Biodegradable pyridinium ionic liquids: design, synthesis and evaluation
Jitendra R. Harjania, Robert D. Singerb, M. Teresa Garciac and Peter J. Scammells*a
aMedicinal Chemistry and Drug Action, Monash Institute of Pharmaceutical Sciences, Monash University, Parkville, 3052, Victoria, Australia. E-mail: peter.scammells@vcp.monash.edu.au; Fax: +61 3 99039582; Tel: +61 3 9903 9542
bDepartment of Chemistry, Saint Mary's University, Halifax, Nova Scotia B3H 3C3, Canada
cDepartment of Surfactant Technology, IIQAB-CSIC, Jordi Girona 18–26, 08034, Spain. E-mail: mtgbet@iiqab.csic.es; Fax: +34 93 204 5904; Tel: +34 93 400 6100
First published on 7th November 2008
Abstract
A range of ionic liquids (ILs) with a pyridinium cation were synthesised and their biodegradability was evaluated using the CO2 Headspace test (ISO 14593). ILs bearing an ester side chain moiety were prepared from either pyridine or nicotinic acid and showed high levels of biodegradation under aerobic conditions and can be classified as ‘readily biodegradable’. In contrast, pyridinium ILs with alkyl side chains showed significantly lower levels of biodegradability in the same test. The utility of the biodegradable IL 6c as a reaction solvent for the Diels–Alder reaction was also investigated.
Introduction
Ionic liquids (ILs) have earned themselves a reputation as excellent reaction media. Earlier applications of ILs were mainly as electrolytes, solvents and extractants. The scope of their application is now broader and covers their use as reaction media, fuel cells, optical fluids, solar cells, energetic materials, heat transfer fluids and lubricants, to name only a few.1 In chemical synthesis, the initial interest in the use of ILs was mainly to realize their advantages for transition metal mediated catalysis in biphasic reaction systems.2 With time, this was further broadened by utilising them for catalysis using supported IL phases,3 designing task-specific (functionalized) ILs for catalysis,4 using ILs as supports for the substrates5 and designing novel materials.6 The properties of ILs, such as broad solvating ability, good thermal and chemical stability, non-volatility and non-flammability under operational conditions, not only adds to their versatility, but may make them a safer substitute to conventional solvents. ILs as reaction media can offer better reaction selectivity, higher reaction yields and reusability. These advantages further add to their green attributes. Although, some of these properties, such as good chemical stability, volatility etc., have been challenged, it is notable that not all the ILs have all these ideal properties.7 By varying the cations and anions that constitute their structure, it is typically possible to design ILs with the desired solvent characteristics for the chemical process of interest. The only principles of green chemistry which may disqualify ILs as a green solvent replacements are the necessity to ‘use safer solvents and reaction conditions’ and ‘design chemicals and products to degrade after use’.8 Designing ILs that are safe to release in to the environment would address both of these issues. As ILs make a transition from academic curiosities to commercial commodities, their environmental impact is certainly concerning.9 Rightfully so, the life cycle assessment of newer chemicals (such as ILs) and designing benign chemicals are now considered as an integral part of green chemistry.10In contrast to the other possible fates of any chemical released into the environment, biodegradation appears to be the cleanest ultimate fate for any compound. This natural process of decay is an intensive collective endeavor of a number of microbes with different metabolic capabilities, ultimately leading to the formation of non-toxic inorganic products such as carbon dioxide, water and/or biomass. The assessment of environmental impact of ILs has mainly centered on the evaluation of their toxicity,10b,11 however it is known that a refractory organic substance, if released in to the environment, would bio-accumulate and may eventually lead to chronic toxic effects.12 Earlier investigations have also indicated that structural features, such as alkyl chain length, that can decrease resistance to microbial attack may also increase the toxicity of the ILs.13 As suitable structural design has helped integrate low toxicity with high biodegradability in some commercial products in the past (e.g. drilling fluids), we believe that appropriately designing the ILs may lead to the solvents with desired attributes that would not pose problems to the environment.12
Our initial investigations on the evaluation of biodegradability of ILs were centered on the most commonly used imidazolium based ILs. The biodegradability of this class of ILs increased if the ester group was integrated in to the structure, however the amide group did not have any significant influence.14 The ester group derived from a C2 acid and C5 linear alcohol on the imidazolium side chain and the octyl sulfate anion together enhances the ready biodegradability of the IL to meet the standard 60% cutoff value in 28 days.15
Our interest in assessing the biodegradability of the pyridinium based ILs (another commonly used class of ILs) was due to their popularity as ionic solvents. Our curiosity was peaked by the results reported by Engberts and Hulst et al.16 Their research on the pyridinium based amphiphiles showed high vulnerability of their products to enzymatic attack. Their results were also suggestive of very low toxicity of these compounds. Around the same time, the results on the study of biodegradability of some cationic surface active compounds with pyridinium head group also indicated the susceptibility of the pyridinium ring towards the microbial attack.17 While our studies were underway, Docherty et al. measured biodegradability of 1,3-dialkylpyridinium bromides using the OECD 301 A (DOC Die-Away Test), and found that pyridinium bromide with a linear C6 side chain mineralized completely after 49 days of incubation, and with a linear C8 side chain, mineralization occurred after 25 days.181H NMR, DNA-PCR-DGGE and TDN analysis established that the pyridinium ring is metabolized in the process of biodegradation. Very recently, Jastorff et al. investigated a family of unsubstituted and substituted 1-n-alkylpyridinium halides for biodegradation.19 The disappearance of the substrates was monitored by HPLC (with UV detection) to calculate their primary biodegradation. However, as respirometric parameters (O2 depletion/CO2 formation) were not used for the measurement of mineralization, the method does not reflect the ready biodegradability of the compounds tested. Being interested by these reports, we sought to systematically investigate the ready biodegradability of the pyridinium based ILs/salts derived from pyridine and nicotinic acid, a relatively inexpensive natural product that can be used to incorporate the pyridinium core in the IL moiety.20
Results and discussion
Target ionic liquids were prepared using standard methodology. This initially involved alkylation of pyridine or the appropriate 3-substituted pyridine with an alkyl halide to afford the corresponding pyridinium halide (Scheme 1). Alternative anions were subsequently introduced by a metathesis reaction with sodium octyl sulfate, potassium hexafluorophosphate or lithium bis(trifluormethanesulfonimide).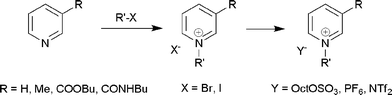 | ||
| Scheme 1 Synthesis of pyridinium ILs. | ||
Pyridinium salts with C4 linear alkyl chains at position 1 (compounds 1a, c and d) did not show high levels of biodegradability (Fig. 1). The presence of the methyl group at the 3-position of the pyridine ring helps induce the asymmetry in the structure, rendering the salt 2a liquid at room temperature, but certainly does not help improve the biodegradability of the IL. The 1-butylpyridinium salts based on Br−, PF6− and NTf2− showed only 1–3% biodegradability over a 28 day period, unless octyl sulfate was employed as the anion. The biodegradability as high as 37–40% was attained by the incorporation of an octyl sulfate counter ion, which is obviously an effect induced by the anion. These observations on the resistance of 1-butylpyridinium salts towards biodegradation are consistent with the earlier results documented by Docherty18 and Jastorff et al.19 1-Alkylpyridinium bromides with linear C10 and C16 substituents (3 and 4) showed poor biodegradability at 9% and 0%, respectively.
 | ||
| Fig. 1 Percentage biodegradability of the alkyl pyridinium based quaternary salts 1a (○), 1b (□), 1c (+), 1d (△), 2a (×), 2b (◊), 3 (■) and 4 (✦). | ||
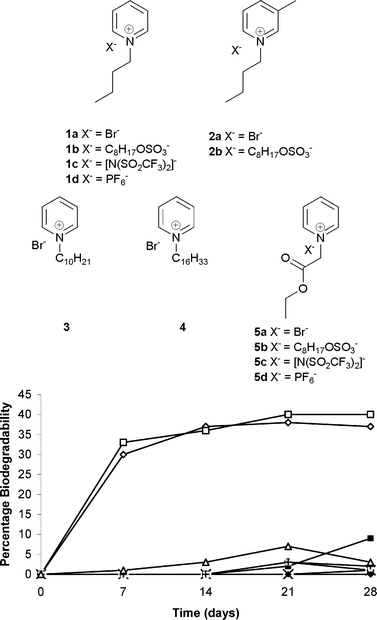
The introduction of the ester group seems to have a profound effect on the biodegradability of these salts.21 The quaternary salt 5a derived from two simple starting materials, pyridine and ethyl bromoacetate and the corresponding octyl sulfate IL, 5b, both showed a comparable biodegradability at 87 and 89% respectively (Fig. 2). The biodegradability attained by these compounds after the first 7 days was 71 and 74% respectively (Fig. 2). As the halide ion is not likely to have significant influence on biodegradability, these results indicate that the pyridinium nucleus is not refractory and that appropriate structural manipulations of this core can lead to compounds with good ready biodegradability values. It is also worth noting that the presence or absence of the octyl sulfate anion (which is generally known to have positive influence on the ready biodegradability) seems to have only a meagre influence on the overall high biodegradability values. The exchange of bromide ion with NTf2− produces a low viscosity IL 5c and with PF6− produces a solid 5d which is sparingly solubility in water. Both 5c and 5d contain fluorine in the anion component and show ready biodegradability at 64 and 86% respectively, both above the 60% threshold (Fig. 2). It is also notable that in this series the NTf2− based IL offers lower (by 23% on average) biodegradability than the corresponding Br−, OctOSO3− and PF6− counterparts. This may, in part, be because of the lack of water miscibility of the NTf2− based IL. The good biodegradability of ILs with fluorine containing anions such as NTf2− and PF6− is particularly significant as the ILs with these anions possess good solvent characteristics and these anions are amongst the most commonly encountered in ILs.
 | ||
| Fig. 2 Percentage biodegradability of alkyl pyridinium based quaternary salts with ester group appended on the side chain; 5a (×), 5b (+), 5c (▲) and 5d (□). | ||
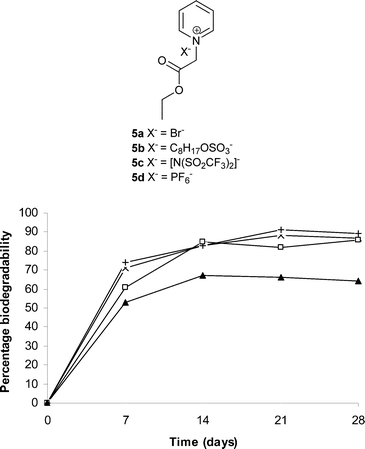
ILs derived from nicotinic acid in which the ester moiety was incorporated into the 3-position of the pyridinium cation were also investigated. Synthesizing the ILs from bio-renewable chemicals such as nicotinic acid, as opposed to petrochemicals such as pyridine is acknowledged as a good argument in favor of sustainable development of products.22 Butyl nicotinate was synthesized from nicotinic acid and was subjected to quaternization with alkyl iodides, mainly because of the reduced nucleophilicity of the nitrogen on the pyridine ring. The quaternization of butyl nicotinate with methyl iodide produced 6a. The N-alkylation of butyl nicotinate with butyl iodide followed by anion metathesis yields 7b. Both 6a and 7b offer promising biodegradability at 72 and 84%, respectively (Fig. 3). The octyl sulfate containing 6b, the triflimide based 6c and the hexafluorophosphate containing IL 6d were synthesized from quaternary iodide 6a showed biodegradability at 75, 68 and 75% respectively (Fig. 3). It is notable that 6a–d showed greater than 60% biodegradability within the first seven days of incubation. The increased biodegradability of 7b over 6b is perhaps because of an increase in the lipophilicity of cation. This finding is consistent with previous studies in which cations with longer alkyl side chains were found to be more biodegradable.14,19 The simple primary amide N-butyl nicotinamide derived from nicotinic acid and butyl amine was used to synthesize the nicotinamide based IL 8b. The ready biodegradability of 30% (Fig. 3) after a standard 28 day period perhaps indicates the recalcitrance of the nicotinamide derived IL under the test conditions. This inertness of amide containing IL is consistent with our earlier observations on the imidazolium series under similar test conditions.14 The resistance of amides towards biodegradation has also been documented earlier in the design of biodegradable imidazolium ILs.12
 | ||
| Fig. 3 Percentage biodegradability of alkyl pyridinium based quaternary salts derived from nicotinic acid; 6a (△), 6b (×), 6c (✦), 6d (+), 7b (■) and 8b (○). | ||
Compounds which undergo 60% mineralization in 28 days using methods based on respirometry are defined as “readily biodegradable”. Levels of 60% mineralization also may indicate the use of test substances for the production of biomass (which is eventually but not immediately used as a source of energy) and thus this value may equate to higher levels of ultimate biodegradation (around 90%) and even higher values of primary biodegradation (at least > 90%). It is important to note that the tests for ready biodegradability are generally performed under the most stringent conditions of lower microbial population and with the test substance as the sole source of carbon. Also, the concentration of the test substance is unrealistically higher than what would normally be observed in a practical scenario in the environment or a typical waste water treatment plant. Therefore 60% biodegradability obtained by these methods indicates that the test substance should undergo speedy and complete biodegradation upon release into an aquatic environment. Not surprisingly, the tests for ready biodegradability have had a long history of successful applications.
A preliminary investigation into the use of the biodegradable ionic liquid (BIL), 3-(butoxycarbonyl)-1-methylpyridinium triflimide (6c), as a reaction solvent has also been conducted. We chose the Diels–Alder reaction for this purpose as this cycloaddition is one of the most extensively used methods to construct six membered carbo- and heterocyclic compounds.23 This reaction has been investigated well not only in the Lewis acidic ILs,24 but also in the ‘second generation’ neutral ILs.25 In the neutral ILs the rates of this reaction are increased by doping the IL with Lewis acid catalysts26 or mineral acid catalysts.27 The combination of neutral ILs with ultrasound28 or microwave29 has also proved useful in enhancing the rate of this reaction. It has been demonstrated that ILs not only offers an advantage as a reaction media, but also exert effects on the rates30 and selectivity31 of Diels–Alder reaction when compared with the conventional solvents. The reaction has also been investigated in the imidazolium based BILs by our group.32 Accordingly, the plethora of comparative data available for Diels–Alder reactions in covalent solvents and ionic liquids made it an obvious choice for the evaluation of the novel ionic liquids reported herein.
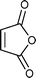
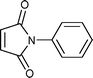
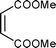
Cyclopentadiene was used as the diene component, as it is one of the most commonly used reactive diene in Diels–Alder reactions. The reaction of cyclopentadiene with methyl acrylate afforded 70% product after 24 h and 97% after 72 h (Table 1, Entry 1). A range of other dienophiles, including methyl vinyl ketone (Entry 2), dimethylacetylene dicarboxylate (Entry 3), maleic anhydride (Entry 4), N-phenylmaleimide (Entry 5) and dimethyl maleate (Entry 6) were also investigated. In all cases, the dienophiles reacted readily with cyclopentadiene in the BIL 6c to produce the corresponding Diels–Alder adduct in good to nearly quantitative yields. It is notable that the reactivity is dictated by the electron deficient character of the dienophile, which also indirectly influences the reaction time and selectivity. The reactive dienophiles also offer excellent endo selectivity. The crude products in all the cases were isolated by extracting the reaction mixture with diethyl ether. The purity of IL was ascertained by 1H NMR and can be reused after evaporating the traces of diethyl ether under reduced pressure. The products in all the cases were purified by column chromatography and the endo:exo ratio in all the cases was determined by 1H NMR.
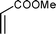
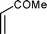
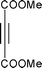
Conclusions
It can be concluded that ionic liquids with a pyridinium cation bearing an ester containing substituent at positions 1 or 3 show excellent biodegradability. The biodegradability does not seem to significantly depend on the anion, although some effects are apparent. The high ready biodegradability values attest the earlier claims of tendency of the pyridine ring to mineralize.16–19 In contrast, 1-alkylpyridinium ILs with linear C4, C10 and C16 showed relatively low levels of biodegradability. Although the presence of linear alkyl chains is an important factor in designing biodegradable compounds as they can act as potential sites for attack by oxygenases,10b,14b no significant improvement was detected for these 1-alkylpyridinium bromides. However, as ionic liquids with longer alkyl chains have been proven to be more toxic,15b,33 further studies are underway to clarify if their resistance towards biodegradation in the ready biodegradability tests could be related to their toxic effects on the biological activity of the aerobic micro-organisms.The biodegradable ionic liquid (BIL), 3-(butoxycarbonyl)-1-methylpyridinium triflimide (6c), was demonstrated to be a viable reaction medium as exemplified with the Diels–Alder reaction. In general, high yields and endo selectivities were observed.
Experimental
Synthesis
Nicotinic acid (Sigma-Aldrich, 98%), lithium triflimide (Aldrich, 97%), potassium hexafluorophosphate (Fluka, 98%), sodium octyl sulfate (Sigma, ∼95%), methyl iodide (Sigma-Aldrich, 99%), butyl iodide (Fluka ≥ 98%), 1-butanol (BDH, 99%), thionyl chloride (Fluka, ≥ 99%), ethyl bromoacetate (Merck, > 98%), toluene (Merck, GR, ≤ 0.03% H2O), acetonitrile (Merck, Chromatography grade, ≤ 0.02% H2O), diethyl ether (Merck, GR, ≤ 0.03% H2O), butyl amine (Sigma-Aldrich, 99.5%), pyridine (BDH, 98%) and triethylamine (Sigma-Aldrich, 99%) were procured from the commercial supplier and used without any pre-treatment. The 1H and 13C NMR spectra of the purified products were recorded in CDCl3 (Cambridge Isotope Laboratories Inc., 99.8% D) or DMSO d6 (Cambridge Isotope Laboratories Inc., 99.9% D) on a Bruker Avance DPX 300 spectrometer at 300 and 75.4 MHz, respectively. Low resolution mass spectra using electrospray ionisation (ES-MS) were obtained using a Micromass Platform II instrument. Unless otherwise stated, cone voltage was 20 eV.Butyl nicotinate
Thionyl chloride (81.30 mmol) was gradually added to the suspension of nicotinic acid (40.65 mmol) in toluene (100 mL) maintained at room temperature. The reaction mixture was heated at 100 °C for 24 h, to assure the quantitative formation of the nicotinic acid chloride (as a hydrochloride salt). After decanting the solution of unreacted thionyl chloride in toluene, the acid chloride was washed with diethyl ether (4 × 50 mL) and then treated with dichloromethane (100 mL). The reaction mixture was cooled under dry ice and gradually treated with triethylamine (81.30 mmol) followed by 1-butanol (81.30 mmol). The reaction mixture was stirred at room temperature for 24 h and then treated with water and stirred vigorously to yield a clear dichloromethane layer. Butyl nicotinate, being a water insoluble Brønsted base, was separated and purified by treating the organic layer with 6 N HCl solution (20 mL). The acidic aqueous layer was first extracted with dichloromethane (2 × 20 mL), and treated with an aqueous base, ammonium hydroxide, to release the pure ester. The ester was separated by extraction of the aqueous layer with dichloromethane (2 × 20 mL), followed by drying and evaporation at reduced pressure. Yield 50%. 1H NMR (CDCl3): δ 0.97–1.02 (t, J = 7.4 Hz, 3H), 1.43–1.55 (m, 2H), 1.73–1.83 (m, 2H), 4.36–4.40 (t, J = 6.6 Hz, 2H), 7.40–7.44 (m, 1H), 8.31–8.35 (m, 1H), 8.77–8.79 (m, 1H), 9.23–9.24 (m, 1H). 13C NMR (CDCl3): δ 13.8, 19.3, 30.8, 65.4, 123.4, 126.5, 137.1, 151.0, 153.4, 165.4. ES-MS (+ ve): m/z, 180.0 [M + 1]+N-Butyl nicotinamide
The acid chloride was prepared and purified as per the procedure described above. The reaction mixture was cooled under dry ice and gradually treated with butyl amine (122 mmol). After stirring at room temperature for 3 h, the reaction mixture was treated with water and stirred vigorously to yield a clear dichloromethane layer. The isolation of product was carried out by quenching the reaction mixture with water, followed by drying and evaporation of the dichloromethane at reduced pressure. Yield 52%. 1H NMR (CDCl3): δ 0.96–1.01 (t, J = 7.2 Hz, 3H), 1.38–1.50 (m, 2H), 1.59–1.69 (m, 2H), 3.46–3.53 (m, 2H), 6.15 (bs, 1H), 7.37–7.41 (m, 1H), 8.10–8.14 (m, 1H), 8.72–8.74 (m, 1H), 8.95–8.96 (m, 1H). 13C NMR (CDCl3): δ 13.8, 20.2, 31.6, 40.0, 123.4, 130.7, 135.1, 148.2, 151.9, 165.9. ES-MS (+ ve): m/z, 179.0 [M + 1]+.General procedures for the synthesis of pyridinium halides (1a, 2a, 3, 4, 5a and 6a)34
To the solution of pyridine/3-substituted pyridine (33.3 mmol) in toluene (20 mL), was added alkyl halide (40.0 mmol) at room temperature, followed by stirring at room temperature for 1 h under nitrogen. The reaction mixture was then stirred at 110 °C for 24 h. In the case of 5a and 6a, the reactions were heated at 70 °C and 45 °C, respectively. In all the cases, the completion of the reaction was marked by the separation of either the dense IL layer or solid from the initially obtained clear and homogenous mixture of amine and alkyl halide in toluene. The product was isolated by decanting the toluene layer to remove the unreacted starting materials and solvent. Subsequently, the quaternary salt was washed with diethyl ether (4 × 20 mL) and each time the ether layer was separated from the quaternary halide by decantation. In some cases, the ether washings resulted in the formation of a solid. The solid was purified by dissolving in acetonitrile (20 mL) and precipitation with diethyl ether (60–70 mL). In each case, the IL/salt was finally dried at a reduced pressure to get rid of all the volatile organic compounds. These quaternary salts (except 6a) being hygroscopic, were handled under nitrogen at each stage of synthesis and purification.General procedure for the synthesis of pyridinium octyl sulfates (1b, 2b, 5b, 6b, 7b and 8b)35
The quaternary salt (2 mmol) was dissolved in water to obtain a clear solution. The aqueous solution of quaternary halide was treated with the aqueous solution of sodium octyl sulfate (1.54 mmol in case of 1b, 2b and 5b or 2 mmol in case of 6b, 7b and 8b in 10 mL of water) and the resultant was stirred for 10 min. The product was isolated by solvent extraction using dichloromethane (3 × 10 mL). The extracts were dried over anhydrous MgSO4 and evaporated under reduced pressure to yield the required product.General procedure for the synthesis of pyridinium triflimides (1c, 5c and 6c)
The quaternary halide (2 mmol) was dissolved in water (10 mL) to obtain a clear solution. The aqueous solution of quaternary halide was treated with the aqueous solution of bis(trifluormethanesulfonimide) lithium salt (2.2 mmol, 0.631 g in 10 mL water) and the resultant was stirred for 10 min. The product was isolated by solvent extraction using dichloromethane (2 × 10 mL). The extracts were dried over anhydrous MgSO4 and evaporated under reduced pressure to yield the required product. As expected, all triflimide based ILs were low viscosity liquids at room temperature.General procedure for the synthesis of pyridinium hexafluorophosphates (1d, 5d and 6d)36
The quaternary salt (2 mmol) was dissolved in water (10 mL) to obtain a clear solution. The aqueous solution of quaternary halide was treated with the aqueous solution of potassium hexafluorophosphate (2.4 mmol, 0.405 g in 10 mL water) and the resultant was stirred for 10 min. The product was isolated by solvent extraction using dichloromethane (2 × 10 mL). The extracts were dried over anhydrous MgSO4 and evaporated under reduced pressure to yield the desired product.General procedure for Diels–Alder reactions in BIL 6c
The dienophile (1.66 mmol) was added to 3-(butoxycarbonyl)-1-methylpyridinium triflimide (6c) (0.5 mL) and the mixture was stirred well for 1–3 min. Cyclopentadiene (2.33 mmol) was added and the reaction mixture was stirred at room temperature for the time specified in the Table 1. Diels–Alder adducts were extracted with diethyl ether (3 × 20 mL), the extracts were dried over anhydrous MgSO4 and evaporated under reduced pressure to yield the crude products. All crude products were purified using column chromatography with ethyl acetate/petroleum spirits (ranging from 5% to 30% ethyl acetate) as an eluent. The 1H and 13C NNMR spectra of these products were identical to those previously reported in the literature.ISO 14593-Carbon dioxide head space test
To evaluate the biodegradability of the test ILs, the ‘‘CO2 Headspace’’ test (ISO 14593) was also applied. This method allows the evaluation of the ultimate aerobic biodegradability of an organic compound in an aqueous medium at a given concentration of microorganisms by analysis of inorganic carbon. The test IL, as the sole source of carbon and energy, was added at a concentration of 40 mg/L to a mineral salt medium. These solutions were inoculated with activated sludge collected from an activated sludge treatment plant, washed and aerated prior to use and incubated in sealed vessels with a headspace of air. Biodegradation (mineralization to carbon dioxide) was determined by measuring the net increase in the total organic carbon (TOC) levels over time compared with unamended blanks. Sodium n-dodecyl sulfate (SDS) was used as reference substance. The tests ran for 28 days. The extent of biodegradation was expressed as a percentage of the theoretical amount of inorganic carbon (ThIC) based on the amount of test compound added initially.Acknowledgements
The authors thank Pfizer Global R & D, the Spanish Ministerio de Educación y Ciencia (CTQ2007–60364/PPQ) and the Australian Research Council (LX0561094) for financial support.Notes and References
- M. Smiglak, A. Metlen and R. D. Rogers, Acc. Chem. Res., 2007, 40, 1182–1192 CrossRef CAS.
- (a) P. Wasserscheid and W. Keim, Angew. Chem., Int. Ed., 2000, 39, 3772–3789 CrossRef; (b) J. Dupont, R. F. de Souza and P. A. Z. Suarez, Chem. Rev., 2002, 102, 3667–3691 CrossRef CAS.
- M. H. Valkenberg, C. de Castro and W. F. Hoelderich, Green Chem., 2002, 4, 88–93 RSC.
- (a) Z. Fei, T. J. Geldbach, D. Zhao and P. J. Dyson, Chem. Eur. J., 2006, 12, 2122–2130 CrossRef CAS; (b) S.-g. Lee, Chem. Commun., 2006, 1049–1063 RSC.
- W. Miao and T. H. Chan, Acc. Chem. Res., 2006, 39, 897–908 CrossRef CAS.
- T. Fukushima and T. Aida, Chem. Eur. J., 2007, 13, 5048–5058 CrossRef CAS.
- (a) P. Wasserscheid, Nature, 2006, 439, 797 CrossRef CAS; (b) S. Chowdhury, R. S. Mohan and J. L. Scott, Tetrahedron, 2007, 63, 2363–2389 CrossRef.
- T. Welton, Green Chem., 2008, 10, 483 RSC.
- (a) J. H. Davis, Jr. and P. A. Fox, Chem. Commun., 2003, 1209–1212 RSC; (b) D. Adam, Nature, 407, 938–940 Search PubMed.
- (a) K. Kümmerer, Green Chem., 2007, 9, 899–907 RSC; (b) J. Ranke, S. Stolte, R. Stöermann, J. Arning and B. Jastorff, Chem. Rev., 2007, 107, 2183–2206 CrossRef CAS.
- (a) M. Matsumoto, K. Mochiduki and K. Kondo, J. Biosci. Bioeng., 2004, 98, 344–347 CAS; (b) C. Pretti, C. Chiappe, D. Pieraccini, M. Gregori, F. Abramo, G. Monni and L. Intorre, Green Chem., 2006, 8, 238–240 RSC; (c) S. Stolte, M. Matzke, J. Arning, A. Boeschen, W.-R. Pitner, U. Welz-Biermann, B. Jastorff and J. Ranke, Green Chem., 2007, 9, 1170–1179 RSC.
- R. S. Boethling, E. Sommer and D. DiFiore, Chem. Rev., 2007, 107, 2207–2227 CrossRef CAS.
- (a) K. M. Docherty, and C. Kulpa, Abstracts of Papers, 231st ACS National Meeting, Atlanta, GA, United States, March 26–30, 2006, 2006, IEC-174; (b) J. Ranke, K. Moelter, F. Stock, U. Bottin-Weber, J. Poczobutt, J. Hoffmann, B. Ondruschka, J. Filser and B. Jastorff, Ecotoxicol. Environ. Safe., 2004, 58, 396–404 Search PubMed.
- (a) N. Gathergood and P. J. Scammells, Aust. J. Chem., 2002, 55, 557–560 CrossRef CAS; (b) N. Gathergood, M. T. Garcia and P. J. Scammells, Green Chem., 2004, 6, 166–175 RSC.
- (a) N. Gathergood, P. J. Scammells and M. T. Garcia, Green Chem., 2006, 8, 156–160 RSC; (b) M. T. Garcia, N. Gathergood and P. J. Scammells, Green Chem., 2005, 7, 9–14 RSC.
- (a) A. Roosjen, J. Šmisterová, C. Driessen, J. T. Anders, A. Wagenaar, D. Hoekstra, R. Hulst and J. B. F. N. Engberts, Eur. J. Org. Chem., 2002, 1271–1277 CrossRef CAS; (b) D. Pijper, E. Bulten, J. Šmisterová, A. Wagenaar, D. Hoekstra, J. B. F. N. Engberts and R. Hulst, Eur. J. Org. Chem., 2003, 4406–4412 CrossRef CAS.
- E. Grabińska-Sota and J. Kalka, Environ. Int., 2003, 28, 687–690 CrossRef CAS.
- K. M. Docherty, J. K. Dixon and C. F. Kulpa, Jr., Biodegradation, 2007, 18, 481–493 CrossRef CAS.
- S. Stolte, S. Abdulkarim, J. Arning, A.-K. Blomeyer-Nienstedt, U. Bottin-Weber, M. Matzke, J. Ranke, B. Jastorff and J. Thoeming, Green Chem., 2008, 10, 214–224 RSC.
- Commercial grade nicotinic acid is priced at ∼9.716 AU $/mole (79 $/kg) in Sigma-Aldrich catalogue.
- J. R. Harjani, R. D. Singer, M. T. Garcia and P. J. Scammells, Green Chem., 2008, 10, 436–438 RSC.
- (a) S. T. Handy, Chem. Eur. J., 2003, 9, 2938–2944 CrossRef CAS; (b) G. Imperato, B. Koenig and C. Chiappe, Eur. J. Org. Chem., 2007, 1049–1058 CrossRef CAS.
- J. Sauer and R. Sustmann, Angew. Chem., Int. Ed. Engl., 1966, 5, 211–230 CrossRef CAS.
- (a) C. W. Lee, Tetrahedron Lett., 1999, 40, 2461–2464 CrossRef CAS; (b) A. P. Abbott, G. Capper, D. L. Davies, R. K. Rasheed and V. Tambyrajah, Green Chemistry, 2002, 4, 24–26 RSC.
- (a) D. A. Jaeger and C. E. Tucker, Tetrahedron Lett., 1989, 30, 1785–1788 CrossRef CAS; (b) T. Fischer, A. Sethi, T. Welton and J. Woolf, Tetrahedron Lett., 1999, 40, 793–796 CrossRef CAS; (c) P. Ludley and N. Karodia, Tetrahedron Lett., 2001, 42, 2011–2014 CrossRef CAS.
- (a) G. Silvero, M. J. Arevalo, J. L. Bravo, M. Avalos, J. L. Jimenez and I. Lopez, Tetrahedron, 2005, 61, 7105–7111 CrossRef CAS; (b) I. Hemeon, C. DeAmicis, H. Jenkins, P. Scammells and R. D. Singer, Synlett, 2002, 1815–1818 CAS.
- A. Kumar and S. S. Pawar, J. Org. Chem., 2007, 72, 8111–8114 CAS.
- J. L. Bravo, I. Lopez, P. Cintas, G. Silvero and M. J. Arevalo, Ultrason. Sonochem., 2006, 13, 408–414 CrossRef CAS.
- I. Lopez, G. Silvero, M. J. Arevalo, R. Babiano, J. C. Palacios and J. L. Bravo, Tetrahedron, 2007, 63, 2901–2906 CrossRef.
- (a) C. E. Song, E. J. Roh, S.-g. Lee, W. H. Shim and J. H. Choi, Chem. Commun., 2001, 1122–1123 RSC; (b) Y. Xiao and S. Malhotra, Tetrahedron Lett., 2004, 45, 8339–8342 CrossRef CAS.
- M. J. Earle, P. B. McCormac and K. R. Seddon, Green Chem., 1999, 1, 23–25 RSC.
- S. Bouquillon, T. Courant, D. Dean, N. Gathergood, S. Morrissey, B. Pegot, P. J. Scammells and R. D. Singer, Aust. J. Chem., 2007, 60, 843–847 CrossRef CAS.
- (a) R. S. Boethling, Designing Safer Chemicals, ACS Symp. Ser., 1996, 640, 156 CAS; (b) P. H. Howard, R. S. Boethling, W. Stiteler, W. Meylan and J. Beauman, Sci. Total Environ., 1991, 109/110, 635 CrossRef; (c) R. S. Boethling, Cationic Surfactants, Surfactant Science Series Vol. 53, Marcel Dekker, New York, 1994, pp. 95-135 Search PubMed.
- P. J. Dyson, M. C. Grossel, N. Srinivasan, T. Vine, T. Welton, D. J. Williams, A. J. P. White and T. Zigras, J. Chem. Soc. Dalton Trans., 1997, 3465–3469 RSC.
- P. Wasserscheid, R. van Hal and A. Boesmann, Green Chem., 2002, 4, 400–404 RSC.
- J. G. Huddleston, A. E. Visser, W. M. Reichert, H. D. Willauer, G. A. Broker and R. D. Rogers, Green Chem., 2001, 3, 156–164 RSC.
| This journal is © The Royal Society of Chemistry 2009 |