Alkylgold complexes by the intramolecular aminoauration of unactivated alkenes†
Rebecca L.
LaLonde
a,
William E.
Brenzovich, Jr.
a,
Diego
Benitez
b,
Ekaterina
Tkatchouk
b,
Kotaro
Kelley
a,
William A.
Goddard, III
b and
F. Dean
Toste
*a
aDepartment of Chemistry, University of California Berkeley, Berkeley, CA, USA. E-mail: fdtoste@berkeley.edu; Fax: +1 510-666-2504; Tel: +1 510-642-2850
bMaterials and Process Simulation Center, California Institute of Technology, Pasadena, CA, USA
First published on 10th June 2010
Abstract
Alkylgold(I) complexes were formed from the gold(I)-promoted intramolecular addition of various amine nucleophiles to alkenes. These experiments provide the first direct experimental evidence for the elementary step of gold-promoted nucleophilic addition to an alkene. Deuterium-labeling studies and X-ray crystal structures provide support for a mechanism involving anti-addition of the nucleophile to a gold-activated alkene, which is verified by DFT analysis of the mechanism. Ligand studies indicate that the rate of aminoauration can be drastically increased by use of electron-poor arylphosphines, which are also shown to be favored in ligand exchange experiments. Attempts at protodeauration lead only to recovery of the starting olefins, though the gold can be removed under reducing conditions to provide the purported hydroamination products.
Introduction
The addition of nucleophiles to alkenes promoted by π-complexation to an electrophilic metal salt has been known for over a century. While this chemistry was initially dominated by the formation of organomercurials using mercuric salts,1 it was subsequently discovered that platinum2 and palladium3 were capable of effecting related alkene addition reactions as well. Moreover, the reaction of alkene complexes with amine nucleophiles was shown to give rise to β-aminoalkylmercury,4 -platinum5 and -palladium6 complexes, though relatively few of these complexes have been characterized by X-ray crystallography.7,8 These discoveries formed the basis for the development of late transition metal-catalyzed hydroamination reactions,9 in which the catalyst may be regenerated from the alkylmetal intermediate by protonolysis of the alkyl–metal bond. This mechanistic paradigm has been proposed and studied in platinum-10 and palladium11-catalyzed hydroamination reactions of alkenes; however, it was subsequently shown that a Brønsted acid was the likely catalyst when sulfonamides were employed as nucleophiles in the platinum-catalyzed reaction.12Gold-catalyzed reactions that proceed through the activation of alkynes and allenes are now well-established,13 though there have been significantly fewer reports of related additions to alkenes.14 Moreover, the majority of these reported gold-catalyzed additions require elevated temperatures and extended reaction times; conditions under which the analogous Brønsted acid-catalyzed reactions also occur.12,15 Therefore, the role of the gold in alkene activation has been closely scrutinized. Recently, several vinylgold intermediates derived from the gold-promoted addition of nucleophiles to alkynes and allenes have been isolated and characterized.16 While gold(I)–alkene complexes have been isolated,17 direct experimental evidence for the mechanism of the elementary step of gold-promoted nucleophilic addition to an alkene is lacking. Herein we report the isolation and characterization of the first compounds derived from the gold-activated nucleophilic addition to an olefin.
Results and discussion
Synthesis and isolation of alkylgold complexes
Mechanisms involving either the gold-promoted anti- or syn-addition of nucleophiles to π-bonds have been previously proposed (Scheme 1). Both of these pathways necessitate the intermediacy of an alkylgold species (A) that subsequently undergoes protodeauration to provide product and regenerate the gold catalyst. We reasoned that the addition of an exogenous base might prevent protodemetallation, allowing for the observation and isolation of intermediates of type A; however, previous reports have suggested that basic amines might “bind gold(I) and thus inhibit the addition step”.14a Thus, we initially chose proton sponge, a hindered, non-nucleophilic base, and a gold(I) complex stabilized by a strongly coordinating p-nitrobenzoate counterion. We were pleased to find that under these conditions alkene 1 was converted to the desired alkylgold complex 2, as indicated by 31P and 1H NMR, in modest yields (Table 1, entry 1). Replacing the starting gold complex with the gold-oxo trimer, [(Ph3PAu)3O]BF4, resulted in near quantitative conversion of 1 to alkylgold 2 (entry 2). Surprisingly, even excess amounts of other less-hindered amine bases (entries 3, 4) were competent at sequestering protons without hindering the activity of the cationic gold complex. 2,6-Di-tert-butylpyridine, however, was not sufficiently basic, yielding only 33% of the desired alkylgold complex and 59% of the pyrrolidine resulting from hydroamination (entry 5). Ultimately, we identified triethylamine as the optimal base, as isolation of alkylgold complex 2 was simplified by performing an aqueous work up.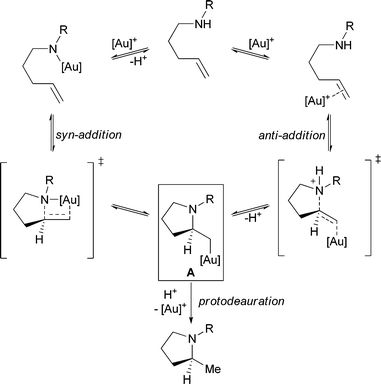 | ||
| Scheme 1 Proposed syn- and anti-mechanisms for the gold(I)-catalyzed hydroamination of alkenes. | ||
|
|
|||
|---|---|---|---|
| Entry | Ph3PAuX | Base | % conversionb |
| a Reaction conditions: to a solution of alkene (1 equiv.) and base (2 equiv.) in CDCl3 was added a molar equivalent of gold. b Determined by 1H and 31P NMR spectroscopy. c Yield in parentheses is protodemetallated product. | |||
| 1 | Ph3PAuOPNB | Proton sponge | ∼10 |
| 2 | [(Ph3PAu)3O]BF4 | Proton sponge | 100 |
| 3 | [(Ph3PAu)3O]BF4 | DABCO | 92 |
| 4 | [(Ph3PAu)3O]BF4 | NEt3 | 98 |
| 5 | [(Ph3PAu)3O]BF4 | 2,6-Di-tert-butylpyridine | 33 (59)c |

Under these optimized reaction conditions, a variety of urea substrates underwent the intramolecular aminoauration reaction (Table 2). The conversions, as judged by 1H and 31P NMR spectroscopy, are generally high and the reported yields are reflective of difficulties in the purification step. 1,1-Disubstituted alkenes also proved to be competent substrates, forming tertiary amine products (entries 6, 7); however, 1,2-disubstituted olefins (entries 8, 9) did not form the desired alkylgold complex in any appreciable amount, even at elevated temperatures. The aminoauration to produce piperidine 20 in 30% yield required an excess of gold and extended reaction time (entry 10). The conversion could be improved by increasing the reaction temperature, although purification was then complicated by the formation of intractable by-products.
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Substrate | n | R1 | R2 | R3 | R4 | Time/h | Product | % yieldb |
| a Reaction conditions: to a solution of alkene (1 equiv.) and base (2 equiv.) in CDCl3 was added 0.4 equiv. of [(Ph3PAu)3O]BF4. b Isolated yield. c 1.0 equiv. of [(Ph3PAu)3O]BF4 used. | |||||||||
| 1 | 1 | 1 | t-Bu | Ph | H | H | 2 | 2 | 80 |
| 2 | 3 | 1 | Me | Ph | H | H | 2 | 4 | 59 |
| 3 | 5 | 1 | Et | Ph | H | H | 2 | 6 | 63 |
| 4 | 7 | 1 | Ph | Ph | H | H | 2 | 8 | 66 |
| 5 | 9 | 1 | Et | –C5H10– | H | H | 2 | 10 | 49 |
| 6 | 11 | 1 | Me | Ph | Me | H | 14 | 12 | 60 |
| 7 | 13 | 1 | Et | Ph | Me | H | 14 | 14 | 40 |
| 8 | 15 | 1 | Et | Ph | H | Me | 14 | 16 | NR |
| 9 | 17 | 1 | Et | Ph | H | Ph | 14 | 18 | Trace |
| 10 | 19 | 2 | Me | Ph | H | H | 48 | 20 | 30c |

Previous reports of gold-catalyzed hydroamination reactions using carbamate nucleophiles generally required elevated temperatures (60–100 °C).16 Therefore, we were surprised to find that, even at room temperature, substrates protected as carbamates underwent significant conversion to the corresponding alkylgold complexes (Table 3). More electron-withdrawing protecting groups, such as tosylamide 31 and trifluoroacetamide 32, primarily led to the formation of gold(I)amide complexes instead of cyclizing to form the alkylgold (Scheme 2).
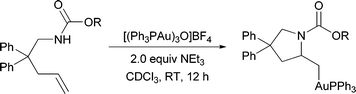
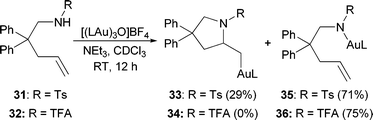 | ||
| Scheme 2 Aminoauration of acidic protected amines (L = PPh3). | ||
With the recent reports of hydroaminations with secondary ammonium salts,16 we were interested in attempting the reaction with amines. We were intrigued to find that benzylamine 37 readily reacted with the trinuclear gold-oxo complex to provide 38 (eqn (1)). Pyrrolidine 38 proved to be highly unstable, and we were therefore unable to successfully isolate and purify this alkylgold complex, though the diagnostic peaks were identified in both the crude 1H and 31P NMR spectra. Interestingly, alkylgold complex 38 was formed even in the absence of an added base, as the amine was capable of sequestering the proton to prevent protodeauration. This observation suggests that catalytic hydroamination with amines is not necessarily precluded by coordination of the gold to the amine as previously predicted,14a but rather by the prevention of efficient protodeauration of the β-aminoalkylgold complex.
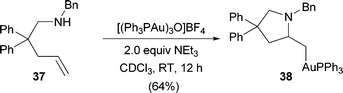 | (1) |
In order to determine the effect of the ligand on the formation of the alkylgold complex, we utilized several trinuclear gold-oxo complexes with various monophosphine ligands (Table 4). We were gratified to find that the reaction proceeded smoothly for a variety of electron-rich and electron-poor phosphine ligands. Electron poor phosphines, such as (p-F3CC6H4)3P (entry 1), appear to enhance the formation of the gold complex, while the analogous reaction with the electron-rich (p-MeOC6H4)3P (entry 6) failed to go to completion, providing only about 80% conversion by 1H NMR.
Recrystallization of 2 from DME and diethyl ether provided crystals suitable for X-ray analysis (Fig. 1). The structure displays the characteristic linear, two-coordinate geometry around gold (P–Au–C19 176°). The phosphine–gold bond length of 2.29 Å is typical for a phosphine gold(I) complex.18a The C19–Au bond length (2.07 Å) is comparable to that of MeAuPPh3 (2.12 Å).18b The N–C–C–Au dihedral angle is an almost perfect antiperiplanar arrangement. The crystal structure of piperidine alkylgold complex, 20, displayed similar characteristics with a C–Au–P angle of 177° and bond lengths of 2.08 and 2.28 Å for the C–Au and Au–P bonds, respectively (Fig. 2).
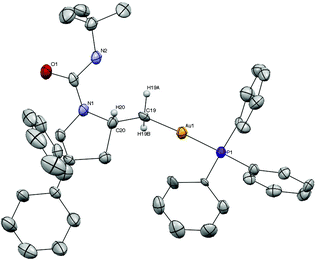 | ||
| Fig. 1 ORTEP of alkylgold 2. Thermal ellipsoids shown at 50% probability. Hydrogens (except H19A, H19B, and H20) and DME solvent molecule omitted for clarity. | ||
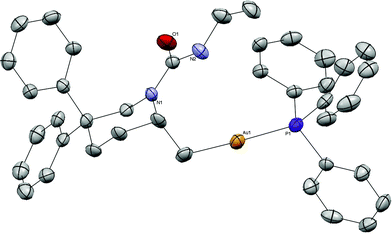 | ||
| Fig. 2 ORTEP of alkylgold 20. Thermal ellipsoids shown at 50% probability. Hydrogens omitted for clarity. | ||
Mechanism of aminoauration
As mentioned previously, activation of the alkene could lead to addition of a nucleophile either syn- or anti- to the forming gold–carbon bond. The stereochemical course of the aminoauration was examined experimentally through the use of deuterated olefins 45 and 46 (eqn (2)).19,20 Upon treatment with [(Ph3PAu)3O]BF4 under standard conditions, trans-deuterated alkene 45 cyclized to form exclusively the anti-addition product 47 (methylene proton, J3 = 8.4 Hz).13 As expected, the cis-deuterated alkene 46 underwent aminoauration reaction to furnish 48 (methylene proton, J3 = 2.0 Hz), confirming the anti-addition of the nucleophile relative to the activating gold complex.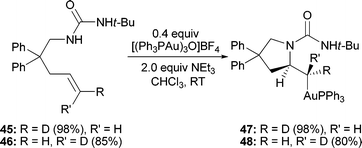 | (2) |
Further mechanistic insight into the aminoauration reaction was gained through the use of a density functional theory (DFT) computational study employing the M06 functional.21 As shown in Scheme 3, we hypothesized that during the course of the reaction, the active gold species could bind to, and therefore activate, either the alkene (Y) or nitrogen nucleophile (X). We find that the gold is preferentially coordinated to the olefin by ΔH = 2.8 kcal mol−1 relative to N for the urea (R = NH2), and ΔH = 5.2 kcal mol−1 relative the N for the carbamate (R = OMe). A series of relaxed coordinate scans found a single pathway for hydroamination: one which involves anti-addition of the nucleophile following activation of the alkene. For the case where the gold is bound to the nitrogen (X), calculations indicate that the metal shifts to bind the alkene, due to proximity. This leads to the same pathway on the potential energy surface as the Au–alkene complex (Y), indicating that a syn-addition pathway is not available for aminoauration.
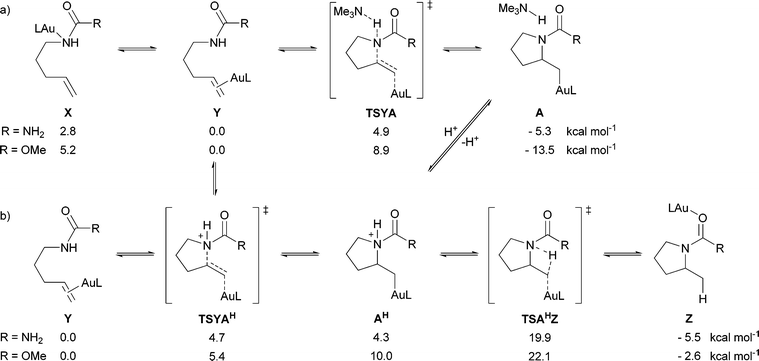 | ||
| Scheme 3 DFT computed potential enthalpies (ΔH) at 298 K for key hydroamination intermediates and transition structures (L = PH3) (a) in basic media, and (b) in acidic media. | ||
Having established the stereochemical course of the aminoauration reaction, we next investigated the role of the base. We were able to find a concerted transition state for the simultaneous nucleophilic addition and deprotonation by an exogenous base (TSYA) with an activation enthalpy of 4.9 kcal mol−1 for the model urea and 8.9 kcal mol−1 for the model carbamate. Another step-wise process where aminoauration precedes proton abstraction (TSYAH) was also discovered, having similar energies to the concerted process. We found that the cationic (non-deprotonated) product of aminoauration (AH) is a moderately unstable intermediate, where its stability may be determined by the electronic nature of L and R.
Electron-withdrawing phosphine ligands and protecting groups (urea vs. carbamate) may lower the barrier for hydroamination and stabilize the intermediate. These two related mechanisms could be in competition depending on the electronic nature of L and R, the concentration, and the relative strength of the base. Our results predict that for the carbamate (R = OMe), the concerted pathway might be lower in energy, while for the urea (R = NH2) the step-wise pathway might be lower in energy. We believe that a key characteristic of this transformation is the difference in acidity of AH before and after (or during) hydroamination.
During the course of our initial investigations, we discovered that the addition of a cationic gold complex to the isolated alkylgold complex 2 leads to a rapid equilibrium of the gold species. Surprisingly, gold(I) chloride complexes were shown to participate in this exchange reaction as well (eqn (3)). When 2 was treated with a stoichiometric amount of (p-F3CC6H4)3PAuCl, the mixture rapidly equilibrated to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3.5 mixture of 2:39. The equilibrium with an electron-donating ligand (such as MeO) favored the starting material. We found a linear relationship between the pKa′ of the arylphosphine and the log(Keq) of the exchange reaction with the isosteric ligand set (4-XC6H4)3P (Fig. 3), with the alkylgold complex favoring the more electron-poor ligand. Interestingly, the more sterically encumbered (o-tolyl)3P ligand favored the formation of the alkylgold complex, indicating a large steric component to the reaction as well. Attempts to utilize more electron rich ligands, such as NHCs and alkylphosphines, led to no significant exchange from 2.
:3.5 mixture of 2:39. The equilibrium with an electron-donating ligand (such as MeO) favored the starting material. We found a linear relationship between the pKa′ of the arylphosphine and the log(Keq) of the exchange reaction with the isosteric ligand set (4-XC6H4)3P (Fig. 3), with the alkylgold complex favoring the more electron-poor ligand. Interestingly, the more sterically encumbered (o-tolyl)3P ligand favored the formation of the alkylgold complex, indicating a large steric component to the reaction as well. Attempts to utilize more electron rich ligands, such as NHCs and alkylphosphines, led to no significant exchange from 2.
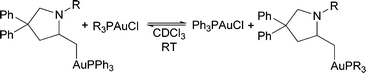 | (3) |
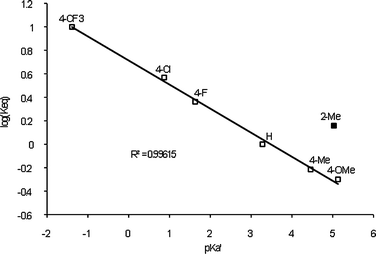 | ||
| Fig. 3 Hammett plot for the ligand exchange reaction of Ar3PAuCl with 2 for the isosteric ligand set (4-XC6H4)3P. Reactions were run at 0.1 M in CDCl3 at room temperature for 12 h. | ||
We surmised that the more electrophilic gold(I) complexes should also enhance the rate of cyclization. To explore this effect we observed the course of the reaction for five electronically differentiated arylphosphine ligands (Fig. 4). We noted a clear induction period in which the gold-oxo complex was presumably transformed into the active cationic species. This induction period was absent when an electron-withdrawn ligand was employed. For example, p-CF3, showed little to no induction period as well as an enhanced rate, reaching completion in under 30 min. After the induction period, the rate of alkylgold formation for PPh3, –OMe, and p-tolyl phosphines was similar, reaching equilibrium in 3–5 h. In accord with our exchange experiments the equilibrium concentrations of alkylgold complexes are greatest for electron withdrawing ligands (p-CF3), although the sterically bulky o-Me also exhibits enhanced stability.
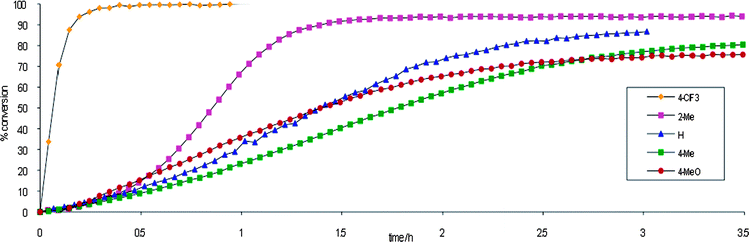 | ||
| Fig. 4 Time course of the aminoauration reaction with various arylphosphine ligands. | ||
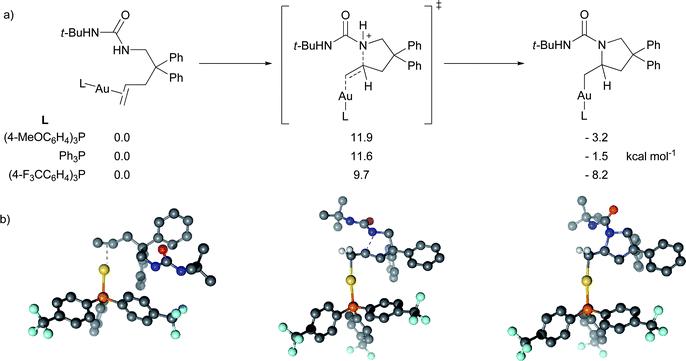 | ||
| Fig. 5 Comparison of aminoauration between p-substituted triphenylphosphine Au(I) species. (a) Enthalpic comparison and (b) representative structures for the (p-CF3Ph)3PAu(I) aminoauration reaction. | ||
Our calculations support the experimentally observed stabilization of the alkylgold complexes by electron poor ligands (Fig. 5). More electron-donating phosphines will populate the 6s orbital of the gold to a higher degree, thereby reducing the interaction with the substrate.22 This results in a less activated alkene and a higher barrier to the generation of sp3 C-bound alkylgold(I) complex. In our calculations we found that the activation barrier for the cyclization with the electron-withdrawing phosphine (p-F3CC6H4)3P is 1.9 kcal mol−1 lower than with PPh3. On the other hand, the barrier with the electron-donating (p-MeOC6H4)3P is predicted to be 0.3 kcal mol−1 higher than for PPh3. Even though the differences in the barriers are close to the limit of the accuracy of the method, the trend is consistent with our experiments. Minor structural differences, especially in the Au–C and N–C bond distances, suggest a later transition state for the more electron donating phosphine. This is a consequence of the 5d10 configuration of Au(I) and the relative population of the 6s orbital in each of the complexes.
To help determine the origin of the increased rate with the more electron withdrawing phosphine, we performed a natural bond orbital (NBO) study on the charge distribution of the transition state TSYAH.23 We find that the charge on L for Ph3P is +0.35, while it is only +0.31 for (p-F3CC6H4)3P, consistent with the higher electronegativity of the CF3 substituted ligand. Analogously, the charge on the substrate shows +0.40 for Ph3P and +0.38 for (p-F3CC6H4)3P. The higher charge on the substrate for the electron-withdrawing phosphine is consistent with the destabilization of the cyclized intermediate and its increased acidity.
Protonation of alkylgold complexes
Protonation of an alkylgold(I) intermediate has been proposed in the mechanism of gold-catalyzed alkene hydroamination reactions. We were therefore surprised to find that upon treatment with p-toluenesulfonic acid, 2 and 30 initially reverted to the starting alkenes 1 and 29, respectively (Table 5). Moreover, while the hydroamination product from 2 was formed on prolonged exposure to acid, the product from carbamate cyclization was not observed even after 18 h. A screen of various Brønsted and Lewis acids led almost exclusively to reversion of the alkylgold complex to the alkene precursor.24 As the crystal structures of the aminoauration products confirm a perfect antiperiplanar arrangement of the amine nucleophile to the gold, activation of the nitrogen by protonation would therefore allow for a facile E2 reaction to provide the starting olefin. Recent reports on the kinetics of protodeauration have indicated that the reaction can be surprisingly slow,25 allowing the elimination ample time to occur.26|
|
|||||
|---|---|---|---|---|---|
| Entry | R | Time/h | A | % yield B | % yield C |
| a Yields based on 1H NMR spectroscopy. | |||||
| 1 | NHt-Bu (2) | 1 | 40 | 0 | 60 |
| 2 | NHt-Bu (2) | 15 | 0 | 100 | 0 |
| 3 | OPh (30) | 1 | 79 | 0 | 19 |
| 4 | OPh (30) | 15 | 64 | 0 | 33 |

Our calculations showed that upon protonation of alkylgold(I) complex A, complex AH is formed, which is 4.3 kcal mol−1 higher in energy than the gold-substrate complex Y, with virtually no barrier (∼0.3 kcal mol−1AH to TSYAH) for R = NH2 and 10.0 kcal mol−1 with no barrier for R = OMe. This is consistent with the apparent reversibility of the aminoauration reaction in which upon protonation of A, starting material is observed initially. We then examined the protodeauration step; we find an internal barrier to protodeauration of 19.9 kcal mol−1 for R = NH2 and 22.1 kcal mol−1 for R = OMe. We envision that the internal proton transfer could be operative, although external protodeauration may be possible from a weak acid that does not readily protonate A. While it is difficult to draw conclusions on the validity of these alkylgold(I) complexes as intermediates in the reported alkene hydroamination reactions, these results are in accord with the hypothesis that the gold(I)-promoted addition of amines to alkenes is reversible. It is possible that under catalytic conditions the formation of an alkylgold complex merely serves to liberate an equivalent of Brønsted acid,27 which catalyzes the observed hydroamination reactions.
Given our inability to deaurate the alkylgold complexes by protonation, we examined other means to functionalize the carbon–gold bond.28,29 In early studies with related metals, such as platinum, palladium, and mercury, it was demonstrated that the metal could be removed under reductive or hydrogenation conditions.6 No reaction was observed when alkylgold 2 was treated to hydrogenation conditions (H2, 1 atm); however, treatment with NaBH4 in THF led to 27% of the protodeauration product after 12 h with no indication of reversion to starting olefin. The conversions were improved through the use of a protic solvent, such as EtOH, which provided 81% yield of the formal hydroamination product (eqn (4)). Similar yields were observed for the formation of 50 from alkylgold 22. While reductive removal of the metal is useful for stoichiometric reactions, it may be difficult to use in a catalytic sense, due to the formation of Au(0) clusters from the reduction of the gold, which prevents catalyst turnover.
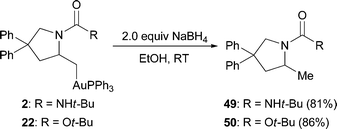 | (4) |
Conclusions
In conclusion, we have provided the first direct crystallographic evidence for electrophilic activation of alkenes for nucleophilic addition by isolation of the products of olefin aminoauration. A variety of protected amine nucleophiles proved competent, and the use of electron deficient ligands on the gold center accelerated the formation of the aminoauration product. In addition, we have provided the first experimental verification of an anti-addition mechanism for alkene aminoauration, which is supported by DFT calculations. While these complexes potentially provide support for gold-catalyzed hydroamination reactions, all attempts to complete the catalytic cycle by means of protodeauration of the purported hydroamination intermediates led only to reversion to the starting alkenes. Furthermore, taking into account the high energy barrier calculated for protodeauration, the precise involvement of gold complexes in catalytic hydroamination reactions remains unclear. Ongoing studies in the laboratory are aimed at discerning the importance of this intermediate in the related catalytic reactions.Notes and references
- (a) K. A. Hofmann and J. Sand, Chem. Ber., 1900, 1340 CrossRef CAS; (b) K. A. Hofmann and J. Sand, Chem. Ber., 1900, 1353; (c) for an early review see: J. Chatt, Chem. Rev., 1951, 48, 7 Search PubMed.
- J. Chatt, L. M. Vallarino and L. M. Venanzi, J. Chem. Soc., 1957, 2496 RSC.
- J. Chatt, L. M. Vallarino and L. M. Vananzi, J. Chem. Soc., 1957, 3413 RSC.
- For selected examples from Hg-promoted intramolecular hydroamination see: (a) J. J. Perie, J. P. Laval, J. Roussel and A. Lattes, Tetrahedron, 1972, 28, 675 CrossRef CAS; (b) S. Danishefsky, E. Taniyama and R. R. Webb II, Tetrahedron Lett., 1983, 24, 11 CrossRef CAS.
- (a) A. Panunzi, A. De Renzi, R. Palumbo and G. Paiaro, J. Am. Chem. Soc., 1969, 91, 3879 CrossRef CAS; (b) D. Hollings, M. Green and D. V. Claridge, J. Organomet. Chem., 1973, 54, 399 CrossRef CAS; (c) J. K. K. Sarhan, M. Green and I. M. Al-Najjar, J. Chem. Soc., Dalton Trans., 1984, 771 RSC; (d) for an example of Pt-promoted intermolecular hydroamination see: J. Ambühl, P. S. Pregosin, L. M. Venanzi, G. Ughetto and L. Zambonelli, Angew. Chem., Int. Ed. Engl., 1975, 14, 369 Search PubMed.
- (a) B. Åkermark, J. Bäckvall, L. S. Hegedus, K. Zetterberg, K. Siirala-Hansén and K. Sjöberg, J. Organomet. Chem., 1974, 72, 127 CrossRef CAS; (b) L. S. Hegedus and J. M. McKearin, J. Am. Chem. Soc., 1982, 104, 2444 CrossRef CAS; (c) for a recent review of aminopalladation reactions, see: A. Minatti and K. Muñiz, Chem. Soc. Rev., 2007, 36, 1142 Search PubMed.
- Hg: (a) R. Pathak, P. Naicker, W. A. Thompson, A. Fernandes, C. B. de Konig and W. A. L. van Otterol, Eur. J. Org. Chem., 2007, 5337 CrossRef CAS. Pt: (b) J. M. Hoover, J. Freudenthal, F. E. Michael and J. M. Mayer, Organometallics, 2008, 27, 2238 CrossRef CAS. Pd: (c) B. M. Cochran and F. E. Michael, J. Am. Chem. Soc., 2008, 130, 2786 CrossRef CAS.
- Late transition metal alkyl complexes readily undergo β-hydride elimination: L. Hegedus, Angew. Chem., Int. Ed. Engl., 1988, 27, 1113 Search PubMed.
- For general reviews of transition metal catalyzed hydroamination, see: (a) T. E. Müller, K. C. Hultsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795 CrossRef. For a review of Pt-catalyzed hydroamination see: (b) J.-J. Brunet, N.-C. Chu and M. Rodriguez-Zubiri, Eur. J. Inorg. Chem., 2007, 4711 CrossRef CAS.
- (a) X. Wang and R. A. Widenhoefer, Organometallics, 2004, 23, 1649 CrossRef CAS; (b) C. F. Bender and R. A. Widenhoefer, J. Am. Chem. Soc., 2005, 127, 1070 CrossRef CAS; (c) C. Liu, C. F. Bender, X. Han and R. A. Widenhoefer, Chem. Commun., 2007, 3607 RSC.
- F. E. Michael and B. M. Cochran, J. Am. Chem. Soc., 2006, 128, 4246 CrossRef CAS.
- J. L. McBee, A. T. Bell and T. D. Tilley, J. Am. Chem. Soc., 2008, 130, 16562 CrossRef CAS.
- For recent reviews on gold-catalyzed reactions see: (a) A. Fürstner, Chem. Soc. Rev., 2009, 38, 3208 RSC; (b) Z. Li, C. Brouwer and C. He, Chem. Rev., 2008, 108, 3239 CrossRef CAS; (c) D. J. Gorin, B. D. Sherry and F. D. Toste, Chem. Rev., 2008, 108, 3351 CrossRef CAS; (d) H. C. Shen, Tetrahedron, 2008, 64, 3885 CrossRef CAS.
- For examples of gold-catalyzed hydroamination of alkenes, see: (a) J. Zhang, C.-G. Yang and C. He, J. Am. Chem. Soc., 2006, 128, 1798 CrossRef; (b) X. Han and R. A. Widenhoefer, Angew. Chem., Int. Ed., 2006, 45, 1747 CrossRef CAS; (c) X.-Y. Liu, C.-H. Li and C.-M. Che, Org. Lett., 2006, 8, 2707 CrossRef CAS; (d) C. F. Bender and R. A. Widenhoefer, Chem. Commun., 2006, 4143 RSC; (e) C. F. Bender and R. A. Widenhoefer, Org. Lett., 2006, 8, 5303 CrossRef CAS. For examples of gold(I)-catalyzed additions of other nucleophiles to alkenes, see: (f) C.-G. Yang and C. He, J. Am. Chem. Soc., 2005, 127, 6966 CrossRef CAS; (g) C.-Y. Zhou and C.-M. Ming, J. Am. Chem. Soc., 2007, 129, 5828 CrossRef CAS; (h) M.-Z. Wang, M.-K. Wong and C.-M. Che, Chem.–Eur. J., 2008, 14, 8353 CrossRef CAS; (i) A. Iglesias and K. Muñiz, Chem.–Eur. J., 2009, 15, 10563 CrossRef CAS; (j) G. Zhang, L. Cui, Y. Wang and L. Zhang, J. Am. Chem. Soc., 2010, 132, 1474 CrossRef CAS. For a review, see: (k) R. A. Widenhoefer and X. Han, Eur. J. Org. Chem., 2006, 4555.
- (a) D. C. Rosenfeld, S. Shekhar, A. Takemiya, M. Utsunomiya and J. F. Hartwig, Org. Lett., 2006, 8, 4179 CrossRef CAS; (b) B. Schlummer and J. F. Hartwig, Org. Lett., 2002, 4, 1471 CrossRef CAS; (c) Z. Li, J. Zhang, C. Brouwer, C.-G. Yang, N. W. Reich and C. He, Org. Lett., 2006, 8, 4175 CrossRef CAS. For a review, see: (d) J. G. Taylor, L. A. Adrio and K. K. Hii, Dalton Trans., 2010, 39, 1171 RSC.
- For isolation of vinylgold(I) intermediates from additions to alkynes and allenes see: (a) J. A. Akana, K. X. Bhattacharyya, P. Muller and J. P. Sadighi, J. Am. Chem. Soc., 2007, 129, 7736 CrossRef CAS; (b) L.-P. Liu and G. B. Hammond, Chem.–Asian J., 2009, 4, 1230 CrossRef CAS; (c) L.-P. Liu, B. Xu, M. A. Mashita and G. B. Hammond, J. Am. Chem. Soc., 2008, 130, 17642 CrossRef CAS; (d) Y. Shi, S. D. Ramgren and S. A. Blum, Organometallics, 2009, 28, 1275 CrossRef CAS; (e) A. S. K. Hashmi, A. M. Schuster and F. Rominger, Angew. Chem., Int. Ed., 2009, 48, 8247 CrossRef CAS; (f) X. Zeng, R. Kinjo, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 942 CAS.
- For X-ray structures of L-gold(I)-alkene complexes see: (a) N. D. Shapiro and F. D. Toste, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2779 CrossRef CAS; (b) T. J. Brown, M. G. Dickens and R. A. Widenhoefer, J. Am. Chem. Soc., 2009, 131, 6350 CrossRef CAS; (c) T. N. Hooper, M. Green, J. E. McGrady, J. R. Patel and C. A. Russell, Chem. Commun., 2009, 3877 RSC; (d) T. J. Brown, M. G. Dickens and R. A. Widenhoefer, Chem. Commun., 2009, 6451 RSC; (e) T. N. Hooper, C. P. Butts, M. Green, M. F. Haddow, J. E. McGrady and C. A. Russell, Chem.–Eur. J., 2009, 15, 12196 CrossRef CAS.
- (a) N. C. Baenziger, W. E. Bennett and D. M. Soborofe, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1976, 32, 962 CrossRef; (b) P. D. Gavens, J. J. Guy, M. Mays and G. M. Sheldrick, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1977, 33, 137 CrossRef.
- For D-labelling studies probing the stereochemistry of alkyne addition, see: (a) J. J. Kennedy-Smith, S. T. Staben and F. D. Toste, J. Am. Chem. Soc., 2004, 126, 4526 CrossRef CAS; (b) A. S. K. Hashmi, J. P. Weyrauch, W. Frey and J. W. Bats, Org. Lett., 2004, 6, 4391 CrossRef CAS.
- On the basis of the dihedral angles in the X-ray crystal structure of alkylgold 2, we predicted J3 values of 1.2, and 9.1 Hz, for H19A and H19B protons, respectively.
- Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157 CrossRef CAS.
- D. Benitez, N. D. Shapiro, E. Tkatchouk, Y. Wang, W. A. Goddard III. and F. D. Toste, Nat. Chem., 2009, 1, 482 Search PubMed.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. B. Carpenter, J. A. Bohmann, C. M. Morales and F. Weinhold, NBO 5.0, Theoretical Chemistry Institute, University of Wisconsin, Madison, 2001 Search PubMed.
- For the reaction of 2 and 30 with alternative Brønsted acids, see ESI†.
- K. E. Roth and S. A. Blum, Organometallics, 2010, 29, 1712 CrossRef CAS.
- Reaction of a 1:1 mixture of alkylgold complex 2 and PhAuPPh3 with HCl, TsOH, or BzOH, showed that the rate of reversion of 2 to olefin 1 far exceeds protodeauration of PhAuPPh3 to form benzene.
- E. Szuromi and P. R. Sharp, Organometallics, 2006, 25, 558 CrossRef CAS.
- M. Murakami, M. Inouye, M. Suginome and Y. Ito, Bull. Chem. Soc. Jpn., 1988, 61, 3641.
- We were able to find conditions for the palladium-catalyzed coupling of MeAuPPh3 with PhI (see ESI†); however, application of these conditions to various alkylgold(I) complexes also returned the starting alkene. For Pd-catalyzed coupling of vinylgold(I) complexes, see: ref. 16d.
Footnote |
| † Electronic supplementary information (ESI) available: Data for new compounds, experimental procedures, and computational data. CCDC reference numbers 772397 and 772398. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0sc00255k |
| This journal is © The Royal Society of Chemistry 2010 |