Iron-catalyzed selective oxidation of sulfides to sulfoxides with the polyethylene glycol/O2 system†
Bin
Li
,
An-Hua
Liu
,
Liang-Nian
He
*,
Zhen-Zhen
Yang
,
Jian
Gao
and
Kai-Hong
Chen
State Key Laboratory and Institute of Elemento-Organic Chemistry, Nankai University, Tianjin, 300071, People's Republic of China. E-mail: heln@nankai.edu.cn; Fax: +86 22 2350 1493; Tel: +86 22-2350 1493
First published on 28th October 2011
Abstract
Readily available iron compounds were found to be active catalysts for the selective oxidation of sulfide to sulfoxide with molecular oxygen as the oxidant in polyethylene glycol (PEG). As an indispensable component, PEG had a great promotive effect on the reaction. Notably, high conversion (>99%) along with excellent chemo-selectivity of up to 94% could be attained by using Fe(acac)2 as the catalyst at 100 °C. This methodology was proved to be applicable for the transformation of various aromatic and aliphatic sulfides into the corresponding sulfoxides with high selectivity. PEG is considered to play a crucial role in stablizing the Fe(IV)-oxo species formed in situ which is supposed to be responsible for the sulfide oxidation.
Introduction
Organic sulfoxides have wide applications in the synthesis of chemically useful and biologically active molecules such as drugs, flavors, germicides as well as catabolism regulators.1 The common synthetic method for the formation of sulfoxide is the oxidation of organic sulfides (Scheme 1). However, a stoichiometric amount of organic or inorganic oxidant is required in those preparative processes and thus a large amount of toxic waste would be generated.2 With the increasing environmental and economical concerns in recent years, the procedures employing hydrogen peroxide3 or molecular oxygen4 as terminal oxidants would be promising because only water is produced as the sole by-product. In this aspect, those catalytic processes involving dioxygen, which is an abundant, inexpensive and easily available oxidant are particularly attractive. However, few successful reaction systems have been established.4a,5 Still, toxic organic solvents such as toluene,4bmethanol,6 and acetonitrile7 as well as ligands and/or metal additives would be inevitably needed to perform the reaction smoothly.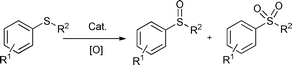
So far, a large number of metal catalysts have been developed for the sulfide oxidation, including V,8 W,9Ti,10Mn,11Co,3aCu,12 and Ag.13 In particular, iron would be an ideal candidate to be an environmentally benign catalyst because of its easy availability, low toxicity and low price as well as exceptional catalytic activity.14 In recent years, significant advances in sulfide oxidation15 has been accomplished by applying iron catalysts, such as Fe(NO3)3/FeBr3,5a,16Fe(acac)3/Schiff base,17 and Fe(Salen)18 oxidative systems. However, those iron-catalyzed processes would either involve complicated ligands to stabilize iron reactive species, or need toxic solvents to promote the reaction. In this aspect, an environmentally benign process remains in great demand.
With respect to such aspects of innocuous chemistry, significant effort has been devoted to the development of sulfide oxidation in green solvents, such as supercritical carbon dioxide (scCO2)19 and water.18a While water would probably be the most desired reaction medium, the inherent hydrophobic nature of the sulfides often prohibits its aqueous reactivity, and the sensitivity of many catalysts in hydrous conditions can not be neglected.20 PEG and its derivatives are commonly known to be inexpensive, thermally stable, and environmentally benign media for chemical reactions and phase transfer catalysts as well as having an almost negligible vapor pressure.21 In particular, Neumann found that PEG-200 could act as an excellent solvent for H5PV2Mo10O40-catalyzed aerobic oxidation reactions including the oxidation of sulfides.20 PEG also can be utilized as a support, leading to recycling of homogeneous catalysts with remarkable synergic effect.22 An environmentally benign scCO2/PEG biphasic system was successfully used in the aerobic oxidation of styrene.23 Recently, we have found that the PEG radicals from its oxidative degradation24 could be used to induce oxidation of benzylic alcohols and benzylic C–H bond oxygenation in compressed CO2.25 In brief, PEG could effectively immobilize and stabilize the metal catalysts, and may provide a more active oxidant by incorporating oxygen into the peroxide species.
Herein, we wish to report an efficient process for the selective synthesis of sulfoxides through the Fe(acac)2-catalyzed aerobic oxidation of sulfides with molecular oxygen as an oxidant in PEG. This approach represents a more environmentally friendly method for the sulfoxidation with potential economic benefits.
Results and discussion
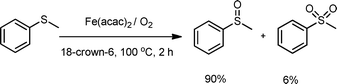
Thioanisole was chosen as a model substrate for a preliminary study. The results are summarized in Table 1. The oxidation was first examined with 0.5 mmol of PEG-1000 (molecular weight: 1000 Da) and 2.0 MPa of O2 at 100 °C for 12 h. Under such conditions, no oxidation product was detected and only intact substrate was recovered (entry 1, Table 1), indicating that PEG/O2 can not initiate the sulfide oxidation. This is different from oxidation reactions initiated by PEG radicals in compressed CO2.25c Fortuitously, a catalytic amount of Fe(acac)2 could remarkably promote the reaction. As a result, the sulfone product (methylsulfonyl benzene) was obtained in 96% yield, along with 4% yield of the sulfoxide product i.e.methyl sulfinyl benzene (entry 2, Table 1). Furthermore, the reaction did not occur in the absence of PEG (entry 3, Table 1). The experiment using 1 atm of air showed almost inactive under otherwise identical reaction conditions (entry 4, Table 1). As a consequence, PEG, molecular oxygen and an iron catalyst are prerequisites for performing the reaction smoothly.| Entry | Cat. (2 mol%) | Time (h) | Conv (%)b | Yield (%)b | |
|---|---|---|---|---|---|
| Sulfoxide | Sulfone | ||||
| a All the experiments were carried out with 0.5 mmol (62 mg) of thioanisole, PEG-1000 (0.3 mmol, 0.3 g), O2 (2 MPa), 100 °C, unless otherwise noted. b Determined by GC using area normalization. c Without PEG. d Air (balloon). e 80 °C. f 120 °C. g Fe(acac)2 (1 mol%). h TEMPO (0.05 mmol) was added. i Fe(acac)2 was purchased from Sigma–Aldrich with 99.95% purity. | |||||
| 1 | None | 12 | 0 | 0 | 0 |
| 2 | Fe(acac)2 | 12 | 100 | 4 | 96 |
| 3c | Fe(acac)2 | 12 | 0 | 0 | 0 |
| 4d | Fe(acac)2 | 12 | 4 | 4 | 0 |
| 5e | Fe(acac)2 | 12 | 28 | 28 | 0 |
| 6f | Fe(acac)2 | 12 | 100 | 3 | 97 |
| 7 | Fe(acac)2 | 2 | 100 | 93 | 7 |
| 8 | Fe(acac)3 | 2 | 98 | 91 | 7 |
| 9 | FeCl2 | 2 | <1 | — | — |
| 10 | FeCl3 | 2 | <1 | — | — |
| 11 | FeBr2 | 2 | <1 | — | — |
| 12 | FeBr3 | 2 | <1 | — | — |
| 13 | Fe(NO3)3·9H2O | 2 | 51 | 51 | 0 |
| 14g | Fe(acac)2 | 2 | >99 | 94 | 6 |
| 15h | Fe(acac)2 | 2 | 0 | 0 | 0 |
| 16i | Fe(acac)2 | 2 | 97 | 94 | 3 |
| 17 | Cu2O | 2 | 0 | 0 | 0 |
| 18 | CuO | 2 | 0 | 0 | 0 |
Subsequently, we found that the reaction temperature had a great influence on the reaction outcome. The reaction gave only 28% conversion at 80 °C (entry 5, Table 1), suggesting that an elevated temperature (100 °C) is required for the reaction. When the temperature was further increased to 120 °C, the same result as that at 100 °C was obtained (entry 6 vs. 2, Table 1). It is also worth mentioning that shortening the reaction time to 2 h gave rise to selective formation of the sulfoxide product as the main product in 93% yield rather than the sulfone product (entry 7 vs. 2, Table 1), suggesting that aerobic oxidation could proceed through an initial oxidation of the sulfide to the sulfoxide, and further oxidization of the sulfoxide to the sulfone as the reaction progressed.
Next we screened various iron catalysts, including Fe(acac)3, FeCl2, FeCl3, FeBr2, FeBr3, Fe(NO3)3·9H2O (entries 8–13, Table 1) with an aim to investigate anion effect on the iron’s catalytic performance. Fe(acac)3 and Fe(acac)2 gave almost the same results (entries 7 and 8, Table 1). However, in the case of chloride or bromide as the anion, neither Fe(III) or Fe(II) can catalyze the oxidation reaction resulting in only starting material being recovered (entries 9–12, Table 1). In addition, Fe(NO3)3·9H2O gave a relatively low yield of sulfoxide as the main product (51%) compared with Fe(acac)2 (entry 7 vs. 13, Table 1). These results could suggest that oxygen-donor anions are effective for iron-catalyzed oxidation of sulfides, presumably because the electron richness of the iron catalyst is an important factor in stabilizing the iron species generated in situ to achieve oxygen transfer from molecular oxygen to the sulfide.26Fe(acac)2 as a catalyst, proved effective for the reaction even when the catalyst loading was as low as 1 mol% (entry 14, Table 1).
This kind of oxygenation reaction was completely suppressed by adding 10 mol% of TEMPO (2,2,6,6-tetramethyl-piperidine-1-oxyl) (entry 15, Table 1), which supports the free radical reaction pathway.25c
Furthermore, to evaluate the role of trace amounts of metal impurities or contaminants (typically copper oxide in iron salts),27Fe(acac)2 with 99.95% purity was employed for this sulfide oxidation. The results show that almost the same results were obtained (entry 7 vs. 16, Table 1). On the other hand, Cu(I) oxide and Cu(II) oxide can not catalyze the reaction, leaving the thioanisole intact (entries 17–18, Table 1).
In order to understand the effect of PEG in the sulfide oxidation, we further examined the reaction in conventional organic solvents3a,28 for the sulfide oxidation, including CH3CN, CH2Cl2, CH3OH, and ethereal solvents like diethyl ether and dioxane. Notably, ethereal solvents were supposed to facilitate the formation of the Fe(IV)-oxo species from Fe(II) and O2.26 However, those solvents were proved to be ineffective, leaving the thioanisole intact (Table S1, see ESI†). Therefore, PEG might be able to play a crucial role in stabilizing the in situ formation of the Fe(IV)-oxo oxidative center under the reaction conditions. Indeed, PEG proved to be indispensable for the oxidation reaction of thioanisole (entries 1–4, Table 1).
Influence of PEG molecular weight
As an indispensable component, PEG had a great promotive effect on the reaction outcome. Therefore, the influence of PEG with different molecular weights on the reaction was investigated. When the reaction was conducted in PEG-200, the sulfoxide was attained in merely 2% yield (entry 1, Table 2). However, as the PEG molecular weight increased to 400, the yield of sulfoxide was dramatically enhanced (entry 2, Table 2). When PEG-1000 was employed, the reaction took 2 h for completion with 94% yield of sulfoxide together with sulfone (6%). We assumed that the chain length of PEG-200 was not sufficient to wrap around the active oxoiron species derived in situ from Fe(acac)2. PEG-1000 was presumably suitable to wrap around the active iron complex and thus, could stabilize the metal intermediate through rounding coordinations. The sulfide conversion gradually decreased with the PEG molecular weight increasing from 1000 to 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (entries 4–7, Table 2), being tentatively ascribed to the increasing mass transport limitation of gaseous oxygen in highly viscous long chained PEG.25a,29
000 (entries 4–7, Table 2), being tentatively ascribed to the increasing mass transport limitation of gaseous oxygen in highly viscous long chained PEG.25a,29
| Entry | PEG | PO2 (MPa) | Conv.b (%) | Yieldb (%) | |
|---|---|---|---|---|---|
| Sulfoxide | Sulfone | ||||
| a Reaction conditions: thioanisole (0.5 mmol, 62 mg), Fe(acac)2 (1 mol%, 1.2 mg), PEG (0.3 g, [OCH2CH2] unit 6.82 mmol), 100 °C, 2 h. b Determined by GC using area normalization. | |||||
| 1 | 200 | 2 | 2 | 2 | 0 |
| 2 | 400 | 2 | 73 | 65 | 8 |
| 3 | 600 | 2 | 92 | 90 | 2 |
| 4 | 1000 | 2 | >99 | 94 | 6 |
| 5 | 2000 | 2 | 79 | 76 | 3 |
| 6 | 6000 | 2 | 61 | 61 | 0 |
| 7 | 20000 |
2 | 3 | 3 | 0 |
Influence of PEG amounts and O2 pressure
Subsequently, the effects of the amount of PEG and the O2 pressure on the reaction were evaluated. As shown in Table 3, the conversion was sensitively affected by the amount of PEG. The suitable molar ratio of thioanisole and PEG-1000 could be 1.67; namely, the proper PEG amount could be 0.3 g when 0.5 mmol of thioanisole was used. Otherwise, the conversion would be restrained by altering PEG amount (entries 1–4, Table 3). Obviously, a higher O2 concentration is more favourable for the oxidation reaction (entry 2, Table 3). While a deficient yield of sulfoxide was observed under 1 MPa of O2 (entry 5, Table 3), and further reducing the O2 pressure to 1 bar resulted in only 2% conversion (entry 6, Table 3). As a consequence, the reaction conditions could be finally set to Fe(acac)2 (1 mmol%), PEG-1000 (0.3 g), O2 (2 MPa), at 100 °C for 2 h.| Entry | PEG-1000 amount (g) | PO2 (MPa) | Conv.b (%) | Yieldb (%) | |
|---|---|---|---|---|---|
| Sulfoxide | Sulfone | ||||
| a Reaction conditions: thioanisole (0.5 mmol), Fe(acac)2 (1 mol%, 1.2 mg), 100 °C, 2 h. b Determined by GC using area normalization. | |||||
| 1 | 0.1 | 2 | 2 | 2 | 0 |
| 2 | 0.3 | 2 | >99 | 94 | 6 |
| 3 | 0.5 | 2 | 59 | 59 | 0 |
| 4 | 1 | 2 | 11 | 11 | 0 |
| 5 | 0.3 | 1 | 37 | 37 | 0 |
| 6 | 0.3 | 0.1 | 2 | 2 | 0 |
Substrate scope
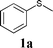
In order to explore the generality of this procedure, a series of sulfides were evaluated by performing the reaction under the given conditions. The results are summarized in Table 4. As expected, the catalyst used with the PEG/O2 system worked well for various aromatic and aliphatic sulfides, thus providing easy access to valuable symmetric and asymmetric sulfoxides. Aromatic substrates 1a–1d gave good to excellent yields of the sulfoxide product (entries 1–4, Table 4). Whereas, a strong electron-withdrawing group at the para-position showed a negative effect on the reaction. In particular, a much longer reaction time (16 h) was needed for 1e to complete the reaction (entry 5, Table 4), presumably due to the strong electron-withdrawing effects of the nitrile group. On the other hand, the influence of steric hindrance was also found in the case of 1f (entry 6, Table 4). To our delight, this procedure also proved applicable to the oxidation of aliphatic sulfides (1g and 1h) (entries 7 and 8, Table 4), giving the corresponding isolated yield of sulfoxide of 83% and 74%, respectively.| Entry | Substrate | Time (h) | Conv.b (%) | Yieldb (%) | |
|---|---|---|---|---|---|
| Sulfoxide (2a–2h) | Sulfone | ||||
| a All the experiments were carried out with the substrate (0.5 mmol), PEG-1000 (0.3 g), Fe(acac)2 (1 mol%, 1.2 mg), O2 (2 MPa) at 100 °C. b Determined by GC using area normalization and data in parentheses refer to isolated yields. | |||||
| 1 |
|
2 | 100 | 94(89) | 6 |
| 2 |
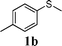
|
2 | 97 | 91(77) | 9 |
| 3 |
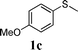
|
2 | 98 | 95(93) | 3 |
| 4 |
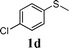
|
3 | 100 | 96(94) | 4 |
| 5 |
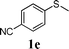
|
16 | 98 | 96(94) | 2 |
| 6 |
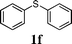
|
4 | >99 | 97(91) | 2 |
| 7 |
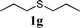
|
6 | 96 | 90(83) | 6 |
| 8 |
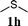
|
3 | 97 | 92(74) | 5 |
Proposed mechanism
A detailed mechanism of iron-catalyzed sulfoxidation is not clear at the present stage. Recently, existence of Fe(IV)-oxo species has been evidenced as the active oxidizing species in enzymatic and biomimetic reactions (Scheme 2)30 Furthermore, mononuclear non-heme Fe(IV)-oxo complexes, generated in situ during the reaction of Fe(II) complexes with O2, were reported to be capable of oxidizing PPh3 to Ph3PO.31 We also found that the PEG radical alone could not initiate the oxidation of thioanisole (entry 1, Table 1), implying that the iron catalyst plays an essential role in the sulfoxidation. Effect of TEMPO also supports the free radical pathway (entry 14, Table 1).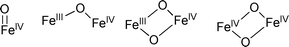 | ||
| Scheme 2 Fe(IV)-oxo species observed in non-heme iron proteins and model complexes. | ||
Based on the understanding of Fe(IV)-oxo species, a mechanism for the iron-catalyzed selective oxidation of sulfides to sulfoxides was proposed as shown in Scheme 3. Firstly, Fe(II) 1 reacts with O2 to form a Fe(III)-superoxo intermediate 2.32 Then the μ-oxo bridged diiron species, Fe(III)–(O)4–Fe(III) 333 could form to further generate 4, Fe(III)–(O)2–Fe(III)4, after releasing one molecule of oxygen. Subsequent O–O bond homolysis of 4 results in the generation of the Fe(IV)-oxo species 5 which are involved in oxygen atom transfer reactions. Simultaneously, Fe(II) 1 is regenerated and could participate in the next catalytic cycle. Alternatively, this Fe(IV)-oxo complex 5 could also be generated through formation of the intermediate 7 from Fe(III) 6 and oxygen. Regeneration of Fe(III) 6viaoxidation of the formed Fe(II) 1 could complete the catalytic cycle. In this context, Fe(II) and Fe(III) gave almost the same catalytic activity (entries 7 and 8, Table 1).
 | ||
| Scheme 3 Proposed mechanism. | ||
On the other hand, PEGs are acyclic polyethers that have flexible polyethylene oxide chains, which render it possible to have ion-dipole interactions with metal salts.34 It has been reported that the PEG-1000 matrix could effectively stabilize and immobilize the catalytically active Pd particles in the aerobic oxidation of alcohols.35 In this regards, we found that 18-crown-6 could play the same role in this process as PEG (Table S2, see ESI†), as depicted in Scheme 4.
 | ||
| Scheme 4 Fe(II) catalyzed aerobic oxidation of sulfide in 18-crown-6. | ||
Accordingly, PEG-1000 like crown ether, could wrap around Fe(acac)2 containing a more oxygen-donor anions, and subsequently create a microenvironment around the iron center that aids in stabilizing the Fe(IV)-oxo unit.34
Conclusions
In summary, we have developed a highly selective sulfoxidation system with molecular oxygen as the oxidant in PEG, using cheap and readily available Fe(acac)2 as the catalyst. Meanwhile, this process has been proven to be applicable for the selective oxidation of the aromatic and aliphatic sulfides. Notably, PEG-1000, which can provide an electron-rich environment, could play a key role in stabilizing the catalytic Fe(IV)-oxo species formed in situ, thus promoting the oxidation of sulfides to sulfoxides. Further studies will be aimed at ultimately disclosing the mechanistic details for this highly chemo-selective sulfoxidation and extending the PEG/O2/Fe(acac)2 system to other oxidative transformations.Experimental Section
Materials
Sulfides were purchased from Sigma-Aldrich and Aladdin. The various iron catalysts were purchased from Aladdin with >98% purity (Cu content was determined to be 0.013% w/w by using the ICP method).General procedure for the iron-catalyzed selective oxidation of sulfide to sulfoxide
A mixture of the substrate (0.5 mmol), PEG-1000 (0.3 mmol) and Fe(acac)2 (1 mol%, 1.2 mg) was placed in a 25 mL autoclave, equipped with an inner glass tube. 2 MPa of O2 was then introduced into the autoclave and heated to 100 °C. The mixture was stirred continuously for the designed reaction time, then the reactor was placed into ice water and the O2 was released slowly. After depressurization, the products were then extracted by diethyl ether (10 mL × 3), and analyzed by a gas chromatograph (SHIMADZU-2014) equipped with a capillary column (RTX-17, 30 m × 0.25 μm) using a flame ionization detector. Then the solvents were distilled off and the products were purified by silica gel column chromatography (200–300 mesh, 1:2 v/v ethyl acetate:petroleum as eluant). The structure and the purity of the products were further identified using NMR (BRUKER-400 MHz), GC–MS (Thermo Finnigan Polaris Q) and GC by comparing retention times and fragmentation patterns with those of authentic samples.
Synthesis of sulfoxide products
Acknowledgements
We are grateful to the National Natural Science Foundation of China (No 20872073), Research Fellowship for International Young Scientists from NSFC (21150110105), and the Committee of Science and Technology of Tianjin for financial support.References
- (a) P. Metzner and A. Thuillier, Sulfur Reagents in Organic Synthesis, Academic Press, London, 1994 Search PubMed; (b) The chemistry of sulfones, sulfoxides and cyclic sulfides, ed. S. Patai, Z. Rappoport, C. Stirling, Wiley, Chichester, 1994 Search PubMed.
- (a) D. H. R. Barton, W. Li and J. A. Smith, Tetrahedron Lett., 1998, 39, 7055–7058 CrossRef CAS; (b) M. Hirano, S. Yakabe, J. H. Clark and T. Morimoto, J. Chem. Soc., Perkin Trans. 1, 1996, 2693–2698 RSC.
- (a) M. D. A. Maria, P. Mastrorilli and C. F. Nobile, J. Mol. Catal. A: Chem., 1996, 108, 57–62 CrossRef; (b) H. Fakhraian and F. Valizadeh, J. Mol. Catal. A: Chem., 2010, 333, 69–72 CrossRef CAS; (c) J. Fujisaki, K. Matsumoto, K. Matsumoto and T. Katsuki, J. Am. Chem. Soc., 2011, 133, 56–61 CrossRef CAS; (d) F. Shi, M. K. Tse, H. M. Kaiser and M. Beller, Adv. Synth. Catal., 2007, 349, 2425–2430 CrossRef CAS.
- (a) T. Punniyamurthy, S. Velusamy and J. Iqbal, Chem. Rev., 2005, 105, 2329–2363 CrossRef CAS; (b) I. Klement, H. Liitjens and P. Knochel, Angew. Chem., Int. Ed. Engl., 1997, 36, 1454–1456 CrossRef CAS; (c) A. M. Khenkin and R. Neumann, Catal. Lett., 2000, 68, 109–111 CrossRef CAS; (d) H. Tanaka, H. Nishikawa, T. Uchida and T. Katsuki, J. Am. Chem. Soc., 2010, 132, 12034–12041 CrossRef CAS; (e) H. Zhang, C. Y. Chen, R. H. Liu, Q. Xu and W. Q. Zhao, Molecules, 2010, 15, 83–92 CrossRef CAS; (f) S. Colonna, N. Gaggero, F. Montanari, G. Pozzi and S. Quici, Eur. J. Org. Chem., 2001, 181–186 CrossRef CAS.
- (a) S. E. Martín and L. I. Rossi, Tetrahedron Lett., 2001, 42, 7147–7151 CrossRef; (b) T. Chinnusamy and O. Reiser, ChemSusChem, 2010, 3, 1040–1042 CrossRef CAS.
- I. Gamba, S. Palavicini, E. Monzani and L. Casella, Chem.–Eur. J., 2009, 15, 12932–12936 CrossRef CAS.
- T. Iwahama, S. Sakaguchi and Y. Ishii, Tetrahedron Lett., 1998, 39, 9059–9062 CrossRef CAS.
- For the vanadium-catalyzed oxidation of sulfide, see: (a) C. Bolm and F. Bienewald, Angew. Chem., Int. Ed. Engl., 1996, 34, 2640–2642 CrossRef; (b) M. Al-Hashimi, E. Fisset, A. C. Sullivan and J. R. H. Wilson, Tetrahedron Lett., 2006, 47, 8017–8019 CrossRef CAS; (c) M. R. Maurya, A. K. Chandrakar and S. Chand, J. Mol. Catal. A: Chem., 2007, 263, 227–237 CrossRef CAS; (d) I. Khedher, A. Ghorbel, J. M. Fraile and J. A. Mayoral, J. Mol. Catal. A: Chem., 2006, 255, 92–96 CrossRef CAS; (e) Y. Jeong, S. Choi, Y. D. Hwang and K. H. Ahn, Tetrahedron Lett., 2004, 45, 9249–9252 CrossRef CAS; (f) J. Sun, C. Zhu, Z. Dai, M. Yang, Y. Pan and H. Hu, J. Org. Chem., 2004, 69, 8500–8503 CrossRef CAS; (g) A. M. Khenkin, G. Leitus and R. Neumann, J. Am. Chem. Soc., 2010, 132, 11446–11448 CrossRef CAS.
- For wolframium-catalyzed oxidation of sulfide: (a) N. N. Mahamuni, P. R. Gogate and A. B. Pandit, Ultrason. Sonochem., 2007, 14, 135–142 CrossRef CAS; (b) B. Karimi, M. Ghoreishi-Nezhad and J. H. Clark, Org. Lett., 2005, 7, 625–628 CrossRef CAS; (c) K. Sato, M. Hyodo, M. Aoki, X. Q. Zheng and R. Noyori, Tetrahedron, 2001, 57, 2469–2476 CrossRef CAS; (d) R. Noyori, M. Aoki and K. Sato, Chem. Commun., 2003, 1977–1986 RSC.
- J. S. Gao, H. C. Guo, S. Z. Liu and M. Wang, Tetrahedron Lett., 2007, 48, 8453–8455 CrossRef CAS.
- (a) F. Hosseinpoor and H. Golchoubian, Tetrahedron Lett., 2006, 47, 5195–5197 CrossRef CAS; (b) J. Brinksma, R. L. Crois, B. L. Feringa, M. I. Donnoli and C. Rosini, Tetrahedron Lett., 2001, 42, 4049–4052 CrossRef CAS.
- R. Das and D. Chakraborty, Tetrahedron Lett., 2010, 51, 6255–6258 CrossRef CAS.
- R. Das and D. Chakraborty, Synthesis, 2011, 2, 277–280 Search PubMed.
- General reviews: (a) C. Bolm, J. Legros, J. Le Paih and L. Zani, Chem. Rev., 2004, 104, 6217 CrossRef CAS; (b) “Iron Catalysis in Organic Chemistry: reactions and applications”, (Ed.: B. Plietker) WILEY-VCH Verlag, Weinheim, 2008 Search PubMed; (c) S. Enthaler, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 3317 CrossRef CAS; (d) A. Correa, O. Garcia Mancheno and C. Bolm, Chem. Soc. Rev., 2008, 37, 1108 RSC; (e) B. D. Sherry and A. Fürstner, Acc. Chem. Res., 2008, 41, 1500 CrossRef CAS; (f) W. M. Czaplik, M. Mayer, J. Cvengros and A. Jacobi von Wangelin, ChemSusChem, 2009, 2, 396 CrossRef CAS; (g) G. Cahiez and S. Marquais, Pure Appl. Chem., 1996, 68, 53 CrossRef CAS; (h) C. L. Sun, B. J. Li and Z. J. Shi, Chem. Rev., 2011, 111, 1293–1314 CrossRef CAS; (i) W. M. Czaplik, M. Mayer, S. Grupe and A. Jacobi von Wangelin, Pure Appl. Chem., 2010, 82, 1545 CrossRef CAS; (j) K. Junge, K. Schrder and M. Beller, Chem. Commun., 2011, 47, 4849–4859 RSC; (k) R. H. Morris, Chem. Soc. Rev., 2009, 38, 2282–2291 RSC.
- (a) L. Chen, Y. Yang and D. L. Jiang, J. Am. Chem. Soc., 2010, 132, 9138–9143 CrossRef CAS; (b) C. Duboc-Toia, S. Ménage, R. Y. N. Ho, L. Que, Jr., C. Lambeaux and M. Fontecave, Inorg. Chem., 1999, 38, 1261–1268 CrossRef CAS.
- C. O. Kinen, L. I. Rossi and R. H. De Rossi, J. Org. Chem., 2009, 74, 7132–7139 CrossRef CAS.
- (a) J. Legros and C. Bolm, Angew. Chem., Int. Ed., 2004, 43, 4225–4228 CrossRef CAS; (b) J. Legros and C. Bolm, Chem.–Eur. J., 2005, 11, 1086–1092 CrossRef CAS; (c) J. Legros and C. Bolm, Angew. Chem., Int. Ed., 2003, 42, 5487–5489 CrossRef CAS.
- (a) H. Egami and T. Katsuki, J. Am. Chem. Soc., 2007, 129, 8940–8941 CrossRef CAS; (b) H. Egami and T. Katsuki, Synlett, 2008, 10, 1543–1546 Search PubMed.
- (a) S. Campestrini and U. Tonellato, J. Mol. Catal. A: Chem., 2000, 164, 263–272 CrossRef CAS; (b) S. K. Karmee, L. Greiner, A. Kraynov, T. E. Müller, B. Niemeijer and W. Leitner, Chem. Commun., 2010, 46, 6705–6707 RSC; (c) R. Mello, A. Olmos, A. Alcalde-Aragonés, A. Díaz-Rodríguez, M. E. G. Núñez and G. Asensio, Eur. J. Org. Chem., 2010, 6200–6206 CrossRef CAS; (d) Q. H. Chen and E. J. Beckman, Green Chem., 2007, 9, 802–808 RSC.
- A. Haimov and R. Neumann, Chem. Commun., 2002, 876–877 RSC.
- (a) J. Chen, S. K. Spear, J. G. Huddleston and R. D. Rogers, Green Chem., 2005, 7, 64–82 RSC; (b) D. J. Heldebrant and P. G. Jessop, J. Am. Chem. Soc., 2003, 125, 5600–5601 CrossRef CAS; (c) Z. S. Hou, N. Theyssen, A. Brindmann and W. Leitner, Angew. Chem., 2005, 117, 1370–1373 CrossRef.
- (a) Y. Du, Y. Wu, A. H. Liu and L. N. He, J. Org. Chem., 2008, 73, 4709–4712 CrossRef CAS; (b) Y. Du, J. Q. Wang, J. Y. Chen, F. Cai, J. S. Tian, D. L. Kong and L. N. He, Tetrahedron Lett., 2006, 47, 1271–1275 CrossRef CAS; (c) X. Y. Dou, J. Q. Wang, Y. Du, E. Wang and L. N. He, Synlett, 2007, 19, 3058–3062 Search PubMed; (d) D. L. Kong, L. N. He and J. Q. Wang, Synlett, 2010, 8, 1276–1280 Search PubMed.
- J. Q. Wang, F. Cai, E. Wang and L. N. He, Green Chem., 2007, 9, 882–887 RSC.
- For typical examples on thermal/oxidatvie degradation of PEG, see: (a) J. Glastrup, Polym. Degrad. Stab., 1996, 52, 217–222 CrossRef CAS; (b) J. R. Conder, N. A. Fruitwala and M. K. Shingari, J. Chromatogr., A, 1983, 269, 171–178 CrossRef CAS; (c) E. A. Altwicker, Chem. Rev., 1967, 67, 475–531 CrossRef CAS.
- (a) J. Q. Wang, L. N. He and C. X. Miao, Green Chem., 2009, 11, 1013–1017 RSC; (b) J. Q. Wang and L. N. He, New J. Chem., 2009, 33, 1637–1640 RSC; (c) J. Q. Wang, L. N. He, C. X. Miao and J. Gao, ChemSusChem, 2009, 2, 755–760 CrossRef CAS.
- S. O. Kim, C. V. Sastri, M. S. Seo, J. Kim and W. Nam, J. Am. Chem. Soc., 2005, 127, 4178–4179 CrossRef CAS.
- S. L. Buchwald and C. Bolm, Angew. Chem., Int. Ed., 2009, 48, 5586 CrossRef CAS.
- (a) P. Veerakumara, Z. Z. Lu, M. Velayudhamb, K. L. Lub and S. Rajagopala, J. Mol. Catal. A: Chem., 2010, 332, 128–137 CrossRef; (b) H. M. Shi, C. G. Yu and J. He, J. Catal., 2010, 271, 79–87 CrossRef CAS; (c) E. Baciocchi, M. F. Gerini and A. Lapi, J. Org. Chem., 2004, 69, 3586–3589 CrossRef CAS.
- T. Seki, J. D. Grunwaldt and A. Baiker, Chem. Commun., 2007, 3562–3564 RSC.
- (a) I. V. Korendovych, S. V. Kryatov and E. V. Rybak-Akimova, Acc. Chem. Res., 2007, 40, 510–521 CrossRef CAS; (b) M. Costas, M. P. Mehn, M. P. Jensen and L. Que, Jr., Chem. Rev., 2004, 104, 939–986 CrossRef CAS; (c) L. D. Slep and F. Neese, Angew. Chem., Int. Ed., 2003, 42, 2942–2945 CrossRef CAS; (d) D. A. Proshlyakov, T. F. Henshaw, G. R. Monterosso, M. J. Ryle and R. P. Hausinger, J. Am. Chem. Soc., 2004, 126, 1022–1023 CrossRef CAS; (e) P. J. Riggs-Gelasco, J. C. Price, R. B. Guyer, J. H. Brehm, E. W. Barr, J. M. Bollinger, Jr. and C. Krebs, J. Am. Chem. Soc., 2004, 126, 8108–8109 CrossRef CAS; (f) J. U. Rohde, J. H. In, M. H. Lim, W. W. Brennessel, M. R. Bukowski, A. Stubna, E. Münck, W. Nam and L. Que, Jr., Science, 2003, 299, 1037–1039 CrossRef CAS; (g) M. H. Lim, J. U. Rohde, A. Stubna, M. R. Bukowski, M. Costas, R. Y. N. Ho, E. Münck, W. Nam and L. Que, Jr., Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3665–3670 CrossRef CAS; (h) J. Kaizer, E. J. Klinker, N. Y. Oh, J. U. Rohde, W. J. Song, A. Stubna, J. Kim, E. Münck, W. Nam and L. Que, Jr., J. Am. Chem. Soc., 2004, 126, 472–473 CrossRef CAS.
- (a) D. H. Chin, G. N. L. Mar and A. L. Balch, J. Am. Chem. Soc., 1980, 102, 5945–5947 CrossRef CAS; (b) A. L. Balch, Y. W. Chan, R. J. Cheng, G. N. L. Mar, L. Latos-Grazynski and M. W. Renner, J. Am. Chem. Soc., 1984, 106, 7779–7785 CrossRef CAS; (c) R. A. Ghiladi, R. M. Kretzer, I. Guzei, A. L. Rheingold, Y. M. Neuhold, K. R. Hatwell, A. D. Zuberbühler and K. D. Karlin, Inorg. Chem., 2001, 40, 5754–5767 CrossRef CAS.
- (a) S. W. Hong, Y. M. Lee, W. Shin, S. Fukuzumi and W. Nam, J. Am. Chem. Soc., 2009, 131, 13910–13911 CrossRef CAS; (b) Y. M. Lee, S. W. Hong, Y. Morimoto, W. Shin, S. Fukuzumi and W. Nam, J. Am. Chem. Soc., 2010, 132, 10668–10670 CrossRef CAS; (c) A. Thibon, J. England, M. Martinho, V. G. Young, Jr., J. R. Frisch, R. Guillot, J. J. Girerd, E. Münck, L. Que, Jr. and F. Banse, Angew. Chem., Int. Ed., 2008, 47, 7064–7067 CrossRef CAS; (d) S. V. Kryatov, S. Taktak, I. V. Korendovych and E. V. Rybak-Akimova, Inorg. Chem., 2005, 44, 85–99 CrossRef CAS; (e) M. Martinho, G. Blain and F. Banse, Dalton Trans., 2010, 39, 1630–1634 RSC.
- C. E. MacBeth, A. P. Golombek, V. G. Young, Jr., C. Yang, K. Kuczera, M. P. Hendrich and A. S. Borovik, Science, 2000, 289, 938–941 CrossRef CAS.
- (a) S. P. Luo, S. Zhang, Y. F. Wang, A. B. Xia, G. C. Zhang, X. H. Du and D. Q. Xu, J. Org. Chem., 2010, 75, 1888–1891 CrossRef CAS; (b) G. W. Gokel, D. M. Goli and R. A. Schuitz, J. Org. Chem., 1983, 48, 2837–2842 CrossRef CAS; (c) H. K. Frensdorff, J. Am. Chem. Soc., 1971, 93, 600–606 CrossRef CAS.
- (a) Z. S. Hou, N. Theyssen, A. Brinkmann and W. Leitner, Angew. Chem., Int. Ed., 2005, 44, 1346–1349 CrossRef CAS; (b) Z. S. Hou, N. Theyssen and W. Leitner, Green Chem., 2007, 9, 127–132 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental methods, experimental procedures, characterization products and copies of the NMR spectra. See DOI: 10.1039/c1gc15821j |
| This journal is © The Royal Society of Chemistry 2012 |