely a result of unspecific interactions between charged residues, but instead requires an ordered peptide with specifically positioned chemical features.
Fluo-IL8RPLoops was shown to be displaced by unlabeled IL8RPLoops in a competition experiment (see ESI Fig. S4†). With 8.8 ± 3.7 μM the Ki of the competition experiment was found to be slightly higher than expected, given that the competitor was structurally equivalent to the displaced ligand except for the fluorescein label. Therefore the impact of the fluorescein label on the IL8RPLoops:CXCL8 interaction was investigated. In a control binding experiment we found that fluorescein alone has low affinity for CXCL8 (Kd > 100 μM; see ESI Fig. S5†). A comparison of the CD spectra of labelled and unlabeled IL8RPLoops showed that the unlabeled peptide has a similar secondary structure, although with less pronounced minima (see ESI Fig. S6†). The fluorescein label therefore probably has a minor effect on binding by most likely helping to stabilize the peptide fold.
To confirm that IL8RPLoops is an effective CXCL8 capture agent, it was synthesized on 300 μm TentaGel MB HMBA resin and treated with fluorescently labelled chemokine. CXCL8 with a fluorescence label at the C-terminus was produced by the reaction of a CXCL8 mutant possessing a C-terminal cysteine residue (CXCL8S72C) with thiol-reactive DyLight550 fluorescent dye. The resulting variant CXCL8S72C-DL550 possesses almost equivalent activity to CXCL8 in biological assays.62 Labelled CXCL8 was effectively captured by IL8RPLoops on TentaGel resulting in significantly increased fluorescence of the beads in the RHO channel (see Fig. 5). A hydrophobic control peptide (FWLDFW) shows no unspecific interaction with CXCL8S72C-DL550. To further validate the affinity of the peptide for CXCL8, a reverse capture assay with immobilized CXCL8S72C-DL550 was conducted. An elegant method for the immobilization of proteins on glass is projection lithography based on photobleaching, which leads to a covalent attachment of the protein at the site of its label.63,64 With this technique, CXCL8S72C-DL550 was immobilized on BSA-coated glass slides to give two identical, characteristic protein patterns on one slide. One pattern was stained by FITC-labeled anti-human-CXCL8 antibody, the other was stained with Fluo-IL8RPLoops. After washing, both patterns were clearly visible (see Fig. 6). Interestingly, CXCL8-binding peptide CXCR1–p1 reported by Attwood et al.17 failed to stain the protein pattern on glass in our experiments.
 | ||
| Fig. 5 On-bead fluorescence assay, scale bar 300 μm. CXCL8S72C-DL550 is captured by IL8RPLoops immobilized on TentaGel beads. From left to right: untreated TentaGel MB HMBA, hydrophobic peptide FWLDFW on TentaGel and IL8RPLoops on TentaGel were treated with (a) ls-PBS and (b) 10 μM CXCL8S72C-DL550 in ls-PBS and imaged in the RHO channel, exposure time 40 ms. Untreated beads show no unspecific interactions with CXCL8S72C-DL550. Hydrophobic control peptide FWLDFW shows no unspecific interactions, either, but some minor autofluorescence in the RHO channel. | ||
 | ||
| Fig. 6 CXCL8 immobilized on BSA-coated glass by protein patterning stained with Fluo-IL8RPLoops (left) and FITC-anti-CXCL8 (right), scale bar: 500 μm. Projection of the Mona Lisa image by a digital mirror display led to the immobilization of CXCL8S72C-DL550 by photobleaching of the fluorophore.63,64 The Mona Lisa by Leonardo da Vinci is openly accessible through Wikimedia commons (public domain, http://en.wikipedia.org/wiki/Mona_Lisa-#mediaviewer/File:Mona_Lisa,_by_Leonardo_da_Vinci,_from_C2RMF_retouched.jpg, accessed 2012). | ||
The immobilization by photobleaching of the fluorophore shown in Fig. 6 covalently attaches the fluorescent label at the C-terminus of CXCL8S72C-DL550 to the BSA-coated glass slide. This orientation is similar to the spatial arrangement of the chemokine in vivo, where the C-terminal α-helix interacts with glycosaminoglycans (GAG) on the endothelium to form stable CXCL8 gradients.31–34 With its C-terminus tethered, the N-terminus of the chemokine is accessible for CXCR1 and CXCR2 binding that induces cell migration. It can therefore be speculated that with the CXCL8 C-terminus tied to the glass slide, the interaction of IL8RPLoops most likely occurs at the accessible N-terminal side of CXCL8 facing away from the glass surface. As the proposed binding mode of CXCL8 with CXCR1 shows that the chemokine N-terminus makes specific contacts residues near ECDs 3 and 4 of CXCR1, it suggests IL8RPLoops elicits its function through binding in the same way, possibly in the same location.
Indeed, if the GPCR and IL8RPLoops binding sites are close or overlapping, IL8RPLoops could likely compete with CXCR1 for CXCL8 binding. To test this hypothesis, fluorescence-activated cell sorting (FACS) and fluorescence microscopy experiments were conducted to investigate the effect of IL8RPLoops on CXCL8:CXCR1 interaction on the cell surface. CXCL8 labelled with CF633 thiol reactive dye for FACS measurements (CXCL8S72C-CF633) was obtained analogously to CXCL8S72C-DL550 as previously reported.62 When HEK293 cells stably transfected with CXCR1 (see ESI Fig. S7†) were treated with a 300 nM solution of CXCL8S72C-CF633 and analyzed by FACS, cells showed fluorescence at an excitation wavelength of 640 nm indicating a CXCL8:CXCR1 interaction (see Fig. 7). No fluorescence was observed when co-incubating the cells with 300 nM CXCL8S72C-CF633 and 390 nM anti-human-CXCL8 antibody (Sigma #I2519, monoclonal anti-interleukin-8, mouse). Co-incubation of CXCL8S72C-CF633 with 500 nM IL8RPLoops also led to reduced cell fluorescence (Fig. 7). No quenching occurred upon mixing of CXCL8-CF633 with different concentrations of CXCL8RPLoops (see ESI Fig. S8†) and no unspecific interactions of Fluo-IL8RPLoops with HEK 293 cells were observed in the FITC channel (see ESI Fig. S9†). In addition, human neutrophil granulocytes were used in a similar staining experiment with CXCL8S72C-DL550 and analyzed under the microscope (see Fig. 8). Co-incubation of neutrophils with 100 nM CXCL8S72C-DL550 and 500 nM Fluo-IL8RPLoops reduced CXCL8S72C-DL550-induced fluorescence of the cells to a minimum. Taken together, these results suggest that IL8RPLoops interacts with CXCL8 to prevent CXCL8:CXCR1 association.
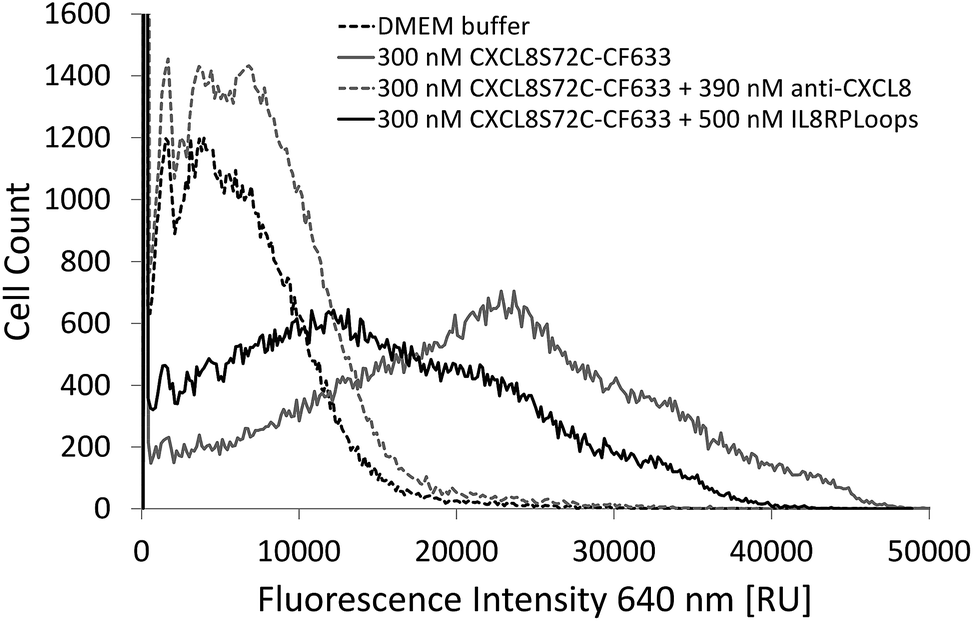 | ||
| Fig. 7 FACS experiment with HEK293 cells stably transfected with CXCR1 and stained with CXCL8S72C-CF633. Incubation of cells with a mixture of 500 nM Fluo-IL8RPLoops and 300 nM CXCL8S72C-CF633 (solid black line) significantly reduces fluorescence of the population mean compared to cells treated with CXCL8S72C-CF633 alone (solid gray line). No fluorescence was observed when CXCL8S72C-CF633 was incubated with 390 nM specific antibody (dashed gray line) or buffer (dashed black line) as a control. | ||
 | ||
| Fig. 8 Inhibition of CXCL8 binding to its receptors on human neutrophil granulocytes, scale bar: 20 μm. Human neutrophil granulocytes were incubated with (a) 100 nM CXCL8S72C-DL550, (b) a mixture of 100 nM CXCL8S72C-DL550 and 500 nM Fluo-IL8RPLoops and (c) ls-PBS buffer. Cells were analysed with a fluorescence microscope in the brightfield (BF) and red fluorescence channel (RHO). Significantly reduced fluorescence intensity was detected for cells treated with a mixture of IL8RPLoops and CXCL8S72C-DL550 compared to treatment with CXCL8S72C-DL550 alone. A light background fluorescence of CXCL8S72C-DL550 can be observed in (a) and (b). | ||
We then investigated the effect of Fluo-IL8RPLoops on neutrophil migration (Fig. 9). Upon stimulation of cells with 10 nM CXCL8, an average of 31% of cells migrated into the lower cavity of the assay plate. When stimulated with a mixture of 10 nM CXCL8 and 2.5 μM IL8RPLoops, the number of migrated cells was reduced by 50%. This inhibitory effect was not observed for fMLP-induced migration thus demonstrating the specificity of the Fluo-IL8RPLoops:CXCL8 interaction. To ensure results were not a result of toxic effects, a cell viability assay was conducted. No toxic effects of IL8RPLoops on human neutrophils were observed in the relevant concentration range (see ESI Fig. S10†).
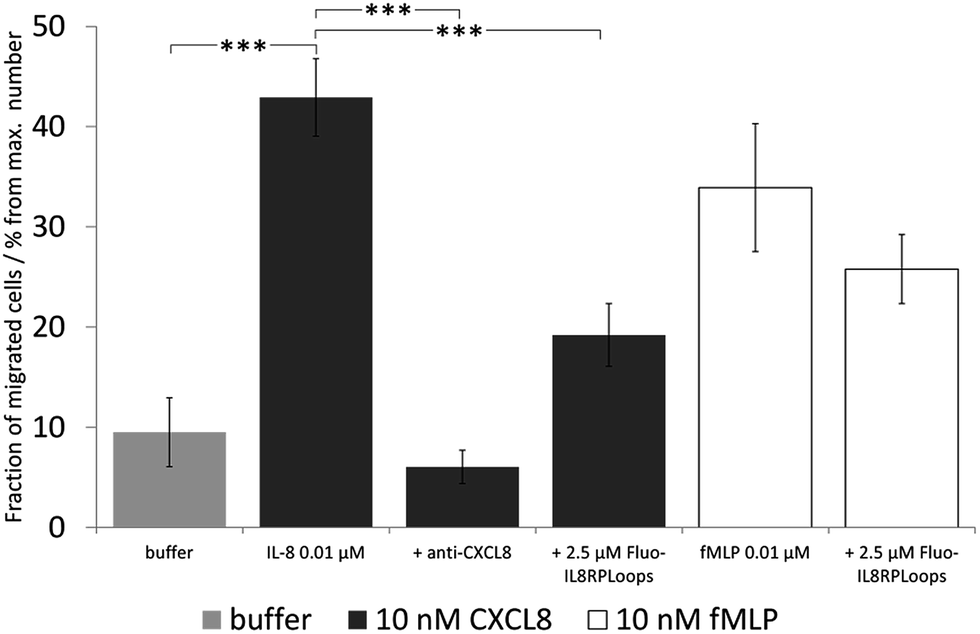 | ||
| Fig. 9 Neutrophil migration assay with human neutrophil granulocytes, the graph shows one experiment with STD. Statistical differences between the CXCL8 treated cells and the cells treated with CXCL8 and inhibitors are indicated (***P < 0.001) and the experiments were repeated three times. Similar results were obtained in three experiments with cells of independent donors (see ESI Fig. S11†). Negative control (buffer, light gray): chemotaxis buffer (RPMI medium with 0.2% bovine serum albumin BSA). Positive control: 10 nM CXCL8 in chemotaxis buffer (dark gray) and 10 nM fMLP in chemotaxis buffer (white). Inhibitor control: chemoattractant with anti-CXCL8 mouse monoclonal antibody. CXCL8-induced migration decreases by ca. 50% upon co-incubation with 2.5 μM Fluo-IL8RPLoops. fMLP induced migration is not influenced. | ||
Finally, we investigated the binding mode of IL8RPLoops to CXCL8 by flexible docking in a constraint-free molecular dynamics simulation. The model complex of CXCR1 with CXCL8 was used as a starting point for initially orientating the peptide in the vicinity of CXCL8 in Cartesian space. During the course of the 200 ns simulation, the peptide and CXCL8 were free to explore alternative conformations. The peptide largely retained its helical conformation and formed stable interactions with E4 and R6 of the ELR-motif of CXCL8 (see Fig. 10), reminiscent of the chemokine's interaction with CXCR1 in the model complex (Fig. 1). In this way, the peptide core wraps around the 30S-loop, binding next to the N-terminus of CXCL8. We postulate that this is characteristic of the functional binding mode of IL8RPLoops to CXCL8 and consequently prevents CXCL8 binding to CXCR1 as shown in cell experiments.
 | ||
| Fig. 10 Proposed binding mode of IL8RPLoops to CXCL8. Residues of IL8RPLoops are marked by an asterisk. The interaction of residues as suggested by the CXCL8:CXCR1 protein–protein docking (Fig. 1) are stably formed and largely maintained over a 200 ns molecular dynamics simulation. | ||
Conclusion
By taking advantage of the wealth of experimental functional data regarding CXCL8:CXCR1 interaction, and by using state-of-the-art tools for flexible protein–protein docking, we constructed a novel structural model of the CXCL8:CXCR1 complex, which shows a high level of consistency with reported functional data. Furthermore, we describe the role of four residues on CXCR1 (R199, R203, D265, and E275) and three residues on CXCL8 (E4 and R6 of the ELR motif and E29) that are important for complex formation. Furthermore, a new model of a CXCR4/chemokine complex52 that was published during the review process of this paper is strikingly similar to our model, further validating the relevance of our CXCR1/CXCL8 complex structure. We demonstrated the rational design of a peptide CXCL8-capture agent called IL8RPLoops, that was designed from parts of ECD3/TMH5 and TMH6/ECD4 of CXCR1 containing the functionally important residues R199 (ECD3), R203 (TMH5) and D265 (TMH6). IL8RPLoops shows an affinity of 0.5 ± 0.3 μM for CXCL8. The affinity of IL8RPLoops for CXCL8 was successfully exploited for CXCL8-capture on TentaGel beads and for a reverse capture assay on glass. Thus, IL8RPLoops may be a promising tool for the capture and identification of CXCL8 in the field of diagnostic microarrays. In addition to being a useful capture agent, it was shown that IL8RPLoops is also capable of inhibiting CXCL8 binding to CXCR1 on the surface of cells and of CXCL8-induced migration of human neutrophils.Theoretical computational simulations confirm the ELR motif of CXCL8 to be a likely interaction site of IL8RPLoops. Furthermore, the IL8RPLoops peptide appears to be a valid starting point for the development of a new class of specific inhibitors for chemokines containing the ELR motif. On a more general note, we believe the overall strategy of this study is potentially extendable to a wider group of GPCR ligands. By first studying the interactions of peptide ligands with their respective GPCRs, novel peptides may be designed based on relevant GPCR ECDs/TMH fragments. In particular, as it is known that TMH5 and TMH6 are highly relevant to GPCR agonistic function and conformational change in general,46 the design of peptides based on parts of these two helices as well as associated ECD3 and ECD4, may prove to be a fruitful technique for inhibiting the ligands of other GPCRs.
Methods and materials
N,N-Dimethylformamide (DMF), dichloromethane (DCM), acetonitrile, trifluoroacetic acid (TFA), piperidine, and ethanol were purchased at VWR. N,N-Diisopropylcarbodiimide and methanol were purchased at TH Geyer, N,N-dimethylaminopyridine, 5(6)-carboxyfluorescein, 1,8-diazabicyclo[5.4.0]undec-7-en (DBU), diisopropylethylamine (DIPEA), triisopropylsilane, bromoacetic acid, α-cyanohydroxy cinnamic acid, monoclonal antibody anti-interleukin-8 mouse, CF-TM-633 maleimide and RPMI chemotaxis medium were purchased at Sigma-Aldrich. Antibody FITC-anti human IL8, mouse was purchased at BioLegend. Amino acids were purchased at Iris Biotech and Orpegen. 1-Hydroxybenzotriazole (HOBt) and N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate (HBTU) were also purchased at Iris Biotech. Chlorotritylchloride resin was purchased at PL-Laboratories. H2N-Gly(OtBu) was purchased at Carbolution Chemicals. N-Methyl-2-pyrrolidone (NMP), diethyl ether, dextran 500 and bovine serum albumin were purchased at Carl Roth. TCEP reduction gel and fluorescent dye DyLight550-maleimide were purchased at Thermo Scientific. Lymphoprep was purchased at Axis-Shield PoC AS. 384-well (low volume) flat transparent bottom, black wall microtiter plates, cell culture plates and 96-well clear/black microtiter plates were purchased from Corning. Transwell migration plates were purchased from Corning.Molecular dynamics simulations
IL8RPLoops starting conformation was defined by extracting the two sequence parts AKWRMVLRI (ECD 3) and ADTLMRTQ (ECD 4) from the two CXCR1 receptor domains and linking them with 6-aminohexanoic using Maestro,66 causing minimal overall perturbation to the peptide structure. The peptide was then energy minimized and simulated in explicit solvent for 100 ns molecular dynamics (production run) using MOE-2012 in the Amber12-EHT force-field at 300° NVT.67 In order to probe the binding mode of IL8RPLoops to CXCL8, the structure of the peptide after 100 ns MD was superimposed onto the respective domains of CXCR1 in the docked conformation with CXCL8 (see below) using CHIMERA.65 The coordinates of the peptide with CXCL8 were then extracted from the ensemble to give a starting conformation for docking by molecular dynamics. The CXCL8:IL8RPLoops complex was then energy minimized and simulated in explicit solvent for 200 ns MD (production run). This protocol yielded a final docked CXCL8:IL8RPLoops conformation.Protein–protein docking
The NMR structure of CXCR1 (2LNL) lacks the N-terminus of CXCR1 (residues 1–28).38 These missing residues were modelled using MODELLER68 based on the structure of the N-terminus-CXCR1-derived peptide in the template CXCL8:CXCR1–p1 complex (PDB id: 1ILQ).18 Modelling was performed so as to cause minimal perturbation to the rest of the CXCR1 structure. This enabled the presentation of the N-terminus of CXCR1 in an experimentally established conformation. The X-ray crystal structure of CXCL8 (PDB id: 3IL8) lacks the first four residues of its sequence and these were added with MODELLER. Prior to docking CXCL8 and CXCR1 were initially orientated by superimposing their respective structures onto the established CXCL8:p1 complex using CHIMERA. This allowed the observed interactions in the CXCL8:p1 complex to be maintained as closely as possible. This protocol resulted in the re-location of the N-terminus of CXCL8 within the proximity of ECD 3 and ECD 4 of CXCR1. ROSETTA43 was then used to flexibly dock CXCL8 with CXCR1 and optimize the docked complex with iterative cycles of relaxation (with CXCR1 in an implicit membrane) until convergence was reached (three cycles in total).Peptide synthesis
Peptides IL8RPLoops and FWLDFW were synthesized on TentaGel MB HMBA (MB300140) resin with an average diameter of 300 μm with a resin loading of 0.24 mmol g−1 for on-bead capture assay. IL8RPLoops was additionally synthesized on a 2-chlorotritylchloride resin (1.5 mmol g−1). The resin was swollen in DCM and manually functionalized with the first amino-acid. 3 eq. of Fmoc-protected amino acid were dissolved in DMF and added to the resin. 4.0 eq. of DIC and 0.1 eq. of DMAP were added (stock solution: 15 mg mL−1 in DMF). The suspension was shaken overnight at 600 rpm. The resin was washed five times with 3 mL of DMF. The resin loading was determined by Fmoc-removal.69 A standard Fmoc/tBu SPPS protocol was used for the peptide synthesizer (Liberty, CEM, USA) equipped with a microwave reactor (Discover, CEM, USA). Amino acids were prepared as 0.2 M solutions in DMF. The Fmoc-protecting group was removed with 20 vol% piperidine in DMF: activating solution was prepared with 0.25 M HOBT and 0.25 M HBTU in DMF. Activator base was prepared as 1 M DIPEA in NMP.Removal of side chain protecting groups and cleavage from chlorotritylchloride resin
For manual removal of side chain protecting groups and cleavage from solid phase the resin was washed thoroughly with DCM and methanol and dried under vacuum overnight. The resin was incubated with 2 mL of 95% TFA, 2.5% triisopropylsilane and 2.5% water for 4 hours. The resin was washed two times with 1 mL of TFA and the solution was combined with the cleavage solution. Peptide was precipitated from the cleavage solution with cold diethyl ether and centrifuged. The supernatant was discarded and the peptide was washed with diethyl ether before drying in vacuo. The same TFA-mixture was employed for removal of side chain protecting groups of peptides on TentaGel-HMBA resin.Cleavage of peptides from TentaGel MB HMBA resin
The resin was washed with DCM and dried under vacuum overnight. The resin was placed in a desiccator filled with ammonia vapor. The resin was left inside the desiccator for 4 h or overnight. Peptides were solubilized with acetonitrile:water = 1:1 (v/v).
Determination of resin loading by Fmoc removal
The resin was washed with DCM. Three times ten TentaGel MB HMBA beads each were manually transferred into three microfuge tubes. The beads were centrifuged to the bottom of the tube and incubated for 30 min with 8 μL of 2 vol% DBU in DMF. The bead suspension was diluted with 992 μL acetonitrile and absorption was measured at 304 nm. The extinction coefficient of dibenzofulvene in DBU/DMF/acetonitrile is ε = 7624 M−1 cm−1,69 resin mass was calculated according to information from Rapp Polymere: 15.7 μg per bead TentaGel MB HMBA. A correction factor of 1.17 was introduced by comparing results from this method with the method from Gude.69 Resin loading was calculated according to the following equation: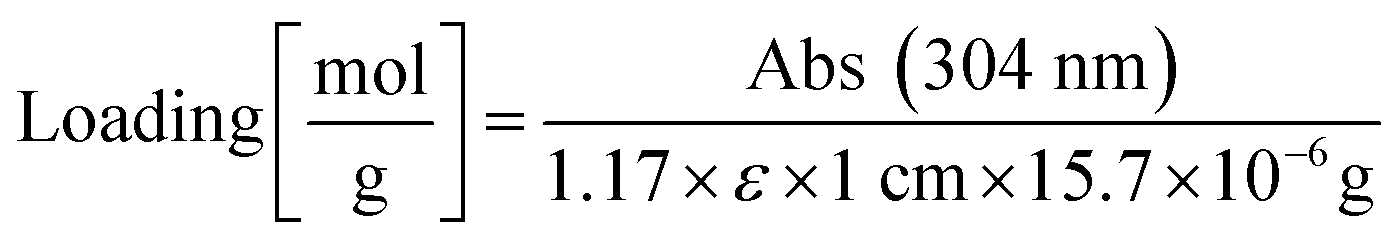
Attachment of 5(6)-carboxyfluorescein to IL8RPLoops peptide
Fluorescein was coupled manually to IL8RPLoops on chlorotritylchloride resin as follows: a mixture of 3 eq. of 5(6)-carboxyfluorescein in DMF, 3 eq. of DIC and 3 eq. of hydroxybenzotriazole (HOBt, 30 mg mL−1 in DMF) was added to the resin and shaken for at least 2 h at room temperature. The resin was washed five times with DMF.Peptide purification by HPLC
HPLC was conducted on a LC20-AD Shimadzu two-pump system with a PDA equipped with a Supelco C18 RP column (250 × 10 mm Discovery HS C18, Supelco, Sigma-Aldrich). The column was equilibrated for 10 min with eluent A (95% water/5% acetonitrile/0.1% TFA). After injection of the crude peptide solution eluent B (95% acetonitrile/5% water/0.1% TFA) was increased to 5% over 1 CV. Then the acetonitrile concentration was elevated continuously to reach 80% eluent B after 5 CV at a flow rate of 1 mL min−1. Absorption was monitored at 254 nm and 495 nm. Fractions were collected manually and lyophilized. Fluo-IL8RPLoops eluted after 68 min.CD spectroscopy
CD-spectra were measured with a Jasco J-810 at room temperature (20 °C) in 0.1 cm fused silica cuvettes. 10 scans per measurement, 190–260 nm (50 nm min−1).Expression and purification of CXCL8 and CXCL8S72C
E. coli BL21 DE3 RIL were transformed with a pET-22b vector containing the codon optimized sequence of human CXCL860 or with pET-22b vector containing the sequence of human CXCL8 with a C-terminal cysteine residue.62 The proteins were expressed and purified by a procedure modified from Wiese et al.60 Briefly, the transformed cells were grown in LB-medium containing ampicillin (60 μg mL−1). At an optical density OD600 of 0.6–0.8 expression was induced by addition of IPTG (0.1 mM final concentration). Expression was continued for 3 h at 30 °C. Cells were harvested by centrifugation (45 min at 5000 × g) and resuspended in buffer A (40 mM Na2HPO4, 90 mM NaCl, pH 7.4) supplemented with 1 mM EDTA, 0.2 mg mL−1 lysozyme, 0.1 mg mL−1 DNase I and incubated on ice for 1.5 h. After addition of 0.5% Triton X-100, the suspension was sonified three times for 30 s at 50% intensity (Sonopuls, Bandelin Electronics). After additional incubation with DNase I the sample was heated to 70 °C for 10 min to precipitate host cell proteins. The lysate was centrifuged at 4 °C and 4500 × g for 45 min. The protein was purified from the supernatant via HPLC on an ÄKTA purifier 10 system (GE Healthcare) using a 5 mL HiTrap SP FF column (GE Healthcare) with a gradient of 0–30% buffer B (2 M NaCl and 40 mM sodium phosphate, pH 7.4) in buffer A. Protein fractions were lyophilized and desalted on a centricon (Vivaspin20; Sartorius) using water. Concentrations were determined by absorption spectroscopy at 280 nm based on a standard curve recorded with purified CXCL8.Fluorescence labelling of CXCL8S72C
150 μL TCEP resin was transferred into a microfuge tube and washed once with 200 μL low-salt-PBS (ls-PBS, 40 mM Na2HPO4·2H2O, 35 mM NaCl at pH 7.41). 2.1 mg of CXCL8S72C in 1 mL ls-PBS were incubated with the reduction gel for 45 min, centrifuged, and the supernatant was transferred into a microfuge tube. The gel was washed twice with 50 μL ls-PBS and the CXCL8S72C supernatants were combined. 1 mg of DyLight550-maleimide or CF633-maleimide was dissolved in 100 μL of peptide grade DMF and 50 μL of this solution was added to the CXCL8S72C in ls-PBS and incubated for 2.5 h on an orbital shaker at room temperature protected from light. The fluorescently labelled protein was purified on an LC20-AD Shimadzu two-pump system with a PDA equipped with a C8 RP column (VP250/10 Nucleosil 300-7, Macherey-Nagel) employing a multistep gradient: the column was equilibrated for 5 min with 10% eluent B (95% acetonitrile/5% water/0.1% TFA). The concentration of eluent B was constantly elevated to 40% over 25 min. The concentration was held constant for 5 min and was then elevated to reach 95% eluent B in one minute. Absorption was monitored at 280 nm and 495 nm. CXCL8S72C eluted after 27 min, while labelled variants eluted after 28 min. Fractions were collected manually and lyophilized. Products were analyzed by MALDI TOF MS.Fluorescence anisotropy measurements
Fluorescence anisotropy measurements were conducted on a Tecan Infinite M1000 microplate reader (Tecan). Measurements were conducted according to Moerke in low-volume black wall, transparent flat-bottom 384-well microtiter plates.61 CXCL8 wild type was dissolved in ls-PBS and 15 consecutive 1:2-dilutions of protein in ls-PBS were prepared to give a final volume of 31.5 μL each. To each of these dilutions, 3.5 μL of 2 μM Fluo-IL8RPLoops (in ls-PBS) was added. Additionally, 31.5 μL of ls-PBS with 3.5 μL of Fluo-IL8RPLoops (negative control) and 31.5 μL of plain ls-PBS buffer (blank) were prepared. The components were gently mixed and three times 10 μL of each solution were transferred into three adjacent cavities of the 384-well microtiter plate. For fluorescence anisotropy competition experiments aliquots of 31.5 μL of a stock solution of 2 μM Fluo-IL8RPLoops and 1 μM CXCL8 were added to 3.5 μL of 1:2-dilutions of IL8RPLoops in ls-PBS. Fluorescence anisotropy was measured at an LED excitation wavelength of 470 nm; the emission wavelength was manually set to 520 nm.
CXCL8 capture assay on TentaGel beads
After cleavage of the side-chain protecting groups peptides on TentaGel MB HMBA 300 μm resin were washed three times with ls-PBS and incubated with 10 μM CXCL8S72C-DL550 in ls-PBS. The beads were incubated for 5 h at room temperature and washed three times with ls-PBS, transferred into a 384-well black wall, transparent flat bottom microtiter plate and analyzed under the fluorescence microscope (Zeiss Axio Observer.Z1, FITC channel Ex: 450–490 nm, Em: 500–550 nm; RHO channel Ex: 538–562 nm, Em: 570–640 nm).CXCL8 reverse-capture assay on glass
Clean room glass slides (Nexterion® Slide Glass B) were immersed in 4 mL 3% BSA in PBS and shaken for 10 min at room temperature. Slides were washed by immersion in 4 mL PBS and shaken for 5 min at room temperature. The PBS washing step was repeated three times. Slides were immersed in water and dried by centrifugation. A HybriWell chamber (Grace Bio-Labs) was attached to the slide, filled with 50 μM CXCL8S72C-DL550 in ls-PBS, sealed and exposed to a greyscale image at 550 nm generated by the maskless lithography system developed by Waldbaur et al. for 6 min.64 HybriWell chambers were removed and slides were washed by immersion in PBS (3 × 4 mL), washed with water and dried by centrifugation. New HybriWell chambers were attached to the area previously exposed to light and filled with a staining solution of 5 nM FITC-anti-CXCL8 (BioLegend #514604, FITC-anti human IL8, mouse) or 20 μM Fluo-IL8RPLoops in ls-PBS and sealed. They were incubated for 10 min at room temperature. HybriWell chambers were removed and slides were washed with water and dried by centrifugation. Slides were analyzed by a GenePix 4000B microarray scanner (Molecular Devices).FACS analysis
HEK293 cells stably expressing CXCR1 were detached with PBS containing 0.5 mM EDTA and washed twice with DMEM/0.2% (w/v) bovine serum albumin (BSA). Cells (5 × 106 cells per mL) were incubated with DMEM/0.2% (w/v) BSA (control), 300 nM CXCL872C-CF633, 500 nM IL8RPloops and a mixture of the latter two for 2 h at 37 °C. In addition, a mixture of 390 nM mouse anti-human CXCL8 (Sigma #I2519, monoclonal anti-interleukin-8, mouse) and 300 nM CXCL8S72C-CF633 was prepared. Cells were incubated on ice. After washing once with DMEM/0.2% BSA and twice with HBSS (Hank's Balanced Salt Solution), cells were analyzed by flow cytometry (BD Influx™, BD Biosciences). 100000 events of each sample were recorded.
Isolation of neutrophil granulocytes from human blood
Equal volumes of human blood (roughly 10 mL per experiment), dextran 500 (6% solution in water) and PBS buffer (12 mM Na2HPO4, 137 mM NaCl, 2.7 mM KCl at pH 7.50) were mixed and incubated at 37 °C for 30 min; sedimented erythrocytes were discarded. The supernatant was transferred into a new tube and centrifuged for 10 min at 15 °C with 240 × g. The supernatant was discarded and the pellet was washed with PBS buffer and centrifuged for 10 min at 15 °C with 240 × g. The pellet was suspended in 10 mL PBS buffer and added to 30 mL Lymphoprep. The mixture was centrifuged for 20 min at 4 °C with 600 × g. The white layer formed in the middle of the tube was carefully removed, the pellet was suspended in PBS buffer and centrifuged for 10 min at 4 °C with 240 × g. The pellet was washed in PBS, and then suspended in 5 mL PBS buffer. Erythrocytes were lysed by addition of 25 mL deionized water. After 30 s 17.2 mL 3.6% NaCl solution in water was added. The suspension was centrifuged for 10 min at 4 °C with 240 × g. The pellet was consecutively washed with PBS buffer and chemotaxis buffer (RPMI medium with 0.2% bovine serum albumin). The cells were suspended in chemotaxis buffer and could be used for 2–3 h at maximum.Fluorescence cell assay
1 × 106 human neutrophil granulocytes (2 × 107 cells per mL) in chemotaxis buffer (RPMI medium with 0.2% (w/v) bovine serum albumin) were aliquoted in wells of a 96-well clear flat bottom MTP. Cells were incubated with 100 nM IL8-S72C-DL550, with 500 nM Fluo-IL8RPLoops and a mixture of 100 nM IL8-S72C-DL550 (0.1 μM) and 500 nM Fluo-IL8RPLoops. Cells were incubated for 1 h at room temperature and washed three times with 100 μL PBS (centrifugation at 400 rcf), pipetted onto glass slides and observed under the fluorescence microscope (RHO-channel, Ex: 538–562 nm, Em: 570–640 nm).Neutrophil migration assay
24-well Transwell plates (3 μm) were purchased at Corning. CXCL8 and fMLP were prepared as a 0.01 M solution in chemotaxis buffer. A mix of 0.01 μM CXCL8 with 2.5 μM Fluo-IL8RPLoops and a mix of 0.01 μM fMLP with 2.5 μM Fluo-IL8RPLoops were prepared. The cavities of the 24-well bottom plate were filled with 595 μL of the chemokine/inhibitor solutions (each solution was prepared in triplicate). To determine the maximum cell number, plain cell suspension was added to three cavities. As a negative control, plain buffer was added to three more cavities. Anti-CXCL8 antibody (10.8 μL of mouse anti-human IL8, monoclonal, 500 μg mL−1) in 900 μL chemotaxis buffer served as a positive inhibitory control. The inserts of the top plate were filled with 150 μL of neutrophil suspension in chemotaxis buffer. After incubation for 1.5 h at 37 °C in a CO2 incubator the inserts were removed and 50 μL of cell suspension were mixed with 50 μL trypan blue solution (0.5% (w/v) in PBS). Cells were incubated with trypan blue for 5 min at room temperature. Living cells of each well were counted using an improved Neubauer chamber. Resulting absolute cell numbers were divided by the maximum cell number to obtain relative cell numbers for evaluation. Statistics: comparisons of the transwell assay with more than two data sets were performed by one-way anova and a posthoc test (Bonferroni's Multiple Comparison Test) using the Prism 5.0 program (GraphPad Software Inc, San Diego, CA).Conflicts of interests
The authors declare that there are no conflicts of financial and/or commercial interests.Acknowledgements
The authors thank S. Hörner and H. Fittler for conducting LC-MS measurements and P. Czechowski for measuring CD spectra. We thank the “Fonds der Chemischen Industrie” for financial support. This work received financial support by the ‘Concept for the Future’ of the Karlsruhe Institute of Technology (KIT) within the framework of the German Excellence Initiative (Research Group 26-2). Molecular graphics and analyses were performed with the UCSF Chimera package. Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311), see http://www.cgl.ucsf.edu/chimera.65References
- E. Galikowska, D. Kunikowska, E. Tokarska-Pietrzak, H. Dziadziuszko, J. M. Los, P. Golec, G. Wegrzyn and M. Los, Eur. J. Clin. Microbiol., 2011, 30, 1067–1073 CrossRef CAS PubMed.
- J. Cha, J. Lim, Y. R. Zheng, S. Tan, Y. L. Ang, J. Oon, M. W. Ang, J. J. Ling, M. Bode and S. S. Lee, J. Lab. Autom., 2012, 17, 186–200 CrossRef CAS PubMed.
- K. Bachhawat-Sikder and T. Kodadek, J. Am. Chem. Soc., 2003, 125, 9550–9551 CrossRef CAS PubMed.
- J. L. Naffin, Y. Han, H. J. Olivos, M. M. Reddy, T. W. Sun and T. Kodadek, Chem. Biol., 2003, 10, 251–259 CrossRef CAS PubMed.
- S. Akita, N. Umezawa and T. Higuchi, Org. Lett., 2005, 7, 5565–5568 CrossRef CAS PubMed.
- H. D. Agnew, R. D. Rohde, S. W. Millward, A. Nag, W. S. Yeo, J. E. Hein, S. M. Pitram, A. A. Tariq, V. M. Burns, R. J. Krom, V. V. Fokin, K. B. Sharpless and J. R. Heath, Angew. Chem., Int. Ed., 2009, 48, 4944–4948 CrossRef CAS PubMed.
- E. Bayer, Angew. Chem., Int. Ed., 1991, 30, 113–129 CrossRef.
- M. G. Paulick, K. M. Hart, K. M. Brinner, M. Tjandra, D. H. Charych and R. N. Zuckermann, J. Comb. Chem., 2006, 8, 417–426 CrossRef CAS PubMed.
- T. Yoshimura, K. Matsushima, S. Tanaka, E. A. Robinson, E. Appella, J. J. Oppenheim and E. J. Leonard, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 9233–9237 CrossRef CAS.
- A. Mortier, N. Berghmans, I. Ronsse, K. Grauwen, S. Stegen, J. Van Damme and P. Proost, PLoS One, 2011, 6, e23913 CAS.
- N. Godessart and S. L. Kunkel, Curr. Opin. Immunol., 2001, 13, 670–675 CrossRef CAS PubMed.
- M. C. Kraan, D. D. Patel, J. J. Haringman, M. D. Smith, H. Weedon, M. J. Ahern, F. C. Breedveld and P. P. Tak, Arthritis Res., 2001, 3, 65–71 CrossRef CAS PubMed.
- J. L. Galzi, M. Hachet-Haas, D. Bonnet, F. Daubeuf, S. Lecat, M. Hibert, J. Haiech and N. Frossard, Pharmacol. Ther., 2010, 126, 39–55 CrossRef CAS PubMed.
- S. Hayashi, A. Kurdowska, E. J. Miller, M. E. Albright, B. E. Girten and A. B. Cohen, J. Immunol., 1995, 154, 814–824 CAS.
- C. Ezerzer, M. Dolgin, J. Skovorodnikova and N. Harris, Peptides, 2009, 30, 1296–1305 CrossRef CAS PubMed.
- M. R. Attwood, N. Borkakoti, G. A. Bottomley, E. A. Conway, I. Cowan, A. G. Fallowfield, B. K. Handa, P. S. Jones, E. Keech, S. J. Kirtland, G. Williams and F. X. Wilson, Bioorg. Med. Chem. Lett., 1996, 6, 1869–1874 CrossRef CAS.
- M. R. Attwood, E. A. Conway, R. M. Dunsdon, J. R. Greening, B. K. Handa, P. S. Jones, S. C. Jordan, E. Keech and F. X. Wilson, Bioorg. Med. Chem. Lett., 1997, 7, 429–432 CrossRef CAS.
- N. J. Skelton, C. Quan, D. Reilly and H. Lowman, Structure, 1999, 7, 157–168 CrossRef CAS PubMed.
- L. Rajagopalan and K. Rajarathnam, J. Biol. Chem., 2004, 279, 30000–30008 CrossRef CAS PubMed.
- J. Lee, R. Horuk, G. C. Rice, G. L. Bennett, T. Camerato and W. I. Wood, J. Biol. Chem., 1992, 267, 16283–16287 CAS.
- R. T. Clubb, J. G. Omichinski, G. M. Clore and A. M. Gronenborn, FEBS Lett., 1994, 338, 93–97 CrossRef CAS PubMed.
- S. H. Park, F. Casagrande, L. Cho, L. Albrecht and S. J. Opella, J. Mol. Biol., 2011, 414, 194–203 CrossRef CAS PubMed.
- I. Clark-Lewis, C. Schumacher, M. Baggiolini and B. Moser, J. Biol. Chem., 1991, 266, 23128–23134 CAS.
- C. A. Hebert, R. V. Vitangcol and J. B. Baker, J. Biol. Chem., 1991, 266, 18989–18994 CAS.
- S. R. Leong, R. C. Kabakoff and C. A. Hebert, J. Biol. Chem., 1994, 269, 19343–19348 CAS.
- C. A. Hebert, A. Chuntharapai, M. Smith, T. Colby, J. Kim and R. Horuk, J. Biol. Chem., 1993, 268, 18549–18553 CAS.
- I. Clark-Lewis, K. S. Kim, K. Rajarathnam, J. H. Gong, B. Dewald, B. Moser, M. Baggiolini and B. D. Sykes, J. Leukocyte Biol., 1995, 57, 703–711 CAS.
- L. Rajagopalan and K. Rajarathnam, Biosci. Rep., 2006, 26, 325–339 CrossRef CAS PubMed.
- P. R. Joseph, J. M. Sarmiento, A. K. Mishra, S. T. Das, R. P. Garofalo, J. Navarro and K. Rajarathnam, J. Biol. Chem., 2010, 285, 29262–29269 CrossRef CAS PubMed.
- E. F. Barter and M. J. Stone, Biochemistry, 2012, 51, 1322–1331 CrossRef CAS PubMed.
- A. Rot, Eur. J. Immunol., 1993, 23, 303–306 CrossRef CAS PubMed.
- A. J. Hoogewerf, G. S. V. Kuschert, A. E. I. Proudfoot, F. Borlat, I. ClarkLewis, C. A. Power and T. N. C. Wells, Biochemistry, 1997, 36, 13570–13578 CrossRef CAS PubMed.
- J. Middleton, S. Neil, J. Wintle, I. ClarkLewis, H. Moore, C. Lam, M. Auer, E. Hub and A. Rot, Cell, 1997, 91, 385–395 CrossRef CAS PubMed.
- D. P. Witt and A. D. Lander, Curr. Biol., 1994, 4, 394–400 CrossRef CAS PubMed.
- G. S. V. Kuschert, A. J. Hoogewerf, A. E. I. Proudfoot, C. W. Chung, R. M. Cooke, R. E. Hubbard, T. N. C. Wells and P. N. Sanderson, Biochemistry, 1998, 37, 11193–11201 CrossRef CAS PubMed.
- E. T. Baldwin, I. T. Weber, R. Stcharles, J. C. Xuan, E. Appella, M. Yamada, K. Matsushima, B. F. P. Edwards, G. M. Clore, A. M. Gronenborn and A. Wlodawer, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 502–506 CrossRef CAS.
- G. M. Clore, E. Appella, M. Yamada, K. Matsushima and A. M. Gronenborn, Biochemistry, 1990, 29, 1689–1696 CrossRef CAS PubMed.
- S. H. Park, B. B. Das, F. Casagrande, Y. Tian, H. J. Nothnagel, M. N. Chu, H. Kiefer, K. Maier, A. A. De Angelis, F. M. Marassi and S. J. Opella, Nature, 2012, 491, 779–784 CrossRef CAS PubMed.
- J. W. Liou, F. T. Chang, Y. Chung, W. Y. Chen, W. B. Fischer and H. J. Hsu, PLoS One, 2014, 9, e94178 Search PubMed.
- D. D. Lopes, R. F. F. Vieira, L. Malavolta, E. F. Poletti, S. I. Shimuta, A. C. M. Paiva, S. Schreier, L. Oliveira and C. R. Nakaie, Amino Acids, 2013, 44, 835–846 CrossRef CAS PubMed.
- H. Demene, S. Granier, D. Muller, G. Guillon, M. N. Dufour, M. A. Delsuc, M. Hibert, R. Pascal and C. Mendre, Biochemistry, 2003, 42, 8204–8213 CrossRef CAS PubMed.
- K. Mobius, R. Durr, C. Haussner, U. Dietrich and J. Eichler, Chem.–Eur. J., 2012, 18, 8292–8295 CrossRef PubMed.
- K. M. Misura, D. Chivian, C. A. Rohl, D. E. Kim and D. Baker, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 5361–5366 CrossRef CAS PubMed.
- E. D. Nguyen, C. Norn, T. M. Frimurer and J. Meiler, PLoS One, 2013, 8, e67302 CAS.
- F. C. Bernstein, T. F. Koetzle, G. J. Williams, E. F. Meyer Jr, M. D. Brice, J. R. Rodgers, O. Kennard, T. Shimanouchi and M. Tasumi, Eur. J. Biochem., 1977, 80, 319–324 CrossRef CAS PubMed.
- B. G. Tehan, A. Bortolato, F. E. Blaney, M. P. Weir and J. S. Mason, Pharmacol. Ther., 2014, 143, 51–60 CrossRef CAS PubMed.
- F. Xu, H. Wu, V. Katritch, G. W. Han, K. A. Jacobson, Z. G. Gao, V. Cherezov and R. C. Stevens, Science, 2011, 332, 322–327 CrossRef CAS PubMed.
- C. A. Hébert, A. Chuntharapai, M. Smith, T. Colby, J. Kim and R. Horuk, J. Biol. Chem., 1993, 268, 18549–18553 Search PubMed.
- I. Y. Torshin, I. T. Weber and R. W. Harrison, Protein Eng., 2002, 15, 359–363 CrossRef CAS PubMed.
- E. N. Baker and R. E. Hubbard, Prog. Biophys. Mol. Biol., 1984, 44, 97–179 CrossRef CAS PubMed.
- C. Bissantz, B. Kuhn and M. Stahl, J. Med. Chem., 2010, 53, 6241 CrossRef CAS.
- L. Qin, I. Kufareva, L. G. Holden, C. Wang, Y. Zheng, C. Zhao, G. Fenalti, H. Wu, G. W. Han, V. Cherezov, R. Abagyan, R. C. Stevens and T. M. Handel, Science Express, 2015, 1–11 Search PubMed.
- E. T. Baldwin, I. T. Weber, R. Stcharles, J. C. Xuan, E. Appella, M. Yamada, K. Matsushima, B. F. P. Edwards, G. M. Clore, A. M. Gronenborn and A. Wlodawer, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 502–506 CrossRef CAS.
- I. U. Schraufstatter, M. Ma, Z. G. Oades, D. S. Barritt and C. G. Cochrane, J. Biol. Chem., 1995, 270, 10428–10431 CrossRef CAS PubMed.
- M. E. Hammond, V. Shyamala, M. A. Siani, C. A. Gallegos, P. H. Feucht, J. Abbott, G. R. Lapointe, M. Moghadam, H. Khoja, J. Zakel and P. Tekamp-Olson, J. Biol. Chem., 1996, 271, 8228–8235 CrossRef CAS PubMed.
- G. Williams, N. Borkakoti, G. A. Bottomley, I. Cowan, A. G. Fallowfield, P. S. Jones, S. J. Kirtland, G. J. Price and L. Price, J. Biol. Chem., 1996, 271, 9579–9586 CrossRef CAS PubMed.
- G. N. Prado, K. Suetomi, D. Shumate, C. Maxwell, A. Ravindran, K. Rajarathnam and J. Navarro, Biochemistry, 2007, 46, 11660 CrossRef CAS.
- H. Suzuki, G. N. Prado, N. Wilkinson and J. Navarro, J. Biol. Chem., 1994, 269, 18263–18266 CAS.
- G. B. Fields and R. L. Noble, Int. J. Pept. Protein Res., 1990, 35, 161–214 CrossRef CAS PubMed.
- D. Wiese and K. Schmitz, J. Immunol. Methods, 2011, 364, 77–82 CrossRef CAS PubMed.
- N. Moerke, Curr. Protoc. Chem. Biol., 2009, 1, 1–15 Search PubMed.
- J. Inglese, P. Samama, S. Patel, J. Burbaum, I. L. Stroke and K. C. Appell, Biochemistry, 1998, 37, 2372–2377 CrossRef CAS PubMed.
- M. A. Holden and P. S. Cremer, J. Am. Chem. Soc., 2003, 125, 8074–8075 CrossRef CAS PubMed.
- A. Waldbaur, B. Waterkotte, K. Schmitz and B. E. Rapp, Small, 2012, 8, 1570–1578 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- L. N. Y. Maestro Version 9.4, Schrödinger, NY, 2013 Search PubMed.
- C. C. G. I., Molecular Operating Environment (MOE), 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2013.
- A. Sali and T. L. Blundell, J. Mol. Biol., 1993, 234, 779–815 CrossRef CAS PubMed.
- M. Gude, J. Ryf and P. D. White, Lett. Pept. Sci., 2002, 9, 203–206 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra13749c |
| This journal is © The Royal Society of Chemistry 2015 |