Solvent-free Diels–Alder reactions of in situ generated cyclopentadiene†
David Huertas, Melinda Florscher and Veljko Dragojlovic*
Wilkes Honors College of Florida Atlantic University, 5353 Parkside Drive, Jupiter, FL 33458, USA. E-mail: vdragojl@fau.edu; Fax: 561-799-8602; Tel: 561-799-8012
First published on 7th November 2008
Abstract
A solvent-free Diels–Alder reaction was carried out by heating a mixture of dicyclopentadiene and a dienophile. Cyclopentadiene, formed in situ, reacted with the dienophile in a thermodynamically controlled reaction. Besides being solvent-free, the described procedure allows for almost complete utilization of dicyclopentadiene and avoids handling of noxious and hazardous cyclopentadiene. The reaction worked well with dienophiles such as maleic anhydride and unsaturated esters. However, unsaturated acids were not suitable dienophiles, yielding Diels–Alder adducts in a low yield.
Introduction
The Diels–Alder reaction is one of the most important methods for formation of six-membered rings.1 Cyclopentadiene is one of the dienes frequently used in Diels–Alder reactions. It is unstable under ordinary conditions and, at room temperature, dimerizes into dicyclopentadiene. Usually it is obtained by cracking of dicyclopentadiene and is used immediately in a reaction. Cyclopentadiene has a rather strong and disagreeable odor and cracking has to be done in a fumehood. Furthermore, while retro Diels–Alder reactions are well known and have been extensively used in organic synthesis,2 with numerous examples of an in situ generated diene being trapped by a more reactive dienophile,3 other than a report of a solvent-free reaction of dicyclopentadiene with ethylene under high temperarature and pressure in an autoclave4 and a similar process described in a patent,5 dicyclopentadiene as an in situ source of cyclopentadiene has received relatively little attention. That is surprising given both its importance and safety implications. Pure cyclopentadiene has to be stored at a low temperature (−20 °C). Under ordinary conditions it undergoes a spontaneous and highly exothermic dimerization into dicyclopentadiene6 and has been implicated in a fatal accident.7 Moreover, only about 2/3 of dicyclopentadiene can be cracked.8 The rest of it forms oligomers, which require considerably higher temperature for cracking. Thus, when cyclopentadiene is obtained by cracking of dicyclopentadiene, one can expect, at best, an overall 65–70% reaction yield with respect to dicyclopentadiene. Therefore, a procedure that allows use of dicyclopentadiene directly would have important practical advantages. Finally, such a procedure may allow isolation of stereoisomers that are not commonly produced in a Diels–Alder reaction of cyclopentadiene. Recently, Diels–Alder reactions under solvent-free conditions in the presence of a catalyst9 or under microwave irradiation10 have been reported. A retro Diels–Alder reaction of oxanorbornenes under solvent-free conditions and microwave irradiation afforded the corresponding products in high yields.11 A patent that describes preparation of 5-norbornene-2-carboxylic acid by heating dicyclopentadine with acrylic acid at ambient pressure in the presence of a free radical inhibitor12 will be discussed in detail in the Results and Discussion section.We have developed a solvent-free procedure in which cyclopentadiene generated in situ is used in a Diels–Alder reaction (Scheme 1). Advantages of this procedure are that cyclopentadiene reacts as it is generated and thus there are neither safety problems associated with use of cyclopentadiene nor problems with formation of oligomers. As a result, most of the starting dicyclopentadiene is utilized. Furthermore, as the reaction is done at a relatively high temperature (162–206 °C), thermodynamically preferred reaction products are obtained. Ordinarily, Diels–Alder reactions involving cyclopentadiene proceed under kinetic conditions to give predominantly or exclusively the endo isomer.13 By adjusting the reaction times, one can obtain either the exo or endo isomer as the major reaction product. Finally, by avoiding use of a reaction solvent this is a “greener” procedure compared to traditional Diels–Alder reactions.
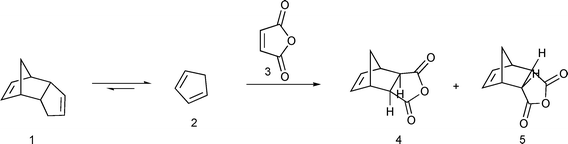 | ||
| Scheme 1 | ||
Results and discussion
Reactions of dicyclopentadiene with dienophiles were relatively fast (5–25 min). In addition, none, or very little, of the byproducts such as tri- and tetracyclopentadienes, which are formed in the course of an ordinary cracking, were observed and one did not have to isolate or handle noxious and unstable cyclopentadiene.Usually, in a Diels–Alder reaction the diene is used in an excess. However, in this case, use of the diene precursor, dicyclopentadiene, in an excess resulted in lower yields and lower purity of the products. In particular, a larger amount of cyclopentadiene oligomers were formed. Therefore, we opted for use of a small excess of the dienophile. Thus, reaction of dicyclopentadiene with 2.30 equivalents (1.15 equivalents with respect to in situ generated cyclopentadiene) of maleic anhydride gave 5-norbornene-2,3-dicarboxylic anhydride cleanly and in good yield (Table 1, entry 1). Very little of the cyclopentadiene oligomers were formed (<5% according to GC-MS analysis). It was important to keep the reaction times within the optimal range. The reaction should be run until the reaction mixture turns dark yellow and the reflux stops. Shorter reaction times resulted in an incomplete reaction, while longer reaction times gave charred products and lower yields. The reaction was successfully scaled up to 0.6 mol scale (67.6 g maleic anhydride and 49.8 mL dicyclopentadiene in a 300 mL round bottom flask). As there was some foaming in the course of the reaction, it may be tempting to use larger flasks. Furthermore, a larger reaction flask would allow for a more efficient heat transfer. Interestingly, use of larger reaction vessels resulted in charred products and lower yields. Use of smaller reaction vessels (10 mL round bottom flask for 75 mmol, 200 mL for 0.3 mol and 300 mL for 0.6 mol reaction) gave better results. Overall, our experience was that it was best to use the smallest possible flask, which should be ∼1/3 filled.
| Entry | Dienophile | Reaction time/min | Products | Yield (exo/endo) | Procedure |
|---|---|---|---|---|---|
| a Isolated yield.b GC ratio.c Product was 95:5 endo/exo mixture according to GC analysis.d Estimated yield based on the amount of recovered material and GC analysis. | |||||
| 1 | 3 | 15–22 | 4/5 | 37%a : 35%a,c | A |
| 2 | 3 | 8–10 | 5 | 32%a (1:6)b | A |
| 3 | 6 | 25 | 7/8 | 78%a (57:35)b | A |
| 4 |  | 15 | 9 | 83%a | A |
| 10 | |||||
| 5 |  | 12 |  | 88%a | A |
| 11 | 12 | ||||
| 6 |  | 5 | 4/5 | <10%d | B |
| 13 | |||||
| 7 | 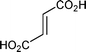 | 5 | 4/5 | ∼20%d | B |
| 14 | |||||
| 8 |  | 5 | 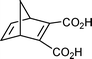 | 0 | B |
| 15 | 16 | ||||
| 9 |  | 5 | 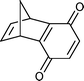 | ∼5%d | B |
| 17 | 18 |
Both dicyclopentadiene and cyclopentadiene react with maleic anhydride in the presence of a free radical catalyst to give polymers.14 However, while a solvent-free reaction of cyclopentadiene with ethylene under high pressure and high temperature gave norbornene in an acceptable yield,4 to our knowledge reaction of neat dicyclopentadiene with maleic anhydride has not been investigated. In contrast to a Diels–Alder reaction conducted at ambient temperature,15 the reaction was thermodynamically controlled and a mixture of exo and endo isomers in approximately equilibrium amounts was obtained. The reaction time was only 15–22 minutes which compares favorably with the standard procedure for obtaining a thermodynamic mixture of exo and endo isomers by equilibrating the endo isomer at 190 °C for 1.5 hours.16 A small excess of maleic anhydride ensured complete utilization of cyclopentadiene and simplified the work up. The isolated yield of a mixture of exo- and endo-5-norbornene-2,3-dicarboxylic anhydride was ∼72% (average of five runs, Table 1, entry 1). Remaining endo-isomer can be re-equilibrated according to a literature procedure16 to give an overall yield of ∼60% of the exo-compound. It has been reported that the thermodynamic mixture contains 57% and 43% of exo and endo isomers by Craig,16 or 54% and 46% of exo and endo isomers by Pincock et al.17 Our results were similar, and the equilibrium mixtures contained a small excess of the exo isomer. These results are in contrast to recent calculations, according to which the endo isomer is more stable by 1.9 kJ/mol.18
The endo isomer was obtained as the major reaction product when the conversion was kept low by keeping the reaction time short. Attempts to increase the amount of endo isomer by slow addition of dicyclopentadiene, either manually or by use of syringe drive, failed. Reproducibility was very poor, with both yields and exo/endo ratios varying considerably between the individual trials. The amount of the starting maleic anhydride did not appear to play a role. Thus, the reaction times, yields of the reaction products and exo/endo ratio were about the same when maleic anhydride was used in 2–6 equivalent amounts (1–3 equivalents with respect to in situ generated cyclopentadiene). The best yield (32% of a 6:1 mixture of endo and exo isomers) of the endo product was obtained when dicyclopentadiene was added to an excess (4.50 equivalents) of boiling maleic anhydride (206 °C) and the reaction time was kept short (Table 1, entry 2). Craig reported that a reaction of maleic anhydride and cyclopentadiene at 190 °C for 10 minutes gave an 82:18 ratio of endo and exo isomers.16
When the reaction was done at 206 °C on a mixture that corresponded to 50% conversion (37.5 mmol of 5-norbornene-2,3-dicarboxylic anhydride, 37.5 mmol of maleic anhydride and 32.5 mmol of dicyclopentadiene), an approximately thermodynamic ratio of exo and endo isomers was obtained in only 5 minutes (Table 2). Interestingly, the reaction outcome was about the same regardless of whether the added 5-norbornene-2,3-dicarboxylic anhydride was the exo-isomer, the endo-isomer, or a 1:1 mixture of the exo and endo isomers. Yields were slightly better compared to a reaction of maleic anhydride and cyclopentadiene (Table 1, entry 1). Under the same conditions, equilibration of a pure endo isomer took 15 minutes.
| Entry | Added adducta | Time | Isolated Yield (exo/endo) |
|---|---|---|---|
| a Reaction conditions: 37.5 mmol of Diels–Alder adduct and 37.5 mmol of maleic anhydride were heated for 2 minutes followed by addition of 16.25 mmol of dicyclopentadiene. Heating was continued for the time indicated. | |||
| 1 | exo | 4′30″ | 44% : 42% |
| 2 | endo | 5′30″ | 36% : 35% |
| 3 | exo/endo (1:1) | 5′ | 41% : 39% |
Esters (Table 1, entries 3–5) worked very well and yields were very good. The reaction products were those expected for a Diels–Alder reaction run under a thermodynamic conditions. As the Diels–Alder reaction usually gives a kinetic (endo) product, this is a way to obtain a thermodynamic product. Dimethyl maleate gave exo and endo isomers in a 57:35 ratio (Table 1, entry 3). There was some isomerization of dimethyl maleate into dimethyl fumarate and a small amount (∼5%, GC analysis, not isolated) of dimethyl trans-5-norbornene-2,3-dicarboxylate was observed (GC analysis, Scheme 2). Both dimethyl fumarate and dimethyl acetylenedicarboxylate gave the corresponding Diels–Alder adducts cleanly and in high yields (Table 1, entries 4 and 5).
 | ||
| Scheme 2 | ||
Carboxylic acids were not suitable substrates for this reaction (Table 1, entries 6–8). Complex mixtures that consisted mainly of insoluble, presumably polymeric, material were obtained accompanied by small amounts of Diels–Alder adducts. Reaction of dicyclopentadiene with maleic acid gave very little (<10% according to GC-MS analysis) of a ∼1:1 mixture of exo and endo isomers of 5-norbornene-2,3-dicarboxylic anhydride (Table 1, entry 6). It is known that upon heating, maleic acid isomerizes into fumaric acid, which in turn isomerizes into maleic anhydride.19 Fumaric acid gave a somewhat better yield of the anhydrides (Table 1, entry 7) and it may be possible to optimize the reaction conditions to improve the yield. However, since maleic anhydride itself works quite well there was no apparent reason to attempt such an exercise. According to the previously mentioned patent, a solvent-free reaction between dicyclopentadiene and acrylic acid works well in the presence of a free radical inhibitor such as hydroquinone.12 While it was stated that 5-norbornene-2-carboxylic acid was isolated in 89% yield, there was no mention of the exo/endo ratio and very few experimental details were provided. In our hands, the reaction was rather slow and heating of dicyclopentadiene prior to addition of a dienophile resulted in formation of a large amount of tri- and tetracyclopentadienes. As expected, addition of a free radical inhibitor prevented polymerization of acrylic acid, but had no effect on oligomerization of in situ generated cyclopentadiene. Thus, the best result we obtained was ∼40% yield of 5-norbornene-2-carboxylic acid. We found that a slow reverse addition (dicyclopentadiene to a boiling acrylic acid in the presence of hydroquinone) gave better results and 5-norbornene-2-carboxylic acid was obtained in ∼60% yield (∼1:1 endo/exo mixture isolated as the corresponding methyl esters). As addition of a free radical inhibitor only prevents polymerization, such modification does not address thermal instability of acids, which is the main problem when using maleic and fumaric acids.
Reaction of dicyclopentadiene with an excess of molten 1,4-benzoquineone was extremely vigorous. Within ∼5 minutes most of the reaction mixture was a charred solid. Extraction yielded the Diels–Alder adduct and dehydrogenated product 18 in a low yield accompanied by a considerable amount of hydroquinone (Table 1, entry 9).
Conclusion
We have developed an environmentally friendly methodology for Diels–Alder reactions of cyclopentadiene by using dicyclopentadiene directly in the reaction, without previous cracking, and by conducting the reactions under solvent-free conditions. Reaction products were obtained in multi-gram quantities. Limitations of the described procedure include use of thermally unstable dienophiles, such as benzoquinone and unsaturated acids. The Meinwald and Hudak article, as well as previously published patents, provide complementary procedures, which may be useful for reactions involving low boiling and polymerizable dienophiles.4,5,12 Somewhat less reactive dienophiles, such as esters and maleic anhydride, worked very well and gave a thermodynamic mixture of exo and endo isomers. This is a “point reaction”– there is a relatively narrow range of optimal conditions and deviation from them reduces the yield.20 Although there are some limitations to the choice of suitable dienophiles, advantages of the process are that it generates no waste, maximizes incorporation of starting materials into products, is done under ambient pressure, is solvent-free, utilizes no other reagents nor catalysts, avoids safety hazards associated with handling of cyclopentadiene and thus conforms to most of the twelve green chemistry principles.21 Finally, and perhaps most importantly, it allows for a fast and convenient preparation of thermodynamic products of Diels–Alder reactions.Experimental
1H NMR spectra were recorded on a Bruker Avance 400 spectrometer. GC-MS analyses were performed by means of Agilent 6890 N Gas Chromatograph equipped with HP-5MS 30 m × 0.25 mm column (Cat. No. 19091S-433) and Agilent 5973 N MSD. Dicyclopentadiene, maleic anhydride, maleic acid, fumaric acid, acetylene dicarboxylic acid, dimethyl maleate, dimethyl fumarate, dimethyl acetylene dicarboxylate and 1,4-benzoquinone were purchased from Acros Organic and used without further purification. Acetone, hexanes and ethyl acetate were purchased from Fisher Scientific Company and used without further purification. Deuterated solvents and silica gel were purchased from Aldrich Chemical Company and used without further purification. Separations were done either by column chromatography or by preparative radial thin layer chromatography (Harrison Chromatotron). All of the isolated products were known compounds and gave satisfactory 1H NMR and GC-MS data.Procedure A
Dienophile (75 mmol) was placed in a 10 mL round bottom flask equipped with a condenser. It was heated with stirring until it began to boil. Dicyclopentadiene (5.35 mL, 32.5 mmol) was added in a single portion and the reaction was continued until the reflux stopped and the reaction mixture turned yellow. Exo and endo isomers were separated by radial thin layer chromatography (Harrison Chromatotron) eluting with ethyl acetate:hexanes (1:4 by volume) in the case of dimethyl cis-5-norbornene-2,3-dicarboxylate or ethyl acetate:acetone:hexanes (1:2:6 by volume) in the case of 5-norbornene-2,3-dicarboxylic anhydride. Other products were purified by column chromatography eluting with ethyl acetate:hexanes (1:3 by volume).Procedure B
Diene (75 mmol) and dicyclopentadiene (5.35 mL, 32.5 mmol) were placed in a 10 mL round bottom flask equipped with a condenser. The mixture was heated to reflux for the time indicated (Table 1, entries 6–9). Products were extracted from the solid residue with ethyl acetate and analyzed by GC-MS.Acknowledgements
We thank Salvatore Lepore, Department of Chemistry, Florida Atlantic University, for helpful discussions and Nicole Windmon from Department of Chemistry, Florida Atlantic University, for recording 1H NMR spectra. Partial financial support from the Wilkes Honors College of Florida Atlantic University is gratefully acknowledged.Notes and references
- K. Takao, R. Munakata and K. Tadano, Chem. Rev., 2005, 105, 4779–4807 CrossRef CAS; E. J. Corey, Angew. Chem., Int. Ed., 2002, 41, 1650–1667 CrossRef CAS; K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2002, 41, 1668–1698 CrossRef CAS; U. Pindur, G. Lutz and C. Otto, Chem. Rev., 1993, 93, 741–761 CrossRef CAS.
- B. Rickborn, Organic Reactions (New York), 1998, 52, 1–393 Search PubMed; A. Ichihara, Synthesis, 1987, 207–222 CrossRef CAS; J. L. Ripoll, A. Rouessac and F. Rouessac, Tetrahedron, 1978, 34, 19–40 CrossRef CAS.
- J. H. M. Lange, A. J. H. Klunder and B. Zwanenburg, Tetrahedron Lett., 1988, 29, 2365–2368 CrossRef CAS; D. Mackay, D. Papadopoulos and N. J. Taylor, J. Chem. Soc. Chem. Commun., 1992, 325–327 RSC; R. N. Warrener, M. A. Houghton, A. C. Schultz, F. R. Keene, L. S. Kelso, R. Dash and D. N. Butler, Chem. Commun., 1996, 1151–1152 RSC.
- J. Meinwald and N. J. Hudak, Organic Syntheses Coll., 1963, 4, 738 Search PubMed , http://www.orgsyn.org/orgsyn/pdfs/CV4P0738.pdf, accessed on April 13, 2008.
- U. Takashi and K. Shoichi, Jpn. Kokai Tokkyo Koho (1974), JP, 49–048650 A (in Japanese), Chem. Abstr., 1974, 81, 135555.
- D. J. am Ende, D. C. Whritenour and J. W. Coe, Org. Process Res. Dev., 2007, 11, 1141–1146 Search PubMed.
- A. Starkie and S. Rowe, Chem. Br., 1996, 32(2), 34–8 Search PubMed.
- R. B. Moffett, Organic Syntheses Coll., 1963, 4, 238 Search PubMed , http://www.orgsyn.org/orgsyn/pdfs/CV4P0238.pdf, accessed on April 13, 2008.
- F. Fringuelli, R. Girotti, F. Pizzo and L. Vaccaro, Org. Lett., 2006, 8, 2487–2489 CrossRef CAS; S. Imachi and M. Onaka, Tetrahedron Lett., 2004, 45, 4943–4946 CrossRef CAS; J. Long, J. Hu, X. Shen, B. Ji and K. Ding, J. Am. Chem. Soc., 2002, 124, 10–11 CrossRef CAS; D. Bradley, Chemistry in Britain, 2002, 38, 42–45 Search PubMed; M. Avalos, R. Babiano, J. L. Bravo, P. Cintas, J. L. Jimenez, J. C. Palacios and B. C. Ranu, Tetrahedron Lett., 1998, 39, 2013–2016 CrossRef CAS.
- A. Loupy, F. Maurel and A. Sabatie-Gogova, Tetrahedron, 2004, 60, 1683–1691 CrossRef CAS.
- M. Bortolussi, R. Bloch and A. Loupy, J. Chem. Res. (S), 1998, 34–35 RSC.
- Y. Murakami, M. Asagai, and I. Yamane, Jpn. Kokai Tokkyo Koho ( 2000), JP, 2000–319222 A (in Japanese). A mechanical translation is available on the Japanese Patent Office web site: http://www.jpo.go.jp/.
- D. Suárez and J. A. Sordo, Chem. Commun., 1998, 385–386 RSC; R. Gleiter and M. C. Böhm, Pure Appl. Chem., 1983, 55, 237–244 CAS; M. Kakushima, Can. J. Chem., 1979, 57, 2564–2568 CAS.
- N. G. Gaylord and A. B. Deshpande, Journal of Macromolecular Science, Part A, 1977, 11, 1795–1807 Search PubMed; N. G. Gaylord, A. B. Deshpande and M. Martan, J. Polym. Sci. Polym. Letters Ed., 1976, 14, 679–682 Search PubMed.
- W. J. Sheppard, J. Chem. Educ., 1963, 40, 40–41 CrossRef CAS.
- D. Craig, J. Am. Chem. Soc., 1951, 73, 4889–4892 CrossRef CAS.
- R. E. Pincock, K. R. Wilson and T. E. Kiovsky, J. Am. Chem. Soc., 1967, 89, 6890–6897 CrossRef CAS.
- L. Rulíšek, P. Šebek, Z. Havlas, R. Hrabal, P. Čapek and A. Svatoš, J. Org. Chem., 2005, 70, 6295–6302 CrossRef CAS.
- W. D. Robinson and R. A. Mount, Kirk-Othmer Encycl. Chem. Technol., 1981, 14, 778–779 Search PubMed.
- S. Turner, Design of Chemical Synthesis, Elsevier, Amsterdam, 1976, 21-22 Search PubMed.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, Oxford, 1998 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details and 1H NMR spectra of the isolated compounds. See DOI: 10.1039/b813485e |
| This journal is © The Royal Society of Chemistry 2009 |