
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly emissive hybrid mesoporous organometallo-silica nanoparticles for bioimaging†
Cintia
Ezquerro
a,
Icíar P.
López
b,
Elena
Serrano
 c,
Elvira
Alfaro-Arnedo
b,
Elena
Lalinde
a,
Ignacio M.
Larráyoz
*de,
José G.
Pichel
bf,
Javier
García-Martínez
*c and
Jesús R.
Berenguer
*a
c,
Elvira
Alfaro-Arnedo
b,
Elena
Lalinde
a,
Ignacio M.
Larráyoz
*de,
José G.
Pichel
bf,
Javier
García-Martínez
*c and
Jesús R.
Berenguer
*a
aDepartamento de Química-Centro de Investigación en Síntesis Química (CISQ), Universidad de La Rioja, Madre de Dios 53, Logroño, E-26006, La Rioja, Spain. E-mail: jesus.berenguer@unirioja.es; Web: https://investigacion.unirioja.es/grupos/24/detalle
bUnidad de Cáncer de Pulmón y Enfermedades Respiratorias, Centro de Investigación Biomédica de La Rioja (CIBIR), Fundación Rioja Salud, Logroño, 26006, La Rioja, Spain Web: http://www.cibir.es
cLaboratorio de Nanotecnología Molecular, Departamento de Química Inorgánica, Universidad de Alicante, Carretera San Vicente s/n, Alicante, E-03690, Spain. E-mail: j.garcia@ua.es; Web: http://www.nanomol.es
dUnidad de Biomarcadores y Señalización Molecular, Centro de Investigación Biomédica de La Rioja (CIBIR), Fundación Rioja Salud, Logroño, 26006, La Rioja, Spain. E-mail: ilarrayoz@riojasalud.es; Web: http://www.cibir.es.
eGRUPAC, Unidad Predepartamental de Enfermería, Universidad de La Rioja, Duquesa de la Victoria 88, Logroño, E-26006, La Rioja, Spain
fSpanish Biomedical Research Networking Centre in Respiratory Diseases (CIBERES), ISCIII, Madrid, E-28029, Spain Web: https://www.ciberes.org
First published on 5th March 2022
Abstract
Production of mesoporous silica nanoparticles (MSNs) with uniform textural characteristics and imaging properties on a large scale is still a challenge. Thus, the design of simple and scalable methods to obtain reproducible functionalized MSNs has become even more relevant. Herein, we describe an in situ strategy for the synthesis and surface functionalization of highly luminescent mesoporous organometallo-silica nanoparticles. Using the [Ir(dfppy)2(dasipy)]PF6 chromophore and TEOS as sol–gel precursors and different capping agents, such as DMDES or APTES, three different emissive MSNs were prepared (NPOH_IS, NPMe_IS and NPNH2_IS), each containing hydroxyl, methyl and amine groups on their surfaces, respectively. All three were tested on human tumor A549 (lung carcinoma) and HeLa (cervix carcinoma) cell lines, showing intense and stable yellow phosphorescence, biocompatibility and efficient internalization. Moreover, NPMe_IS nanoparticles showed excellent colloidal stability, both in water and biological media, and a BET area of 1120 m2 g−1, making them not only luminescent biomarkers, but potentially also controlled delivery vectors.
Introduction
The development of nanostructured materials for biomedical applications has become a leading technology, yielding a wide variety of therapeutic and diagnostic agents over the last decade.1 The ever-increasing demand for efficient monitoring of specific biological processes has called for probes that exhibit many properties including biocompatibility, high stability, good solubility and appropriate emission features. In this context, different types of nanomaterials, including selenium- or cadmium-containing quantum dots (QDs),2 carbon-based nanomaterials,3 noble metal nanoparticles,4 magnetic nanoparticles2a,5 and metal–organic frameworks (MOFs),6 have been used as probes for bioimaging. Among these nanomaterials, mesoporous silica nanoparticles (MSNs) have aroused great interest primarily due to their straightforward synthesis, the versatility of both their morphology and size, which can be appropriately adjusted to target purposes,7 and their relatively low-toxicity in a biological environment.8 Moreover, unlike other nanoparticles, MSNs offer advantages such as outstanding chemical modularity, inertness and biodegradability.9 In this field, particle sizes play an important role in determining their cellular uptake behavior. It is well known that endocytosis is the main mechanism of cellular uptake for silica nanoparticles with sizes smaller than 200 nm.8,10 This is one of the reasons why the synthesis of monodisperse MSNs with diameters of approximately 100 nm has recently gained much attention. Another relevant factor is their easy surface functionalization. Unmodified MSNs possess a negative zeta potential due to dissociation of superficial silanol groups at pH > 2–3. The cellular membrane is also mostly negatively charged, but silanol groups allow the cell to recognize these MSNs. Nevertheless, in order to enhance cellular uptake, there are many examples of surface functionalization of MSNs with positive charges.11 The benefits of this are not completely clear, as positive charges on the surface can induce an immune response or cytotoxicity when compared with anionic or neutral materials.12 Also, shell formation around nanoparticles using polymers or other biocompatible molecules to enhance biocompatibility can be highly desirable.10a,11a,13The optical transparency of silica in the visible region and chemical inertness make these MSNs exceptional substrates for the design of luminescent probes.14 The design of fluorescent silica nanoparticles based on non-covalent inclusion or superficial grafting of conventional organic dyes, such as fluorescein or rhodamine, has attracted much research interest due to their remarkable properties.15 However, these organic fluorescent molecules usually present aggregation-caused quenching (ACQ), and many of these inclusion approaches use reverse microemulsion or Stöber synthetic methods, leading, inevitably, to aggregation of the chromophores to produce MSNs with low emission efficiencies. To overcome this problem, several groups have doped MSNs with molecules featuring aggregation-induced emission (AIE) fluorescence or designed encapsulation methods to separate the chromophores.16 Nevertheless, organic fluorophores still present several limitations such as photobleaching or small Stokes shifts and short lifetimes, which make the discrimination of their emissions from cell auto-fluorescence more difficult. In addition, non-covalently included or even superficially grafted chromophores can experience leaking in biological media, which causes an additional decrease of the emission and can induce biotoxicity.16c
In this context, chromophores that meet some important requirements, such as chemical- and photo-stability, large Stokes shifts or long lifetimes, would be desirable.17 To meet this goal, phosphorescent transition metal complexes with a d6 (RuII, ReI, and IrIII) or d8 (PtII) electronic configuration18 and, predominantly, lanthanide derivatives (EuIII, TbIII, and YbIII)15b,19 have been investigated for the preparation of emissive silica nanoparticles. Among them, IrIII compounds can be considered an interesting alternative to organic fluorophores, as they display, in addition to all the above mentioned properties, high quantum yields (ϕ) and an easy adjustment of the emission wavelength by controlling the coordination ligands.20 In fact, cyclometalated IrIII complexes have already been extensively studied over the last few years in bioimaging and biosensing,21 although many of them show considerable cytotoxicity.22 This fact, very promising when using these complexes as therapeutic agents, is not desirable for their exclusive use as intracellular biomarkers. The design of MSNs for biomedical purposes by a combination of cyclometalated IrIII complexes with silica nanoparticles is not very widespread23 and, as far as we know, only two examples have been studied in vivo.23c,e Most of the reported studies involve incorporation of IrIII complexes via encapsulation (non-covalent interactions) or through post-synthetic methods (grafting). Hence, it seems judicious to design a synthetic strategy to obtain highly emissive MSNs with a better integration of the IrIII complex in the silica matrix and, if possible, employ only small amounts of the expensive chromophore in a more effective way.
In the last few years, our group has studied the “sol–gel coordination chemistry” approach based on the in situ co-condensation of a silica source with small amounts of organometallic complexes bearing terminal alkoxysilane groups.24 Following this method, we have recently prepared white-light emitting discrete MSNs containing three different emissive cyclometalated IrIII derivatives,25 which have been used in lighting applications. Herein, we report a modification of this synthetic approach to fabricate stable, monodisperse, precisely size-controllable and brightly phosphorescent MSNs, in which the amount of dyes has been extremely reduced (less than 1 wt% of the MSN). We chose the bis(cyclometalated) complex [Ir(dfppy)2(dasipy)]PF6 (1; dfppy = 2-(2,4)-difluorophenyl-pyridinyl; dasipy = N,N′-dipropyltriethoxysilane-2,2′-bipyridine-4,4′-dicarboxamide), which is highly emissive in solution. In an attempt to control the aggregation tendency of the nanoparticles, as well as their morphology and surface charge, in situ functionalization was carried out using two different capping agents, diethoxydimethylsilane (DMDES), which should make the particles more hydrophobic, and 3-aminopropyltriethoxysilane (APTES), with hydrophilic amine groups, which should give a positive charge to the nanoparticle surface. For comparison purposes, related MSNs with the chromophore grafted on their surface were also prepared. All of the in situ hybrid MSNs showed internalization into cells with excellent biocompatibility and high photoluminescence efficiency. Moreover, these MSNs also exhibit excellent textural properties with high surface areas (up to 1000 m2 g−1), paving the way for their future use not only as phosphorescent biomarkers, but also in controlled intramolecular delivery.
Results and discussion
Synthesis and characterization of the organometallic precursor and organometallo-silica nanoparticles
Highly emissive complex 1 was prepared by reacting the solvate precursor [Ir(dfppy)2(NCMe)2]PF626 and a stoichiometric amount of the dasipy ligand,27 using dichloromethane as a solvent (Scheme S1, ESI†). The complex was fully characterized by elemental analysis, mass spectrometry and the usual spectroscopic means (IR, absorption, multinuclear NMR; see the Experimental section in the ESI†). The NMR data of complex 1 show the presence of two equivalent cyclometalated groups and one symmetric dasipy ligand, thus confirming the proposed structure, with both cyclometalated nitrogen atoms in a mutually trans disposition. The FTIR spectrum of complex 1 exhibits characteristic vibration bands of the N–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O units of the amide groups at ca. 3327 and 1670 cm−1, respectively. The strong intensity features in the high energy region correspond to the ν(C–H) of the aromatic systems (>3000 cm−1) and aliphatic groups (<3000 cm−1),28 while the absorption observed between 1600 and 1400 cm−1 is characteristic of stretching vibrations within aromatic rings.29 The spectrum also features intense peaks at 1163 and 1074 cm−1 corresponding to ν(Si–O–C) stretching, which is commonly assigned to terminal trialkoxysilyl groups.24a
O units of the amide groups at ca. 3327 and 1670 cm−1, respectively. The strong intensity features in the high energy region correspond to the ν(C–H) of the aromatic systems (>3000 cm−1) and aliphatic groups (<3000 cm−1),28 while the absorption observed between 1600 and 1400 cm−1 is characteristic of stretching vibrations within aromatic rings.29 The spectrum also features intense peaks at 1163 and 1074 cm−1 corresponding to ν(Si–O–C) stretching, which is commonly assigned to terminal trialkoxysilyl groups.24a
Complex 1 was used as a precursor for preparing three series of pale-yellow hybrid mesoporous organometallo-silica nanoparticles (NPMe_IS, NPOH_IS and NPNH2_IS) with three different surface functionalities. The synthetic one-pot route of all these nanoparticles is illustrated in Scheme 1 (see also the Experimental section), while their metal content and textural properties are shown in Table 1. In all cases, the amount of each reactant was calculated to achieve a nominal metal content of ca. 0.2 wt%. In detail, NPOH_IS was synthesized by direct co-condensation of complex 1 and TEOS, by mixing a solution containing both reactants in ethanol with a basic aqueous solution of CTAB and triethanolamine (TEA) at 80 °C. After two hours of stirring at this temperature, spherical discrete emissive nanoparticles with an average diameter of ca. 180 nm were obtained (Scheme 1, i). NPMe_IS and NPNH2_IS nanoparticles, containing methyl or propylamine superficial groups, respectively, were prepared following a similar procedure, but adding the corresponding capping agent (DMDES for NPMe_IS and APTES for NPNH2_IS) after the first 10 minutes of reaction at 80 °C (Scheme 1, ii). Discrete emissive nanoparticles of ca. 80 (NPMe_IS) or 60 nm (NPNH2_IS) were obtained after a total reaction time of two hours or one hour, respectively (Scheme 1, iii, iv). Besides their different textural properties, the three hybrid in situNPs contain the chromophore homogeneously dispersed within the silica matrix and show the mesoporosity characteristic of the surfactant (CTAB) employed, which was removed by ionic exchange with a saturated ammonium nitrate solution in all cases. The molar ratio of the synthesis gel was 1.00 TEOS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6.6 × 10−4 complex 1:0.060 CTAB:0.026 TEA:80.0 H2O (0.135 DMDES or 0.023 APTES, when appropriate).
:6.6 × 10−4 complex 1:0.060 CTAB:0.026 TEA:80.0 H2O (0.135 DMDES or 0.023 APTES, when appropriate).
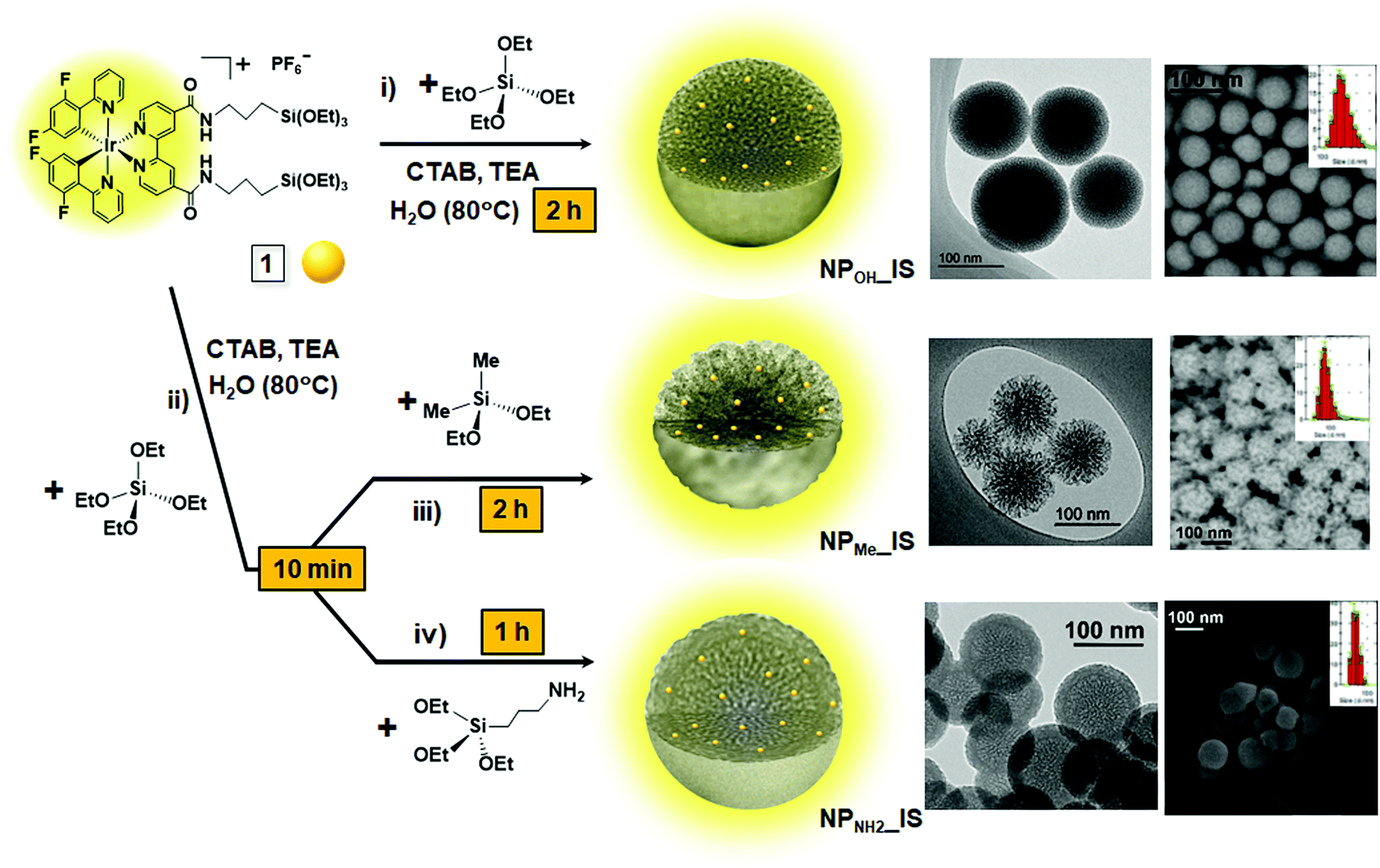 | ||
| Scheme 1 Schematic representation of the synthesis of in situ hybrid mesoporous organometallo-silica nanoparticles (NPOH_IS, NPMe_IS and NPNH2_IS), including TEM and SEM images of each, showing their textural differences, as well as their DLS size distributions. | ||
| Sample | Ira (wt%) | A BET (m2 g−1) | V p 0.99 (cm3 g−1) | d p (nm) | ZPe (mV) | DLS sizef (nm) |
|---|---|---|---|---|---|---|
| a Iridium content calculated from high resolution ICP mass spectroscopy analyses after treatment of the samples with diluted HF. Values in brackets represent the nominal metal content. b BET surface area estimated by the multipoint BET method using adsorption data in the relative pressure (P/P0) range of 0.05–0.3. c Total pore volume read directly from the adsorption branch of the isotherm at 0.99. d Average mesopore diameter determined from the isotherm using the NLDFT equilibrium model. Samples were centrifuged and air dried before gas adsorption measurements. e ZP is zeta potential measured from Milli-Q H2O colloidal suspensions. f Dynamic light scattering (DLS) sizes were determined based on the intensity distribution. g Not determined. | ||||||
| NPOH | — | 760 | 1.2 | 3.0 | n.d.g | n.d.g |
| NPOH_IS | 0.1 (0.2) | 780 | 1.0 | 3.1 | −13.1 ± 0.4 | 180 |
| NPOH_G | 0.2 (0.2) | 390 | 1.2 | 4.3 | −19.0 ± 0.6 | 40–200 |
| NPMe | — | 980 | 1.6 | 4.8 | n.d.g | n.d.g |
| NPMe_IS | 0.1 (0.2) | 1120 | 2.2 | 5.0 | −22.6 ± 1.7 | 80 |
| NPMe_G | 0.2 (0.2) | 950 | 1.5 | 5.1 | −21.6 ± 0.8 | 40–200 |
| NPNH2 | — | 230 | 1.2 | 4.0 | n.d.g | n.d.g |
| NPNH2_IS | 0.2 (0.2) | 100 | 0.4 | 5.2 | 26.3 ± 1.0 | 60 |
| NPNH2_G | 0.1 (0.2) | 100 | 0.9 | 5.5 | 35.6 ± 2.5 | 60 |
For comparative purposes, three types of related pale-yellow grafted MSNs (NPMe_G, NPOH_G and NPNH2_G) were obtained, with the chromophore conventionally attached to the surface of the NPs. To this end, the corresponding white complex-free silica nanoparticles (NPMe, NPOH and NPNH2) were prepared, following the same procedures described for each of the in situ materials, but without the addition of the precursor complex 1 (Scheme S2, i–iv, ESI†). These complex-free nanoparticles were reacted in ethanol with an appropriate amount of complex 1 to yield a material with the same nominal silica/complex molar ratio as that obtained for hybrid in situ materials. These reactions were carried out using NaF as the catalyst to ensure hydrolysis and condensation of most of the alkoxysilane groups of complex 1 (Scheme S2, v and see the Experimental section, ESI†).
Incorporation of complex 1 into all types of MSNs and preservation of its molecular structure were clearly verified by absorption and emission studies (see the Photophysical properties section), ICP mass spectroscopy analyses and FTIR analysis. ICP analyses of these solids (see Table 1) gave incorporation yields between 50% and 100%, depending on the type of the NP. Thus, incorporation yields of the NPOH and NPMe samples were 50% and 100% for the in situ (IS) and grafted (G) materials, respectively. Nevertheless, capping with the APTES amine seems to favor the incorporation of the cationic complex 1 into the in situ material and to hinder grafting of the complex on the surface of the previously synthesized NPNH2, thus leading to the opposite trend (100% incorporation yield for NPNH2_IS and 50% for NPNH2_G). Also, the FTIR spectra of all hybrid NPs, both in situ and grafted, show, in addition to the bands corresponding to the Si–O and Si–O–Si bonds (see Fig. S1, ESI†), the weak absorptions characteristic of complex 1 at ca. 2900 cm−1 and 1500–1400 cm−1, which are due to the ν(C–H) of aliphatic groups and the ν(ring) of aromatic groups, respectively. Moreover, the FTIR spectra of the NPMe and NPNH2 samples (IS and G) also feature characteristic bands due to the ν(Si–CH3) (1267 cm−1 and 850 cm−1) and ν(N–H) (3285 cm−1 and 1390 cm−1) vibration modes, respectively, which correspond to the presence of their respective capping agents (NPMe, DMDES; NPNH2, APTES). The absence of intense peaks in the area between 1470 and 1390 cm−1, related to the C–H vibrations of CTAB, confirms the complete removal of the surfactant, which ensures a high biocompatibility when exposing the NPs to biological media.
As commented previously, all the materials are formed of well-defined spherical nanoparticles, although with clear distinctive textural properties given by the differential surface functionalization (Table 1 and Fig. S2–S4, ESI†). Regarding the hybrid in situNPs (Scheme 1), those obtained in the absence of capping agent (NPOH_IS) displayed the characteristic morphology and mesoporosity of discrete MSNs prepared using CTAB as the surfactant. Through DLS analyses (Table 1 and Fig. S5, ESI†), it was found that NPOH_IS is the largest one with an average diameter of 180 nm, although inspection by electronic microscopy (TEM and FESEM, Fig. S2a, b and S4a, ESI†) seems to point to a lower average diameter of between 100 and 120 nm. NPMe_IS, capped with DMDES, displayed an average diameter of 80 nm and a stellate morphology with an open mesoporosity (Fig. S2c, d and S4b, ESI†), while NPNH2_IS, which is the smallest one with an average diameter of 60 nm, clearly shows a core–shell morphology formed by a mesoporous core and a disordered organosilica shell of ca. 16 nm in diameter (Fig. S2e, f and S4c, ESI†). This fact, due to surface functionalization with APTES, would explain the blocking of their mesoporosity detected by gas adsorption measurements in these materials as described below.
The three types of complex-free control MSNs (NPMe, NPOH and NPNH2; Fig. S3, ESI†) show similar sizes to those described for their related hybrid in situ materials (Fig. S2, ESI†). Nevertheless, as observed in the TEM and DLS results (Fig. S3 and S5, ESI†), the procedure for the synthesis of the grafted nanoparticles produces a wide dispersion of sizes for NPOH_G and NPMe_G (from 40 to 200 nm) with respect to the complex-free starting materials (NP). This fact can be associated with the presence of NaF in the reaction media, which could modify both the size and morphology of the silica nanoparticles. It is well known that the fluoride anion has been revealed as a versatile catalyst for sol–gel processes, indeed at nearly neutral pH and moderate temperatures,30 although the kinetics of the reactions are still under study.31 In contrast, NPNH2_G maintains a similar size distribution to that shown for NPNH2_IS, perhaps due to the presence of their organosilica shell.
With respect to physisorption studies (Table 1 and Fig. 1), all materials yielded type IV isotherms, typical of mesoporous materials. Capping of the nanoparticles with APTES provokes a partial blocking of the mesoporosity in NPNH2, which shows the smallest surface area compared to those in NPOH and NPMe (230 m2 g−1vs. 760 m2 g−1 and 980 m2 g−1, respectively). The hybrid organometallo-silica nanoparticles NPOH_IS and NPMe_IS show very similar textural parameters to those observed for their control complex-free related MSNs (Table 1), which is consistent with the homogeneous integration of the organometallic complex in the silica matrix.24a Regarding their mesoporosity, NPMe_G also presents excellent textural parameters, whereas NPOH_G has half the BET surface area compared with the related NPOH_IS sample. A marked decrease in the mesopore volume is observed after incorporation of the chromophore into both the in situ and grafted amine functionalized MSNs (ABET, 230 m2 g−1NPNH2vs. 100 m2 g−1NPNH2_IS,G).
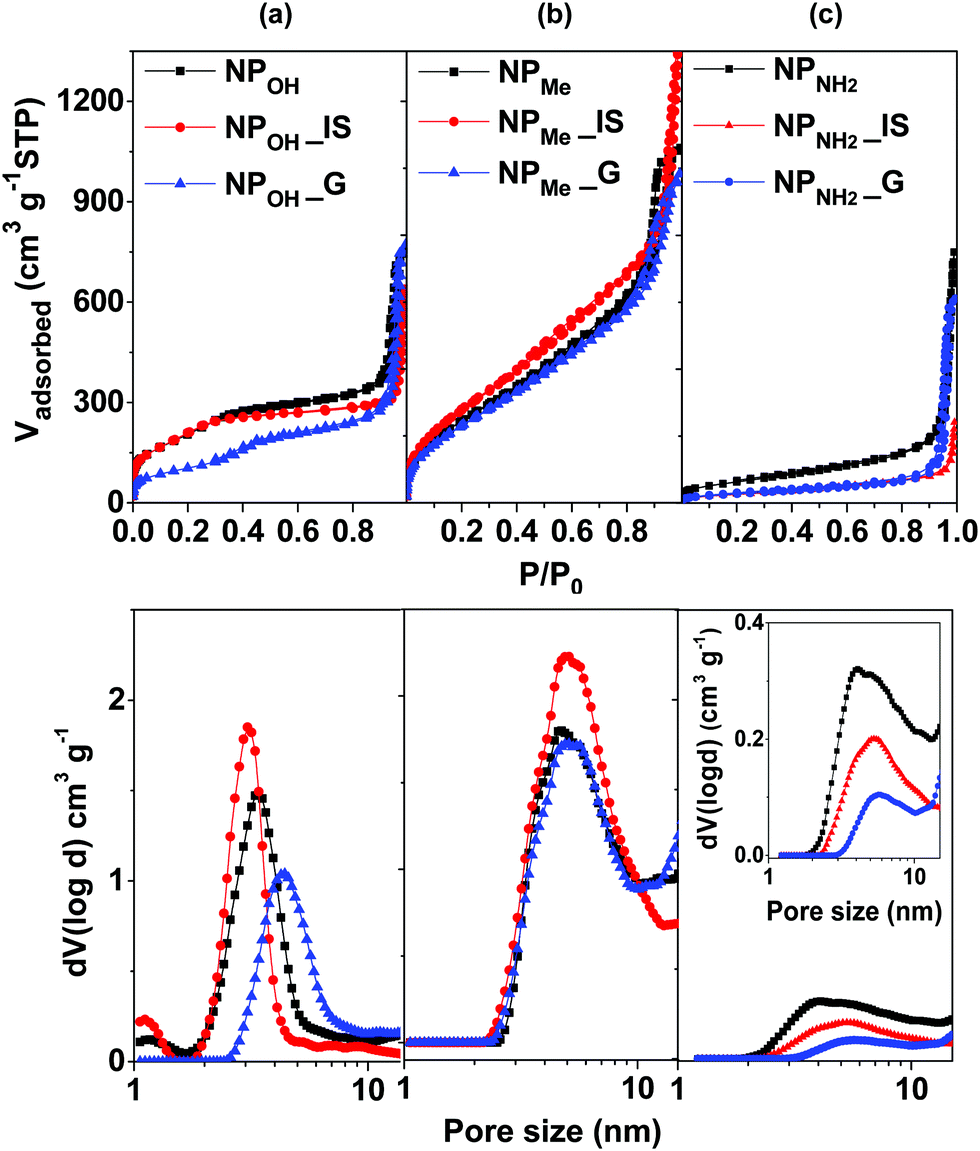 | ||
| Fig. 1 N2 adsorption/desorption isotherms at 77 K (above) and the corresponding pore size distribution calculated using the NLDFT method (below) of the hybrid MSNs, both IS and G, in comparison to the related complex-free silica nanoparticles (NP): (a) NPOH, (b) NPMe, and (c) NPNH2. | ||
Zeta potentials of the six types of hybrid nanoparticles were measured from Milli-Q H2O colloidal suspensions (Table 1). As expected, in these media, both unmodified NPOH and methyl capped NPMe show negative zeta potentials, while amine functionalized NPNH2 features positive values; nevertheless, a clear trend between the values of the in situ and related grafted materials cannot be established. Finally, all the materials can be easily resuspended in water or biological media (see biological activity), showing good (NPOH_IS,G) to excellent (NPMe_IS,G) colloidal stability, except for NPNH2_IS,G, which forms large aggregates of up to 4 µm in water within a few hours.
To further study the stability of MSNs in the physiological medium used for the cell culture, hybrid organometallo-silica MSNs, both in situ and grafted, were suspended in a complete medium for six months (RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) and 2.0 mM L-glutamine, penicillin (100 U mL−1) and streptomycin (100 µg mL−1)). After this time, nanoparticles were centrifuged and redispersed in EtOH, still displaying their yellow emission under UV illumination. Inspection by microscopy (see Fig. S4 for FESEM and S6, ESI† for TEM) shows that all the nanoparticles were highly stable in this biological media and only the organosilica shells of NPNH2_IS and NPNH2_G were partially damaged. Moreover, the centrifuged mother liquors were analyzed by high resolution ICP–MS, showing an iridium metal concentration ranging from 1 to 10 ppb in all the cases, which indicates that there was a negligible transference of the chromophore from the nanoparticles to the biological media in this period.
Photophysical properties and theoretical calculations
The absorption and emission data of complex 1 and those of the different organometallo-silica nanoparticles (NPs) in several media are collected in Tables S1 and S2 (ESI†). Selected spectra are shown in Fig. 2, 3 and Fig. S7–S10 (ESI†). Also, TD-DFT calculations based on the optimized cation model 1+ in THF have been carried out (see Fig. 2b and 4 and Fig. S11–S14 and Tables S3–S5 in the ESI†). The experimental UV-vis spectrum in tetrahydrofuran solution of complex 1 (Fig. 2a and Table S1, ESI†) is in good qualitative agreement with the calculated allowed absorptions (Fig. S13 and Table S5, ESI†). According to calculations, the intense high-energy band below 300 nm is ascribed to spin allowed π–π* transitions located on the C^N and N^N aromatic ligands with minor metal-to-ligand contributions. The shoulder at 305 nm might be related to the transition calculated at 308 nm (S19), which corresponds to a combination of intraligand and ligand-to-ligand charge transfer transitions (1IL/1LL′CT) (L: C^N, L′: N^N), with a certain metal-to-ligand charge transfer (1ML′CT) character. The low energy band at 360 nm agrees with the calculated transitions at 367 (S7) and 369 (S6) nm, which are mainly associated with HOMO→L+1 and H−4→LUMO transitions (Fig. 2b and Table S4, ESI†). The HOMO and H−4 are located at the iridium center (ca. 40%) and the phenyl groups of both phenylpyridine ligands (ca. 60%), whereas the LUMO and L+1 are centered on the bipyridine ligand (96%). Therefore, this low energy transition is attributed to a mixture of 1ML′CT/1LL′CT transitions. On the other hand, due to the strong spin–orbit coupling (SOC) associated with iridium, complex 1 also shows additional weak absorptions (>400 nm) ascribed to spin-forbidden 3ML′CT/3LL′CT transitions. These assignments also agree with those found for other related cyclometalated Ir(III) complexes.25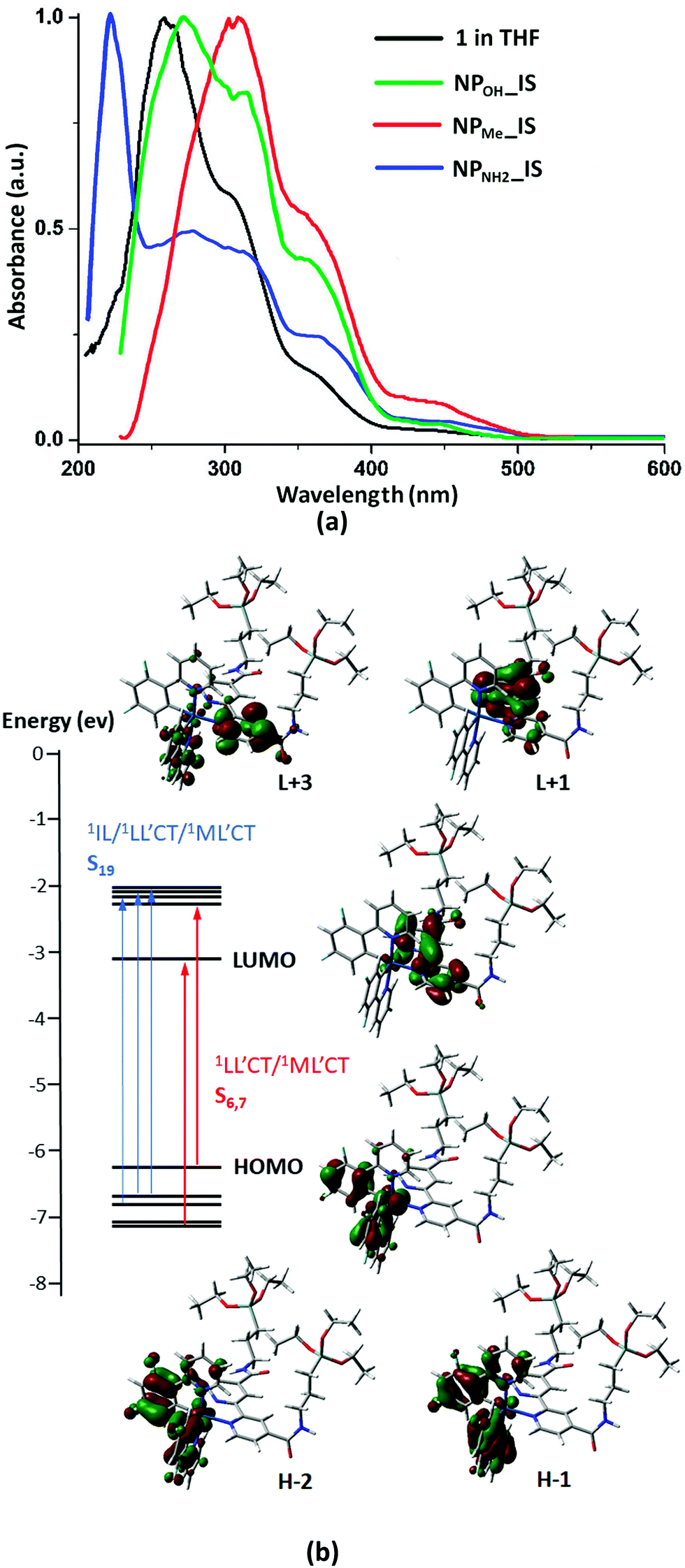 | ||
| Fig. 2 (a) Comparison of the absorption spectrum of complex 1 in THF (5 × 10−5 M) with those of the in situ NPs in the solid state. (b) Selected frontier molecular orbitals and transitions involved in the absorption spectra for complex 1. | ||
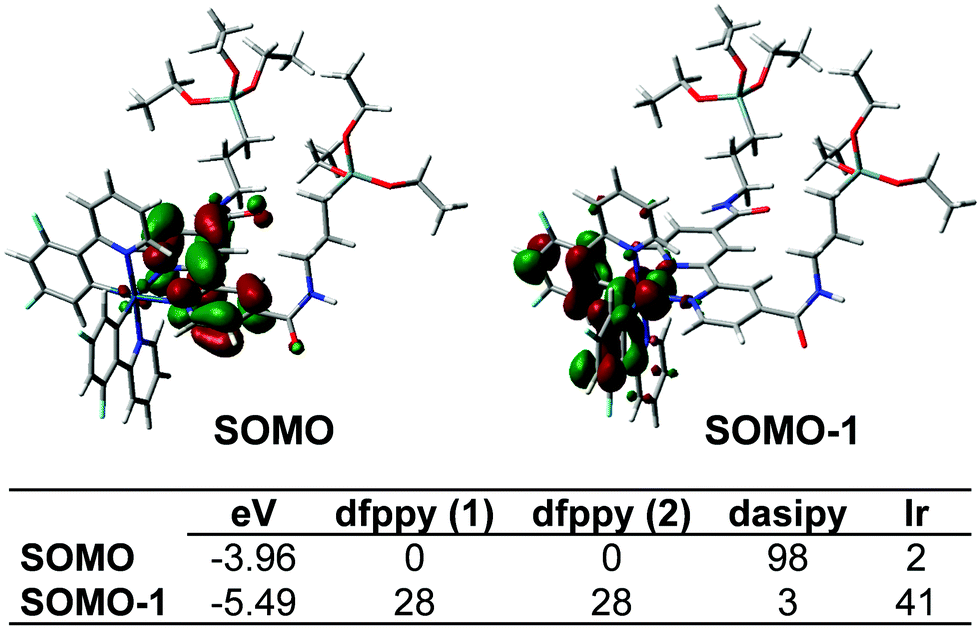 | ||
| Fig. 3 SOMO and SOMO−1 orbitals and their composition (%) for the lowest excited triplet state in complex 1. | ||
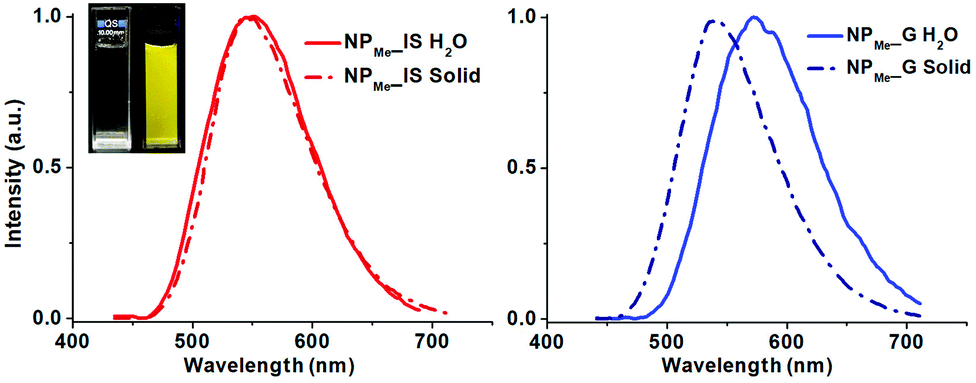 | ||
| Fig. 4 Emission spectra at room temperature of NPMe_IS (left, red) and NPMe_G (right, blue) in the solid state (dotted line) and in water suspension (solid line). Inset: photograph of the aqueous suspension of NPMe_IS under visible and UV λexc 370 nm illumination. | ||
The solid state diffuse reflectance UV-vis (DRUV) spectra of all the organometallo-silica NPs (IS, Fig. 2a and G, Fig. S7, ESI†) display essentially a similar profile to that observed for complex 1 in solution, confirming the preservation of the molecular structure of the complex during its incorporation into the silica matrix.
In degassed THF solution, complex 1 emits brightly (ϕ 56%) in the yellow spectral region, featuring a long-life (0.7 µs) structureless emission band with λmax at 550 nm (Fig. S8, ESI†). As shown in Fig. 3, the SOMO and SOMO−1 are analogous to the LUMO and HOMO in the ground state. Therefore, this emission is attributed to a mixture of 3ML′CT (Ir→N^N)/3LL′CT (dfppy→N^N) with a remarkable metal character. According to this assignment, the emission is notably blue-shifted at 77 K (510 nm; Fig. S8, ESI†), and also markedly red shifted in a more polar solvent like MeOH (λem 585 nm, 298 K; 530 nm 77 K; ϕ 66.4%), features that are commonly observed in cyclometalated iridium(III) polypyridine systems. There is good correlation with the calculated emission values in both solvents [ΔE(T1–S0) 575 nm (THF) and 587 nm (MeOH)]. The observed bathochromic shift (positive solvatochromism) indicates that the excited state is more polar than the ground state as it is stabilized by the solvent polarity and consequently has a decrease in the emission energy.32 In the solid state, complex 1 emission exhibits a slight red-shift (λem 560 nm, 298 K; 550 nm 77 K; Fig. S8, ESI†), but with a shorter life time and a lower quantum yield (0.35 µs and ϕ 29.4%), which are indicative of faster non-radiative decay compared to what occurs in solution. These features can be tentatively attributed to the presence of π··π interactions in the solid state, which favor quenching due to triplet–triplet annihilation.
The photophysical properties of all the organometallo-silica NPs have been measured at room temperature in the solid state and in aqueous suspensions (5 × 10−4 M) (Table S2, ESI†). The three in situ materials (NP_IS) feature, in the solid state and in aqueous suspension, a non-structured emission band with λmax and lifetime values (λem 550–558 nm, τ 0.70–0.80 µs in the solid state) similar to those found for complex 1 in the fluid THF (Fig. 4 for NPMe_IS and see also Fig. S9, S10, ESI†). It is noteworthy that in spite of the small amount of chromophore incorporated into the nanoparticles (less than 1 wt% of complex 1), the materials are highly emissive, exhibiting in the solid state comparable emission quantum yields to those found for the pure complex in THF solution (ϕ 41.2% NPOH_IS, 50.1% NPMe_IS and 52.0% NPNH2_ISvs. 56.0% complex 1) and slightly lower values in aqueous suspension (ϕ 25.8% NPOH_IS, 39.6% NPMe_IS and 27.5% NPNH2_IS). This is consistent with homogeneous and well-dispersed incorporation of the chromophore throughout the silica matrix during the in situ formation of the actual NP_IS hybrid, as well as with the presence of a rigid environment around the cyclometalated iridium complex. Thus, incorporation of the organometallic dye into the silica framework not only increases its rigidity, decreasing non-radiative deactivation due to molecular distortion, but also precludes the occurrence of non-covalent interactions between the chromophores. It is noteworthy that in both media the capped NPs (NPMe_IS and NPNH2_IS), which are smaller in size, demonstrated better emissive performance than NPOH_IS.
On the other hand, the three grafted materials (NP_G) show a similar broad emission (λem 545–550 nm, Table S2, ESI†) in the solid state, but with shorter life times and lower quantum yields (ϕ 28–41%; τ 0.50–0.65 µs) than those observed for their related in situ nanoparticles (NP_IS). This fact can be attributed to a decreased protection of superficially grafted molecules of the chromophore, which are therefore exposed to the external media. Interestingly, in aqueous suspensions, all NP_G display a clearly red-shifted emission maxima (λem ∼570 nm, Table S2, ESI†), a feature attributable to the likely stabilization of the excited state of the chromophore by easier interaction with the polar solvent.
Biological properties
Given the advantageous properties shown by all the nanoparticles, such as high quantum yields, long lifetimes and good dispersion and stability in aqueous and ionic media, we decided to investigate their biocompatibility and possible use as biological markers.Cell viability assay
To find any possible cytotoxic effect of the organometallo-silica nanoparticles, IC50 values (µg mL−1) were determined against human tumor A549 (lung carcinoma) and HeLa (cervix carcinoma) cell lines. IC50 values for each of the NPs were measured in vitro using the MTT assay33 after cellular exposure for 72 h, as we have previously described for other compounds.34 All silica nanoparticles tested showed very low cytotoxicity values towards both cell lines (Table 2 and Fig. S15, S16, ESI†). Actually, both in situ and grafted NPMe and NPOH_G nanoparticles did not affect cell viability, since their IC50 values could not be determined at any concentration tested. The negligible cytotoxicity shown by NPNH2 (IC50, µg mL−1 ≈ 90 IS, 80 G) could be related to the presence of amino groups on the nanoparticle surface or to the release of these groups into the cellular environment, as previously observed in other similarly functionalized MSNs.35 Interestingly, regardless of whether the iridium chromophores were primarily located in the silica matrix or on the nanoparticle surface, each nanoparticle type (OH, Me or NH2) presents similar IC50 values in spite of being IS or G, respectively. Moreover, IC50 values did not change significantly after six months of storage in the culture medium (Fig. S15 and S16, ESI†), corroborating the above-mentioned nanoparticle stability and the fact that they do not release any derivative with cytotoxic activity into the medium.| Sample | A549 | HeLa |
|---|---|---|
| IC50 values are presented as mean ± standard error of the mean of three different experiments. NT, non-toxic (IC50 values could not be determined). | ||
| NPOH_IS | 166.00 ± 23.93 | 219.30 ± 7.64 |
| NPOH_G | 184.30 ± 32.93 | NT |
| NPMe_IS | NT | NT |
| NPMe_G | NT | NT |
| NPNH2_IS | 93.36 ± 5.70 | 94.36 ± 11.82 |
| NPNH2_G | 74.28 ± 11.33 | 80.23 ± 14.29 |
Cellular uptake and cytolocalization
Once the favorable non-toxic properties of the organometallo-silica NPs were observed, their possible use as luminescent biomarkers was studied by fluorescence and confocal microscopy. To this aim, NPOH_IS and NPMe_IS were first examined in living cells by fluorescence microscopy, after being incubated for 24 h with A549 and HeLa cells in the presence of the dye Hoechst 33258 nucleic acid stain. Living cell preparations and documentation were performed as previously reported.36 Superimposition of images obtained by alternate imaging with Nomarski visualization, Hoechst staining of nuclear DNA, and fluorescence emission of nanoparticles enabled us to decipher the fluorescence emission site inside the cells (Fig. S17, ESI†). Both types of nanoparticles seemed to be successfully internalized in the living cells and emissions were restricted to the cytoplasm, with a tendency to accumulate in perinuclear areas, excluding the nucleus in both cell lines (Fig S17, ESI†).Next, in order to ascertain the cytoplasmic accumulation of NPs inside the cells, we analyzed cellular uptake using confocal fluorescence imaging in fixed cells immunostained for tubulin. This stain labels the microtubules and allows visualization of the cytoskeleton and cell body outline. As expected, in the absence of organometallo-silica nanoparticles, cells did not show any emissive signals (data not shown). Incubation of cells with NPs for 24 h resulted in cellular uptake in all cases. As seen in Fig. 5, the NPs appeared aggregated in the cytoplasm, forming granules with sizes ranging from 200 nm to several microns. Intracellular distribution seems to be random, although with a clear preference for perinuclear zones, as seen in cells in vivo (Fig. S17, ESI†). These organometallo-silica nanoparticles are mostly excluded from the cell nucleus, a behavior previously reported with other MSNs sized > 50 nm.16a,33,37 Thus, as previously suggested and based on their good cellular internalization, intensive fluorescence and low cytotoxicity, we can state that our organometallo-silica MSNs are biocompatible enough for bioimaging applications, keeping in mind that the dosage used for in vivo fluorescence imaging is only 50 µg mL−1.16a
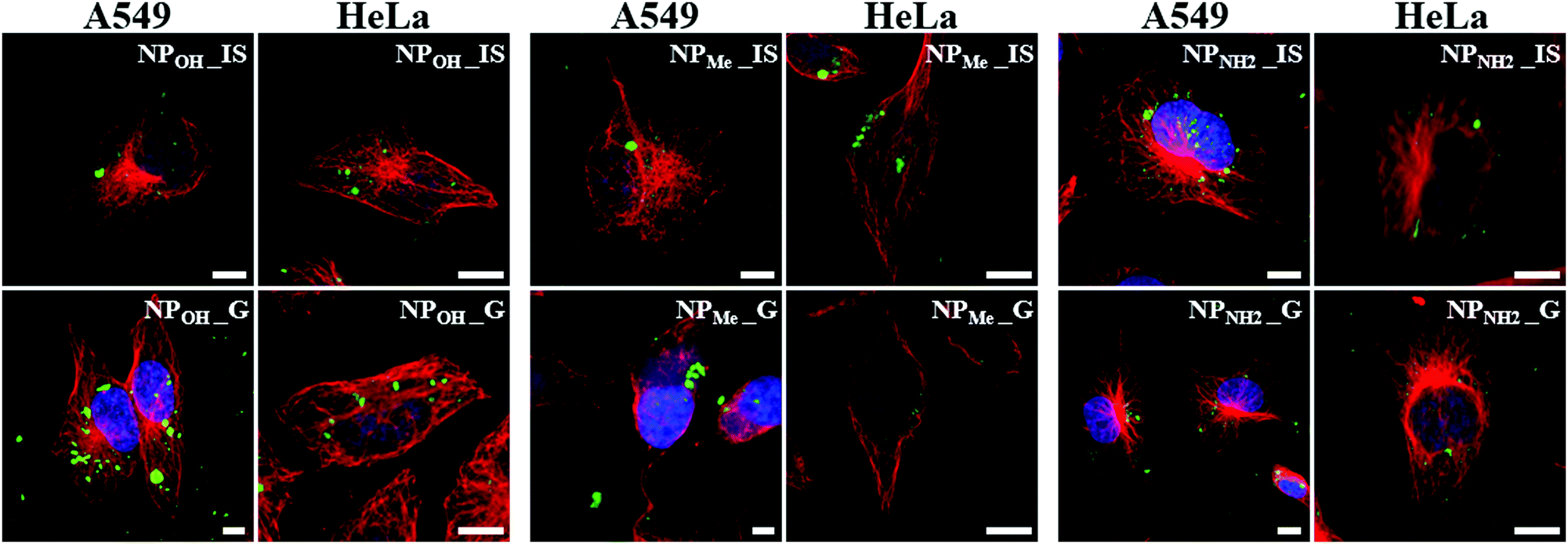 | ||
| Fig. 5 Cellular uptake and intracellular localization of silica nanoparticles in A549 and HeLa cells. Laser scanning confocal microscopy images of fixed cells incubated with nanoparticles (green) for 24 h and immunostained for β-tubulin (red). Blue: Nuclei marker (DAPI). Scale bars: 10 µm. | ||
Extracellular aggregation
The relationship between the silica nanoparticles in biological media and their colloidal stability is still a matter of discussion.10b,11a,38 Given the size of the NP aggregates found intracellularly, the question arises whether they were internalized as discrete particles and aggregated intracellularly, or if they aggregated extracellularly and were then internalized as larger agglomerates.To determine which scenario was the case, we studied phosphorescent NPs found on the extracellular surface of the fixed culture media analyzed by confocal microscopy. As seen in Fig. 6a, all types of NPs located surrounding the cells were found to be forming extracellular aggregates to some degree. Morphological analyses showed that the mean size of the aggregated particles was in the range of 1–3 µm. Nevertheless, the degree of aggregation varied between NP types. Thus, NPMe_IS, NPMe_G and NPOH_IS showed lower levels of aggregation, while NPOH_G, and especially NPNH2_IS and NPNH2_G, showed the highest levels (Fig. 6). This result is not surprising because MSN aggregation in cell culture medium and inside cells has been reported elsewhere.39 In line with the previously noted behavior observed in aqueous media, methyl functionalization seems to enhance the colloidal stability of NPMe (both for in situ and grafted NPs) in biological media, hindering the formation of extracellular aggregates. Previous studies have shown that long chain PEG grafting increases the colloidal stability of silica nanoparticles,11a,38a,40 but as far as we know there are no similar results using aliphatic groups as short as methyl. On the other hand, the presence of amine groups (NPNH2_IS,G) at the surface of the NPs favors aggregation. This fact has previously been reported for short alkyl chain aminosilanes such as APTES and can also be related to the interaction of the –NH2 groups with the components of the culture media, and easy formation of the protein corona around the nanoparticle.35a,38a,39d Finally, the greatest difference in behavior between in situ and grafted NPs is observed for the unmodified NPOH, with NPOH_IS showing better colloidal stability than NPOH_G in extracellular media. In any case, the impact of certain NP aggregation on cytotoxicity and cellular uptake is controversial and not clear,39b which indicates that it may depend on each NP structure and composition.
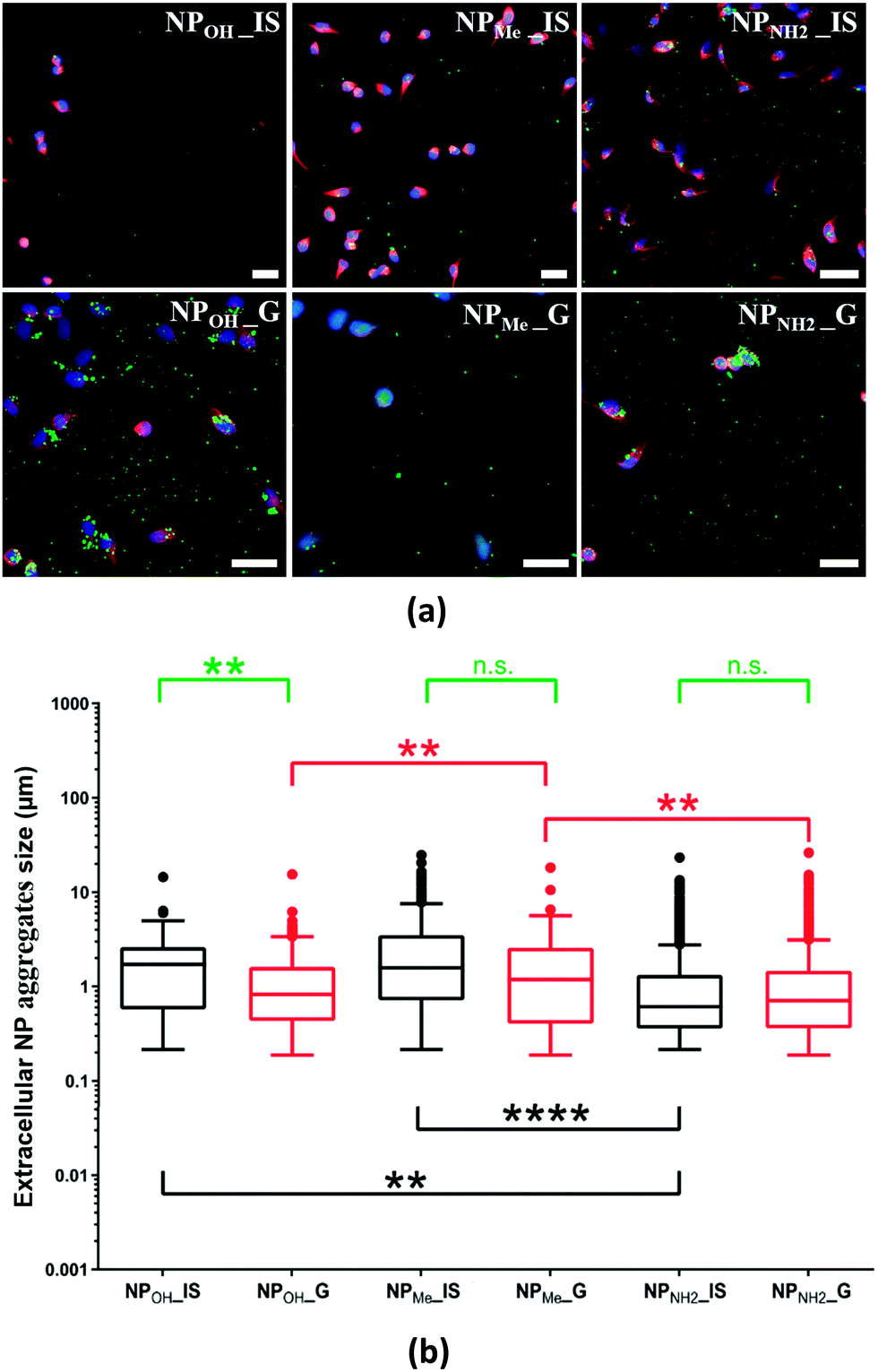 | ||
| Fig. 6 Distribution and size of extracellular NP aggregates on coverslips containing growing A549 cells analyzed by laser scanning microscopy. (a) Confocal microscopy images of cells in culture showing the NP distribution in extracellular spaces. Green: NPs; red: tubulin antibody staining; and blue: nuclei marker (DAPI). Scale bars: 40 µm. (b) Box plot representation of extracellular NP aggregate size distribution. Black boxes: NP_IS and red boxes: NP_G. Statistical comparisons were performed between NP_IS with different surface functionalization (black asterisks), NP_G with different surface functionalization (red asterisks) or between NP_IS and NP_G with the same functionalization (green asterisks). **: p > 0.01, ****: p > 0.0001, n.s.: non-significant. | ||
Conclusions
In summary, beginning with [Ir(dfppy)2(dasipy)]PF6 (1) and TEOS as precursors, we describe herein a one-pot synthetic strategy for preparing chemically inert and well-defined spherical hybrid organometallo-silica nanoparticles (NPs), which are not only highly emissive but also can be easily surface functionalized during the in situ procedure. Following this methodology, we prepared three types of MSNs with different surface functionalization (NPOH_IS, NPMe_IS and NPNH2_IS), which exhibit better emissive, textural and biological properties than those observed for their respective grafted counterparts (NPOH_G, NPMe_G and NPNH2_G). Despite the low chromophore complex load (less than 1 wt%), both in situNPs display an intense and photostable yellow phosphorescence (λemca. 550 nm; ϕ 40–50%), which can be easily observed in biological media. Also, these in situNPs show mesoporosity with surface areas up to 1100 m2 g−1 for NPMe_IS and can be easily resuspended in water and biological media, where NPMe_IS and NPOH_IS show a good colloidal stability. Notwithstanding, every type of NP has been found to form extracellular aggregates in the range of 1–3 µm in cultured media, although NPMe_IS and NPOH_IS clearly exhibit lower levels of aggregation than NPNH2_IS.Independently of the zeta potential displayed for the nanoparticles in water (negative for NPMe_IS and NPOH_IS, and positive for NPNH2_IS), all are efficiently taken up by cells from the surrounding extracellular space as aggregates that primarily localize in the cytoplasm. This fact, combined with the absence of cytotoxicity toward cells cultured in vitro, even after long-term storage, demonstrates that MSNs prepared in situ can be used as excellent luminescent probes for cell imaging.
Finally, among all the materials described, NPMe_IS seems to be of particular interest. It is well established that long chain PEGylated MSNs show excellent colloidal stability, but in this case, this important property is obtained simply by using diethoxydimethylsilane (DMDES) as the capping agent in the in situ synthesis procedure, which also allows the efficient and homogeneous incorporation of the chromophore into the silica matrix to form discrete nanoparticles of ca. 80 nm with a stellate morphology and a BET area of 1120 m2 g−1. Moreover, NPMe_IS nanoparticles can be further functionalized if necessary, as observed in the behavioural study of their related grafted nanoparticle (NPMe_G).
Further studies will be needed to determine the viability of the synthetic method using other chromophores, the behavior of these hybrid organometallo-silica NPs in controlled intramolecular delivery and how they perform in in vivo applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Spanish MCIN/AIE/10.13039/501100011033, the “ERDF A way of making Europe”, the “European Union” (projects CTQ2015-74494-JIN, RTI2018-099504-B-C21 and PID2019-109742GB-I00) and the Agencia de Desarrollo Económico de La Rioja (Gobierno de la Rioja. Project 2017-I-IDD-00031). E. S. also thanks the University of Alicante through the “Programa de Retención de Talento” (ref. UATALENTO16-03). I. M. L. is supported by a Miguel Servet contract (CPII20/00029) from the Instituto de Salud Carlos III, co-funded by European Social Fund (ESF) “Investing in your future”. E. A.-A. is grateful to the Spanish Association Against Cancer (AECC) for her Ph.D. fellowship. We want to thank Dr. Ana García Fontecha for her help with the DLS measurements.References
- (a) J. I. Joo, M. Choi, S.-H. Jang, S. Choi, S.-M. Park, D. Shin and K.-H. Cho, Adv. Mater., 2020, 32, 1906783 CrossRef CAS PubMed; (b) S. Parveen, N. Yadav and M. Banerjee, in Nanomaterials and Environmental Biotechnology, ed. I. Bhushan, V. K. Singh and D. K. Tripathi, Springer International Publishing, Cham, 2020, pp. 189–199 Search PubMed; (c) A. Acharya, Nanomaterial - Based Biomedical Applications in Molecular Imaging, Diagnostics and Therapy, Springer, Singapore, 2020 CrossRef; (d) A. Pallotta, I. Clarot, J. Sobocinski, E. Fattal and A. Boudier, ACS Appl. Bio Mater., 2019, 2, 1–13 CrossRef CAS PubMed; (e) C. Fornaguera and M. J. García-Celma, J. Pers. Med., 2017, 7, 12 CrossRef PubMed; (f) S. Mukherjee and C. R. Patra, Nanoscale, 2016, 8, 12444–12470 RSC; (g) W. Yucai, S. M. Suk, L. S. Nathanael, S. Hsing-Wen and X. Younan, Adv. Funct. Mater., 2014, 24, 4206–4220 CrossRef PubMed.
- (a) G. A. Marcelo, C. Lodeiro, J. L. Capelo, J. Lorenzo and E. Oliveira, Mater. Sci. Eng., C, 2020, 106, 110104 CrossRef CAS PubMed; (b) F. Yang, Q. Zhang, S. Huang and D. Ma, J. Mater. Chem. B, 2020, 8, 7856–7879 RSC; (c) M. A. Cotta, ACS Appl. Nano Mater., 2020, 3, 4920–4924 CrossRef CAS.
- (a) C.-L. Shen, Q. Lou, K.-K. Liu, L. Dong and C.-X. Shan, Nano Today, 2020, 35, 100954 CrossRef CAS; (b) D. Maiti, X. Tong, X. Mou and K. Yang, Front. Pharmacol., 2019, 9, 1401 CrossRef PubMed.
- (a) M. Azharuddin, G. H. Zhu, D. Das, E. Ozgur, L. Uzun, A. P. F. Turner and H. K. Patra, Chem. Commun., 2019, 55, 6964–6996 RSC; (b) W. Chen, S. Zhang, Y. Yu, H. Zhang and Q. He, Adv. Mater., 2016, 28, 8567–8585 CrossRef CAS PubMed.
- (a) A. Avasthi, C. Caro, E. Pozo-Torres, M. P. Leal and M. L. García-Martín, Top. Curr. Chem., 2020, 378, 40 CrossRef CAS PubMed; (b) S. Palanisamy and Y.-M. Wang, Dalton Trans., 2019, 48, 9490–9515 RSC.
- (a) X. Ren, Y. Han, Y. Xu, T. Liu, M. Cui, L. Xia, H. Li, Y. Gu and P. Wang, Coord. Chem. Rev., 2020, 213676 Search PubMed; (b) R. F. Mendes, F. Figueira, J. P. Leite, L. Gales and F. A. Almeida Paz, Chem. Soc. Rev., 2020, 9121–9153 RSC; (c) A. Bieniek, A. P. Terzyk, M. Wiśniewski, K. Roszek, P. Kowalczyk, L. Sarkisov, S. Keskin and K. Kaneko, Prog. Mater. Sci., 2020, 100743 Search PubMed.
- (a) M. Manzano and M. Vallet-Regí, Adv. Funct. Mater., 2020, 30, 1902634 CrossRef CAS; (b) Q. Lei, J. Guo, A. Noureddine, A. Wang, S. Wuttke, C. J. Brinker and W. Zhu, Adv. Funct. Mater., 2020, 30, 1909539 CrossRef CAS; (c) B. G. Cha and J. Kim, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2019, 11, e1515 Search PubMed; (d) L. Travaglini and L. De Cola, Chem. Mater., 2018, 30, 4168–4175 CrossRef CAS; (e) C. Caltagirone, A. Bettoschi, A. Garau and R. Montis, Chem. Soc. Rev., 2015, 44, 4645–4671 RSC.
- J. G. Croissant, K. S. Butler, J. I. Zink and C. J. Brinker, Nat. Rev. Mater., 2020, 5, 886–909 CrossRef CAS.
- (a) J. G. Croissant, Y. Fatieiev and N. M. Khashab, Adv. Mater., 2017, 29, 1604634 CrossRef PubMed; (b) J. L. Paris, M. Colilla, I. Izquierdo-Barba, M. Manzano and M. Vallet-Regí, J. Mater. Sci., 2017, 52, 8761–8771 CrossRef CAS.
- (a) R. K. Singh, K. D. Patel, K. W. Leong and H.-W. Kim, ACS Appl. Mater. Interfaces, 2017, 9, 10309–10337 CrossRef CAS PubMed; (b) S. Behzadi, V. Serpooshan, W. Tao, M. A. Hamaly, M. Y. Alkawareek, E. C. Dreaden, D. Brown, A. M. Alkilany, O. C. Farokhzad and M. Mahmoudi, Chem. Soc. Rev., 2017, 46, 4218–4244 RSC; (c) I.-Y. Kim, E. Joachim, H. Choi and K. Kim, Nanomedicine, 2015, 11, 1407–1416 CrossRef CAS PubMed.
- (a) M. Gisbert-Garzarán and M. Vallet-Regí, Nanomaterials, 2020, 10, 916 CrossRef PubMed; (b) M. Martínez-Carmona, Q. P. Ho, J. Morand, A. García, E. Ortega, L. C. S. Erthal, E. Ruiz-Hernandez, M. D. Santana, J. Ruiz, M. Vallet-Regí and Y. K. Gun’ko, Inorg. Chem., 2020, 59, 10275–10284 CrossRef PubMed.
- (a) S. Jafari, H. Derakhshankhah, L. Alaei, A. Fattahi, B. S. Varnamkhasti and A. A. Saboury, Biomed. Pharmacother., 2019, 109, 1100–1111 CrossRef CAS PubMed; (b) F. Tang, L. Li and D. Chen, Adv. Mater., 2012, 24, 1504–1534 CrossRef CAS PubMed.
- T. T. Hoang Thi, V. D. Cao, T. N. Q. Nguyen, D. T. Hoang, V. C. Ngo and D. H. Nguyen, Mater. Sci. Eng., C, 2019, 99, 631–656 CrossRef CAS PubMed.
- L. Li, W. Wang, J. Tang, Y. Wang, J. Liu, L. Huang, Y. Wang, F. Guo, J. Wang, W. Shen and L. A. Belfiore, Nanoscale Res. Lett., 2019, 14, 190 CrossRef CAS PubMed.
- (a) S. Yang and Y. Li, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2020, 12, e1603 Search PubMed; (b) D. Yuan, C. M. Ellis and J. J. Davis, Materials, 2020, 13, 3795 CrossRef CAS PubMed.
- (a) L. Huang, S. Yu, W. Long, H. Huang, Y. Wen, F. Deng, M. Liu, W. Xu, X. Zhang and Y. Wei, Microporous Mesoporous Mater., 2020, 308, 110520 CrossRef CAS; (b) N. Song, Z. Zhang, P. Liu, Y.-W. Yang, L. Wang, D. Wang and B. Z. Tang, Adv. Mater., 2020, 32, 2004208 CrossRef CAS PubMed; (c) C. Liu, H. Yu, Q. Li, C. Zhu and Y. Xia, ACS Appl. Mater. Interfaces, 2018, 10, 16291–16298 CrossRef CAS PubMed; (d) L. Yan, Y. Zhang, B. Xu and W. Tian, Nanoscale, 2016, 8, 2471–2487 RSC.
- (a) H. Jin, X. Jiang, Z. Sun and R. Gui, Coord. Chem. Rev., 2020, 213694 Search PubMed; (b) K. Y. Zhang, Q. Yu, H. Wei, S. Liu, Q. Zhao and W. Huang, Chem. Rev., 2018, 118, 1770–1839 CrossRef CAS PubMed.
- (a) A. Zanut, F. Palomba, M. Rossi Scota, S. Rebeccani, M. Marcaccio, D. Genovese, E. Rampazzo, G. Valenti, F. Paolucci and L. Prodi, Angew. Chem., Int. Ed., 2020, 59, 21858–21863 CrossRef CAS PubMed; (b) N. Soliman, G. Gasser and C. M. Thomas, Adv. Mater., 2020, 32, 2003294 CrossRef CAS PubMed; (c) K. Zanoni, R. Vilela, I. Silva, N. Iha, H. Eckert and A. Camargo, Inorg. Chem., 2019, 58, 4962–4971 CrossRef CAS PubMed; (d) Y. Atoini, E. A. Prasetyanto, P. Chen, S. Silvestrini, J. Harrowfield and L. De Cola, Chem. – Eur. J., 2018, 24, 12054–12060 CrossRef CAS PubMed; (e) S. Kesarkar, E. Rampazzo, G. Valenti, M. Marcaccio, A. Bossi, L. Prodi and F. Paolucci, ChemElectroChem, 2017, 4, 1690–1696 CrossRef CAS; (f) M. Galli, E. Moschini, M. V. Dozzi, P. Arosio, M. Panigati, L. D’Alfonso, P. Mantecca, A. Lascialfari, G. D’Alfonso and D. Maggioni, RSC Adv., 2016, 6, 38521–38532 RSC.
- (a) Y. Wang, S. Song, S. Zhang and H. Zhang, Nano Today, 2019, 25, 38–67 CrossRef CAS; (b) A. M. G. Mutti, J. A. O. Santos, D. G. S. M. Cavalcante, A. S. Gomes, A. E. Job, G. R. Teixeira, A. M. Pires and S. A. M. Lima, Mater. Today Chem., 2019, 14, 100204 CrossRef CAS; (c) K. Binnemans, Chem. Rev., 2009, 109, 4283–4374 CrossRef CAS PubMed.
- (a) R. Bai, X. Meng, X. Wang and L. He, Adv. Funct. Mater., 2020, 30, 1907169 CrossRef CAS; (b) E. Fresta, M. A. Monclús, M. Bertz, C. Ezquerro, J. M. Molina-Aldareguia, J. R. Berenguer, M. Kunimoto, T. Homma and R. D. Costa, Adv. Opt. Mater., 2020, 8, 2000295 CrossRef CAS; (c) D. R. Martir and E. Zysman-Colman, Coord. Chem. Rev., 2018, 364, 86–117 CrossRef; (d) I. N. Mills, J. A. Porras and S. Bernhard, Acc. Chem. Res., 2018, 51, 352–364 CrossRef CAS PubMed; (e) M. Dongxin, T. Taiju, Q. Yong and D. Lian, Adv. Mater., 2017, 29, 1603253 CrossRef PubMed.
- (a) S. Abbas, I.-U.-D. Din, A. Raheel and A. Tameez ud Din, Appl. Organomet. Chem., 2020, 34, e5413 CrossRef CAS; (b) C. Caporale and M. Massi, Coord. Chem. Rev., 2018, 363, 71–91 CrossRef CAS; (c) Y. You, Curr. Opin. Chem. Biol., 2013, 17, 699–707 CrossRef CAS PubMed.
- (a) P.-Y. Ho, C.-L. Ho and W.-Y. Wong, Coord. Chem. Rev., 2020, 413, 213267 CrossRef CAS; (b) J. Li and T. Chen, Coord. Chem. Rev., 2020, 418, 213355 CrossRef CAS; (c) R. Guan, Y. Chen, L. Zeng, T. W. Rees, C. Jin, J. Huang, Z.-S. Chen, L. Ji and H. Chao, Chem. Sci., 2018, 9, 5183–5190 RSC; (d) A. Zamora, G. Vigueras, V. Rodríguez, M. D. Santana and J. Ruiz, Coord. Chem. Rev., 2018, 360, 34–76 CrossRef CAS.
- (a) X. Li, K. Fan, R. Yang, X. Du, B. Qu, X. Miao and L. Lu, J. Hazard. Mater., 2020, 386, 121929 CrossRef CAS PubMed; (b) X. Li, R. Zeng, C. Xie, D. Tang, Q. Li, B. Zhang and T. Huang, Dyes Pigm., 2019, 165, 128–136 CrossRef CAS; (c) M. Cheng, L. Zhou, J. Ma, J. Mu, C. Yi and M.-J. Li, Mater. Sci. Eng., C, 2019, 104, 109972 CrossRef CAS PubMed; (d) X. Tang, K. Fan, B. Qu and L. Lu, Microchim. Acta, 2019, 186, 841 CrossRef CAS PubMed; (e) Z. Chen, P. Yan, L. Zou, M. Zhao, J. Jiang, S. Liu, K. Zhang, W. Huang and Q. Zhao, Adv. Healthcare Mater., 2018, 7, 1800309 CrossRef PubMed; (f) Q. Zhao, Y. Liu, Y. Cao, W. Lv, Q. Yu, S. Liu, X. Liu, M. Shi and W. Huang, Adv. Opt. Mater., 2015, 3, 233–240 CrossRef CAS.
- (a) C. Ezquerro, A. Sepúlveda, A. Grau-Atienza, E. Serrano, E. Lalinde, J. R. Berenguer and J. Garcia-Martinez, J. Mater. Chem. C, 2017, 5, 9721–9732 RSC; (b) E. Serrano, N. Linares, J. Garcia-Martinez and J. R. Berenguer, ChemCatChem, 2013, 5, 844–860 CrossRef CAS.
- C. Ezquerro, E. Fresta, E. Serrano, E. Lalinde, J. García-Martínez, J. R. Berenguer and R. D. Costa, Mater. Horiz., 2019, 6, 130–136 RSC.
- V. E. Pritchard, D. Rota Martir, E. Zysman-Colman and M. J. Hardie, Chem. – Eur. J., 2017, 23, 8839–8849 CrossRef CAS PubMed.
- G. M. Greenway, A. Greenwood, P. Watts and C. Wiles, Chem. Commun., 2006, 85–87 RSC.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds. Part B Applications in Coordination, Organometallic, and Bioinorganic Chemistry, John Wiley & Sons Inc., New Jersey, 6th edn, 2009 Search PubMed.
- S.-H. Lai, J.-W. Ling, Y.-M. Huang, M.-J. Huang, C. H. Cheng and I. C. Chen, J. Raman Spectrosc., 2011, 42, 332–338 CrossRef CAS.
- U. Díaz, T. García, A. Velty and A. Corma, J. Mater. Chem., 2009, 19, 5970–5979 RSC.
- T.-M. Hu, C.-Y. Lin and M.-J. Wu, RSC Adv., 2019, 9, 28028–28037 RSC.
- (a) C. Sahin, A. Goren, S. Demir and M. S. Cavus, New J. Chem., 2018, 2979–2988 RSC; (b) J. Xu, C. Yang, B. Tong, Y. Zhang, L. Liang and M. Lu, J. Fluoresc., 2013, 23, 865–875 CrossRef CAS PubMed.
- L. Pan, Q. He, J. Liu, Y. Chen, M. Ma, L. Zhang and J. Shi, J. Am. Chem. Soc., 2012, 134, 5722–5725 CrossRef CAS PubMed.
- E. Lalinde, R. Lara, I. P. López, M. T. Moreno, E. Alfaro-Arnedo, J. G. Pichel and S. Piñeiro-Hermida, Chem. – Eur. J., 2018, 24, 2440–2456 CrossRef CAS PubMed.
- (a) C. Graf, Q. Gao, I. Schütz, C. N. Noufele, W. Ruan, U. Posselt, E. Korotianskiy, D. Nordmeyer, F. Rancan, S. Hadam, A. Vogt, J. Lademann, V. Haucke and E. Rühl, Langmuir, 2012, 28, 7598–7613 CrossRef CAS PubMed; (b) E. A. Smith and W. Chen, Langmuir, 2008, 24, 12405–12409 CrossRef PubMed.
- J. R. Berenguer, J. G. Pichel, N. Gimenez, E. Lalinde, M. T. Moreno and S. Pineiro-Hermida, Dalton Trans., 2015, 44, 18839–18855 RSC.
- C.-C. Chou, W. Chen, Y. Hung and C.-Y. Mou, ACS Appl. Mater. Interfaces, 2017, 9, 22235–22251 CrossRef CAS PubMed.
- (a) A. M. Clemments, C. Muniesa, C. C. Landry and P. Botella, RSC Adv., 2014, 4, 29134–29138 RSC; (b) J. G. Croissant, Y. Fatieiev, A. Almalik and N. M. Khashab, Adv. Healthcare Mater., 2018, 7, 1700831 CrossRef PubMed.
- (a) Á. Martínez, E. Fuentes-Paniagua, A. Baeza, J. Sánchez-Nieves, M. Cicuéndez, R. Gómez, F. J. de la Mata, B. González and M. Vallet-Regí, Chem. – Eur. J., 2015, 21, 15651–15666 CrossRef PubMed; (b) V. Rabolli, L. C. J. Thomassen, F. Uwambayinema, J. A. Martens and D. Lison, Toxicol. Lett., 2011, 206, 197–203 CrossRef CAS PubMed; (c) Q. Mu, N. S. Hondow, Ł. Krzemiński, A. P. Brown, L. J. C. Jeuken and M. N. Routledge, Part. Fibre Toxicol., 2012, 9, 29 CrossRef CAS PubMed; (d) R. P. Bagwe, L. R. Hilliard and W. Tan, Langmuir, 2006, 22, 4357–4362 CrossRef CAS PubMed.
- R. Huang, Y.-W. Shen, Y.-Y. Guan, Y.-X. Jiang, Y. Wu, K. Rahman, L.-J. Zhang, H.-J. Liu and X. Luan, Acta Biomater., 2020, 116, 1–15 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full Experimental section, containing general and synthetic methods, textural characterization of NPs, theoretical calculations for complex 1, photophysical properties of complex 1 and NPs, and biological procedures. See DOI: 10.1039/d1ma01243f |
| This journal is © The Royal Society of Chemistry 2022 |