
Iron-catalyzed graphitization of biomass†
E.
Thompson
,
A. E.
Danks
,
L.
Bourgeois
and
Z.
Schnepp
*
School of Chemistry, University of Birmingham, B15 2TT UK. E-mail: z.schnepp@bham.ac.uk
First published on 10th October 2014
Abstract
This paper reports the direct transformation of raw lignocellulosic biomass into nanostructured graphitic carbon in a single step. Catalytic iron carbide nanoparticles are generated in situ by thermal decomposition of absorbed iron nitrate followed by carbothermal reduction. The Fe3C particles then etch through the biomass to generate intertwined graphitic tubules through catalytic graphitization. The materials are mesoporous with the pore size dependant on the iron content. Conversion of raw biomass into stable graphitic carbon at relatively low temperatures (800 °C) offers a promising route to large-scale and sustainable synthesis of carbons for electrode or filtration applications. This facile method can also be used to produce nanocomposites of Fe3C/graphite combined with nanoparticles of metal oxides such as CaO or MgO, again from a one-pot precursor, giving potential for a wide range of applications.
Introduction
Porous carbons are widely used as electrodes,1 as catalyst supports,2 in environmental remediation3 and in filtration and separation technology.4 These diverse applications result from the remarkable properties that carbon can display, such as electrical and thermal conductivity, chemical stability, high porosity and tunable surface chemistry. Carbon is viewed as a sustainable resource for materials as it is one of the most abundant crustal elements. Furthermore, there is increasing interest in ‘fixing’ carbon in functional materials as one route to atmospheric CO2 reduction.5The precise structural, physical and chemical properties of a porous carbon are critical to the application. For example, carbons for key energy technologies such as fuel cells or batteries require a high electrode/electrolyte contact area and short path length for ion transport (i.e. high accessible surface area).6 This must be combined with efficient packing to maximize volumetric energy density and a high level of graphitization for durability and corrosion resistance.7 Graphitic carbon nanostructures such as graphene or carbon nanotubes offer many of the requisite properties. However, these ideal structures are generally expensive to manufacture and/or use complex methods and precursors.8 For widespread and sustainable use of carbon in large-scale applications such as energy devices, environmental remediation and carbon capture, the synthesis must be simple, safe and scalable.
Waste biomass is an attractive resource for materials synthesis since it is produced in large quantities from food, agriculture and industry.9 Many methods have been developed for carbonizing biomass, e.g. pyrolysis10 and hydrothermal carbonization.11 However, there are many challenges that remain in this field, particularly the controlled introduction of porosity and tuning of graphitic content. This structural tailoring becomes particularly difficult when the starting material is raw biomass rather than a purified derivative such as cellulose or starch. Raw biomass is typically a composite of many different macromolecules, small molecules and inorganic components. Wood, for example, is a mixture of cellulose, hemicellulose, lignin, small molecules such as tannins and trace inorganic minerals.12 These components exhibit very different chemical properties and thermal decomposition profiles, which can make controlled carbonization difficult.
One strategy for converting carbon-rich precursors into graphitic products is catalytic graphitization. Carbon nanotubes are widely synthesized using transition metal-containing catalysts such as iron carbide (Fe3C). This is often carried out by thermal decomposition of carbon-rich vapours such as ethylene.13 The gas decomposes and carbon diffuses into the catalyst nanoparticle, followed by either base or float growth of a carbon nanotube from the catalyst.14 Another common synthesis involves carbonizing metal-polymer mixtures.15 In these cases, the catalyst nanoparticles are often produced in situ by thermal decomposition of the iron precursor followed by carbothermal reduction to produce Fe3C.16 Recently, it was demonstrated that Fe3C could be used to catalyze the graphitization of pure cellulose fibres.17 This method is particularly interesting since the Fe3C nanoparticles ‘etch’ into the solid material, leaving graphitic nanotubes behind. Other transition metals can be used for graphitization, for example nickel nanoparticles have been used to convert cellulose into graphitic ‘coils’ after a hydrothermal pretreatment.18
In this paper, we report that catalytic graphitization can be used to produce mesoporous graphitic nanotubes in one step from raw lignocellulosic biomass (Scheme 1) using softwood sawdust as a model system. Softwood acts as a useful model biomass since it is produced on a very large scale (10.5 million green tonnes in 2013 in the UK)19 and is a complex mixture of cellulose (∼50% by weight) lignin, hemicellulose and small molecule/inorganic species. Iron carbide (Fe3C) nanoparticles are produced in situ and convert the biomass into graphitic tubules. The porosity is tunable simply by changing the Fe![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :biomass ratio, without any apparent reduction in level of graphitization. This simple method offers the opportunity to produce complex graphitic nanostructures directly from waste biomass.
:biomass ratio, without any apparent reduction in level of graphitization. This simple method offers the opportunity to produce complex graphitic nanostructures directly from waste biomass.
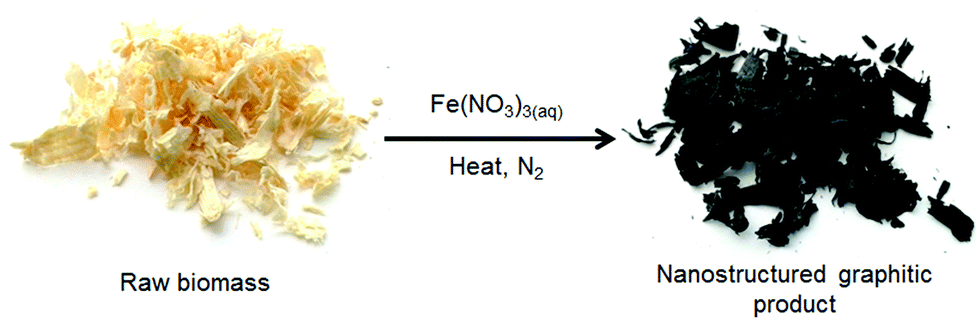 | ||
| Scheme 1 Outline of synthetic process. | ||
The method is also extended so that nanocomposites of Fe3C/nanotubes with metal oxides such as CaO or MgO can be synthesized in a single step. This simultaneous formation of oxide and carbide phases has previously only been achieved in sol–gel matrices.20 The theory behind the method is that different metals require different temperatures to achieve carbothermal reduction from an oxide to a carbide. Therefore by choosing an intermediate temperature it is possible to achieve selective carbothermal reduction of one metal while leaving the other one as an oxide.21 Metal oxide/graphitic carbon nanostructures are investigated in a wide range of applications from supercapacitors and batteries,22 to water treatment.23 The proof of concept of making mesoporous graphitic nanotubes alongside metal oxide nanoparticles in a single step therefore offers a promising future route to synthesizing functional materials for many technologies.
Results and discussion
Calcination of iron-treated sawdust to 800 °C under nitrogen yields black solids, with the individual wood flakes maintaining their shape but shrinking slightly (Fig. S1†). A 5 g sample of sawdust yields 1.5 g of graphitic carbon product. Powder X-ray diffraction (PXRD) of a crushed sample (Fig. 1a i) reveals sharp peaks for Fe3C (ICDD 00-035-0772) and broad peaks for graphite (ICDD 01-071-4630) as well as elemental iron. This does not change over a wide range of heating conditions or with scaling up to a volume of 1 L (Fig. S2†). A control sample, prepared without iron, shows only two very broad humps in the PXRD pattern, indicating amorphous carbon (Fig. 1a ii). Scanning electron microscopy (SEM) reveals that the microstructure of the woody biomass has been retained, giving an open framework of aligned, straight vessels ∼10 μm in diameter (Fig. 1b). SEM of the sample surface and fracture planes shows a complex, rough structure that penetrates through the solid vessel walls (Fig. 1c). After mild acid washing, the structure is more clearly revealed to consist of sinuous, intertwined tubules with a rounded (i.e. closed) ends (Fig. 1d and e). In some places, the tubes appear to have ruptured (Fig. 1f) – a similar observation has been made before when acid-washing graphite-coated Fe3C nanoparticles, producing ruptured graphitic ‘capsules’.24 Transmission electron microscopy (TEM) images show the internal diameter of the carbon tubes to be in the range 20–50 nm (Fig. 1g). The walls are comprised of graphitic layers that are aligned parallel to the long axis of the tubules as in multi-walled carbon nanotubes. | ||
| Fig. 1 (a) PXRD pattern for (i) iron-treated and (ii) control (no iron) carbonized biomass showing peaks for graphite and Fe3C as well as a minor Fe (*) phase. (b) Low-resolution SEM image of the sample showing woody microstructure and (c–e) high-resolution SEM images of the sample showing complex microstructure of nanotubes penetrating through the sample. (f) SEM image showing possible rupture sites of the graphitic tubules after acid washing to remove Fe3C. (g) TEM image of the porous graphitic structure with (inset) high resolution image of graphite sheets. (h) N2 sorption isotherm for graphitized sawdust. | ||
The nitrogen sorption isotherm for the material is type IV, with a hysteresis loop exhibiting both H2 and H3 characteristics (Fig. 1h). The shape of the isotherm is due to capillary condensation, indicating the presence of mesopores (IUPAC definition 2–50 nm), but the lack of an adsorption plateau at relative pressures close to unity suggests a pore size distribution extending into the macropore range (>50 nm). Hysteresis loops that close sharply at p/p0 = 0.42, are normally attributed to the presence of ‘ink-bottle’ pores with a narrow entrance and larger internal cavity which is emptied by cavitation. Hence, no information regarding the size of the pore opening can be obtained from these isotherms. This observation could be consistent with SEM observations of irregular nanotube structures with rounded or closed ends and small rupture sites. Analysis of the data using Brunauer–Emmett–Teller (BET) theory gives a surface area of 220 m2 g−1.
Given the lack of graphite in the control (0% Fe) sample, it is clear that iron is responsible for formation of the graphitic nanotubes. TEM images of the samples show the dark catalyst particles, which have lattice planes that can be indexed to iron carbide (Fig. S3†). This phase (Fe3C) has been proposed as the active catalyst in graphite formation in similar systems. In many of the images, these catalyst particles appear to have been frozen while flowing through the tubules (Fig. 2a). There are also many examples of ‘bamboo-like’ nanotubes, where bridging layers of graphite occur at intervals along the tubes (Fig. 2b). Both straight and bamboo-like carbon nanotubes are observed in vapour-decomposition synthesis techniques. However, it seems unlikely that a vapour decomposition step is occurring in this system since the tubes are observed to penetrate through the whole sample, rather than protrude from the surface. Given the ‘flowing’ appearance of many of the particles, it seems likely that the solid amorphous carbon dissolves directly into a liquid Fe3C catalyst particle. This is consistent with previous in situ observations of iron-catalyzed graphitization of pure cellulose. The bamboo-like structures possibly then form as the moving catalyst particle slows or stops at interfaces between amorphous carbon of different density or structure, allowing graphite sheets to form behind the catalyst. This seems a reasonable conclusion, since it is known that regions of different biopolymers in biomass do carbonize and shrink at different rates during pyrolysis.25 Alternatively, the rings crossing some of the tubes in the TEM images could just represent bends and kinks in the tube, viewed as a silhouette in the 2D TEM image. The reason for both ‘straight’ and ‘kinked’ tubes appearing in this sample could be that both Fe3C and Fe catalyze graphitization and form different types of nanotube. The different phases have previously been proposed to catalyze these different types of nanotube in CVD (chemical vapour deposition) synthesis26 and both exist in the PXRD pattern. However, in the extremely complex biomass system, there are many other possible influences such as regions of different carbon density (from lignin and cellulose) or the presence of trace inorganic species that may dope into the iron phase.
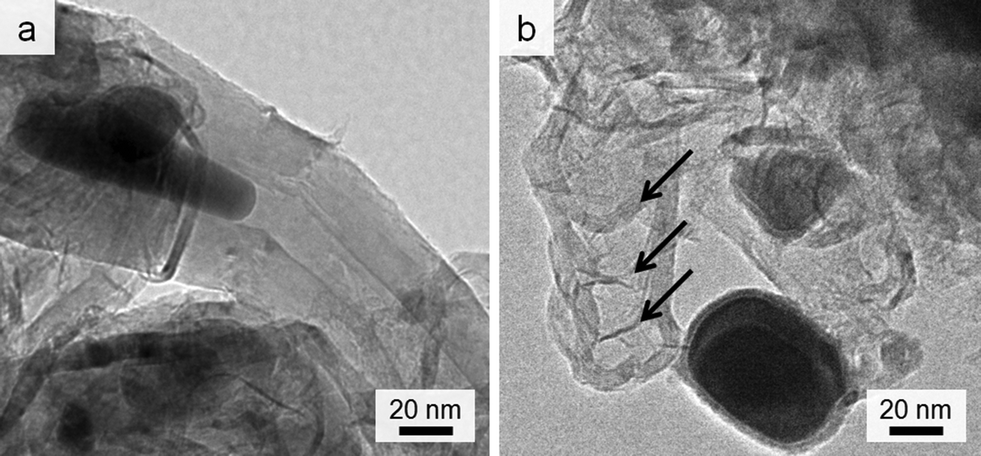 | ||
| Fig. 2 TEM images showing (a) catalyst particle trapped while ‘flowing’ inside nanotube and (b) possible ‘bamboo’-like nanotube. | ||
Interestingly, the amount of graphitization does not appear to diminish with a substantial decrease in iron content. Fig. 3a shows that a reduction of the iron concentration to 10% of the original value results in broader Fe3C peaks of much lower intensity but with the graphite peak intensity maintained. This is more clearly shown in a plot of the relative intensities of the main graphite and Fe3C peaks (Fig. 3a inset). With the graphite peak as the point of maximum intensity, the relative intensity of the main Fe3C peak decreases from ∼100% to ∼40% as the iron concentration reduces from 100% to 10%. Nitrogen porosimetry of the samples shows a change in the sorption isotherm (Fig. 3b), in particular the appearance of a plateau at high p/p0 as the iron content is reduced, giving a more standard type IV isotherm with H2 hysteresis. Type H3 hysteresis can be associated with plate-like aggregates and slit-shaped pores. However, in these systems, microscopy images show very similar structures across the range of different iron concentrations. i.e. the same catalytic graphitization and formation of nanotubes. This would suggest that the change in isotherm shape with decreasing iron content is more likely to be due to a reduction in the number of macropores and a restriction of pore size to the mesopore range. TEM images of two samples with different iron content are shown in Fig. 3c and 3d and clearly indicate a significant difference in Fe3C particle size. Not only does this reflect the broadened peaks in the XRD pattern, but if smaller Fe3C catalyst particles result in smaller graphitic nanotubes, this may explain the apparent change to a smaller pore size distribution indicated by the sorption isotherms. BET analysis shows a corresponding increase in surface area with decreasing iron content (220 m2 g−1, 280 m2 g−1, 270 m2 g−1 and 340 m2 g−1 for 100%, 75%, 50% and 25% iron respectively).
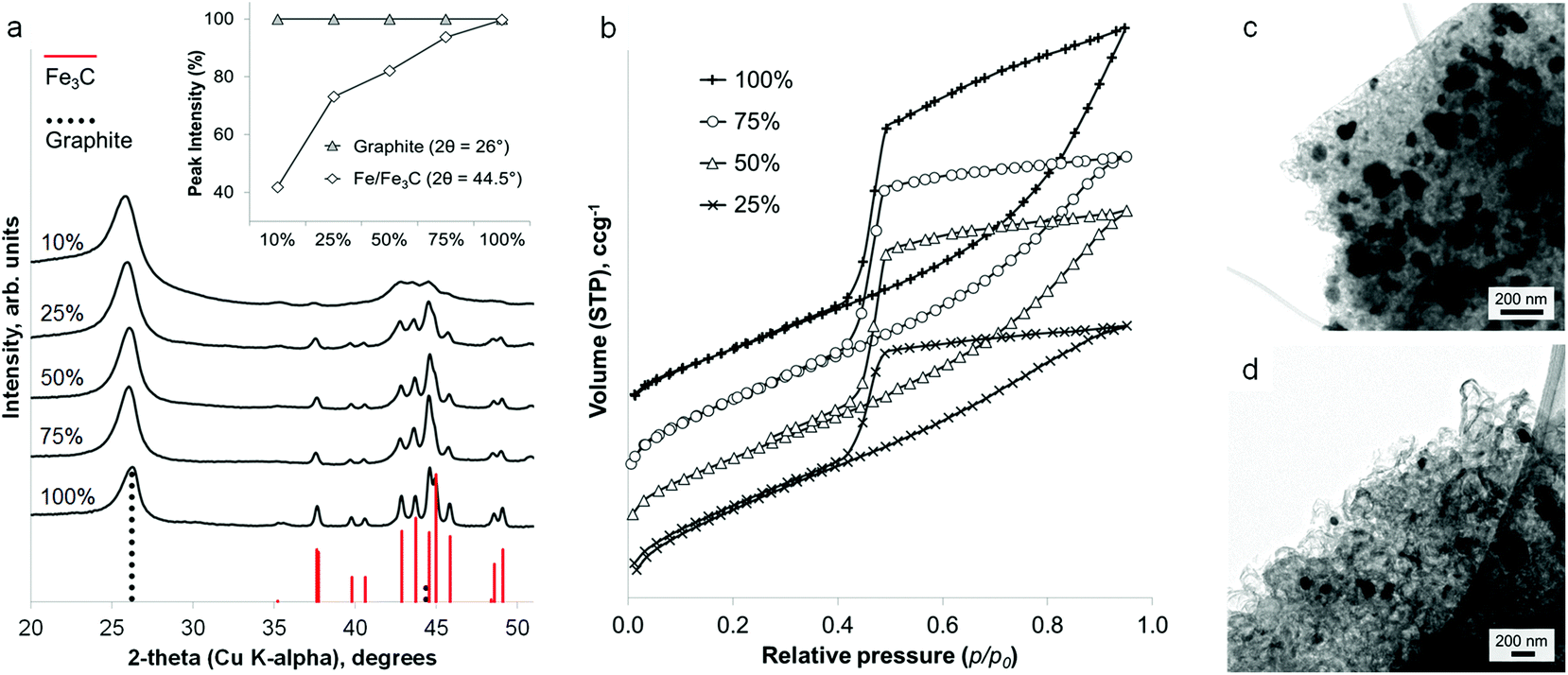 | ||
| Fig. 3 (a) PXRD patterns and (b) N2 sorption isotherms (vertically offset) for sawdust carbonized with a range of iron concentrations (values in mol%), inset plot of relative intensities of main Fe/Fe3C and graphite peaks with changing iron:biomass ratio. Low magnification TEM images of (c) the standard (100 mol%) carbonized Fe-sawdust sample and (d) a sample with iron concentration reduced to 50 mol%. | ||
The iron content of the samples can be removed under mildly acidic conditions (0.1 M HCl) to leave a powder of porous graphitic carbon. PXRD shows the disappearance of the iron carbide peaks, with broad peaks for graphite remaining in the sample (Fig. 4a). At a high iron concentration, some very low intensity peaks remain that can be attributed to a trace of Fe3C. This is probably due to Fe3C particles coated in layers of graphite and embedded deep in the sample. The lack of any peaks for iron oxide phases supports the conclusion of protected Fe3C particles. Porosimetry data shows the effect of removing the iron carbide particles to be very small (Fig. 4b and c), with a slight increase in BET surface area for each sample (220 m2 g−1 to 260 m2 g−1 for 100% Fe and 270 m2 g−1 to 280 m2 g−1 for 50% Fe). This is reasonable if most of the porosity has been generated by moving Fe3C nanoparticles/droplets.
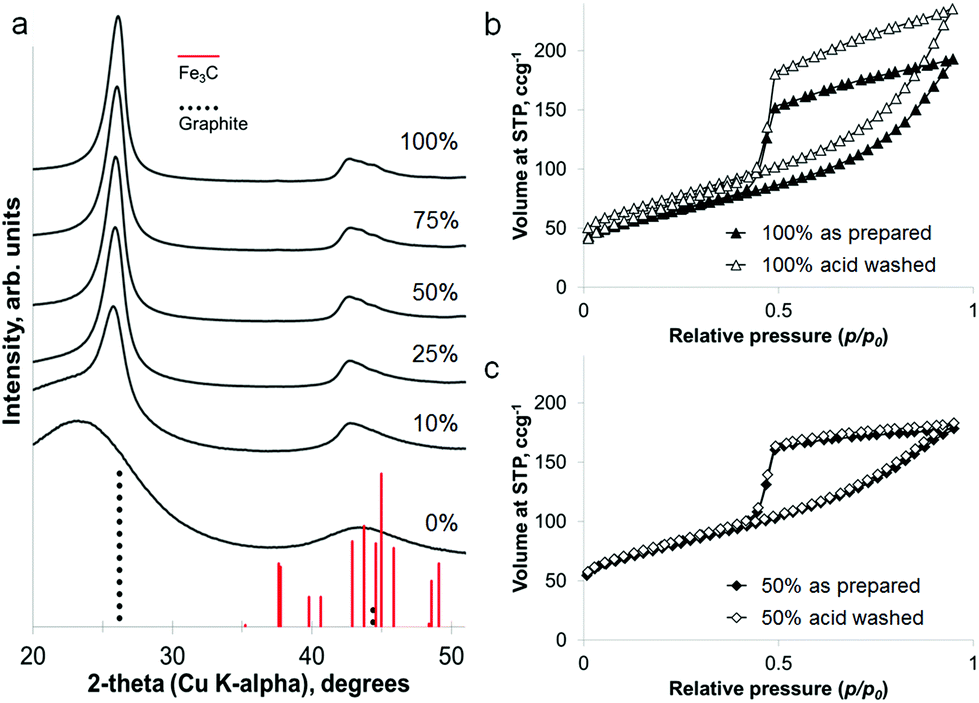 | ||
| Fig. 4 (a) PXRD patterns of graphitized samples prepared at various iron contents, after acid washing. (b) and (c) Nitrogen sorption isotherms showing a very low change in porosity after acid washing for samples prepared at two different iron concentrations. | ||
Remarkably, it is possible to achieve a mixture of crystalline phases in these samples, combining the iron carbide with a separate oxide phase in a single synthetic step, from a homogeneous mixture of metal nitrates. Samples of sawdust were soaked in a solution of iron nitrate mixed with either calcium nitrate or magnesium nitrate, followed by heating under nitrogen to 800 °C. PXRD clearly shows a mixture of Fe3C and graphite with either CaO (ICDD 04-002-6758) or MgO (ICDD 01-089-7746). These carbide/oxide composites can be achieved for a wide range of different Fe:Ca (Fig. 5a) and Fe:Mg (Fig. S4†) molar ratios. The oxide particles can be seen with TEM to be distinct from the Fe3C particles (Fig. 5b), having an approximately cubic geometry. Importantly, the presence of the oxide phase does not significantly hinder graphitization. A direct comparison of samples of the same Fe:biomass ratio, with and without Mg or Ca, shows approximately the same graphite:Fe3C peak ratio in the PXRD pattern (Fig. S5†). This result is mirrored in Raman spectra of the samples, which show almost identical D and G bands for all of the Fe-containing samples across a range of Fe:Ca molar ratios (Fig. 5c). Only the control (100 mol% Ca) sample shows the broad D and G bands and smaller D:G peak height ratio indicating carbon of much lower crystallinity and order. Nitrogen porosimetry does show a smaller gas uptake for calcium-containing samples (Fig. 5d). However, this is probably due to the large mass of non-adsorbing calcium oxide. This can be confirmed by acid washing samples of Fe3C/graphite and Fe3C/CaO/graphite (from the same Fe:biomass ratio). This results in very similar isotherms (Fig. 4e) and comparable BET surface areas (280 m2 g−1 and 240 m2 g−1, respectively), supporting the conclusion that the presence of the alkaline earth metal does not inhibit the formation of nanotubes through Fe-catalyzed graphitization. Acid washing in these cases (MgO/CaO) also removes the oxide phase, since alkaline earth oxides are highly soluble in dilute acid. However, this method could readily be extended to many other metal oxide materials in combination with Fe3C/carbon. In the case of non-soluble oxides (e.g. TiO2), this would offer a quick route to TiO2/graphite nanostructures by acid washing out just the soluble Fe3C phase. A simple synthesis of such oxide/graphite nanostructures may find many applications e.g. as electrode materials in lithium ion batteries.27
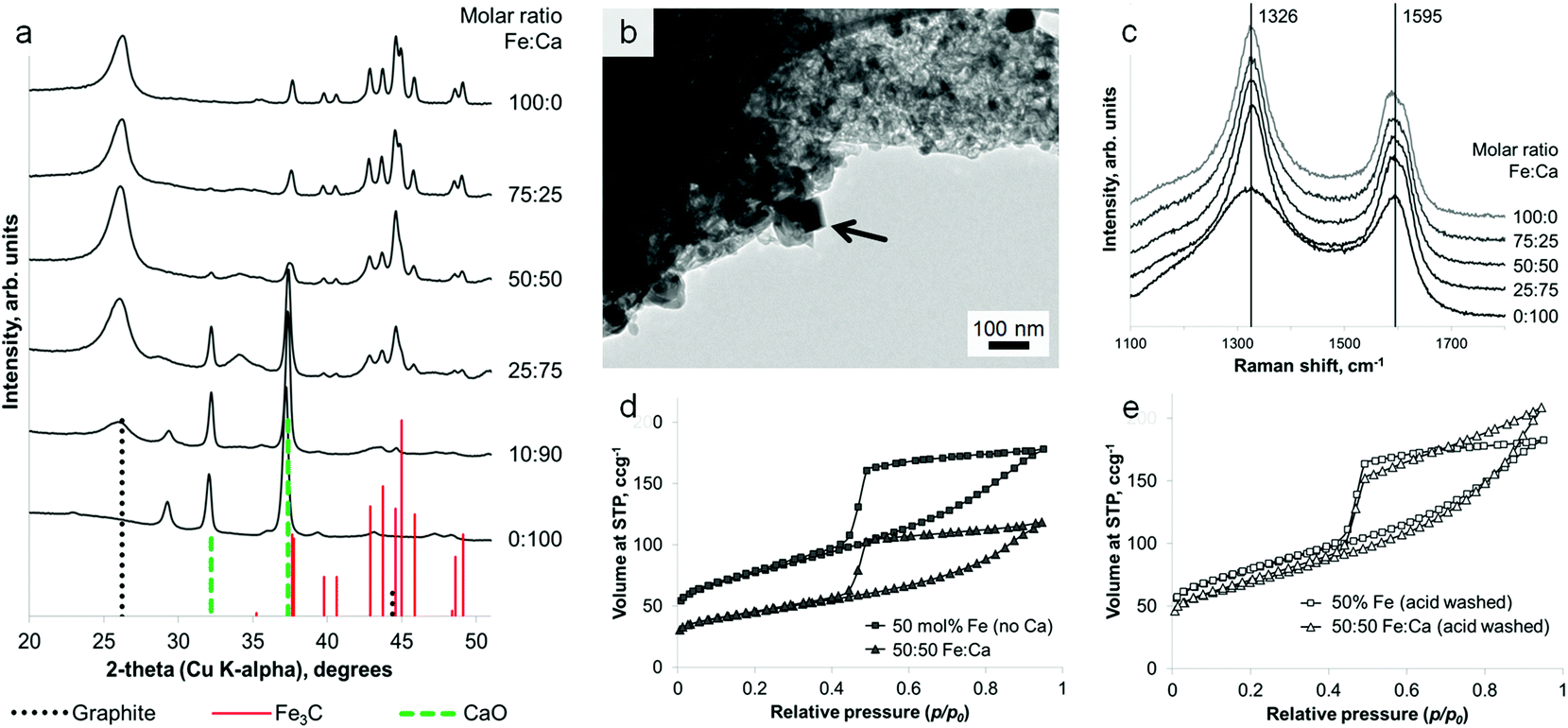 | ||
| Fig. 5 (a) PXRD patterns for a range of graphite/Fe3C/CaO nanocomposites prepared at different Fe:Ca ratios from sawdust. (b) TEM image of a sample prepared from a 50:50 molar ratio of Fe:Ca with arrow showing distinct oxide particle. (c) Raman spectroscopy showing characteristic D and G bands for samples with a range of Fe:Ca molar ratios. Nitrogen sorption isotherms for samples of Fe/sawdust and Fe/Ca/sawdust (d) before and (e) after acid washing. | ||
Conclusions
In summary, we have shown a facile synthesis of nanostructured tubular graphitic carbon in a single step from untreated lignocellulosic biomass. Sawdust is soaked in iron nitrate solution and due to the hydrophilic functional groups of the biomass, the Fe3+ cations can be readily and evenly adsorbed across the surface.28 Heating under nitrogen then decomposes the biomass to amorphous carbon and also drives in situ formation of iron carbide nanoparticles. Both straight and bamboo-like multiwalled graphitic nanotubes are formed by the catalyst particles etching through the biomass, possibly involving dual catalysts of Fe and Fe3C. Varying metal:biomass ratio is a simple route to changing the nanotube diameter.
It is also possible to generate metal oxide (CaO and MgO) nanoparticles alongside the graphite during the single heating step. This is particularly remarkable, since the precursor is a homogeneous mixture of Fe and either Ca or Mg nitrates which then phase separate on heating. Even reducing the Fe:Ca molar ratio to 10:90 still results in separate CaO and Fe3C and substantial graphitization, despite the fact that Fe-doped CaCO3 and CaO are both known phases (e.g. Srebrodolskite Ca2Fe2O5). This in situ phase separation has been demonstrated by us in sol–gel systems20 but to the best of our knowledge it is the first example of simultaneous oxide/carbide formation on a solid template. In our previous work, we showed that Fe3C nanoparticles could be synthesized from a sol–gel alongside a wide range of oxides such as TiO2 and CeO2. The ability to produce Fe3C-catalyzed graphite from sawdust alongside metal oxide nanoparticles therefore offers an exciting and simple new route to graphite/oxide nanocomposites. Such materials are of interest in a wide range of technologies such as battery electrodes, electrocatalysts and water treatment.
Without a full life-cycle analysis, it is not possible to conclude whether this method is more ‘Green’ than the commercial synthesis of carbon nanotubes. There may also be ways to improve the method, for example reducing energy requirements by using microwave heating. However, the use of aqueous precursors and a fast, single-step heating process to convert raw biomass to a desirable porous graphitic carbon certainly addresses a global challenge of simple routes to new materials.29 Considering the simplicity and scalability of the method, we anticipate that it will find widespread application.
Experimental
Samples were prepared by mixing untreated softwood sawdust (5 g) with iron nitrate solution (20 ml), drying at 80 °C in air and carbonizing in a muffle furnace under a flow of N2 at 5 °C min−1 to 800 °C with 1 hour hold. For the standard sample (100% Fe), 0.005 mol Fe was used in the form of 10% (by mass) aqueous Fe(NO3)3·9H2O solution. For lower iron concentrations, the iron nitrate stock solution was diluted to keep the liquid volume constant. Full details of all samples are listed in Table 1.| Mol% of full iron concentration | Moles of iron | Volume of 10% iron nitrate solution (ml) | Volume of water (ml) |
|---|---|---|---|
| 100% | 0.005 | 20 | 0 |
| 75% | 0.0038 | 15 | 5 |
| 50% | 0.0025 | 10 | 10 |
| 25% | 0.0013 | 5 | 15 |
| 10% | 0.0005 | 2 | 18 |
| 0% | 0 | 0 | 20 |
For Fe/Ca and Fe/Mg samples, aqueous metal nitrate solutions (10% by mass) were premixed then added to sawdust, before drying and carbonizing as above. The volume of the mixed nitrate solution was again kept at 20 ml to ensure complete homogeneous coating of the sawdust. The total moles of metal was also kept constant at 0.005 moles. The full details of each sample are listed in Table S1.†
Acid washing was performed by sonicating samples (1 g) for 1 hour in 1 M HCl (10 ml), followed by stirring for a further 23 hours then washing with DI water and ethanol and finally drying in air at room temperature.
Metal salts including Fe(NO3)3·9H2O, Ca(NO3)2·4H2O and Mg(NO3)2·6H2O were purchased from Sigma-Aldrich and used as received. Sawdust was sourced from a pet shop. X-ray diffraction experiments were performed on a Panalytical Empyrean diffractometer equipped with a Pixcel 2D detector. Samples were ground to fine powders. SEM images were recorded using a JEOL JSM-7001F field emission scanning electron microscope Samples were prepared by depositing on conductive carbon substrates and coating with Pt/Pd. TEM images were recorded using a JEOL JEM-2100F transmission electron microscope equipped with CCD camera. Samples were prepared by dispersing in ethanol and dropping onto holey carbon-coated copper grids. Nitrogen porosimetry was performed using a Quantachrome Nova-e porosimeter. Raman microscopy was performed using a Renishaw Raman Microscope.
Acknowledgements
The authors acknowledge the University of Birmingham and EU (SusNano) for funding. Many thanks also to Martin Hollamby and Jens Weber for helpful discussions.Notes and references
- C. Merlet, B. Rotenberg, P. A. Madden, P.-L. Taberna, P. Simon, Y. Gogotsi and M. Salanne, Nat. Mater., 2012, 11, 306–310 CrossRef CAS PubMed.
- Y. Zhang, A. Wang and T. Zhang, Chem. Commun., 2010, 46, 862–864 RSC.
- M. S. Mauter and M. Elimelech, Environ. Sci. Technol., 2008, 42, 5843–5859 CrossRef CAS.
- A. V. Herrera-Herrera, M. A. Gonzalez-Cubelo, J. Hernandez-Borges and M. A. Rodriguez-Delgado, Anal. Chim. Acta, 2012, 734, 1–30 CrossRef CAS PubMed.
- M. M. Titirici, A. Thomas and M. Antonietti, New J. Chem., 2007, 31, 787–789 RSC.
- G. Centi and S. Perathoner, Catal. Today, 2010, 150, 151–162 CrossRef CAS PubMed.
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS PubMed.
- R. Van Noorden, Nature, 2011, 469, 14–16 CrossRef CAS PubMed.
- L. A. Pfaltzgraff, M. De bruyn, E. C. Cooper, V. Budarin and J. H. Clark, Green Chem., 2013, 15, 307–314 RSC.
- V. Budarin, J. H. Clark, J. J. E. Hardy, R. Luque, K. Milkowski, S. J. Tavener and A. J. Wilson, Angew. Chem., Int. Ed., 2006, 118, 3866–3870 CrossRef.
- B. Hu, K. Wang, L. Wu, S. Yu, M. Antonietti and M. M. Titirici, Adv. Mater., 2010, 22, 813–828 CrossRef CAS PubMed.
- D. Fengel and G. Wegener, in Wood: chemistry, ultrastructure, reactions, Walter de Gruyter & Co., Berlin, 1989, pp. 26–27 Search PubMed.
- A. Rinaldi, J.-P. Tessonnier, M. E. Schuster, R. Blume, F. Girgsdies, Q. Zhang, T. Jacob, S. B. Abd Hamid, D. S. Su and R. Schlögl, Angew. Chem., Int. Ed., 2011, 50, 3313–3317 CrossRef CAS PubMed.
- H. Yoshida, S. Takeda, T. Uchiyama, H. Kohno and Y. Homma, Nano Lett., 2008, 8, 2082–2086 CrossRef CAS PubMed.
- A.-H. Lu, W.-C. Li, E.-L. Salabas, B. Spliethoff and F. Schüth, Chem. Mater., 2006, 18, 2086–2094 CrossRef CAS.
- Z. Schnepp, S. C. Wimbush, C. Giordano and M. Antonietti, Chem. Mater., 2010, 22, 5340–5344 CrossRef CAS.
- S. Glatzel, Z. Schnepp and C. Giordano, Angew. Chem., Int. Ed., 2013, 52, 2355 CrossRef CAS PubMed.
- M. Sevilla and A. B. Fuertes, Chem. Phys. Lett., 2010, 490, 63–68 CrossRef CAS PubMed.
- The Forestry Commission, Forestry Statistics 2014, http://www.forestry.gov.uk/website/forstats2014a.nsf/LUContents/824A4E0E2DDEDC858025731B00541EFF (accessed September 2014).
- Z. Schnepp, M. Hollamby, M. Tanaka, Y. Matsushita, Y. Xu and Y. Sakka, Chem. Commun., 2014, 50, 5364–5366 RSC.
- Z. Schnepp, M. J. Hollamby, M. Tanaka, Y. Matsushita, Y. Katsuya and Y. Sakka, Sci. Tech. Adv. Mater., 2012, 13, 035001 CrossRef.
- M. Zhi, C. Xiang, J. Li, M. Li and N. Wu, Nanoscale, 2013, 5, 72–88 RSC.
- V. Chandra, J. Park, Y. Chun, J. W. Lee, I. Hwang and K. S. Kim, ACS Nano, 2010, 4, 3979–3986 CrossRef CAS PubMed.
- Z. Schnepp, Y. Zhang, M. J. Hollamby, B. R. Pauw, M. Tanaka, Y. Matsushita and Y. Sakka, J. Mater. Chem. A, 2013, 1, 13576–13581 CAS.
- E. Senoz and R. P. Wool, J. Appl. Polym. Sci., 2010, 118, 1752–1765 CAS.
- Z. He, J.-L. Maurice, A. Gohier, C. S. Lee, D. Pribat and C. S. Cojocaru, Chem. Mater., 2011, 23, 5379–5387 CrossRef CAS.
- R. Mo, Z. Lei, K. Sun and D. Rooney, Adv. Mater., 2014, 26, 2084–2088 CrossRef CAS PubMed.
- J. Huang and T. Kunitake, J. Am. Chem. Soc., 2003, 125, 11834–11835 CrossRef CAS PubMed.
- C. Stanier and J. Hutchinson, A Sustainable Global Society (White paper), Royal Society of Chemistry, London, 2011 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full sample compositions as well as supplementary images, XRD and porosimetry data. See DOI: 10.1039/c4gc01673d |
| This journal is © The Royal Society of Chemistry 2015 |