The use of environmental metrics to evaluate green chemistry improvements to the synthesis of (S,S)-reboxetine succinate
Georges
Assaf
,
Graham
Checksfield
,
Doug
Critcher
,
Peter J.
Dunn
,
Stuart
Field
,
Laurence J.
Harris†
*,
Roger M.
Howard
,
Gemma
Scotney
,
Adam
Scott
,
Suju
Mathew
,
Geoffrey M. H.
Walker†
and
Alexander
Wilder
Chemical Research and Development, PharmaTherapeutics Pharmaceutical Sciences, Pfizer Worldwide Research and Development, Sandwich Laboratories, Ramsgate Road, Sandwich, Kent CT13 9NJ
First published on 31st October 2011
Abstract
The Pfizer Green Chemistry metrics program is described and exemplified with a case history involving the synthesis of (S,S)-reboxetine succinate. The initial route used a classical resolution approach and generated high levels of waste. This route was replaced by an enantiospecific synthesis which used Sharpless epoxidation chemistry, an enzymatic process to selectively protect a primary alcohol and a new efficient method of chiral morpholine construction as key steps. These improvements reduced the levels of waste produced by the synthesis by more than 90%. Detailed metrics starting from a common starting material (trans-cinnamyl alcohol) for all routes of synthesis are presented.
Introduction
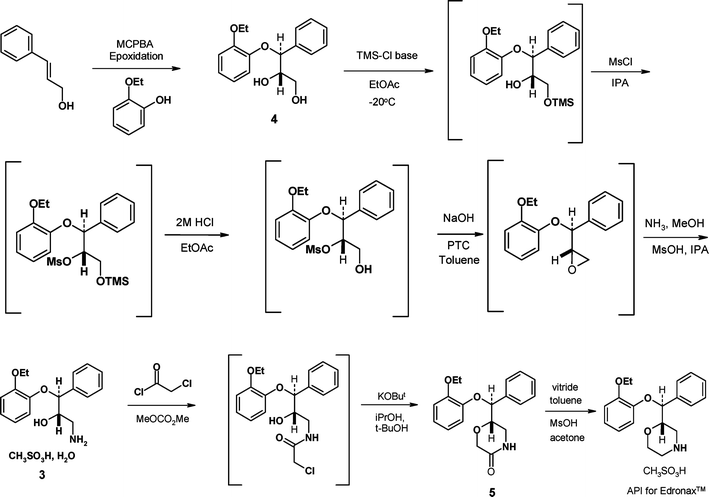
(±)-Reboxetine mesylate 1 is a selective norepinephrine uptake (NRI) inhibitor1 which is marketed as the racemate, under the trade name Edronax™, for the treatment of depression, in more than 60 countries. The (S,S)-enantiomer is significantly more active than the racemate in a number of studies2 and has undergone clinical evaluation as the succinate salt 2 for a number of indications in the pain therapeutic area. The chemical development program for (S,S)-reboxetine succinate was very much influenced by the unusual history of the compound. Pharmacia had manufactured 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 kg of (±)-reboxetine mesylate or late stage intermediates, such as 3 in readiness for a US approval of Edronax™. When this approval did not occur it was clear that only a small proportion of the stock would be required to support the other markets and the remainder was “written off” and made available to the research organisation. Not surprisingly the large stock pile of material was initially used as a starting point for the synthesis of (S,S)-reboxetine succinate, the single enantiomer which was being developed for new indications in the pain area. The development work was being carried out at a time when the discipline of Green Chemistry was becoming more established in the pharmaceutical industry.3 As such, in this article we focus on the environmental (and cost) improvements that were achieved through process development and synthetic route design coupled with rigorous measurement and analysis of Green Chemistry metrics. The synthesis of (±)-reboxetine mesylate is shown in Scheme 1 and its conversion to (S,S)-reboxetine succinate shown in Scheme 2.
000 kg of (±)-reboxetine mesylate or late stage intermediates, such as 3 in readiness for a US approval of Edronax™. When this approval did not occur it was clear that only a small proportion of the stock would be required to support the other markets and the remainder was “written off” and made available to the research organisation. Not surprisingly the large stock pile of material was initially used as a starting point for the synthesis of (S,S)-reboxetine succinate, the single enantiomer which was being developed for new indications in the pain area. The development work was being carried out at a time when the discipline of Green Chemistry was becoming more established in the pharmaceutical industry.3 As such, in this article we focus on the environmental (and cost) improvements that were achieved through process development and synthetic route design coupled with rigorous measurement and analysis of Green Chemistry metrics. The synthesis of (±)-reboxetine mesylate is shown in Scheme 1 and its conversion to (S,S)-reboxetine succinate shown in Scheme 2.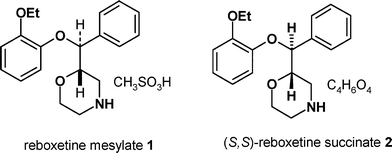
 | ||
| Scheme 1 The synthesis of (±)-reboxetine mesylate,4 the Active Pharmaceutical Ingredient (API) for Edronax™. | ||
 | ||
| Scheme 2 The conversion of (±)-reboxetine mesylate to (S,S)-reboxetine succinate. | ||
Pharmacia intended to commercialise this synthesis based on a very fast development program and a desire to use up the existing stocks and in late 2003 the chemistry was successfully transferred to a manufacturing site. Following the takeover of Pharmacia by Pfizer, the program was reassessed both from a clinical and chemistry point of view and this led to an evaluation of new routes and processes. From a Green Chemistry perspective the chemistry outlined in Schemes 1 and 2 had a number of disadvantages:
• The use of a late stage resolution meant that by definition more than half of the materials used to prepare (±)-reboxetine were wasted.
• The isolation of the mesylate salt (Scheme 1) and breaking that salt back to reboxetine free base (Scheme 2) was clearly historical and produced large quantities of unnecessary chemical waste.
• Differentiation of the two hydroxyl groups in compound 4 required a protection and deprotection strategy which was sub-optimal, as the initial TMS protection was poorly selective for the primary alcohol.
• Chloroacetyl chloride was used as a two carbon fragment to build up the morpholine ring, however this produced intermediate 5 at the wrong oxidation level so necessitating a reduction reaction with a strong metal hydride reagent (Vitride™).
• Dichloromethane was used as a solvent in the processes to break both the mesylate and mandelate salts (Scheme 2).
• In total more than 1098 kg of solvent, 77 kg of reagents and 770 kg of water was used to convert the (±)-diol 4 into (S,S)-reboxetine succinate.
Green Chemistry metrics program
Pfizer started to collect Green Chemistry metrics more than a decade ago5–7 and by 2004 had identified and agreed a global set of metrics to be collected at all of its research sites. This global set of metrics included organic solvent use, reagent use, water use, overall percentage yield, number of synthetic steps, number of catalytic steps, number of isolations, number of solvents used and the mass of solid waste generated. In the early days there was a tendency to collect metrics on projects that showed a story of success but in 2006 the company moved to a position of capturing metrics for all of its development compounds. In 2008 reduction goals were set for the first time (for example a 20% reduction in organic solvent use across the development portfolio by 2012). To help achieve these goals a number of strategies were put in place.• The avoidance of late stage resolutions – An analysis of the portfolio showed that having just one late stage resolution in the portfolio (classical, chromatographic or biocatalytic) would mean that the reduction target would not be met.
• To increase the number of genuine telescoped steps, where the solvent for one reaction is also used for the next reaction, with little or no additional processing.
• To increase the number of reactions carried out in water (especially reactions catalysed by enzymes).
• To run reactions with low heats of reaction in more concentrated solution or suspension after detailed discussion with the process safety laboratory. Process chemists in pharmaceutical and fine chemical companies routinely obtain heat of reaction data for safety reasons before scale up, but this data is not always used to help with efficient process design.
• To introduce more efficient intermediate isolations (e.g. by direct isolation processes).8
• To use technologies to reduce solvent usage (e.g. using solubility data from automated solubility measurements in process design, using centrifugal extraction when appropriate and if solvent replacement (striping one solvent and replacing with another) is required to use the Pfizer distillation optimisation tool.9
• Engage the manufacturing group early in solvent recovery plans especially for expensive solvents with a high life cycle impact such as THF and 2-MeTHF.
In general these strategies were successful and the goal of reducing solvent use by 20% across the development portfolio was achieved two years early. For (S,S)-reboxetine succinate, the waste levels produced via the Edronax™ route (Schemes 1 and 2) were more than 20 times the 2012 target, so there was much work to be done.
Improving the throughput and minimising the environmental impact of the Edronax process
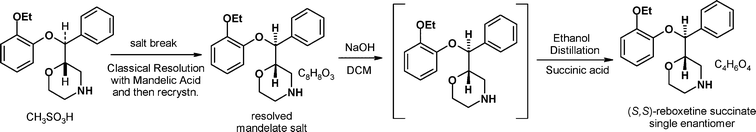
One of the obvious issues with using (±)-reboxetine mesylate 1 as an isolated intermediate en-route to (S,S)-reboxetine was that the end game of the synthesis became very protracted. All of the carbon framework of the API was established after the metal hydride reduction step but this still required mesylate salt formation, resolution, recrystallisation (to upgrade the mandelate salt enantiomeric excess) and finally counter ion exchange to afford the succinate salt of (S,S)-reboxetine. This inefficiency (overall yield 17.8% from 3) could be improved upon by moving the resolution step much earlier in the synthesis. We also envisaged being able to make improvements to the metal hydride reduction step to reduce both the reagent stoichiometry and the amount of solid waste generated by the work-up procedure. We also wanted to improve on the conversion and in situ quality generated in the ring closure reaction to avoid the need for carbon treatments that purged significant process related impurities and color generated in the Pharmacia Edronax™ process.Scheme 3 shows how this early resolution strategy was successfully implemented at pilot scale.
 | ||
| Scheme 3 The Pfizer early resolution route to (S,S)-reboxetine succinate. | ||
Key features of this new process that made a significant contribution to the overall reduction in the amount of materials used to prepare the API were:
• The (±)-amino alcohol 3 was resolved in the first step using a “non-classical” resolution process to establish the absolute stereochemistry.
• The (±)-mesylate salt was no longer generated or isolated as a process intermediate and this simplified the overall synthesis in terms of number of steps and hence reduced waste generation.
• The N-acylation/cyclisation sequence to generate 8 was improved significantly. The use of tert-butanol/tert-butoxide for the ring closing step allowed the chemistry to be run safely in a semi-batch process. Having more control over the addition rate minimised the heat generated and gave a superior in situ impurity profile (>97% vs. 65%) meaning that activated carbon treatment was no longer required to purge process related impurities.
• In the reduction of 8, Vitride™ (MW = 202.16) was replaced by the more atom economical lithium aluminium hydride (LAH, MW = 37.95) and the stoichiometry was reduced from 5 to 1 molar equivalents. This resulted in a >95% reduction in the mass of reducing agent charged to the vessel when carrying out this reaction.
• The work-up of the reduction reaction was improved by implementing an aqueous triethanolamine quench followed by an aqueous NaOH extraction of the complexed aluminium salts, which completely eliminated solid waste from the process. As an additional benefit, the reduction in molar equivalents of metal hydride reagent reduced the energy evolved by the quench, making it much safer to scale-up.
In the resolution step the chiral acid used, (S)-camphanic acid forms an insoluble salt with the undesired enantiomer. After filtration, the resultant toluene solution of the desired camphanate salt 6 was then used to make an insoluble benzoate salt which upgrades in chiral purity from 80% e.e. to 99% e.e. upon isolation. A classical resolution would have crystallised the desired enantiomer and this could be achieved by using (R)-camphanic acid but due to its much higher cost and limited availability on scale the non-classical process was preferred. A great feature of this resolution process was the use of a single organic solvent (toluene) all the way through the process, which made it operationally simple but also made full recovery of this solvent a viable option. On laboratory scale we also identified that the (S)-camphanic acid could be recycled and reused as the resolving agent in subsequent batches.
The overall yield at pilot plant scale for this route was 24.6% from 3 (versus 17.8% for the late resolution process) and all chlorinated solvents were removed from the manufacturing process that was used to generate clinical (phase 3) quality API. Although this represented a significant improvement this route was still limited by the absolute stereochemistry being established by a resolution step that has a maximum theoretical yield of 50%.
A step change in efficiency; changes to the bond forming chemistry
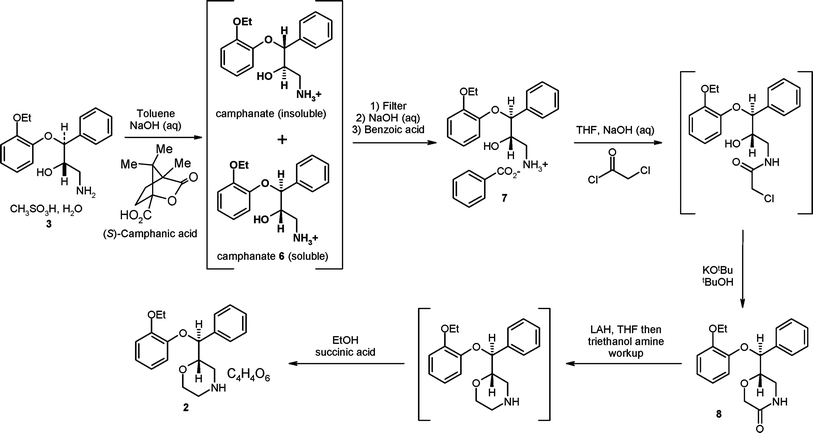
Although good progress had been made with the introduction of the early resolution route it was felt that changes in the bond forming chemistry were required to take the project to the next level of environmental performance and further reduce the API $/kg cost. The team focussed on; establishing the absolute stereochemistry of the molecule via asymmetric synthesis, the protecting group strategy and the construction of the morpholine ring without the need to adjust the oxidation state. We envisaged using the common cinnamyl alcohol as the starting point of the synthetic sequence as it was known to be a good substrate for the Sharpless asymmetric epoxidation reaction (SAE)10 and the subsequent ring opening of the epoxide with 2-ethoxyphenol (to generate 9) had been demonstrated in earlier work by the Pharmacia team.11Scheme 4 shows the route that was developed and intended for use in commercial manufacturing.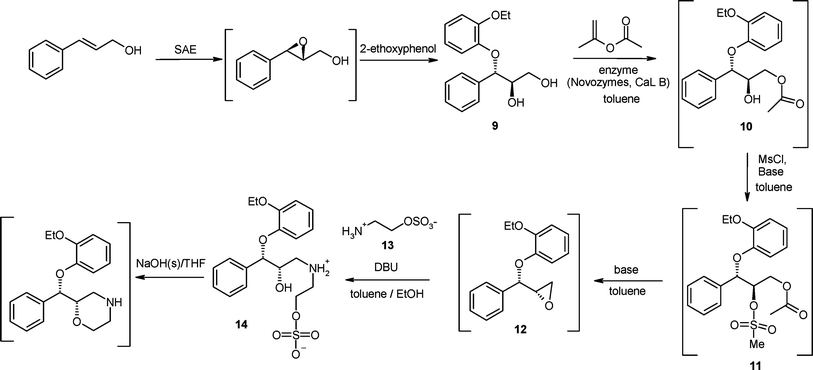 | ||
| Scheme 4 The Pfizer asymmetric synthesis for (S,S)-reboxetine intended for commercialisation. | ||
Once the key chiral diol intermediate 9 had been secured we turned our attention to the protecting group strategy. The Edronax™ process utilised trimethylsilyl (TMS) protection at −20 °C to selectively protect the primary alcohol which was followed by mesylation of the secondary alcohol, acidic cleavage of the O-TMS bond and then base-induced epoxidation under phase transfer conditions, (Scheme 1). The major issues with this chemistry are related to TMS protection step which required cryogenic reaction conditions. In addition TMS migration to the secondary alcohol led to a loss of selectivity and generation of impurities in the downstream steps. In addition it was found that the chiral diol 9 was much higher melting (by some 28 °C)4,11 and much less soluble than the racemic diol 4, and this necessitated the whole sequence was run in a chlorinated solvent (DCM).11 To address these issues we evaluated alternative protecting groups. An initial investigation of chemical methods (e.g.acyl chlorides) lacked the required selectivity, however enzymatic methods proved to be successful in selectively acylating the primary alcohol at 30 °C using iso-propenyl acetate as the acyl source to deliver protected intermediate 10. A nice advantage of this approach was that the by-product is acetone. Activation of the secondary alcohol then afforded the mesylate 11 which was subjected to a one pot, base mediated acetate cleavage and subsequent ring closure to generate the chiral epoxide 12.
The major innovative feature of the chemistry depicted in Scheme 4 was a redesign of the morpholine ring construction. From the Edronax™ process we knew that the epoxide 12 would undergo ring opening at its terminus with ammonia. We hoped to exploit this reactivity towards amines in a ring opening reaction with a bifunctional reagent; that would deliver the two carbon atoms required to complete the skeleton of the API but also embed a leaving group into the molecule to set up a system that would undergo a ring closure reaction to generate the morpholine.
The commercial process (Scheme 4) utilises a low toxicity bi-functional reagent, 2-aminoethylhydrogensulfate 13 (tested AMES negative) that cleanly opens the epoxide 12 in the presence of DBU to generate zwitterion 14, which was insoluble in the toluene/ethanol reaction solvents. The product of this reaction could be filtered and dried to afford an intermediate of very high purity. Finally we found that by careful selection of the reaction conditions (base, solvent, temperature) the zwitterion 14 would cyclise to (S,S)-reboxetine free base via a highly selective and high yielding intramolecular reaction.12
With the bond forming chemistry established the environmental performance was optimised by designing all the reactions into an efficient manufacturing process; telescoping most of the transformations, minimising solvent volumes, optimising reagent stoichiometries and isolating only two intermediates (9 and 14).
Green Chemistry metrics for (S,S)-reboxetine succinate
Throughout the design and development of the (S,S)-reboxetine commercial route the team regularly (at least annually) collected a set of metrics and analysed these for opportunities to improve the environmental performance of the API manufacturing process. We used an in-house metrics tool for data collection to model the effect of individual process changes on the performance of the overall process and our ability to achieve or exceed the goals that we had established at a portfolio level.A comparison of the three routes described in this paper is presented in Table 1. As the key raw material to all routes is trans-cinnamyl alcohol then this data provides a like for like comparison of waste generated by the three different routes.
| Solvent (L kg−1 API) | Aqueous (L kg−1 API) | Reagent (kg kg−1 API) | Number of intermediate isolations | Overall yield | |
|---|---|---|---|---|---|
| Pharmacia Edronax process starting from trans-cinnamyl alcohol (Scheme 1 & 2) | 1237 | 854 | 112 | 6 | 7.3% |
| Pfizer early resolution process starting from trans-cinnamyl alcohol (Scheme 3) | 657 | 272 | 64 | 4 | 10.1% |
| Pfizer process for commercialization starting from trans-cinnamyl alcohol (Scheme 4) | 79 | 61 | 13 | 2 | 35.0% |
| Input material reduction relative to initial Edronax process | 94% | 93% | 88% | ||
This data clearly shows that significant reductions in the amount of process waste can be achieved by reordering steps and eliminating redundant unit operations and isolations. This is supported by the observed 55% reduction in input materials required to run the early resolution process at pilot scale. However by switching to the commercial route that installs the absolute stereochemistry by a catalytic asymmetric reaction, requires fewer intermediate isolations and avoids the lactam reduction step then order of magnitude reductions in the total amount of materials used to make a kilogram of product were realised. The process that was intended for commercialisation generates more than 90% less waste than the original Pharmacia process, with an almost five-fold increase in overall yield.
Key experimental procedures13
General
Ethanol refers to ethanol denatured with 3% cyclohexane. The diol 9 was prepared by the method of Henegar.11NMR spectra were obtained on a Joel ECS 400 spectrometer operating at 400 MHz for proton and 100 MHz for carbon. The number of hydrogens attached to each carbon was assigned using a HSQC experiment. Reactions were monitored using reversed phase HPLC (Fortis C18, 3 μM particle size column, buffer; 10mMol NH4OAc at pH 6.8). The mass spectrum of the zwitterion 14 was recorded on a Waters Micromass ZQ Mass Spectrometer. Optical rotation of the final product 2 was measured on a Perkin Elmer 341 polarimeter. For (S,S)-reboxetine succinate 2 the assay was determined by reversed phase HPLC using a Phenomenex Luna C8(2) 150 × 4.6 mm 5 μm column, MPA = 0.02 M KH2PO4 @ pH 6.8 (0.5 M KOH) and MPB = acetonitrile. Chiral purity was determined by HPLC using a Diacel chiracel OJ 250 × 4.6 mm column, eluting with heptane/ethanol/diethylamine (1500/500/1).Preparation of acetate 10
Toluene (5.0 L) and iso-propenyl acetate (694.43 g, 6.94 mol) were charged to a 25 L fixed rig reactor followed by the diol 9 (1.0 kg, 3.47 moles). The reaction mixture was agitated at 100 rpm and heated to an internal temperature of 30 °C. Novozymes CaL B (20.0 g) was added and the mixture was agitated for 17 h after which time the acylation was complete by HPLC analysis. The enzyme was removed by filtration and the filtrate progressed to the next step.Preparation of the mesylate 11
To a stirred solution of 10 from the previous step, triethylamine (596.6 g, 5.90 mol) was added in one portion at 20 °C. A solution of methanesulfonyl chloride (536.3 g, 4.68 mol) in toluene (1.0 L) was added dropwise over 120 min. The reaction mixture was stirred for a further 60 min after which time analysis by HPLC showed that the reaction was complete.Hydrochloric acid (1 M, 3.0 L, 3.0 mol) was added in one portion. The biphasic mixture was agitated at 100 rpm for 30 min then allowed to separate for 10 min. The phases were separated and the organic (toluene) phase of the mesylate 11 progressed directly to the next step.
Preparation of epoxide 12
A solution of sodium hydroxide (1.44 kg, 36 mol) in water (3.0 L) and methyltributylammonium chloride (81.79 g, 346.8 mmol) was added to toluene solution of the mesylate 11 from the previous step. The resulting mixture was agitated at 100 rpm for 3.5 h after which time analysis by HPLC showed the reaction was complete.The phases were separated, the bottom phase discarded. A thin dark layer was present between the organic (top) and aqueous (bottom) layers and this material was mainly residue from the phase transfer catalyst. To remove the phase transfer catalyst from the vessel, an extra water wash (2.0 L) was applied. The biphasic mixture was agitated at 100 rpm for 15 min then the phases were allowed to separate. The lower aqueous phase was separated and the upper organic phase (containing the epoxide 12) was held at 0 °C before proceeding to next step.
Preparation of the zwitterion 14
Epoxide 12 (937.5 g, 3.47mol) was diluted with toluene (5.0 L) and charged to an addition funnel. 2-Aminoethylhydrogensulfate 13 (1.22 kg, 8.67 mol) was slurried in a mixture of toluene (2.0 L) and ethanol (2.0 L). 1,8-Diazabicyclo[5.4.0]undec-7-ene (1.29 L, 8.6 mol) was added in one portion and the contents were heated to 65 °C and agitated for 60 min. The solution of the epoxide 12 in toluene was added to the activated amine mixture over 1 h and the mixture stirred at 70 °C. Two hours after the end of the addition, the reaction was finished and worked up. The reaction mixture was cooled to room temperature and treated with an aqueous solution of sodium hydroxide (416.1 g, 10.4 mol) in water (8.0 L). The resulting biphasic mixture was stirred at room temperature for 2 h. Stirring was stopped and the phases allowed to separate. The organic layer was discarded and the aqueous layer recharged to the reactor. The aqueous layer was treated with 1.0 M HCl to adjust the pH value to 5.1–5.2 (the isoelectric point of the zwitterion 14). The zwitterion crystallised during the pH adjustment, was isolated by filtration and washed with water (1.225 kg, 86% from diol 9). The zwitterion 14 was purified by reslurrying in ethanol (6.1 L) at 50 °C for 2 h, followed by cooling to 25 °C and isolation by filtration (1.06 kg, 74% from diol 9). 1H NMR (400 MHz, CD3OD) δ = 1.43 (3H, t, J = 7.1 Hz, CH3CH2O), 3.10 (1H, dd J = 12.6, 3.4 Hz, –CHOHCH2NH2-), 3.18 (1H, dd, J = 12.6, 9.8 Hz, –CHOHCH2NH2-), 3.32 (2H, m, –H2NCH2CH2OSO3−), 4.10 (1H, dq, J = 11.8, 7.1, CH3CH2OAr), 4.11 (1H, dq, J = 11.8, 7.1, CH3CH2OAr), 4.21 (2H, m, –H2NCH2CH2OSO3−), 4.26 [1H, ddd, J = 9.8, 5.3, 3.4 Hz, Ph(CHOAr)CHOH–], 5.14 [1H, d, J = 5.3 Hz, Ph(CHOAr)–], 6.68 (1H, ddd, J = 8.1, 7.3, 1.6 Hz, Ar–H), 6.74 (1H, dd, J = 8.1, 1.6 Hz, Ar–H), 6.84 (1H, ddd, J = 8.1, 7.3, 1.6, Ar–H), 6.93 (1H, dd, J = 8.1, 1.6 Hz, Ar–H), 7.28 (1H, m, Ph–H), 7.34 (2H, m, Ph–H), 7.42 (2H, m, Ph–H) ppm: 13C NMR [100 MHz, [CD3)2SO] δ = 15.3 (CH3), 47.7 (CH2), 49.6 (CH2), 61.8 (CH2), 64.6 (CH2), 69.4 (CH), 82.7 (CH), 114.8 (CH), 118.1 (CH), 121.2 (CH), 122.8 (CH), 127.9 (CH), 128.4 (CH), 128.7 (CH), 138.0 (C), 147.6 (C), 149.9 (C) ppm. MS (positive ion) m/z = 412 (100%, M +1), 332 [50%, (M-SO3) + 1].Conversion of the zwitterion 14 to (S,S)-reboxetine succinate 2
To a 25 L fixed rig reactor was introduced tetrahydrofuran (7.14 L) and ethanol (210 ml) followed by the zwitterion 14 (1.05 kg, 2.55 moles). The resulting slurry was stirred at 18 °C. Sodium hydroxide (317.69 g, 7.66 mol) was added in one portion. On completion of the addition, the temperature was increased to reflux (65 °C) by setting the jacket temperature to 80 °C. A stir rate of 100 rpm was used. The reaction was monitored by HPLC until completion (approximately 3 h).The reaction mixture was cooled to room temperature then treated with water (5.25 L) and left to stir for 16 h at room temperature. Cyclohexane (4.20 L; 3.28 kg) was added to improve the phase separation. The biphasic mixture was stirred for 1 h after which time the phases were separated.
The organic layer was washed with water (2.20 L). This water wash was analysed before proceeding [ensuring that the pH > 12.0 and that conductivity (to assess removal of inorganics) to be within 1–2 mS cm−1, actual σ = 1.63mS cm−1].
The organic layer was distilled at atmospheric pressure under Dean–Stark conditions to decrease the water level to 0.2% (Karl-Fischer analysis). The organic phase was concentrated by distillation under atmospheric pressure to approximately 2.0 L (2.0 L/kg of total volume). The concentrate was treated with ethanol (5.10 L) and distilled at atmospheric pressure to remove most of the THF. The operation was repeated until no THF was detected by NMR (typically two strip and replace cycles were required to achieve the target).
After removing the THF, ethanol (4.94 L) was charged to the reaction vessel giving a total volume of 7.0 L/kg input zwitterion 14. A suitable crystallisation process with respect to seeding point occurs between 5.5 and 6.5 L/kg. Succinic acid (301.3 g, 2.55 mol) was added as solid to the reactor and the resulting mixture heated to reflux to ensure full dissolution. Approximately 1.0 L of solvent was removed in the recirculation loop lowering the concentration to the desired range. The temperature was lowered to 65–66 °C (internal) and seeded with 2 (5.51 g, 12.76 mmol). The seeds were allowed to grow over a period of 3 h at 65–66 °C. The mixture cooled at a rate of 0.5 °C per minute to reach 0 °C (internal temperature) and granulated at 0 °C overnight for 16 h.
The reaction product was isolated by filtration. The reactor was washed with cold ethanol (2.10 L) and this solvent was also used to wash the filter cake. Another charge of ethanol (2.10 L) at room temperature (∼20 °C) was also used to wash the filter cake.
The wet solid (0.912 kg) was dried for 24 h under 350 mbars at 50 °C to give (S,S)-reboxetine succinate 2 (897.0 g; 81.5% yield) as a white solid. HPLC assay 72.2% as is, 99.5% salt corrected, no impurities detected above 0.05%. Chiral purity determined by HPLC > 99.9% e.e., [α]32.4D +18.37 (c 0.37, EtOH). 1H NMR data (400 MHz, CDCl3) was in agreement with that reported in the literature.11
Acknowledgements
We thank Stewart Hayes, Wilfried Hoffman, Alan Pettman and Rob Walton for their help and contributions to this project.References
- P. Melloni, A. Della Torre, E. Lazzari, G. Mazzini and M. Meroni, Tetrahedron, 1985, 41, 1393–1399 CrossRef CAS.
- B. Hughes, I. McKenzie and M. J. Stoker WO2006/000903.
- P. J. Dunn, A. S. Wells and M. T. Williams, Green Chemistry in the Pharmaceutical Industry, Wiley-VCH, 2010 Search PubMed.
- K. E. Henegar, C. T. Ball, C. M. Horvath, K. D. Maisto and S. E. Mancini, Org. Process Res. Dev., 2007, 11, 346–353 CrossRef CAS.
- D. J. Dale, P. J. Dunn, C. Golightly, M. L. Hughes, P. C. Levett, A. K. Pearce, P. M. Searle, G. Ward and A. S. Wood, Org. Process Res. Dev., 2000, 4, 17–22 CrossRef CAS.
- P. J. Dunn, S. Galvin and K. Hettenbach, Green Chem., 2004, 6, 43–48 RSC.
- G. P. Taber, D. M. Pfisterer and J. C. Colberg, Org. Process Res. Dev., 2004, 8, 385–388 CrossRef CAS.
- N. G. Anderson, Org. Process Res. Dev., 2004, 8, 260–265 CrossRef CAS.
- The Pfizer distillation optimisation tool predicts the most solvent sparing conditions for the scale up of a stripping and replacement operation. The tool is set against a backdrop of green solvent selection. This predictive tool is available on-line to all Pfizer chemists and chemical engineers and its use can lead to significant reductions in solvent use. A discussion of the tool was given by D. J. Am Ende, 13th Annual Green Chemistry & Engineering Conference, June 23–25, 2009.
- R. M. Hanson and K. B. Sharpless, J. Org. Chem., 1986, 51, 1922–1925 CrossRef CAS; Y. R. M. Hanson, J. M. Klunder, S. Y. Ko, H. Masamune and K. B. Sharpless, J. Am. Chem. Soc., 1987, 109, 5765–570 CrossRef.
- K. E. Henegar and M. Cebula, Org. Process Res. Dev., 2007, 11, 354–358 CrossRef CAS.
- G. Assaf, G. Cansell, D. Critcher, S. Field, S. Hayes, S. Mathew and A. Pettman, Tetrahedron Lett., 2010, 51, 5048–5051 CrossRef CAS.
- For a publication giving a more detailed account of the process development aspects of the (S,S)-reboxetine succinate project see S. Hayes, G. Assaf, G. Checksfield, C. Cheung, D. Critcher, L. Harris, R. Howard, S. Mathew, C. Regius, G. Scotney and A. Scott, Org. Process Res. Dev., 2011, 15 DOI:10.1021/op200181f.
Footnote |
| † Analytical development |
| This journal is © The Royal Society of Chemistry 2012 |