
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advanced development of metal-loaded mesoporous organosilicas as catalytic nanoreactors†
Yucang
Liang
 *
*
Anorganische Chemie, Eberhard Karls Universität Tübingen, Auf der Morgenstelle 18, Tübingen, 72076, Germany. E-mail: yucang.liang@uni-tuebingen.de; Fax: +49 7071 292436
First published on 22nd October 2021
Abstract
Ordered periodic mesoporous organosilicas have been widely applied in adsorption/separation/sensor technologies and the fields of biomedicine/biotechnology as well as catalysis. Crucially, surface modification with functional groups and metal complexes or nanoparticle loading has ensured high efficacy and efficiency. This review will highlight the current state of design and catalytic application of transition metal-loaded mesoporous organosilica nanoreactors. It will outline prominent synthesis approaches for the grafting of metal complexes, metal salt adsorption and in situ preparation of metal nanoparticles, and summarize the catalytic performance of the resulting mesoporous organosilica hybrid materials. Finally, the potential prospects and challenges of metal-loaded mesoporous organosilica nanoreactors are addressed.
1. Introduction
Periodic mesoporous organosilicas (PMOs) feature an exciting class of nanoporous materials, which were first reported in 1999, independently by three groups from Canada, the USA and Japan.1–3 Accordingly, PMOs are accessible via a templating approach using surfactants as structure-directing agents (SDAs) and silsesquioxane of the type (EtO)3Si–R–Si-(OEt)3 (such as 1,2-bis(triethoxysilyl)ethane, BTEE) as the sole organosilica precursor under acidic or basic or even neutral conditions. Therefore, the PMO framework consists of (R)CSiO3 tetrahedra which are interlinked by the organic groups R. Naturally, the silicon-bridging organic (functional) groups are uniformly integrated into the framework, thus accomplishing a perfectly ordered mesoporous inorganic–organic hybrid materials.4 The PMO surface is terminated by the respective organic group R, as well as siloxane and silanol sites. The fabrication of PMOs avoids an inhomogeneous distribution of organic groups within the framework as found in organic–inorganic hybrid mesoporous silicas obtained via (i) post-synthesis strategies of mesoporous siliceous materials (e.g., grafting of surface silylation) or (ii) co-condensation methods involving mixed silica precursors, such as R1Si(OR2)3 or (R2O)3SiRSi(OR2)3 with Si(OR2)4, where R, R1 and R2 represent organic functional groups. Moreover, PMOs are less prone to pore blockage caused by the grafted organic group protruding into the pore channels.Due to the diversity of organo-bridged trialkoxysilane precursors, early investigations of PMOs mainly focused on the preparation of PMOs with single/dual/multiple functional organo-bridge groups,1–3,5–9 controllable pore size10–13 and various mesoporous phases with hexagonal (p6mm) and cubic (Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, Fmm, Imm, and Iad) symmetries.1–3,14–17 But these explorations barely considered effective approaches to improve the inherent chemical properties of PMOs and their practical applications in catalysis, as well as the impact of their crystalline structure and diverse shape and size on reactivity, akin to their zeolite counterparts. Many efforts were then concentrated on promoting the crystallinity and surface modification of PMOs as well as the functionalization of the organo-bridge groups R. These endeavors were facilitated by the semi-crystalline structures reminiscent of zeolites and the natural presence of organic groups as potential carriers of any desired PMO functionality. In the aspect of the fabrication of a crystalline framework (“pore wall”) structure at the molecular level, the first crystal-like pore wall-structured PMO was reported by Inagaki and co-workers in 2002 using 1,4-bis(triethoxysilyl)-benzene (BTEB) as the organosilica precursor and octadecyltri-methylammonium chloride (ODTMA) as the SDA.18 Afterwards, crystalline phenylene-bridged PMOs were widely synthesized, but they didn't indicate special significances and practically improved applications in industry such as crystalline zeolites. Hence, the objectives of PMO research were amended, now focusing on the surface modification of PMOs. Surface-tailoring chemistry included the grafting of additional functional organic groups and the incorporation of organo-bridge groups with unsaturated carbon–carbon bonds, N/S-functional groups, or even more sophisticated organometallic complexes. Such functionalized PMOs revealed, e.g., enhanced catalytic performances and high adsorption abilities for heavy metal ions.19,20 In these PMOs, decaoctahedral, spherical, and truncated rhombic dodecahedral shapes, and faceted rod-, platelet-, pin-, syringe-, stick-, and bullet-like particle morphologies were reported.21–23 However, the targeted development of monodisperse PMOs with a uniform size and regular morphology hasn't attracted much attention.24–26 In contrast, monodisperse mesoporous materials with a uniform size and regular shapes indicated some outstanding advantages: (i) the uniform composition/properties of individual particles make the properties of whole particles strictly controllable compared to bulk materials, (ii) short channel pathways are beneficial for controllable mass transport in confined nanocatalysts, and (iii) a low density, high surface area, well-defined mesoporous wall structures and biocompatibility endow the carrier with high permeability needed as an adsorbent, for targeted drug and gene delivery, as well as for biomolecule encapsulation.
m, Fmm, Imm, and Iad) symmetries.1–3,14–17 But these explorations barely considered effective approaches to improve the inherent chemical properties of PMOs and their practical applications in catalysis, as well as the impact of their crystalline structure and diverse shape and size on reactivity, akin to their zeolite counterparts. Many efforts were then concentrated on promoting the crystallinity and surface modification of PMOs as well as the functionalization of the organo-bridge groups R. These endeavors were facilitated by the semi-crystalline structures reminiscent of zeolites and the natural presence of organic groups as potential carriers of any desired PMO functionality. In the aspect of the fabrication of a crystalline framework (“pore wall”) structure at the molecular level, the first crystal-like pore wall-structured PMO was reported by Inagaki and co-workers in 2002 using 1,4-bis(triethoxysilyl)-benzene (BTEB) as the organosilica precursor and octadecyltri-methylammonium chloride (ODTMA) as the SDA.18 Afterwards, crystalline phenylene-bridged PMOs were widely synthesized, but they didn't indicate special significances and practically improved applications in industry such as crystalline zeolites. Hence, the objectives of PMO research were amended, now focusing on the surface modification of PMOs. Surface-tailoring chemistry included the grafting of additional functional organic groups and the incorporation of organo-bridge groups with unsaturated carbon–carbon bonds, N/S-functional groups, or even more sophisticated organometallic complexes. Such functionalized PMOs revealed, e.g., enhanced catalytic performances and high adsorption abilities for heavy metal ions.19,20 In these PMOs, decaoctahedral, spherical, and truncated rhombic dodecahedral shapes, and faceted rod-, platelet-, pin-, syringe-, stick-, and bullet-like particle morphologies were reported.21–23 However, the targeted development of monodisperse PMOs with a uniform size and regular morphology hasn't attracted much attention.24–26 In contrast, monodisperse mesoporous materials with a uniform size and regular shapes indicated some outstanding advantages: (i) the uniform composition/properties of individual particles make the properties of whole particles strictly controllable compared to bulk materials, (ii) short channel pathways are beneficial for controllable mass transport in confined nanocatalysts, and (iii) a low density, high surface area, well-defined mesoporous wall structures and biocompatibility endow the carrier with high permeability needed as an adsorbent, for targeted drug and gene delivery, as well as for biomolecule encapsulation.
Moreover, the expansion of the pore size is very pivotal for biomolecule adsorption and release in biomedical applications. Very recently, by using silica colloidal crystals as the templates, the PMO pore size could be significantly increased to 47 nm.27
More recently, an increasing number of distinct morphologies have emerged, and different composites and various mesophases were combined.28–32 These new advancements include the anisotropic growth-induced synthesis of dual-compartment Janus mesoporous silica and organosilica nanoparticles (NPs) with hexagonal and cubic symmetries,29 as well as hierarchical mesoporous organosilica-silica core–shell NPs.30 Especially, hybrid multipodal PMO (mp-PMO) NPs with crystal-like architectures were subtly designed and elaborated in a one-pot two-step process, involving the condensation of phenylene-based (B) spherical PMO cores followed by the condensation of ethylene-based (E) rod-shaped PMO pods on these cores.31
Monodisperse hollow PMOs were prepared by soft and hard templating methods or by in situ dissolution and re-assembly approaches.33 The corresponding synthesis and application of hollow PMO nanoparticles were reviewed by Zhao and co-workers.34 Recently, the flash-nano-precipitation technique was successfully developed as a facile and efficient strategy for the rapid fabrication of morphology-controlled nanosized organosilica particles.35 Due to their biocompatibility,36 PMO NPs have been widely used as versatile platforms for the smart delivery of therapeutics,37 photodynamic therapy of cancer,38 drug delivery,39–41 chemo-immuno-therapy42 and oxygen-sensitized radiotherapy.43 It is very obvious that at present the application of PMO NPs mainly deals with the aspects of biomedicine and biotechnology.
Although a great deal of effort has focused on the preparation, morphology and structure control of PMOs, their applications as supports or catalysts are becoming more important and significant.44,45 For PMOs, in terms of catalysis, the homogeneous incorporation of organic groups can effectively enhance the surface hydrophobicity of the materials, which is conducive to the adsorption, diffusion, and access of organic reactants and more likely contact between reactants and catalytically active sites.46 However, for PMO synthesis, a key issue is the fabrication of a functional organic group-bridged organosilane precursor and modification of the physical and chemical properties of the final PMO obtained. The capability of terminal or bridging organosilane functional groups (carboxylic, thio, nitrogenous, chelating, pyridine, amine etc.) in silica frameworks to coordinate to metal species is of extraordinary importance in many areas, especially, in catalysis.47 Although many reviews have addressed the preparation, characterization and applications of PMOs,48–53 a systematic review with focus on the catalytic performance of metal-loaded PMO heterogeneous catalysts is absent except for that reported by Corma and co-workers in 2013.45 In 2013, nanostructured heterogeneous catalysts prepared by the grafting of metal-amide complexes onto silica and organosilica were reviewed by us and the respective catalytic reactions were pointed out.54 In 2016, Copéret and co-workers systematically elucidated surface organometallic and coordination chemistry toward single-site heterogeneous catalysts including designable strategies, preparation methods, surface structures and catalytic activities.55
In general, nanostructured materials with pore cavities or pore channels are termed nanoreactors as they are a type of chemical reactor, providing unique nanoscale chemical environments compartmentalized by the surrounding bulk space (such as the (organo)silica framework).56 Such nanoreactors are crucial for maintaining a confined environment for manufacturing products on a nanotechnological scale, and have particularly proven themselves in the disciplines of nanotechnology and nanobiotechnology. Hence, “small-scaled” nanoreactors do have big implications for chemistry. In recent years, chemists have tremendously improved their knowledge about how fundamental chemical principles change when systems are confined to spaces with nanoscale dimensions or sub-microliter volumes.56 Metal-loaded PMOs contain not only ordered nanospaces as the nanoreactors, but also confined metal species of relevance for catalysis. It is for these reasons that such metal-loaded PMOs were widely applied in catalytic systems be it as nanoreactors and nanocatalysts.
In this review, we focus on the recent advancements in the area of transition metal-loaded PMOs as nanoreactors in catalysis. The contents include novel preparation strategies and the catalytic activities of metal-loaded PMOs. Consideration is also given to the influence of the PMO mesopore structure on catalytic performance. For the convenience of the reader, an illustration of metal-loaded PMOs as nanoreactors is shown in Scheme 1. Moreover, metal or metal-complex-supported PMO heterogeneous catalysts are introduced according to their group appearance in the periodic table of the elements. A key point of this review is to emphasize the great variety of molecularly defined heterogeneous catalysts made accessible via PMOs. This could be achieved via the uniform incorporation of functional organic groups, metal complexes or particles into PMO frameworks. The successfully performed catalytic transformations (addition reactions, coupling reactions, reduction reactions, oxidation reactions, hydrogenation and hydrogen production reactions) also benefitted from the uniform pore size, regular mesopore structure and high hydrophobicity of PMOs. In addition, whether single-atom catalysis can be realized by the heterogenization of homogeneous molecules on PMO carriers will be explored.
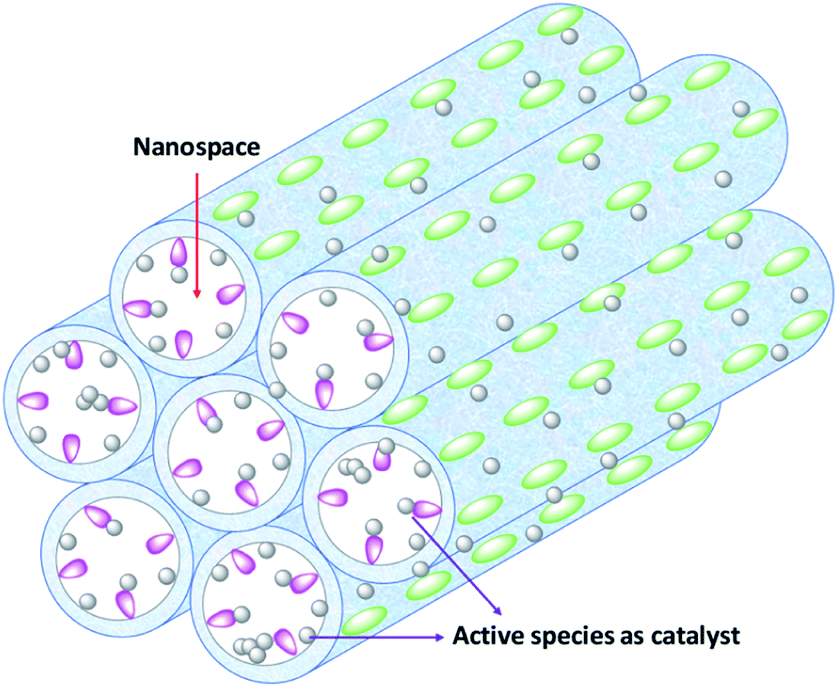 | ||
| Scheme 1 Illustration of metal-loaded PMOs used as nanoreactors and nanocatalysts. Green ellipsoids represent the organo-bridge groups which are integrated into the PMO framework. Red drops represent the functional groups (or linkers) and grey balls represent metal complexes or metal NPs that are stabilized by bonding to the functional group or functionalized organo-bridge group or randomly deposited onto the surface of the PMO. Mesopore channels act as the nanoreactors and metal-loaded PMOs as the nanocatalysts. | ||
2. Transition metal-based PMO heterogeneous catalysts and catalytic activities
In general, metal-loaded PMO heterogeneous catalysts are routinely fabricated by four different approaches. The first method utilizes a one-pot synthesis to directly integrate a metal precursor into (organo)silica frameworks to form Si–O–M–O entities, such as Ti-containing PMO heterogeneous catalysts. Another approach features metal ions or metal complexes linked to the surface of organosilica frameworks with functional organic groups to form metal-loaded PMO catalysts. The third route exploits the direct grafting of a metal complex onto the surface of organosilica frameworks via the surface-terminating silanol/hydroxyl groups or functional groups integrated into the PMO framework. Finally, incipient wetness impregnation applies metal salts which are randomly adsorbed onto the surface of organosilica frameworks by a simple physical adsorption process, which, however, might involve hydrogen- or π-bonding of the metal to the surface. Metal ions or complexes are then reduced with a reductant or by calcination to obtain metal nanoparticle (NP)-loaded PMO catalysts. Generally, the nature of the metal precursors and the structural characteristics of the support are critical for the preparation of different metal-loaded PMO heterogeneous catalysts.2.1. Group 4 (Ti, Zr, Hf)-based PMO nanoreactors and catalyses
Ti-based materials are widely used as photocatalysts in organic synthesis. Herein, we address the preparation and catalytic application of Ti-based mesoporous organosilica catalysts as nanoreactors. Due to the facile hydrolysis of titanium sources (TiCl4, Cl2TiCp2, Ti[OCH(CH3)2]4, etc.), Ti species can effectively integrate into (organo)silica frameworks to form Si–O–Ti–O connectivities. Hence, Ti-containing PMOs can be readily fabricated by using single bis(trialkoxysilyl)alkylsilane [(R′O)3–Si–R–Si–(OR′)3] or a mixture of bis(trialkoxysilyl)alkylsilane and tetraalkoxysilane Si(OR′′)4 as the organosilica source and Ti complexes in the presence of a surfactant under acidic or basic or neutral media. The composition, structure, morphology and pore size can be well adjusted by changing synthetic parameters such as the initial molar ratio, choice and concentration of the surfactant(s), organic or inorganic additives, reaction temperature and time, and so on. As a result, the incorporation of Ti species greatly improved the physical–chemical properties of the PMO, especially, its catalytic activity, compared to pristine PMO. Titanocene dichloride [Cl2TiCp2] was integrated into inorganic-organic silica frameworks via the hydrolysis of a mixture of BTEE and tetraethyl orthosilicate (TEOS) with varying molar ratios ranging from 0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 to 100:0 in the presence of nonionic surfactant Pluronic P123 under acidic conditions to form Ti-PMO materials with large channel-like pores.46 With increasing BTEE, the hydrophobicity of the obtained Ti-PMO gradually increased. Such obtained Ti-PMO was further reacted with hexamethyldisilazane to form a surface-silylated Ti-PMO. The latter increasingly hydrophobic Ti-PMO exhibited the highest catalytic performance in the epoxidation of 1-octene with tert-butyl hydroperoxide (TBHP). This shows the importance of the hydrophobic microenvironment, provided by both the organosilicon precursor and the surface silanol silylation.46 In 2012, an organosilane compound containing a bis(propyliminomethyl)-phloroglucinol moiety was synthesized by the Vilsmeier–Haack formylation of phloroglucinol followed by a Schiff base condensation with 3-aminopropyl-triethoxysilane (APTES).57 The hydrothermal reaction of such an organosilane with tetraethyl orthosilicate and titanium isopropoxide (TIP) in the presence of cetylpyridinium chloride (CPC) formed an inorganic–organic hybrid titanosilicate composite. After removal of CPC, a hybrid Ti-containing PMO catalyst was obtained and used in the epoxidation of alkenes with TBHP under mild liquid phase reaction conditions. The hybrid catalyst revealed excellent catalytic activity, thanks to the existence of multiple active Ti sites, derived from Ti centers simultaneously integrated into the silica framework by strong coordination with the phenolic-OH and imine-N donor atoms.57 A similar procedure was used to prepare a Ti-incorporated phenylene-bridged PMO by the co-condensation of tetrabutyl titanate (TBOT), BTEB and TEOS in the presence of nonionic surfactant Pluronic P123 under acidic conditions via one-pot synthesis. After removal of Pluronic P123 and surface silylation, the surface silylated Ti-PMO showed high conversion and selectivity in cyclohexene epoxidation with TBHP compared to non-silylated Ti-PMO and Ti-periodic mesoporous silica (Ti-PMS).58 By optimizing the ageing time, the Ti-PMO catalyst showed the highest selectivity (97.7%) for cyclohexene epoxidation.59 Furthermore, similarly structured Ti-containing ethylene- or phenylene-bridged PMO catalysts were applied to cyclohexene epoxidation with TBHP, but they exhibited different conversions and selectivities, thanks to different Ti contents and active sites.60 When mixtures of BTEB and TEOS at various molar ratios were used as the silica source and TIP as the Ti precursor, phenylene-bridged Ti-PMO spheres with a wrinkled structure were generated in the presence of cetyltrimethylammonium bromide (CTAB) under basic conditions. The selectivity of Ti-PMO-catalyzed cyclohexene epoxidation could reach 94%.61 However, mesoporous titania–organosilica nanoparticles comprised of anatase nanocrystals crosslinked with organosilica moieties indicated an excellent photoactivity in the degradation of rhodamine 6G and the partial oxidation of propene under UV irradiation, especially calcined samples.62 These mesoporous titania-organosilica NPs were prepared by direct co-condensation of TBOT and BTEB or BTEE in the absence of SDA under mild conditions. Apart from multiple functional bridged groups, an ionic liquid (IL) crystal unit was incorporated into an organosilane to form a new organosilica precursor63 for the preparation of PMO-IL with or without metal content. Briefly, the hydrolysis and (co-)condensation of 1,3-bis(tri-methoxysilylpropyl)-imidazolium halide (Cl, I) with or without tetramethoxysilane (TMOS) or TEOS formed PMO-IL in the presence of SDA under acidic or basic conditions.64 After removal of the SDA, the IL-based ordered PMO material was used as the support. For some metal salts or complexes, the IL fragment (integrated into (organo)silica frameworks) is often beneficial for the physical adsorption of the metal precursor via electrostatic interaction and for the formation of a stable metal complex. In the presence of a metal precursor, metal-containing PMO-ILs were readily accessible. For example, titanium was incorporated into an IL-based PMO to prepare a nanostructured titanium-containing ionic liquid-based PMO catalyst (Ti@PMO-IL) by the hydrolysis and co-condensation of TMOS, TBOT and 1,3-bis(3-trimethoxysilylpropyl)-imidazolium chloride in the presence of Pluronic P123 as the SDA under acidic conditions (Fig. 1).65 After removal of the surfactant, such a Ti@PMO-IL material was employed as a heterogeneous nanocatalyst for the one-pot multi-component synthesis of biologically useful polyhydroquinoline derivatives. By optimizing the synthesis conditions, the yield of a Ti@PMO-IL-catalyzed four-component Hantzsch reaction to afford polyhydroquinoline could be improved to 96%, while indicating high recyclability and reusability.65
:100 to 100:0 in the presence of nonionic surfactant Pluronic P123 under acidic conditions to form Ti-PMO materials with large channel-like pores.46 With increasing BTEE, the hydrophobicity of the obtained Ti-PMO gradually increased. Such obtained Ti-PMO was further reacted with hexamethyldisilazane to form a surface-silylated Ti-PMO. The latter increasingly hydrophobic Ti-PMO exhibited the highest catalytic performance in the epoxidation of 1-octene with tert-butyl hydroperoxide (TBHP). This shows the importance of the hydrophobic microenvironment, provided by both the organosilicon precursor and the surface silanol silylation.46 In 2012, an organosilane compound containing a bis(propyliminomethyl)-phloroglucinol moiety was synthesized by the Vilsmeier–Haack formylation of phloroglucinol followed by a Schiff base condensation with 3-aminopropyl-triethoxysilane (APTES).57 The hydrothermal reaction of such an organosilane with tetraethyl orthosilicate and titanium isopropoxide (TIP) in the presence of cetylpyridinium chloride (CPC) formed an inorganic–organic hybrid titanosilicate composite. After removal of CPC, a hybrid Ti-containing PMO catalyst was obtained and used in the epoxidation of alkenes with TBHP under mild liquid phase reaction conditions. The hybrid catalyst revealed excellent catalytic activity, thanks to the existence of multiple active Ti sites, derived from Ti centers simultaneously integrated into the silica framework by strong coordination with the phenolic-OH and imine-N donor atoms.57 A similar procedure was used to prepare a Ti-incorporated phenylene-bridged PMO by the co-condensation of tetrabutyl titanate (TBOT), BTEB and TEOS in the presence of nonionic surfactant Pluronic P123 under acidic conditions via one-pot synthesis. After removal of Pluronic P123 and surface silylation, the surface silylated Ti-PMO showed high conversion and selectivity in cyclohexene epoxidation with TBHP compared to non-silylated Ti-PMO and Ti-periodic mesoporous silica (Ti-PMS).58 By optimizing the ageing time, the Ti-PMO catalyst showed the highest selectivity (97.7%) for cyclohexene epoxidation.59 Furthermore, similarly structured Ti-containing ethylene- or phenylene-bridged PMO catalysts were applied to cyclohexene epoxidation with TBHP, but they exhibited different conversions and selectivities, thanks to different Ti contents and active sites.60 When mixtures of BTEB and TEOS at various molar ratios were used as the silica source and TIP as the Ti precursor, phenylene-bridged Ti-PMO spheres with a wrinkled structure were generated in the presence of cetyltrimethylammonium bromide (CTAB) under basic conditions. The selectivity of Ti-PMO-catalyzed cyclohexene epoxidation could reach 94%.61 However, mesoporous titania–organosilica nanoparticles comprised of anatase nanocrystals crosslinked with organosilica moieties indicated an excellent photoactivity in the degradation of rhodamine 6G and the partial oxidation of propene under UV irradiation, especially calcined samples.62 These mesoporous titania-organosilica NPs were prepared by direct co-condensation of TBOT and BTEB or BTEE in the absence of SDA under mild conditions. Apart from multiple functional bridged groups, an ionic liquid (IL) crystal unit was incorporated into an organosilane to form a new organosilica precursor63 for the preparation of PMO-IL with or without metal content. Briefly, the hydrolysis and (co-)condensation of 1,3-bis(tri-methoxysilylpropyl)-imidazolium halide (Cl, I) with or without tetramethoxysilane (TMOS) or TEOS formed PMO-IL in the presence of SDA under acidic or basic conditions.64 After removal of the SDA, the IL-based ordered PMO material was used as the support. For some metal salts or complexes, the IL fragment (integrated into (organo)silica frameworks) is often beneficial for the physical adsorption of the metal precursor via electrostatic interaction and for the formation of a stable metal complex. In the presence of a metal precursor, metal-containing PMO-ILs were readily accessible. For example, titanium was incorporated into an IL-based PMO to prepare a nanostructured titanium-containing ionic liquid-based PMO catalyst (Ti@PMO-IL) by the hydrolysis and co-condensation of TMOS, TBOT and 1,3-bis(3-trimethoxysilylpropyl)-imidazolium chloride in the presence of Pluronic P123 as the SDA under acidic conditions (Fig. 1).65 After removal of the surfactant, such a Ti@PMO-IL material was employed as a heterogeneous nanocatalyst for the one-pot multi-component synthesis of biologically useful polyhydroquinoline derivatives. By optimizing the synthesis conditions, the yield of a Ti@PMO-IL-catalyzed four-component Hantzsch reaction to afford polyhydroquinoline could be improved to 96%, while indicating high recyclability and reusability.65
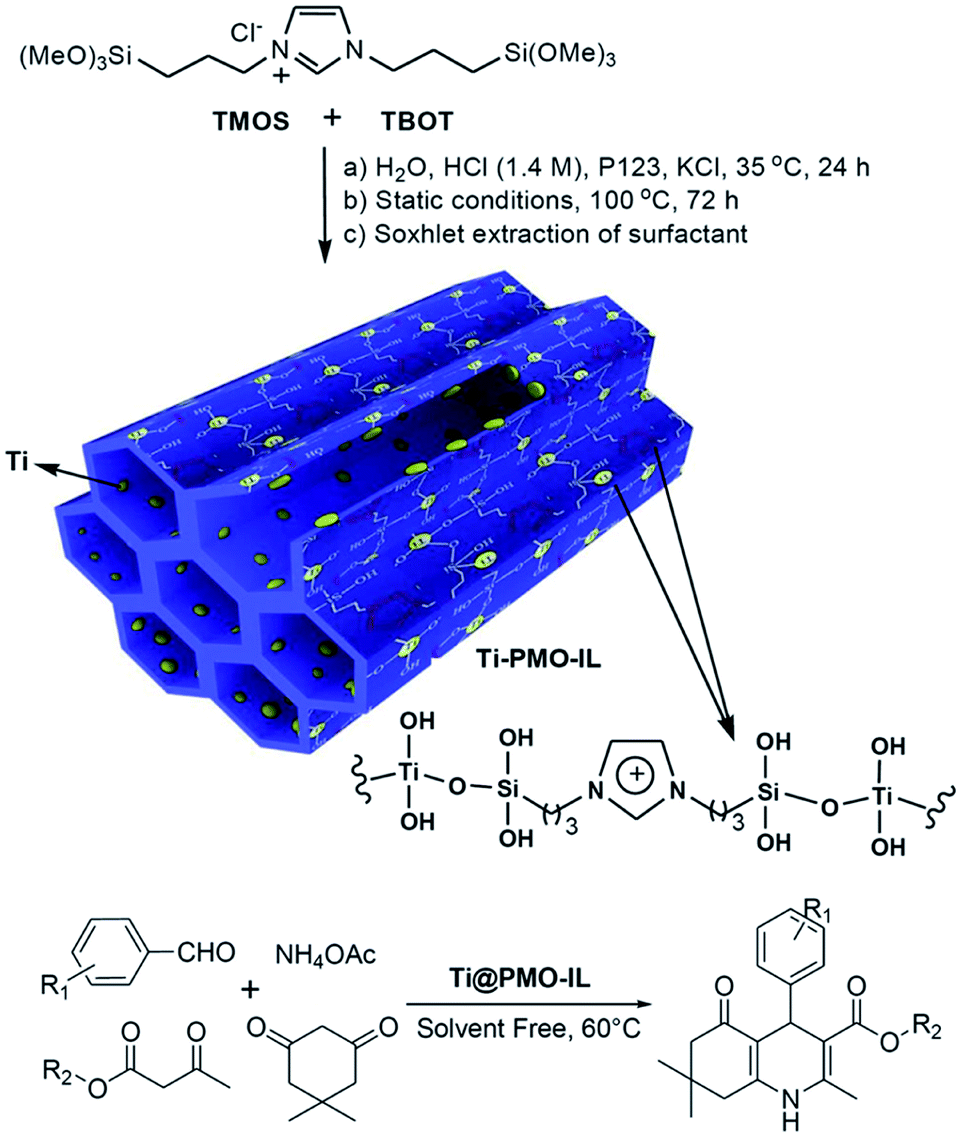 | ||
| Fig. 1 Preparation of the Ti@PMO-IL nanocatalyst and its application in the synthesis of biologically active polyhydroquinolines.65 Reproduced with permission.65 Copyright 2018, Jon Wiley & Sons. | ||
In addition, the incorporation of titanium into a sulfonic acid or thiol group-functionalized PMO material afforded a bifunctional nanocatalyst PrSO3H–SiCH2CH2Si–Ti (Fig. 2) with both Brønsted and Lewis acid sites. It was found that the Ti loading remarkably influenced the catalytic performance in the esterification of levulinic acid with ethanol to produce ethyl levulinate (Scheme 2).66 In the presence of CTAB, the co-condensation of BTEB, TMOS and TBOT on Fe3O4@SiO2 particles formed a core–shell structured magnetic Ti-loaded phenylene-based nanoporous organosilica (Mag@Ti–NOS) nanocomposite. Since the magnetic characteristic is conducive to the separation of the catalyst, substrate and product, Mag@Ti–NOS was employed as an effective, powerful and recyclable catalyst in the synthesis of tetrahydrobenzo[b]pyrans in water under ultrasonic conditions.67
 | ||
| Fig. 2 Illustration of the P123-templated sol–gel co-condensation route for the preparation of periodic mesoporous PrSO3H–SiCH2CH2Si–Ti hybrid catalysts.66 Reproduced with permission.66 Copyright 2019, Elsevier. | ||
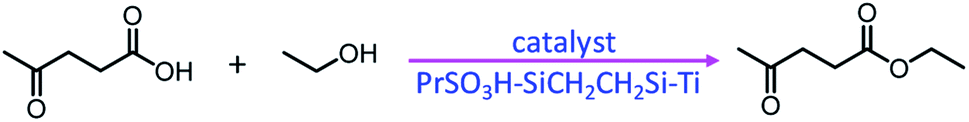 | ||
| Scheme 2 PrSO3H–SiCH2CH2Si–Ti-catalyzed esterification of levulinic acid with ethanol. | ||
Very recently, a novel hybrid core–shell structured TiO2–SiO2-PMO composite (TS@PMO) with a Ti-surface-enriched mesoporous TS core and an amphiphilic PMO shell was reported by Li and co-workers (Fig. 3).68 In such a TS@PMO composite, the active Ti sites were facilely enriched in near-surface layers of the core and extra mesoporous cavities were introduced for substrate reservation (Fig. 4).
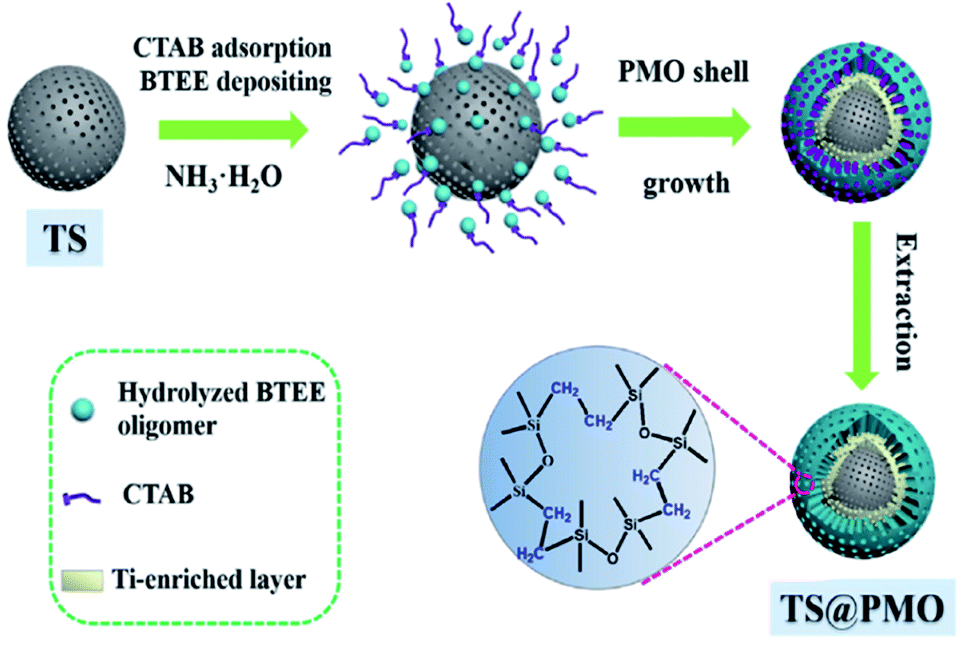 | ||
| Fig. 3 Synthetic process for the core–shell structured TS@PMO microspheres.68 Reproduced with permission.68 Copyright 2021, Elsevier. | ||
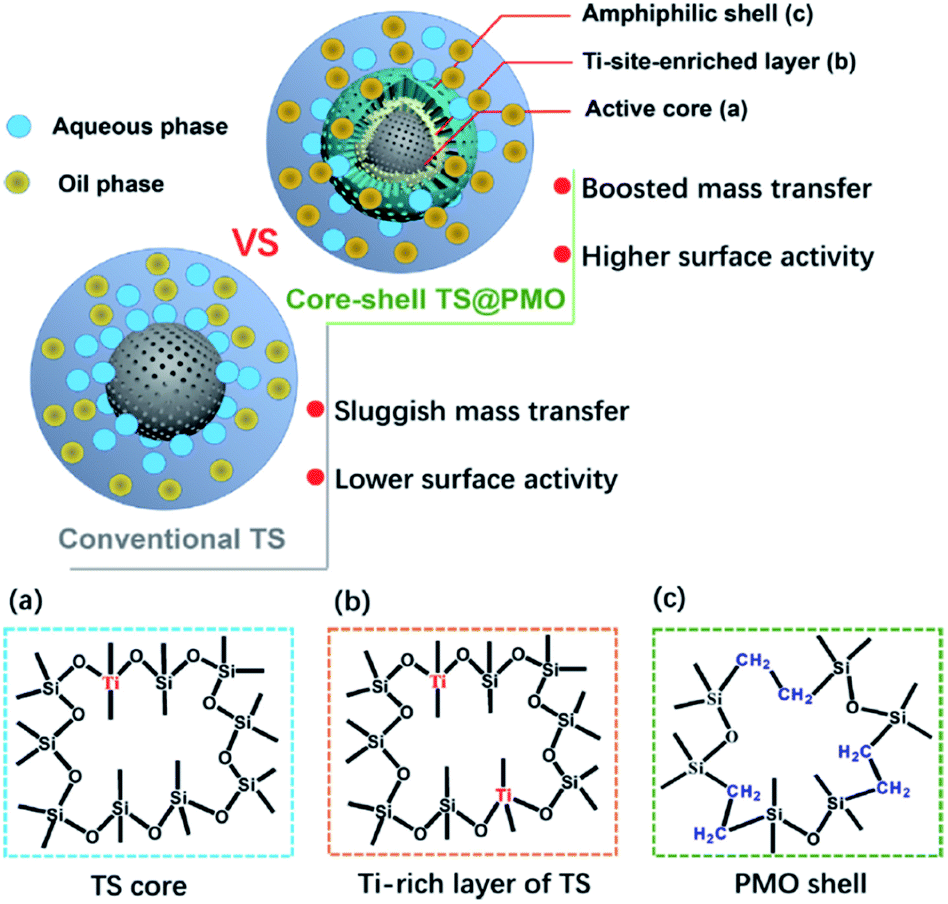 | ||
| Fig. 4 Illustration of TS and TS@PMO showing the merits of material design with an amphiphilic shell and surface-Ti-enriched core ((a), (b), and (c) indicate the skeleton structure of the TS core and PMO shell; note: (b) is the outer layer of (a)).68 Reproduced with permission.68 Copyright 2021, Elsevier. | ||
When TS@PMO was applied for the solvent-free epoxidation of methyl oleate (MO) with H2O2 (Scheme 3), a remarkably boosted catalytic activity with a conversion of 90.2% and epoxide selectivity of 70.2% was found. This performance was markedly superior to the conversion of 63.7% and selectivity of 49.2% of unmodified titanosilicate and the conversion of 39.8% and selectivity of 25.0% of Ti-containing organosilica. This was ascribed to enhanced interphase mass transfer and sufficiently accessible active Ti sites in TS@PMO: (i) the hydrophobic characteristics and pore channels of PMO are conducive to the access and diffusion of hydrophobic substrates/hydrogen peroxide, and (ii) an increased framework Ti(IV) density and surface-enriched active Ti sites in the TS core are beneficial to the improvement of the utilization of active Ti sites. More recently, self-assembled ionic liquid-based organosilica-titania was reported and used as a novel and efficient catalyst for the green epoxidation of alkenes with H2O2, showing high activity and recoverable and reusable features.69 Furthermore, a TiO2 precursor was mixed with amine-functionalized mesoporous organosilica particles in a mass ratio of 1:1, followed by hydrothermal treatment to form water-floatable TiO2-loaded cup-shaped organosilica particles. After calcination at 400 °C, the specific surface area and the growth of anatase crystallites of the material increased. Such a material was used as an active photocatalyst for CO2 evolution from aqueous solutions of formic acid and compared to the as-made TiO2-loaded organosilica.70
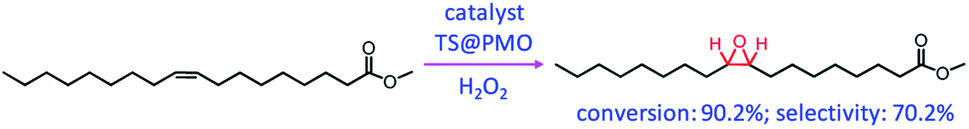 | ||
| Scheme 3 TS@PMO-catalyzed solvent-free epoxidation of methyl oleate with H2O2. | ||
The post-grafting strategy is a vital approach to immobilize homogeneous catalysts on solid supports to form heterogeneous catalysts. The subsequent multi-step grafting can effectively control the metal loading. Titanium tetraisopropoxide Ti(OiPr)4 was grafted onto surfactant-free PMO-ICS, prepared by the hydrolysis and co-condensation of tris-[3-methoxysilylpropyl]isocyanurate (ICS) and TEOS in the presence of Pluronic P123 as a SDA under acidic conditions, to form a PICS-Ti heterogeneous catalyst.71 With the subsequent layer-by-layer grafting,72 a series of PICS-Ti catalysts with various Ti contents as amorphous TiO2 layers coating the pore channels of PMO-ICS were prepared. Such hybrid catalysts were applied to the regeneration of carbonyl compounds from oximes under sunlight irradiation (Fig. 5), indicating excellent photocatalytic activity and high selectivity. This is due to a unique double synergetic effect of the presence of isocyanuric groups in pristine PMO-ICS and the photoactive titania inside the mesopore channels of PMO-ICS with an appropriate Ti loading.71 When titanium(IV) butoxide [Ti(n-OBu)4] was used as the Ti precursor, layer-wise titania grown within a dimeric organic functional group viologen-based PMO was employed as an efficient photocatalyst for oxidative formic acid decomposition.73
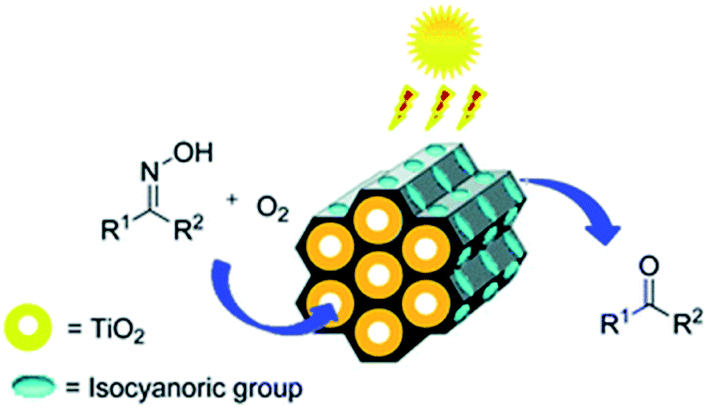 | ||
| Fig. 5 Catalytic reaction illustration promoted by PICS-Ti.71 Reproduced from ref. 71 with permission from the Royal Society of Chemistry. | ||
In addition, titanocene dichloride was directly grafted onto ethylene-bridged PMO, followed by the substitution of the surface cyclopentadienyl ligands with ethoxy groups in ethanol to afford Ti-PMO.46 Although such Ti-PMO contained a high Ti content, its catalytic activity per titanium site in the epoxidation of 1-octene with TBHP was much lower than that of the corresponding Ti-PMO material synthesized by a one-pot synthesis procedure and even lower than that of pure silica Ti-SBA-15 having a similar titanium content, due to the low dispersion of Ti species.46 Moreover, Ti species can also be grafted onto PMO or ionic liquid-based nanoporous organosilica (ILNOS) prepared by surfactant-directed simultaneous hydrolysis and condensation of alkyl-imidazolium ionic liquids (such as 1,3-bis(3-trimethoxysilyl-propyl)imidazolium chloride) and tetramethoxysilane. For example, TBOT was initially reacted with ILNOS in anhydrous toluene under reflux for the expected time to generate Ti(OtBu)4@ILNOS, followed by treatment with a solvent mixture of ethanol and water to form ordered mesoporous organosilica-titania with an ionic liquid framework (ILNOS-Ti). In this case, the active Ti species were located on the surface of ILNOS. Such ILNOS-Ti with an enhanced number of active sites was used as an efficient nanocatalyst for the green oxidation of various primary and second alcohols (benzylic and aliphatic alcohols, 1-phenylethanol, 1-phenyl-1-propanol etc.) with hydrogen peroxide (H2O2) to the corresponding aldehydes or ketones under mild conditions. The results indicated high catalytic activity, high stability, recoverability and reusability.74
Zirconium can also be integrated into PMO to adjust the chemical and physical properties of Zr-PMO. In general, zirconium n-butoxide [Zr(OnBu)4] was used as a Zr precursor and co-condensed with BTEB under acidic conditions in the presence of Pluronic P123 to form as-made Zr-PMO.75 After removal of P123, surfactant-free Zr-PMO was treated with chlorosulfonic acid to generate sulfonic acid-functionalized ZrO2/organosilica bifunctional catalysts (SO42−/ZrO2-PMO-SO3H) for direct catalytic conversion of glucose to ethyl levulinate. With alteration of the initial Si/Zr molar ratio, catalysts SO42−/ZrO2-PMO-SO3H showed various catalytic activities, revealing the synergistic effect of super strong Brønsted acidic and moderate Lewis acidic sites. Especially, SO42−/ZrO2-PMO-SO3H with an initial Si/Zr molar ratio of 1 exhibited the highest yield of ethyl levulinate. The co-hydrolysis and -condensation of 1,2-bis(trimethoxy-silyl)ethane (BTME) with Zr(OnBu)4 in the presence of H3PW12O40, triblock copolymer surfactant F127 and 1,3,5-trimethylbenzene (TMB) formed heteropolyacid and ZrO2 bifunctionalized organosilica hollow nanospheres (H3PW12O40/ZrO2-PMO). Through adjusting the molar ratios of Si/Zr in the initial gel mixture, the morphology transformed from a 3D interconnected mesostructure into a hollow spherical nanostructure. TMB as an expander could effectively tune the inner diameter range of the H3PW12O40/ZrO2-PMO materials from 6 to 12 nm.76 Owing to its strong Brønsted and Lewis acidity, unique hollow nanospherical morphology and hydrophobic surface, H3PW12O40/ZrO2-PMO showed an excellent heterogeneous acid catalytic activity for the esterification of levulinic acid (LA) with methanol to methyl levulinate and transesterification of yellow horn oil with methanol to biodiesel. Similarly, H3PW12O40/ZrO2-PMO nanotubes were shown to efficiently catalyze the esterification of levulinic acid or ethanolysis of furfuryl alcohol to alkyl levulinates with a high yield.77
2.2. Group 5 (V, Nb, Ta)-based PMO nanoreactors and catalyses
In 2004, Corma and co-workers prepared a vanadyl Schiff base complex with two terminal trimethoxysilyl groups and combined this complex with TEOS as a co-organosilica precursor in the presence of CTAB as a SDA to obtain a MCM-41-like mesoporous organosilica.78 After removal of the SDA, VO(salen)∝PMO or VO(salen)∝ChiMO was obtained and used as an efficient heterogeneous catalyst (Fig. 6) for the cyanosilylation of aldehydes showing good activity and high selectivity (Scheme 4). Vanadyl sulfate was immobilized on Schiff base-modified PMO to form an efficient and recyclable oxo-vanadium Schiff base-modified PMO for the selective oxidation of alcohols to carbonyl compounds by using TBHP as the oxidant under mild reaction conditions.79 The resulting heterogeneous catalyst showed high catalytic activity with good to excellent product yields, convenient recoverability by filtration and very good reusability for at least 8 recycles without any significant decrease in catalytic activity and leaching of the metal or ligand. Moreover, vanadyl(IV) acetylacetonate, VO(acac)2, reacted with (3-aminopropyl)triethoxysilane (APTES) via Schiff base condensation to form a vanadyl complex-incorporated organosilica precursor (SB–V) (Fig. 7). The co-condensation of SB-V and TEOS in the presence of CTAB under basic conditions generated a novel V-PMO. After removal of CTAB, a vanadyl complex-incorporated PMO nanocatalyst was obtained (Fig. 7) and used for the hydroxylation of benzene to phenol, showing 100% selectivity and high conversion (>51%) compared to V(acac)2.80 A similar method was applied to the preparation of a mesoporous SiO2-Pro-Sal-VO catalyst, showing even higher selectivity (>95%) and excellent recyclability but low conversion (11.3%) for the hydroxylation of benzene to phenol.81 Moreover, a Schiff base ligand protruding into the mesopore channels of SiO2-Pro-Sal could also coordinate to metal salts (Co, Cu, Fe, Zn, and Mn) to form a metal-Schiff base complex. Such a type of metal-bearing mesoporous composite also showed considerable catalytic activity for the hydroxylation of benzene.81 Additionally, reacting VOSO4·5H2O, TEOS and N-[3-(trimethoxysilyl)propyl]ethylenediamine in a one-pot synthesis in the presence of CTAB under basic conditions generated a V complex-incorporated PMO (V-PMO), in which vanadium contents could be adjusted by changing the initial vanadium salt content and vanadium existed in these V-PMO materials in the form of V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and V–N bonds. After removal of the template, the V-PMO was used in styrene oxidation, showing a high yield and excellent selectivity for benzaldehyde. This was ascribed to the advanced structure of V-PMO and well-dispersed and highly stable oxovanadium(IV) species in the inorganic frameworks.82
O and V–N bonds. After removal of the template, the V-PMO was used in styrene oxidation, showing a high yield and excellent selectivity for benzaldehyde. This was ascribed to the advanced structure of V-PMO and well-dispersed and highly stable oxovanadium(IV) species in the inorganic frameworks.82
 | ||
| Fig. 6 Preparation of the organosilica precursor and V-incorporated PMOs, VO(salen)∝PMO and VO(salen)∝ChiMO.78 Reproduced with permission.78 Copyright 2004, Elsevier. | ||
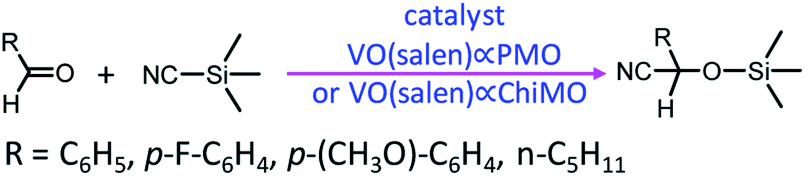 | ||
| Scheme 4 Cyanosilylation of aldehydes mediated by VO(salen)∝PMO or VO(salen)∝ChiMO. | ||
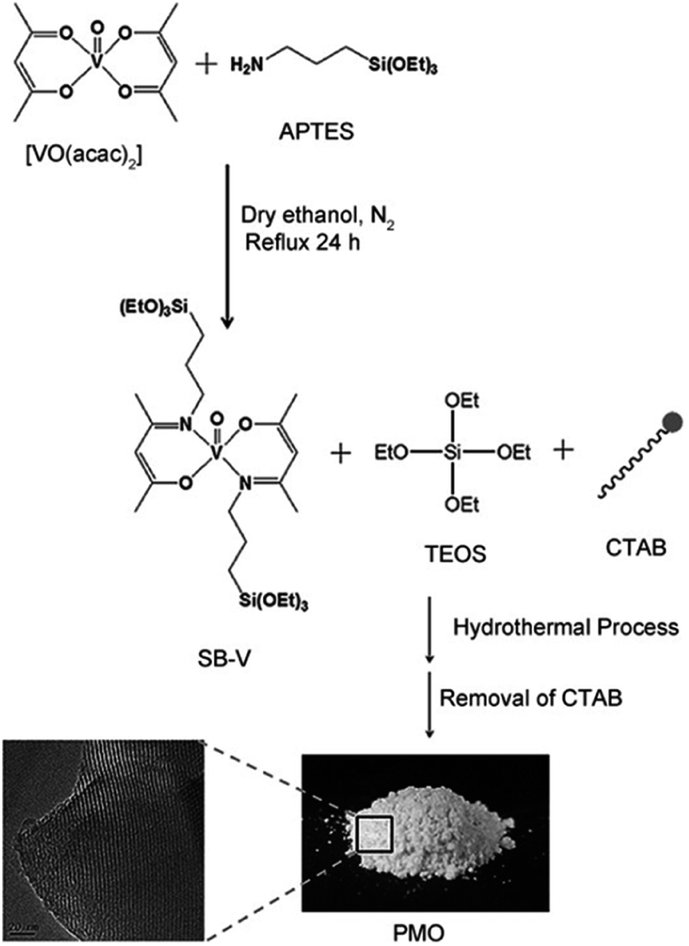 | ||
| Fig. 7 The synthesis of SB-V and periodic mesoporous organosilica materials (PMOs).80 Reproduced with permission.80 Copyright 2012, John Wiley & Sons. | ||
Nb species were integrated into ethylene- or octane-bridged PMO frameworks via a one-pot synthetic sol–gel technique to fabricate Nb-PMO catalysts. With an adjustable molar ratio of Nb/Si, the obtained Nb-PMO catalysts indicated high catalytic activities with a conversion of 89–99% and selectivity of 100% for the epoxidation of methyl oleate, a conversion of 86–96% and a monoepoxy derivative selectivity of 70–79% for the epoxidation of sunflower oil as well as a conversion of 93–99% and ω-hydroxy acid selectivity of 75–88% for the oxidation of stearic acid.83 Moreover, H3PW12O40/Nb2O5-co-integrated phenylene-bridged PMO catalysts [H3PW12O40/Nb2O5–Si(Ph)Si] with different molar ratios of Nb/Si were obtained by co-condensation of NbCl6, H3PW12O40 and BTEB in the presence of Pluronic P123 under acidic conditions and template removal (Fig. 8). Such ordered pore-structured nanocatalysts displayed high catalytic activity compared to their disordered counterparts in the esterification of biomass-derived levulinic acid with methanol to produce methy levulinate. It was revealed that the pore morphologies and porosity properties of the catalysts readily influenced their catalytic activity except for the Brønsted and Lewis acidity as well as surface hydrophobicity.84
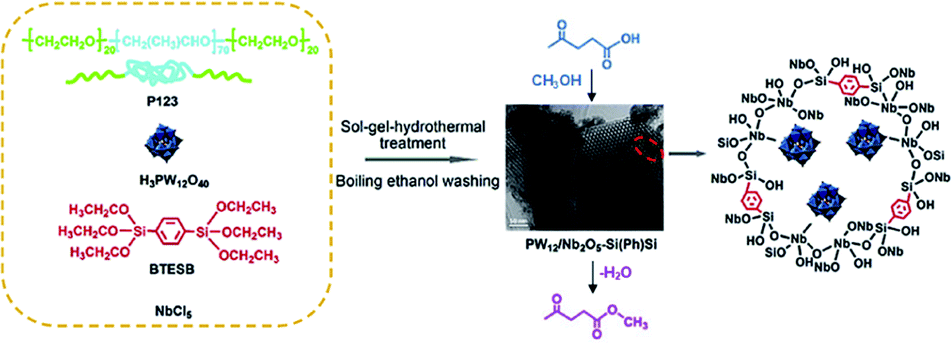 | ||
| Fig. 8 Preparation route for phenylene-bridged 2D ordered mesoporous PW12/Nb2O5–Si(Ph)Si and the catalytic reaction.84 Reproduced with permission.84 Copyright 2016, Elsevier. | ||
For tantalum, its oxide combines with heteropoly acids as robust and strong Brønsted acids to form a three dimensional (3D) interconnected composite catalyst with improved acid strength, which applies to a wide variety of acid-catalyzed reactions. For example, the acid strength of the Ta2O5–H3PW12O40 composite was much higher than that of pure Ta2O5 or H3PW12O40 as revealed by esterification and transesterification reactions.85 Due to the pronounced hydrophilicity of Ta2O5–H3PW12O40, hydrophilic products formed during the catalytic process get easily adsorbed and thereby deactivate the catalyst or inhibit the catalytic transformation. Hence, hydrophilic Ta2O5–H3PW12O40 was incorporated into hydrophobic alkyl-/phenylene-bridged PMO to form Ta2O5–H3PW12O40–PMO with Ta–O–Si–C bonds. Such composite catalysts could markedly improve the catalytic performance, stability and reusability. In 2009, the material Ta2O5/Si(R)Siy/(H3PW12O40)x (R = ethylene or phenylene; x and y represent the H3PW12O40 loading (wt%) and mole percentage (mol%) of Si in the product, respectively) was successfully prepared by effectively tuning the ratio of Si/W via a one-pot co-condensation method in the presence of Pluronic P123. The obtained composite catalysts were applied to the transesterification of soybean oil to biodiesel, exhibiting excellent catalytic activity, thanks to the improved acid strength, increased hydrophobicity and textural properties of the mesoporous material.86
2.3. Group 6 (Cr, Mo, W)-based PMO nanoreactors and catalyses
Chromium as a heavy metal element is not considered as environmentally benign, neither are Cr-PMO-type catalysts. Notwithstanding, Cr salts such as chromium nitrate were incorporated into mesoporous organosilica via one-pot synthesis to prepare a Cr-PMO catalyst. After removal of the surfactant template by solvent extraction, the obtained Cr-incorporated PMO catalyst did not show any catalytic activity for the oxidation of cyclohexane. However, upon calcination, the organic fragment was removed and Cr(III) species were converted into Cr(VI) species. Due to the presence of highly dispersed active chromium sites available for the substrate molecules, the calcined Cr-PMO exhibited an improved catalytic activity in the aerobic oxidation of cyclohexane and ethyl benzene compared to its parent, but conversion was low.87,88Incipient wetness impregnation and post-calcination is a traditional strategy to prepare metal-based catalysts on supports such as oxides and carbon. Chromium(III) nitrate nonahydrate was impregnated into the as-made and calcined mesoporous organosilica (MOS) prepared with TEOS and BTEE, followed by calcination to generate two different catalysts, Cr/MOS and Cr/MOS-cal.89 Both catalysts promote the partial oxidation of cyclohexane with high selectivity (>86%). Cr/MOS-cal synthesized with 10% BTEE displayed a higher surface hydrophobicity, which accelerated the access of cyclohexane to chromate species and thereby showed the highest catalytic activity.
Oxido-peroxido molybdenum compounds (MoOx) have attracted great interest due to their catalytic properties.90 Molecular Mo-based complex catalysts often encounter the problems of separation and reusability as well as insufficient stability. The heterogenization of homogeneous Mo catalysts is a potential strategy to ensure that active Mo species are stable enough toward the substrate. In 2002, the oxodiperoxo molybdenum compound MoO(O2)2·(DMF)2 was linked to pyrazolyl pyridine-functionalized mesoporous silica MCM-41 to form a heterogeneous catalyst for the epoxidation of cyclooctene, showing good catalytic activity and reusability.91 Compared to mesoporous silica, organo-bridged PMO possesses a stronger hydrophobicity, which is greatly conducive to the access and diffusion of organic substrate molecules. MoO(O2)2·(DMF)2 was immobilized on Bipy-PMO-IL to generate MoO(O2)2@Bipy-PMO-IL (Fig. 9) for the oxidation of organic sulfides, such as methyl phenyl sulfide, showing a high recyclability and an excellent catalytic activity with a conversion of up to 100% and a yield of up to 90%.92
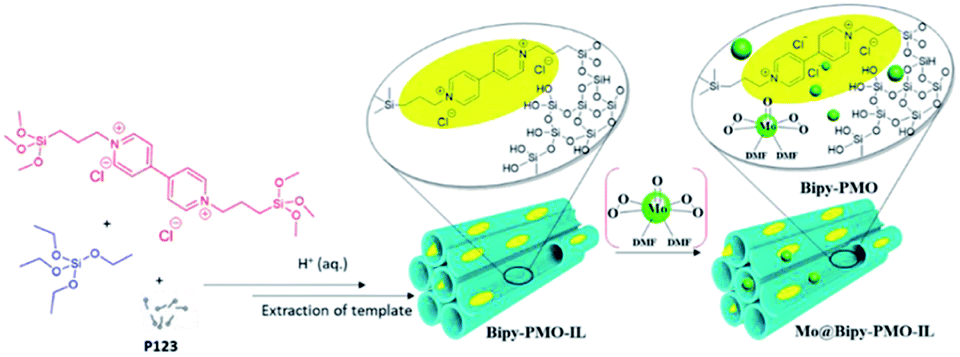 | ||
| Fig. 9 Schematic illustration of the preparation of the catalyst MoO(O2)2@Bipy-PMO-IL.92 Reproduced with permission.92 Copyright 2020, Elsevier. | ||
A molecular catalyst [MoX2(CO)3(NCCH3)2] (X = I, Br) was immobilized onto pyca-integrated periodic mesoporous silica PMO-pycaSi (pycaSi = C5H4NCHN(CH2)2CH2Si(OEt)3), prepared by co-condensation of TEOS and pycaSi through the sol–gel method, to form a hybrid heterogeneous catalyst PMO-pycaSi-MoX2 with 2.72 wt% Mo (X = I) and 3.90 wt% (X = Br).93 Owing to a better-controlled quantity and distribution of the active sites, PMO-pycaSi-MoX2 catalyzed the epoxidation of cyclohexyne (Cy6), cis-cyclooctene (Cy8) and styrene (Sty) and showed a good catalytic performance with conversions of 72% for Cy6, 82% for Cy8 and 100% for Sty, when X = I, and conversions of 75% for Cy6, 100% for Cy8 and 98% for Sty when X = Br.
In 2010, MoO2Cl2 was linked to a phosphanyl-functionalized phenylene-bridged hybrid PMO, which was synthesized via the co-condensation of BTEB and (diphenyl-phosphanyl)propyltriethoxy-silane (TEPPP) in the presence of Pluronic P123 (EO20PO70EO20) under acidic conditions. Surfactant removal and oxidation of the surface phosphane groups with H2O2 afforded a Mo-linked hybrid PMO (Mo-HPMO) heterogeneous catalyst containing surface Mo species [MoCl2(O)2{OP(Ph)2CH2CH2CH2Si–}2] with 0.47 mmol g−1 of Mo and a P/Mo molar ratio of 1.8.94 The catalyst was used for the solvent-free epoxidation of cis-cyclooctene and different olefins with TBHP,94 with both conversion and selectivity up to 100%.
In 2015, mesoporous polyoxometalate cluster–crosslinked organosilica frameworks were first prepared using a surfactant-assisted co-polymerization of organically modified polyoxometalate cluster (NMe4)4[SiW11O39{O–(SiC2H4Si(OH)3)2}] (Fig. 10a) building blocks and ethylene-bridged silsesquioxane BTEE under acidic conditions.95 The structures of the as-made mesoporous surfactant-SiW11O39-ethane-silica composite and mesoporous surfactant-free SiW11O39-ethane-silica (SiW11/MES) are shown in Fig. 10b and c. The SiW11O39 species are located near the edge area of the pore wall or protrude into the pore channels. These new hybrid polyoxometalate-organosilica materials possess an ordered pore structure, a large surface area and a high density of catalytic sites, which should be highly effective in the photocatalytic oxidation of aryl alcohols with molecular oxygen. As a result, 15%SiW11/MES exhibited high catalytic activities in the aerobic oxidation of various para-substituted aryl alcohols with conversions ranging from 41% to 98% in dependency of the substrate. Subsequently, a series of XM11/MES (X = Si, P; M = Mo, W) composites were reported, accessible by a similar approach to SiW11/MES.96 Thus the obtained PW11/MES, SiW11/MES and PMo11/MES had large surface areas, pore volumes and considerable pore diameters ranging from 890 to 930 m2 g−1, from 1.1 to 1.3 cm3 g−1 and from 4.9 to 5.2 nm, respectively. The results of the XM11/MES-promoted photocatalysis for the hydrogen evolution reaction (HER) indicated that the pore surface of XM11/MES was catalytically active and accessible to electrolytes, contributing to a high HER performance. SiW11/MES had a higher HER activity with a H2 evolution rate of about 6.3 mmol h−1 under UV/vis light irradiation and with good reusability. Note that the photocatalytic activity for H2 production occurred at neutral pH in the absence of a precious metal or any co-catalysts and photosensitizers, indicating a promising candidate for water-splitting applications.
 | ||
| Fig. 10 (a) Molecular structure drawing of the complex [SiW11O39{O(SiC2H4Si(OH)3)2}]4− (O: red, C: black, Si: blue and {WO6}: green). (b) Proposed structure of the as-made mesoporous SiW11-modified ethylene-bridged PMO-surfactant composite. (c) Surfactant-free mesoporous SiW11-PMO.95 Reproduced from ref. 95 with permission from the Royal Society of Chemistry. | ||
In 2011, a novel PMO containing phenylpyridine (PPy) moieties (PPy-PMO) was synthesized by the hydrolysis and condensation of 2-(4-triethoxysilylphenyl)-5-triethoxysilylpyridine via a sol–gel process and used for the fabrication of Ru or Ir polypyridine complexes on the pore surfaces.97 In 2014, bipyridine-bridged PMO (BPy-PMO) with a crystalline pore wall was successfully prepared by the hydrolysis and condensation of 5,5′-bis(triisopropoxysilyl)-2,2′-bipyridine in the presence of SDA octadecyltrimethylammonium chloride (C18TMACl) under basic conditions (Fig. 11).98 The surfactant-free BPy-PMO had a specific surface area of 739 m2 g−1, a uniform pore diameter of 3.8 nm and a crystalline pore wall with a 1.4 nm thickness. BPy-PMOs display a very strong coordination ability with different metals to form surface metal complexes, and hence attracted considerable attention as excellent supports for the preparation of heterogeneous catalysts. For example, metal compounds from groups 6–10, MoO2Cl2, Re(CO)5Cl, Ru(bpy)2Cl2·2H2O, [Ir(ppy)2Cl]2, [Ir(OMe)(cod)]2 and Pd(OAc)2 readily react with the chelating ligand incorporated into BPy-PMO to form a variety of bipyridine-based metal complexes on the pore surface of BPy-PMO (Fig. 12), exhibiting distinct optical properties and catalytic activities.98,99
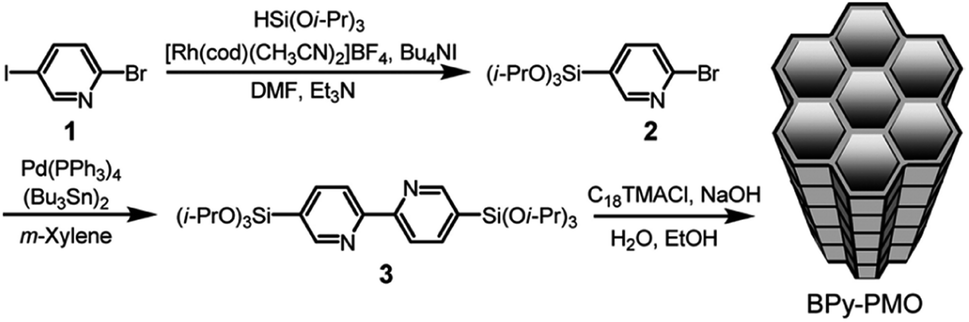 | ||
| Fig. 11 Synthesis of 5,5′-bis(triisopropoxysilyl)-2,2′-bipyridine and preparation of BPy-PMO through hydrolysis and polycondensation under basic conditions.98 Reproduced with permission.98 Copyright 2014, American Chemical Society. | ||
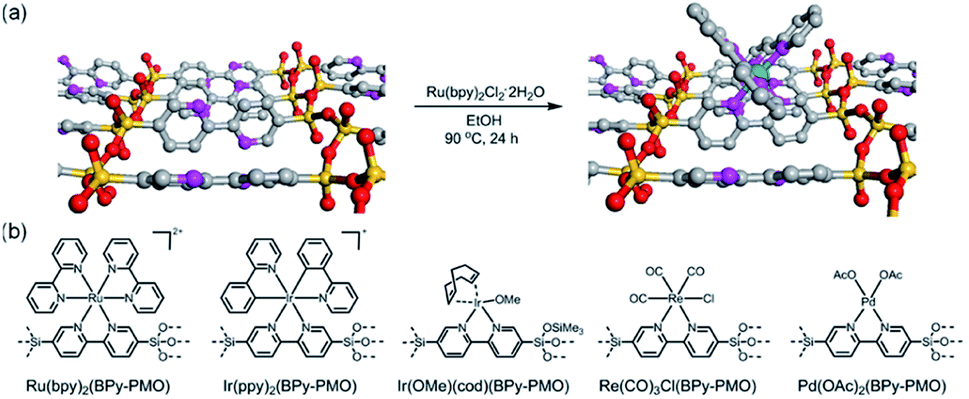 | ||
| Fig. 12 (a) CG image of the direct Ru-complex formation on the pore surface of BPy-PMO. Green, ruthenium; silicon, yellow; oxygen, red; carbon, gray; nitrogen, pink; hydrogen, white. (b) Chemical structures of metal complexes prepared using BPy-PMO as a solid chelating ligand in this study.98 Reproduced with permission.98 Copyright 2014, American Chemical Society. | ||
BPy-PMO reacted with hexamethyldisilazane in dry organic solvent to form trimethylsilyl (TMS) group-caped BPy-PMO (BPy-PMO-TMS) with improved hydrophobicity. MoO2Cl2 reacted with BPy-PMO-TMS in acetonitrile to afford materials Mo-BPy-PMO-TMS with different Mo contents (Fig. 13a).99 Mo-BPy-PMO-TMS included two different Mo species, MoO2Cl2(bpy) and MoO2Cl(OH)(bpy), which were confirmed by FT-IR and Raman spectroscopy as well as XAFS analysis. Mo-BPy-PMO-TMS showed high catalytic performance in the epoxidation of cis-cyclooctene with TBHP under ambient and anhydrous conditions (Fig. 13b). Upon optimizing the reaction conditions, 10%Mo-BPy-PMO-TMS exhibited the highest TOF at 90 °C. The immobilized Mo catalyst could be applied in the epoxidation of a variety of aliphatic and aromatic olefins showing yields up to 100% and good recyclability for at least three reuse cycles in the epoxidation of cis-cyclooctene.
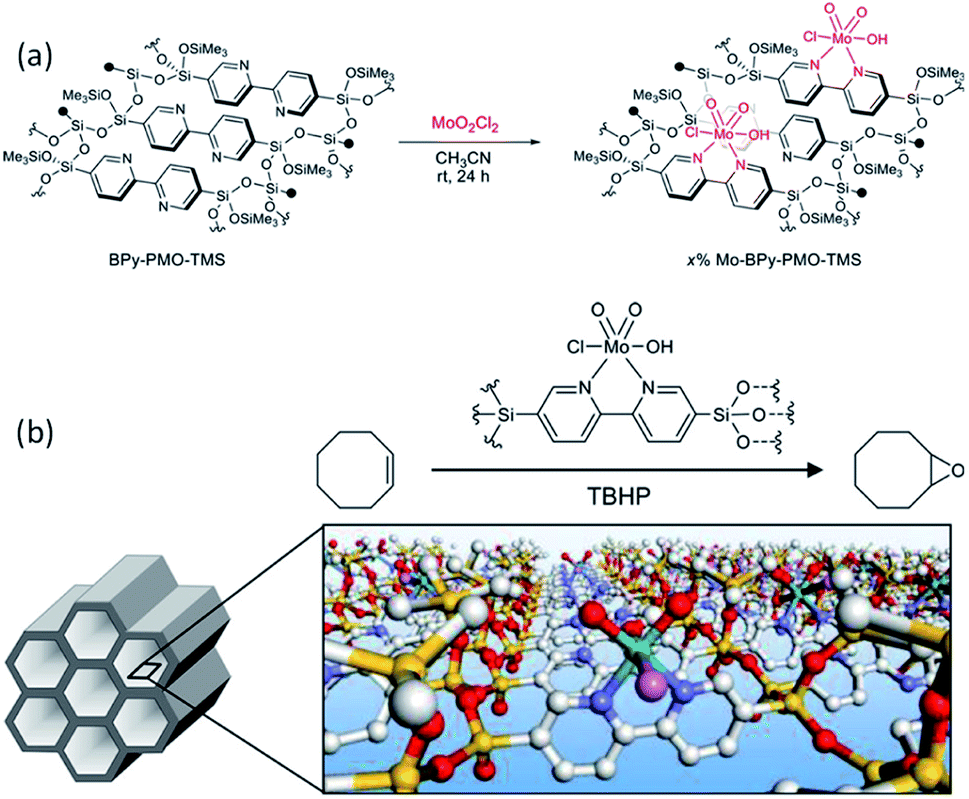 | ||
| Fig. 13 (a) Schematic for direct Mo-complex formation on the pore surface of BPy-PMO-TMS. (b) Illustration of the catalytic epoxidation of cis-cyclooctene.99 Reproduced with permission.99 Copyright 2018, American Chemical Society. | ||
More recently, a BPy-PMO thin film was fabricated on the inner wall of a microreactor by an evaporation-induced self-assembly approach.100 The obtained BPy-PMO thin films exhibited open mesopores with a diameter of 8–12 nm on the outermost surfaces and a thickness of approximately 50 nm. MoO2Cl2 could be immobilized into BPy-PMO thin film layers to form Mo-BPy-PMO inside the microreactor for the epoxidation of cis-cyclooctene with TBHP in a continuous flow manner. The resulting immobilized catalysts showed an excellent catalytic performance with TOFs > 3000 h−1 and TONs > 66000 at 80 °C, while maintaining catalytic activity over 26 h. This corresponds to a considerably higher stability compared to conventional methods.
For tungsten, previous studies have shown that silica-based tungstate interphase catalysts could effectively promote the selective oxidation of sulfides to sulfoxides101,102 and the oxidation of primary and second alcohols103 with 30% hydrogen peroxide, while exhibiting a high recoverability and high reusability. In 2014, tungstate ions (WO42−) were successfully supported on a PMO with an imidazolium framework (PMO-IL) to form WO42−@PMO-IL for the oxidation of alcohols.104 The oxidation of benzyl alcohol with 30% H2O2 was markedly affected by solvent effects as seen in the composition of the final product. Note that when a 1:1 mixture of H2O/CH3CN at 90 °C was used, benzyl alcohol was selectively converted into the corresponding benzaldehyde. Benzoic acid and other products were not detected. Under these optimized conditions, substituted benzyl alcohols bearing either electron-releasing or electron-withdrawing groups were also oxidized selectively and afforded the corresponding aldehydes in good to excellent yields. Primary and second alcohols were oxidized to the corresponding aldehydes and ketones (Scheme 5a) with moderate to excellent yields and 100% selectivity. Moreover, under such conditions, WO42−@PMO-IL as a supported phase transfer catalyst indicated a high stability and high reusability, due to the imidazolium unit stabilized WO42− which inhibited the leaching of the active species. Similarly, WO42−@PMO-IL catalyzed the oxidation of various sulfides to the corresponding sulfones in H2O/CH3CN (1:1), achieving 100% selectivity and moderate to excellent yields.105 When H2O/CH3OH (1:10) was used as a solvent mixture, WO42−@PMO-IL also exhibited a considerable catalytic performance (Scheme 5b). For such highly efficient catalysts WO42−@PMO-IL, a possible mechanism for the catalytic oxidation of sulfide in H2O/CH3CN and H2O/CH3OH solvent was suggested (Fig. 14).
 | ||
| Scheme 5 WO42−@PMO-IL-catalyzed oxidation of various alcohols (a) and sulfides (b). | ||
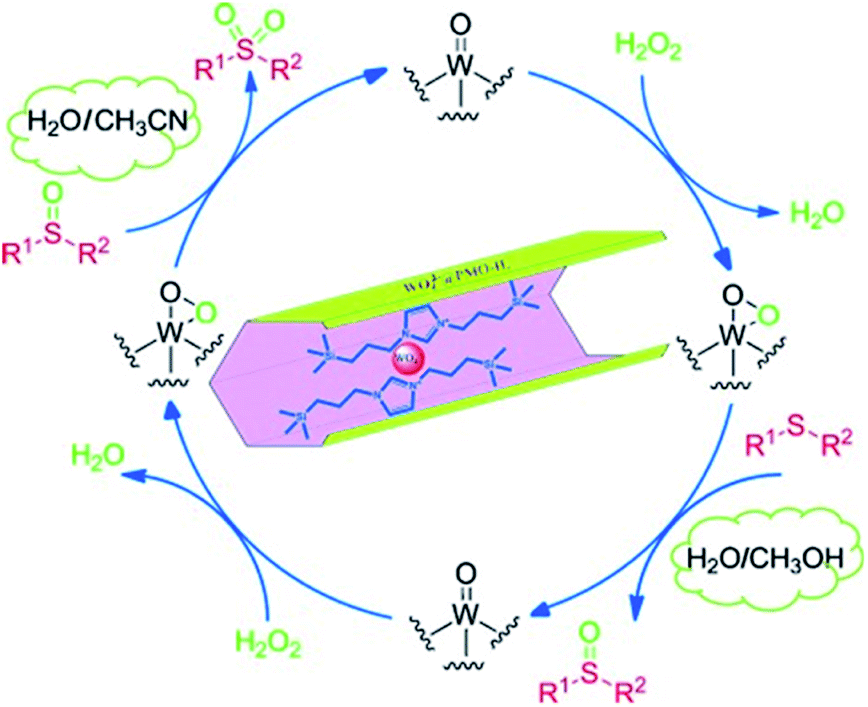 | ||
| Fig. 14 A possible mechanism for the WO42−@PMO-IL-catalyzed oxidation of sulfides.105 Reproduced with permission.105 Copyright 2015, John Wiley & Sons. | ||
2.4. Group 7 (Mn, Re)-based PMO nanoreactors and catalytic applications
In recent years, the heterogenization of homogeneous catalysts has attracted increasing attention due to the ease of separation and reusability of catalysts. Heterogeneous catalysts based on active metals incorporated into/grafted onto supports (carbon, porous materials, metal oxides etc.) with a high surface area are in high demand for industrial applications. A previous review by Li and co-workers has focused on chiral metal complexes, confined in the pore channels of mesoporous (organo)silica nanoreactors, as heterogeneous catalysts for asymmetric reactions and emphasized the pore confinement effect and cooperative activation effect of the nanoreactor.106 At present, the heterogenization of homogeneous metal-based catalysts can be performed by trapping molecular catalyst into support, direct grafting of molecular catalyst onto support and immobilization of molecular catalyst linked by functional group on support.The epoxidation of olefins is an important reaction to produce chemical intermediates for industrial-scale syntheses.107 Metal–salen complex-based homogeneous or heterogeneous catalysts are very versatile and efficient catalysts for alkene epoxidation,108 especially Mn-based catalysts.109 In 2011, a monomeric or dimeric Mn(salen) complex was trapped into the nanocages of a 3D ethylene-bridged PMO with body-centered cubic symmetry. Subsequent surface silylation with organosilane was conducted to prevent the leakage of the composite (Fig. 15).110 The Mn(salen) complexes trapped inside the PMO pore system remained intact and no structural alteration was observed. Such immobilized catalysts were probed in the catalytic epoxidation of olefins. The results indicated that the catalysts derived from dimeric Mn(salen) complexes trapped inside PMOs showed considerably higher catalytic activity and stability towards the epoxidation of alkenes than homogeneous molecular catalysts. The implications of the electron-donating effects of the substituents on the Mn(salen) complexes and steric hindrance for the catalytic activity of both the homogeneous and heterogeneous Mn(salen) complexes were discussed.110 Note that the type of solvent, ratio of oxidant to substrate, and textural structure and morphology of the supports110,111 significantly influenced the catalytic activity of the heterogeneous dimeric Mn(salen) complexes. Compared to the corresponding parent homogeneous catalysts, the immobilized catalysts could be recovered and reused at least three times without obvious loss of activity.110
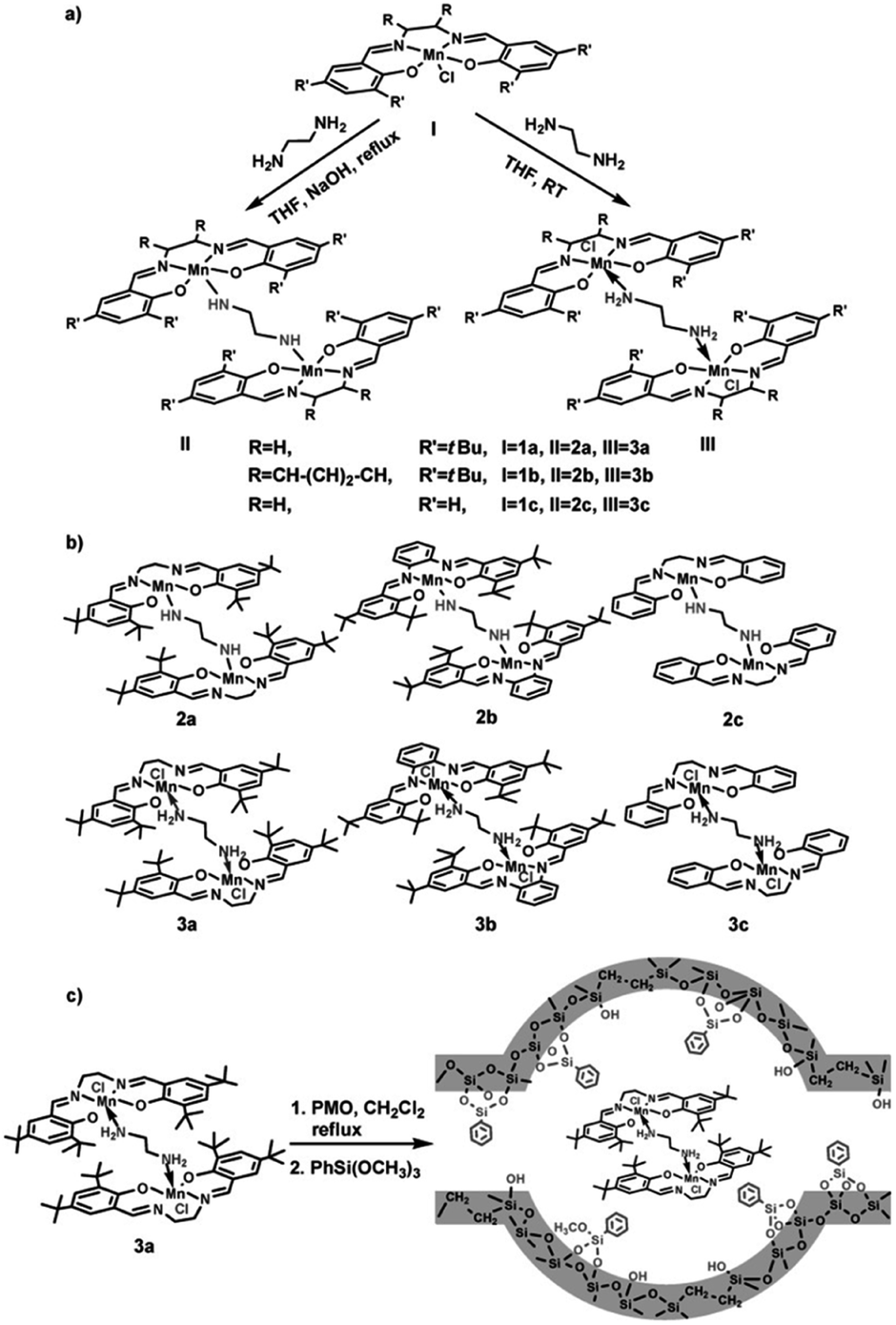 | ||
| Fig. 15 (a) Synthesis of diamine-bridged dimeric Mn(salen) complexes and (b) their corresponding structures. (c) Synthesis of heterogeneous diamine-bridged dimeric Mn(salen) complexes (3a is a representative example).110 Reproduced with permission.110 Copyright 2011, John Wiley & Sons. | ||
The compound Mn(OAc)3·2H2O, when directly reacted with PMO-IL in dimethyl sulfoxide, produced the nanocatalyst Mn@PMO-IL,112 with Mn–IL surface complexes as active species located on pore walls. Mn@PMO-IL catalyzed the one-pot Biginelli condensation of various aldehydes with urea and alkyl acetoacetates under solvent-free conditions. High activity and high to excellent yields and selectivities as well as high recoverability and reusability were achieved with short reaction times for the corresponding dihydropyrimidone derivatives. It was pointed out that a large pore diameter, high pore volume and hydrophobic nanopore channels of the nanocatalyst are greatly conducive to the access and diffusion of substrates and products. Moreover, Mn@PMO-IL can also be used as an efficient and recyclable nanocatalyst for the unsymmetric Hantzsch reaction.113
In order to improve catalyst separation, IL-modified magnetic mesoporous organosilica Fe3O4@OS/IL was prepared and used as the support for the immobilization of Mn(OAc)3·2H2O to generate Fe3O4@OS/IL–Mn with Mn(OAc)3 species.114 The Mn loading reached 0.44 mmol g−1. Nanostructured Fe3O4@OS/IL–Mn effectively catalyzed the multicomponent reaction of benzaldehyde, dimedone and phthalhydrazide to 2-H-indazolo[2,1-b]phthalazines or its derivatives, in a short time under ultrasonic irradiation, exhibiting a high yield, high recoverability and an excellent reusability at least 12 times (last recycle reaction for 13 min, 76% yield). In addition, a 3-aminopropyltrimethoxysilane (APTS)-modified phenylene-bridged PMO (APTS-BPMO or BPMO-Pr-NH2) was prepared by the reaction of APTS with BPMO in toluene. Subsequently, BPMO-Pr-NH2 was reacted with isatin in toluene to form a BPMO-supported isatin-Schiff-base (BPMO@ISB). Finally, Mn(NO3)2·4H2O was immobilized onto BPMO@ISB to obtain the nanostructured mesoporous catalyst BPMO@ISB/Mn for the multicomponent reaction of aldehyde, malononitrile and 4-hydroxycoumarin to dihydro-pyrano[3,2-c]chromenes (Scheme 6). Also this transformation exhibited very good catalytic activity, recoverability and high reusability at least six times (6th recycle, 85% yield).115
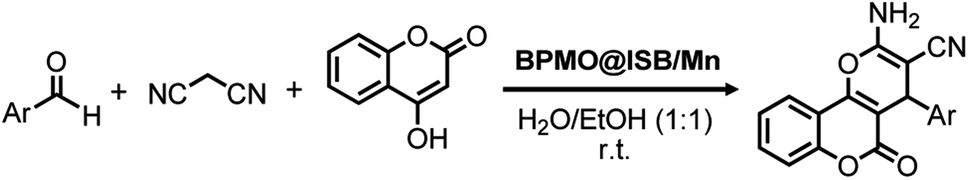 | ||
| Scheme 6 BPMO@ISB/Mn-catalyzed multicomponent reaction of an aldehyde, malononitrile and 4-hydroxycoumarin to dihydro-pyrano[3,2-c]chromenes. | ||
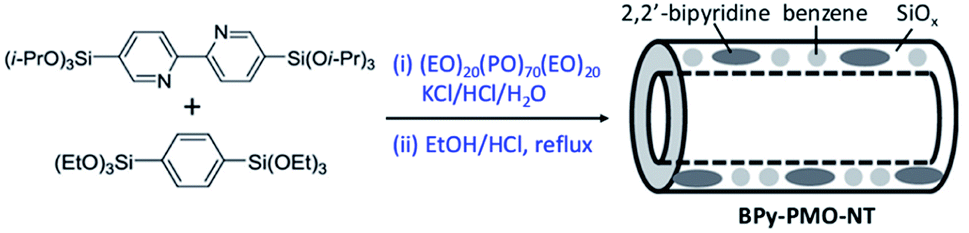 | ||
| Scheme 7 Synthesis process of BPy-PMO-NT by a template-directed sol–gel technique. | ||
Additionally, Mn(CO)5Br was immobilized on BPy-PMO, prepared by hydrolysis and condensation of 5,50-bis(triisopropoxysilyl)-2,20-bipyridine in the presence of the SDA trimethylstearylammonium iodide under basic conditions. The resulting hybrid material [Mn(bpyPMO)(CO)3Br] contained fac-[Mn(bpyPMO)(CO)3Br] as the major and mer-[Mn(bpyPMO) (CO)3Br] as a minor component (Fig. 16).116 By changing the initial amount of Mn(CO)5Br, the Mn content in [Mn(bpyPMO)(CO)3Br] could be adjusted from 1.41 wt% (Mn/bpy = 0.09) to 1.22 wt% (Mn/bpy = 0.08), and to 0.41 wt% (Mn/bpy = 0.09).
 | ||
| Fig. 16 Synthesis pathway to [Mn(bpyPMO) (CO)3Br].116 Reproduced from ref. 116 with permission from the Royal Society of Chemistry. | ||
The components fac-[Mn(bpyPMO)(CO)3Br] and mer-[Mn(bpyPMO)(CO)3Br] were confirmed by FT-IR and UV-diffuse reflectance spectroscopy (UV-DRS). Under light irradiation, UV-DRS verified the formation of 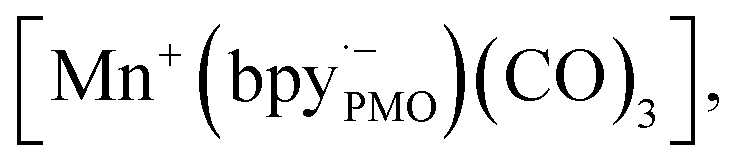 while electron paramagnetic resonance (EPR) measurements further supported the presence of bpy·− and the assignment of
while electron paramagnetic resonance (EPR) measurements further supported the presence of bpy·− and the assignment of  The existence of
The existence of 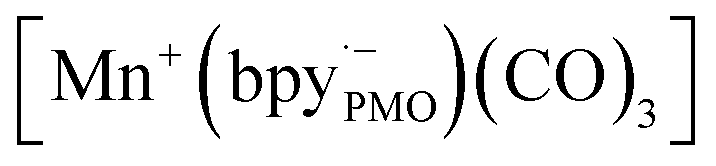 in [Mn(bpyPMO)(CO)3Br] likely initiated the catalytic reaction under light irradiation. Previous investigations have confirmed that radicals play a pivotal role in Mn-bpy complex-catalyzed CO2 reduction.117,118 This result inspired the authors to utilize [Mn(bpyPMO)(CO)3Br] as a catalyst for photocatalytic CO2 reduction to CO and HCOOH in the presence of 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzoimidazole as the electron donor, indicating a high activity up to ca. 720 TON. Site-isolated manganese carbonyl on BPy-PMO inhibited bimolecular processes such as dimerisation and disproportionation and thus allowed the spectroscopic observation of key reaction intermediates, two meridional isomers of the carbonyl complexes and the bipyridine radical anion species.116 The proposed mechanism for the formation of HCO2H and CO from the photocatalytic reaction with [Mn(bpyPMO)(CO)3Br] is shown in Fig. 17.
in [Mn(bpyPMO)(CO)3Br] likely initiated the catalytic reaction under light irradiation. Previous investigations have confirmed that radicals play a pivotal role in Mn-bpy complex-catalyzed CO2 reduction.117,118 This result inspired the authors to utilize [Mn(bpyPMO)(CO)3Br] as a catalyst for photocatalytic CO2 reduction to CO and HCOOH in the presence of 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzoimidazole as the electron donor, indicating a high activity up to ca. 720 TON. Site-isolated manganese carbonyl on BPy-PMO inhibited bimolecular processes such as dimerisation and disproportionation and thus allowed the spectroscopic observation of key reaction intermediates, two meridional isomers of the carbonyl complexes and the bipyridine radical anion species.116 The proposed mechanism for the formation of HCO2H and CO from the photocatalytic reaction with [Mn(bpyPMO)(CO)3Br] is shown in Fig. 17.
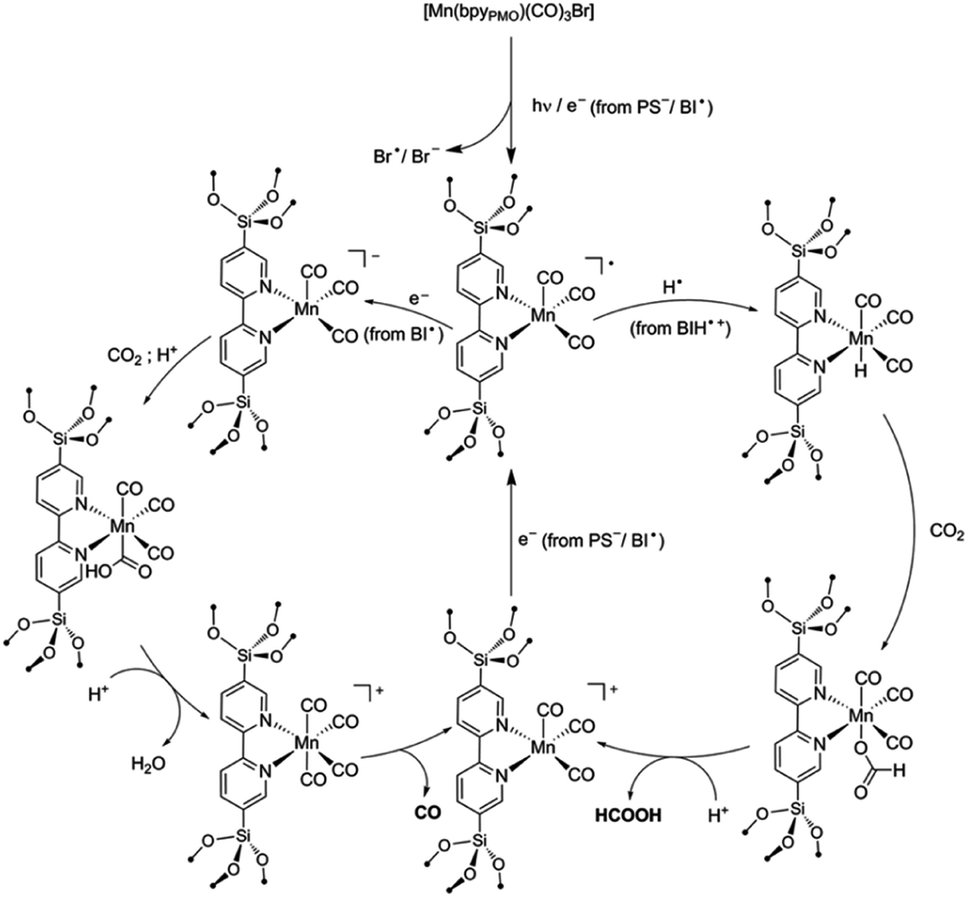 | ||
| Fig. 17 Proposed mechanisms for the formation of HCO2H and CO from the photocatalytic reaction with [Mn(bpyPMO)(CO)3Br].116 Reproduced from ref. 116 with permission from the Royal Society of Chemistry. | ||
Re(bpy)(CO)3X (X= Cl, Br) showed a strong ability for the photochemical reduction of CO2 to CO under visible light irradiation.119,120 When molecular Re(CO)5Cl similar to Re(bpy)(CO)3X was immobilized on ordered bipyridine-bridged mesoporous organosilica BPy-PMO, the Re complex was directly formed on the pore surface to afford Re(CO)3Cl@BPy-PMO.98 The textural structure and chemical composite of Re(CO)3Cl@BPy-PMO were characterized by X-ray diffraction (XRD), physisorption, FT-IR, ultraviolet–visible (UV–vis) spectroscopy, X-ray absorption near edge structure (XANES) spectroscopy, extended X-ray absorption fine structure (EXAFS) spectroscopy, X-ray photoelectron spectroscopy and nuclear magnetic resonance (NMR) spectroscopy, but its physicochemical properties were not explored after preparation. Until 2018, Re(bpy)(CO)3Cl similar to Re(CO)5Cl was immobilized on the pore surface of BPy-PMO to obtain Re(bpy)(CO)3Cl@BPy-PMO (Re-BPy-PMO) (Fig. 18).121
 | ||
| Fig. 18 Immobilization of the rhenium catalyst Re(bpy)(CO)3Cl on BPy-PMO.121 Reproduced with permission.121 Copyright 2018, John Wiley & Sons. | ||
Note that the metal-to-ligand charge transfer (MLCT) absorption band of solid Re-BPy-PMO appeared at almost the same wavelength (actually a slight blue shift) as that of molecular Re(bpy)(CO)3Cl in toluene. However, the phosphorescence lifetime of Re-BPy-PMO was shorter than that of molecular Re(bpy)(CO)3Cl, hence directly resulting in a low photocatalytic performance for CO2 photoreduction. Apparently, the reaction of Re(bpy)(CO)3Cl with solid BPy-PMO completely took place under unusual pore environmental conditions compared to conventionally the bulk liquid–liquid and solid–liquid reactions. The Re-complex in Re-BPy-PMO interacted not only with water molecules adsorbed on the pore surfaces, but also with SiOH groups of the silica framework. Both interactions were responsible for the unexpectedly short MLCT absorption wavelength of Re-BPy-PMO.122 This result was corroborated by a red shift in the in situ UV–vis spectra of Re-BPy-PMO and surface silylated Re-BPy-PMO after vacuum heating, revealing the effects of pore surfaces on the electronic states of metal complexes formed on BPy-PMO. Moreover, it was very interesting to find that when ruthenium photosensitizers like [Ru(bpy)3]2+ and Re(bpy)(CO)3Cl were co-immobilized on BPy-PMO, the photocatalytic activity greatly enhanced. This was ascribed to an efficient transfer of photoinduced electrons from the Ru photosensitizer to the Re catalyst on the BPy-PMO, which was also verified by quantum chemical calculations. Furthermore, BPy-PMO nanotubes (BPy-PMO-NT) were successfully synthesized by the co-condensation of 2,2′-bipyridine-bridged organosilane [(iPrO)3Si–C10H6N2–Si(OiPr)3] and phenylene-bridged organosilane [(EtO)3Si–C6H4–Si(OEt)3] with a suitable ratio in the presence of Pluronic P123 and KCl under acidic conditions, followed by HCl-acidized EtOH solvent extraction (Scheme 7).123 BPy-PMO-NT displayed a high surface area of 961 m2 g−1, pore volume of 0.9 cm3 g−1 and pore diameter of 7.8 nm.
Such a state-of-the-art BPy-PMO-NT material with a high surface area, large pore size and pore volume can be used as an ideal support for the immobilization of molecular catalysts. For example, the reaction of Re(CO)5Cl with BPy0.3-PMO-NT (0.3 represents the mole content of BPy-bridged organosilane in 1 mol total organosilane) gave a heterogenized molecular Re-based catalyst on BPy0.3-PMO-NT (Re-BPy0.3-PMO-NT).124 A high surface area, large pore size and hydrophobic pore channel are conducive to the access and diffusion of reactant(s) and product(s) as well as adsorption of CO2. Meanwhile, in order to further improve the hydrophobicity of the heterogeneous catalyst, the silanol groups on the Re-BPy0.3-PMO-NT surface were passivated with hexamethyldisilazane to form trimethylsilyl-capped Re-BPy0.3-PMO-NT (Re-BPy0.3-PMO-NT-Me) (Fig. 19a). Such a state-of-the-art heterogeneous catalyst Re-BPy0.3-PMO-NT-Me was applied to CO2 photoreduction with high activity (the turnover number (TON) of CO2 reached 30.6 per Re) and tailored selectivity (max. 94%) in an aqueous solution, which was much higher than that from Re-BPy-PMO.121 The Re-BPy0.3-PMO-NT-Me-catalyzed CO2 photo-reduction mechanism was proposed and verified by density functional theory (DFT) calculations (Fig. 19b).124 Very recently, the material resulting from the immobilization of Re(bpy)(CO)3Cl on BPyx-PMO-NT was further investigated for photocatalytic CO2 reduction by adjusting the contents of bpy-bridged organosilane and Re as well as by adding a photosensitizer [Ru(bpy)3]2+, exhibiting an improved catalytic activity.125
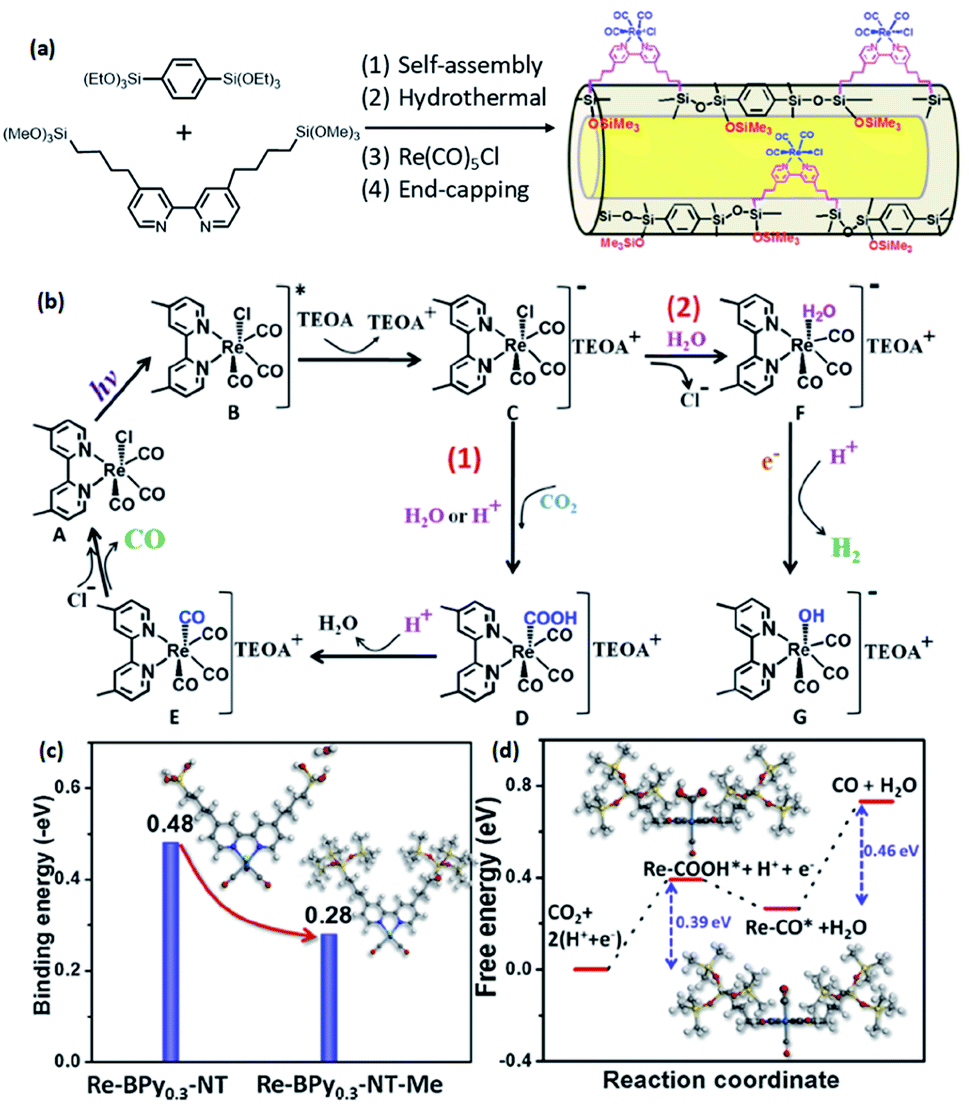 | ||
| Fig. 19 (a) Synthetic routes for the design of heterogeneous molecular Re-based catalysts on organosilica nanotubes (Re-BPy0.3-NT-Me). (b) Possible photocatalytic CO2 reduction mechanism. DFT calculations: (c) the binding energy of H2O onto Re-BPy0.3-NT and Re-BPy0.3-NT-Me and (d) free energy evolution of the reduction of CO2 to CO on Re-BPy0.3-NT-Me.124 Reproduced with permission.124 Copyright 2019, Elsevier. | ||
2.5. Group 8 (Fe, Ru)-based PMO nanoreactors and catalytic applications
Iron especially iron oxides were often used as a magnetic separator to integrate into mesoporous materials and fabricate core–shell or yolk–shell structured materials or magnetic materials.126 The integration of magnetic iron oxides is greatly beneficial to the convenient separation of the product and catalyst after utilization.127 In general, iron oxide itself did not participate the catalytic reaction. Herein, the cases where iron or iron oxide or iron complexes in PMO directly participated in or catalyzed the chemical reaction are focused on. In 2013, (C5H5)Fe(C5H4)Si(CH3)2Cl was anchored on phenylene-bridged PMO (PMO-Ph) to form a heterogeneous catalyst PMO-Si(CH3)2(C5H4)-Fe(C5H5) which was used in the catalytic oxidation of styrene with H2O2 as the oxidant under mild conditions.128 The results showed a considerable conversion and good selectivity for benzaldehyde in comparison with molecular ferrocene. During the catalytic reaction, although active species leaked into the solution, benzaldehyde selectivity was improved up to 100% by using acetonitrile as a solvent and adjusting the molar ratio of the catalyst and substrate.129 Moreover, the organic compound can react with functionalized PMO to form a solid ligand with a strong coordination ability on the surface. For example, the reaction of 2-hydroxybenzaldehyde with the –NH2 group of APTES-modified PMO-IL with the ion liquid unit (PMO-IL-NH2) produced the Schiff base and ionic liquid-based bifunctional PMO (SBIL-BPMO). Finally, SBIL-BPMO further interacted with iron(III) nitrate to yield a novel solid iron-Schiff base nanocatalyst Fe@SBIL-BPMO (Fig. 20).130 This nanostructured catalyst with a high surface area and large pores effectively catalyzed the one-pot, three-component Biginelli condensation of aldehydes, urea/thiourea, and alkyl acetoacetate under solvent-free conditions to 3,4-dihydropyrimidinones/thiones.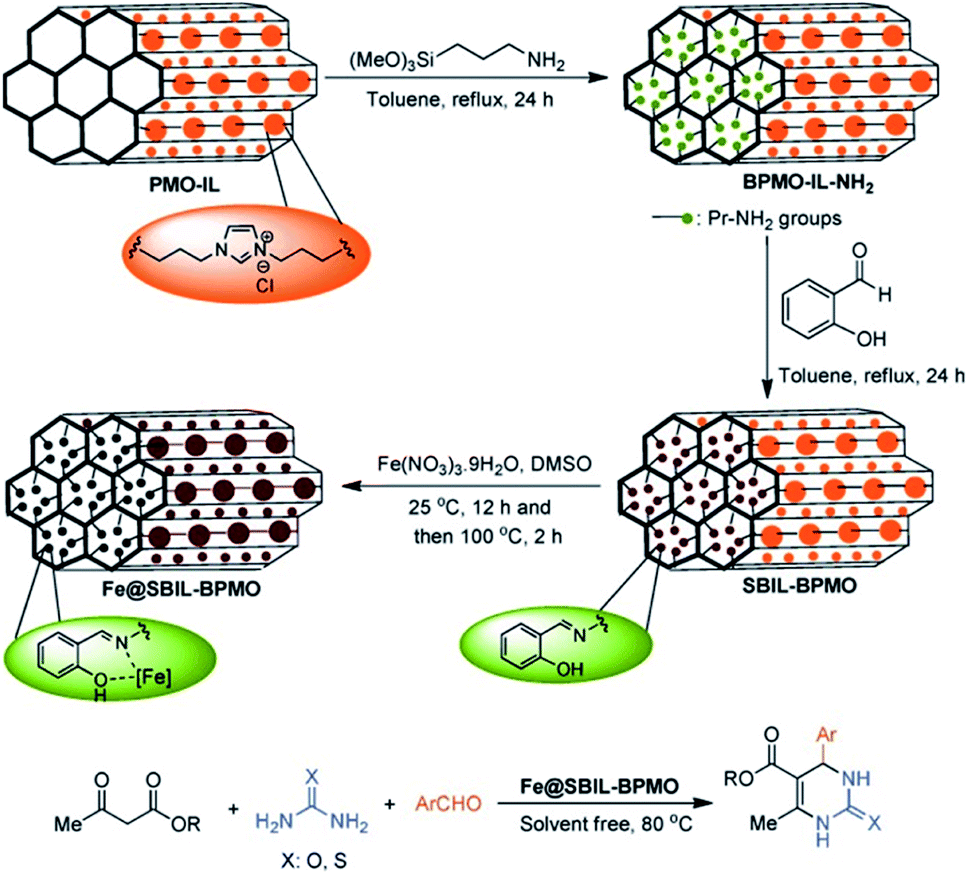 | ||
| Fig. 20 Preparation of the Fe@SBIL-BPMO nanocatalyst and its application in the synthesis of 3,4-dihydropyrimidinones/thiones.130 Reproduced with permission.130 Copyright 2015, John Wiley & Sons. | ||
By optimizing the reaction conditions, the high catalytic activity gave good to excellent yields (83–97%). In addition, after 9 recycles, the Fe@SBIL-BPMO nanocatalyst in the reaction of benzaldehyde, methyl acetoacetate and urea still retained 94% yield, strongly confirming the powerful reactivity, reusability and durability of the catalyst under the applied conditions. This high catalytic performance is directly related to a regular and uniform pore structure and structure-supported stability of the catalyst. In addition, iron(III) complex-incorporated mesoporous organosilicas with various iron contents were synthesized by the hydrolysis and co-condensation of TEOS and iron(III) complexes with multidentate pyridinyl ligands containing propyltrimethoxysilane via a sol–gel method. These materials were used in the direct hydroxylation of benzene to phenol, showing a considerable conversion, yield and selectivity as well as an excellent recyclability compared to molecular Fe precursors.131
More recently, FeCl2·4H2O and FeCl3·6H2O were immersed into PMO-ICS in the presence of ammonia solution, followed by drying at 100 °C, grafting of [(trimethoxysilyl)propyl]thiol in toluene and H2O2 oxidation, to obtain magnetic propylsulfonic acid-anchored isocyanurate-based PMO (iron oxide@PMO-ICS-PrSO3H).132 Such a novel and readily recoverable magnetic nanocomposite could be utilized as a nanoreactor for the sustainable heteroannulation synthesis of imidazopyrimidine derivatives through the Traube-Schwarz multicomponent reaction of 2-aminobenzo-imidazole, C–H acids and diverse aromatic aldehydes (Scheme 8). The results indicated that iron oxide@PMO-ICS-PrSO3H had much high catalytic activity under various conditions (temperature, solvent, no solvent, time etc.) compared to those of catalysts, iron oxide, PMO-ICS, iron oxide@PMO and iron oxide@PMO-PrSH, clearly revealing that the high catalytic activity of iron oxide@PMO-ICS-PrSO3H was contributed to the significant synergic effect of sulfonic acid groups (–SO3H) along with iron oxide in this mesoporous catalyst. Moreover, iron oxide@PMO-ICS-PrSO3H also exhibited some excellent advantages of a low catalyst loading, low cost, short reaction time, high yield, recoverability and reusability.
 | ||
| Scheme 8 Iron oxide@PMO-ICS-PrSO3H-catalyzed Traube-Schwarz multicomponent reaction of 2-aminobenzo-imidazole, C–H acids and diverse aromatic aldehydes to imidazopyrimidine derivatives. | ||
Ru- or Rh-based complexes are very vital in the enantioselective hydrogenation of alkenes and carbonyl compounds in chemistry. The heterogenization of molecular Ru or Rh complexes on supports is an efficient strategy to extend the applications of homogeneous catalysts. In 2012, benzeneruthenium(II) chloride dimer Ru2(C6H6)2Cl4 was immobilized on TMS-capped BINAP-PMO with a high surface area and good pore volume to form the catalyst Ru/5%(R)-BINAP-PMO. This TMS-capped BINAP-PMO support was prepared via several steps: (i) the hydrolysis and co-condensation of TEOS and 2,2′-bis(diphenyl-phosphinyl)-5,5′-bis(triethoxysilyl)-1,1′-binaphthyl (BINAPO) in the presence of SDA, (ii) template removal (5%BINAPO-PMO), (iii) surface silylation with hexamethyldisilazane to generate TMS-capped 5%BINAPO-PMO (TMS-5%BINAPO-PMO) and (iv) the reduction of phosphine oxides. Such a catalyst was applied in asymmetric catalytic hydrogenation methylaceto-acetate with different substitutes in the presence of high-pressure hydrogen gas, and in asymmetric transfer hydrogenation of 4-methoxyaceto-phenone (Scheme 9). This catalyst showed an excellent catalytic efficiency with >99 yield and enantioselectivity >99% enantiomeric excess (ee).133 The Ru complex (η6-p-cymene)RuCl2[4-diphenylphos-phinylbenzenecarboxylic acid-4N-(3-trimethoxysilypropyl)amide] was covalently anchored on the PMO surface to form a heterogeneous catalyst Ru-PMO.134 Owing to the pore confinement effect and high hydrophobicity of the inside pore channel of PMO, Ru-PMO indicated higher catalytic activity and selectivity in the hydrogenation of olefin compared with those of homogeneous molecular catalysts and Ru-MCM-41 (an ordered periodic mesoporous pure siliceous material with a 2D hexagonal symmetry).
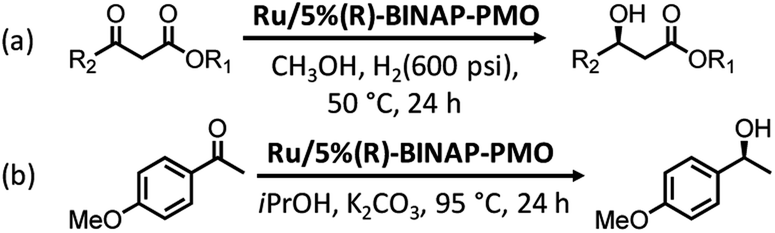 | ||
| Scheme 9 Ru/5%(R)-BINAP-PMO-catalyzed asymmetric catalytic hydrogenation of methylacetoacetate with different substitutes under high-pressure hydrogen gas (a) and asymmetric transfer hydrogenation of 4-methoxyacetophenone (b). | ||
Note that if two or more active species are incorporated into a PMO to generate a heterogeneous catalyst with partitioned sites, perhaps, this special catalyst exhibits multi-step catalytic properties from the substrate to the product. In 2014, Liu and co-workers rationally designed and successfully prepared a site-isolated organoruthenium-/organopalladium-bifunctionalized PMO catalyst (Fig. 21a). This catalyst was used in Suzuki cross-coupling and cascade asymmetric transfer hydrogenation (ATH) (Fig. 21b).135 Such a structural catalyst indicated three aspects of advantages: (i) an isolated and well-defined single molecular characteristic with different catalytic behaviors, (ii) the active sites located inside pore channels or on the surface, and (iii) high hydrophobicity of PMO and uniformly large pore size (beneficial to the access and diffusion of the substrate). As a result, when the substrate passed through or diffused into pore channels, different active species simultaneously promote the occurrence of different reactions in nanopore reactors. By optimizing reaction times, the desirable product with a high yield could be obtained in one-pot synthesis. For example, site-isolated Ru-/Pd-bifunctionalized PMO was used in one-pot cascade ATH/Suzuki cross-coupling of haloacetophenones and arylboronic acids, and a one-pot cascade ATH/Heck reaction. The resulting catalyst exhibited a high catalytic activity with a high yield and a very nice enantioselectivity (ee). Moreover, such a catalyst also showed an excellent recoverability and reusability. As a model reaction, one-pot relay-catalysis of 4-iodoacetophenone and phenylboronic acid still afforded a desired product with a conversion of 95% and 94% ee value for at least 8 recycles.
 | ||
| Fig. 21 (a) Synthesis of catalyst and (b) the expected multiple-steps catalytic reactions.135 Reproduced with permission.135 Copyright 2014, John Wiley & Sons. | ||
Similarly, the hydrolysis and self-assembly co-condensation of (S,S)-4-((trimethoxysilyl)ethyl)phenylsulfonyl-1,2-diphenylethylene-diamine (ArDPEN), 3-mercaptopropyl triethoxysilane and BTEE generated SH@ArDPEN@PMO.136 Pd NPs were loaded onto SH@ArDPEN@PMO by immobilization of PdCl2 and reduction with NaBH4 to afford Pd@ArDPEN@PMO. Afterwards, [RuCl2(mesty-lene)]2 reacted with Pd@ArDPEN@PMO to yield Pd(0)-Ru(III)-/diamine-bifunctionalized PMO (Pd@mestyleneRuArDPEN@PMO). This catalyst can effectively promote an ATH-Sonogashira coupling one-pot enantioselective tandem reaction and indicated an excellent catalytic activity with a high yield and a high ee value as well as high reusability.136
To confirm the role of the hydrophilic/hydrophobic nature of organosilica/silica as supports in heterogeneous catalysis, Ru(II) and Rh(II)-supported aminofunctionalized-organosilica/silica catalysts were prepared by immobilization of RuHCl(CO)(PPh3)3, RuCl2(PPh3)3 and RhCl(PPh3)3 onto aminofunctionalized ethylene(E)-/phenylene (B)-bridged PMO (PrNH2PMOE/PMOB)/silica (PrNH2MCM-41/SBA-15) to generate RuHCl(CO)(PPh3)3-PrNH2PMOE/PMOB/MCM-41/SBA-15, RuCl2(PPh3)3-PrNH2PMOE/PMOB/MCM-41/SBA-15 and RhCl(PPh3)3-PrNH2PMOE/PMOB/MCM-41/SBA-15, respectively.137 RuHCl(CO)-(PPh3)3-PrNH2PMOE/PMOB/MCM-41/SBA-15 and RhCl(PPh3)3-PrNH2PMOE/PMOB/MCM-41/SBA-15 were used for the hydrogenation of olefins (cyclohexene, styrene and α-methyl styrene), and RuCl2(PPh3)3-PrNH2PMOE/PMOB/MCM-41/SBA-15 for the oxidation reaction of mixed sulfides (thioanisole, ethyl methylsulfide and diethyl sulfide), to evaluate their catalytic activities and roles of hydrophobicity and hydrophilicity. By comparing the catalytic results of both hydrogenation and sulfoxidation reactions, Ru(II) and Rh(I) complex-immobilized PrNH2PMOB catalysts exhibited higher activities and selectivities than PrNH2MCM-41/SBA-15/PMOE supported catalysts, neat homogeneous complexes and without catalysts, clearly confirming a key role of the hydrophobic nature and high surface area of PMOB. Similarly, a [Ru(II)Cl2(p-cymene)]2 complex was anchored on PrNH2PMOB to form a heterogeneous catalyst Ru(II)Cym@PMOB. This catalyst was used in transfer hydrogenation of ketones and showed a considerable catalytic activity with up to ∼97% conversion compared with conventional hydrogenation reactions (∼5%) by using H2 and the molecular complex [Ru(II)Cl2(p-cymene)]2.138
A Rh complex-PMO composite could be directly synthesized by (i) the co-condensation of BTEB and RuCl2[PPh2(CH2)2Si(OEt3)3]3 in the presence of Pluronic P123 under acidic conditions and (ii) removal of P123, to afford organometal-/phenylene-bridged PMO with the [RuCl2(PPh2(CH2)2)3] unit, located on the surface of the pore wall.139 Such an intact organometal-bridged integrated PMO could be used in water-medium isomerization of 1-(4-methylphenyl)-3-buten-1-ol and showed a yield of 77%, slightly higher than 75% that of the homogeneous molecular catalyst RuCl2(PPh3)3. Note that such a heterogeneous catalyst was readily recoverable and could be reused several times.
The Ru(bpy)32+ complex is a very useful and vital photosensitizer in photoredox catalysis and photophysics.140 Ru(bpy)32+ incorporated into PMO probably exhibits more stable photocatalytic properties. Zhao and co-workers synthesized 2,2′-bipyridine-amido-bridged organosilane via a reaction of triethoxy(3-isocyanatopropyl)silane with [2,2′-bipyridine]-4,4′-diyldimethanol, and further made it coordinate to ruthenium trichloride RuCl3 in the presence of sodium hypophosphite, to form a Ru(bpy)3 unit-based organosilica precursor. Under basic conditions, the hydrolysis and co-condensation of Ru(bpy)3 unit-based organosilane with TEOS as the silica source in the presence of CTAB generated an as-made ruthenium-bipyridyl-tethered porous organosilica (as-made Ru-POS) (Fig. 22).141 After removal of the template reagent CTAB, the obtained Ru-POS was a characteristic mesoporous material with a high surface area of 620 m2 g−1 and pore diameter of about 3–4 nm (the large Ru-bipyridyl complex between the repeating units). The pore volume of 0.77 cm3 g−1 was large enough for the access and diffusion of organic compounds. Under the optimized photocatalytic conditions, the catalytic activity of the catalyst Ru-POS was evaluated by (i) a three component Hantzsch reaction of ethyl acetoacetate, ammonium acetate and aromatic aldehydes to the corresponding highly substituted pyridine derivatives by both usual and unusual paths, (ii) reductive dehalogenation of alkyl halides, and (iii) functional group interconversion of alcohols to alkyl halides. The three different types of reactions confirmed the high catalytic performances of newly developed Ru-POS photocatalysts in yield, durability and reusability.
 | ||
| Fig. 22 Schematic representation of the synthesis of Ru-POS.141 Reproduced from ref. 141 with permission from the Royal Society of Chemistry. | ||
In 2016, bipyrindine-based Ru complexes Ru(BpyC4SC3Si)3(PF6)2, Ru(BpyC4Si)3(PF6)2 and Ru(BpyC3Si)3(PF6)2 were synthesized and used as metal complex-based organosilica precursors.142 Such a bipyridine–Ru complex precursor co-condensed with the BTEB (the original proportion of Ru-precursors was 20 or 50 wt% [Ru-precursor/(Ru-precursor + BTEB)]) in the presence of Pluronic P123 as a SDA under acidic conditions to form a surfactant-Ru-PMO composite. After removal of the template Pluronic P123, a Ru-PMO material was obtained and characterized by X-ray diffraction analysis, N2 physisorption and scanning/transmission electron microscopy (S/TEM) to confirm a characteristic mesoporous structure. Note that 20 wt% Ru-PMO was a highly ordered PMO, while 50 wt% Ru-PMO with an ordered pore structure was only gained in the presence of polyacrylic acid. For 20 wt% Ru-PMO and 50 wt% Ru-PMO, the Ru contents were 0.15–0.16 mmol g−1 and 0.35–0.36 mmol g−1, respectively. All the Ru complex moieties in Ru-PMO were embedded in the pore walls at high density and showed characteristically photo- and electro-chemical properties as Ru(dmb)3(PF6)2, and hence, such Ru-PMO was used as a photo-sensitizer in heterogeneous photocatalytic systems for hydrogen evolution and water oxidation by respectively loading metal platinum (Pt) and iridium oxide (IrOx) onto the pore surface of Ru-PMO.142
Moreover, other Ru complexes [RuCl2(CO)3]2, RuCl3·xH2O and [RuCl2(bpy)2]·2H2O were immobilized directly on bipyridylene-bridged PMO (BPy-PMO)98 to produce single-site heterogeneous solid molecular catalysts [RuCl2(CO)3]2@BPy-PMO, RuCl3·xH2O@BPy-PMO and [RuCl2(bpy)2]·2H2O@BPy-PMO, respectively. In those catalysts, the coordination structure of Ru species in a single-isolated form was similar to that of the molecular complex RuCl2(bpy)(CO)2.143 Those single-site heterogeneous Ru catalysts were used in the oxidation of tertiary C–H bonds of adamantine with NaClO as an oxidant to the corresponding alcohols and showed 57 times faster than the secondary C–H bonds, thereby exhibiting remarkably high regioselectivity. Using the oxidation of cis-decalin to cis-9-decalol to evaluate the stereospecificity of the catalytic system, the Ru-immobilized catalyst also showed a considerable conversion (70%) and yield of cis-9-decalol (63%) with complete retention of the substrate configuration. This catalyst was easily recoverable by filtration and showed the same catalytic activity with respect to yield and selectivity as the original one, confirming the recoverability and reusability of Ru-immobilized catalysts.
It is interesting to find that two different ruthenium complexes, cis-[Ru(bpy)2(DMSO)Cl]Cl and [Ru(CO)2Cl2]n, were immobilized stepwise (first [Ru(bpy)2(DMSO)Cl]Cl, and then [Ru(CO)2Cl2]n) on BPy-PMO to form two different single-site ruthenium complexes as photosensitizing (PS)x and catalytic (Cat)y sites, respectively, in which [Ru(bpy)2(DMSO)Cl]Cl was used as a precursor of the PS center and [Ru(CO)2Cl2]n as the catalytic precursor.144 In this case, a novel photocatalyst Ru(PS)x-Ru(Cat)y-BPy-PMO was fabricated and used in photocatalytic CO2 reduction to CO and HCOO−. The total turnover frequency of CO and HCOO− strongly depended upon the molar ratio of PS and Cat in the photocatalyst. The product selectivity (CO/HCOO−) became large with increasing the ratio of Ru(PS)-to-Ru(Cat) (x/y). More recently, [Ru(dmbpy)2(dmso)Cl]Cl, Ru(dtbbpy)2Cl2 and Ru(dmcbpy)2Cl2-supported BPy-PMO catalysts were prepared and respectively applied in water oxidation, indicating a considerable catalytic behavior.145
Apart from Ru coordination complexes, KRuO4 salt could also be incorporated into PMOs with an imidazolium ionic liquid network (PMO-IL) to form Ru-PMO-IL catalysts with a Ru content of 0.3 mmol g−1.146 Ru-PMO-IL-catalyzed aerobic oxidation of benzyl alcohol was performed under various conditions including catalyst loading, reaction time, solvent and temperature to investigate the influence of experimental conditions for catalytic activity and further optimize the catalytic reaction conditions. The highest conversion (>99%) and TOF (8.8 h−1) could be reached at 70 °C for 4.5 h for aerobic oxidation of benzyl alcohol with a 2.5 mol% Ru-PMO-IL catalyst in trifluorotoluene (TFT). Under the optimized conditions, the Ru-PMO-IL catalyst was applied in aerobic oxidation of alcohols and showed different catalytic performances due to the difference of substituted groups on the substrate.
Ru-PMO-IL-catalyzed aerobic oxidation was further extended to the oxidation of benzylamine.147 By optimizing reaction conditions including the catalyst dose, reaction time, solvent and temperature, for 2.5 mol% Ru-PMO-IL-promoted aerobic oxidation of benzylamine in anhydrous toluene, the conversion of benzylamine and selectivity of benzonitrile respectively reached >99% and 94% at 85 °C for 6 h. Under optimized conditions, Ru-PMO-IL could effectively catalyze the aerobic oxidation of amines and exhibited various catalytic activities in good to excellent yields (70–99%) for substrates with different substituted groups. Furthermore, ruthenium oxide hydrate (RuO2·xH2O) was impregnated into magnetic ethylene-bridged PMO nanospheres (M-Et-PMO NPs) to form a readily recoverable Rh-M-Et-PMO NPs. This catalyst was used in the hydrogenation of aromatic compounds with H2.148 The results showed an excellent catalytic activity with >99% yield at 80–120 °C for 1–12 h.
Apart from the aforementioned Ru complex-supported PMO with/without functional groups for catalysis, Ru nanoparticles could be dispersed on magnetic yolk–shell nanoarchitectures with an Fe3O4 core and sulfoacid-containing PMO shell (Fe3O4@void@PMO-SO3H) to form bifunctional nanocatalysts Ru/Fe3O4@void@PMO-SO3H with a pore diameter of 4 nm. Such a catalyst promoted the direct conversion of cellulose to isosorbide (Scheme 10).149 In this case, Ru nanoparticle sizes and –SO3H group contents (–SO3H group derived from the oxidation of the thiol group in 3-mercaptopropyltrimethoxysilane (MPTMS) during the preparation of Fe3O4@void@PMO-SO3H) could be adjusted in the range of 0.9–8.8 nm and the MPTMS molar ratio of 0.14–0.50, respectively. The Ru/Fe3O4@void@PMO-SO3H-catalyzed reaction of cellulose to isosorbide at high temperature under high H2 pressure was carried out. The size effect of Ru particles and influence of –SO3H group contents on the yield for isosorbide were investigated in detail. The results showed that with increasing Ru particle size from 0.9 to 8.8 nm the yield for isosorbide decreased. Especially, when the Ru particle size was higher than 4 nm, Ru nanoparticles were mainly located on the external surface and readily aggregated together and became unstable. With gradually increasing –SO3H content, the yield for isosorbide originally increased and then decreased, but cellulose conversion still remained almost constant (98–99%), implying that a suitable size of Ru NPs and an appropriate –SO3H content were necessary for the improvement of the yield for isosorbide (max. 58.1% in this case). In addition, due to the magnetic properties of the nanocatalyst, it was easily separated and reused and showed a stable catalytic activity.
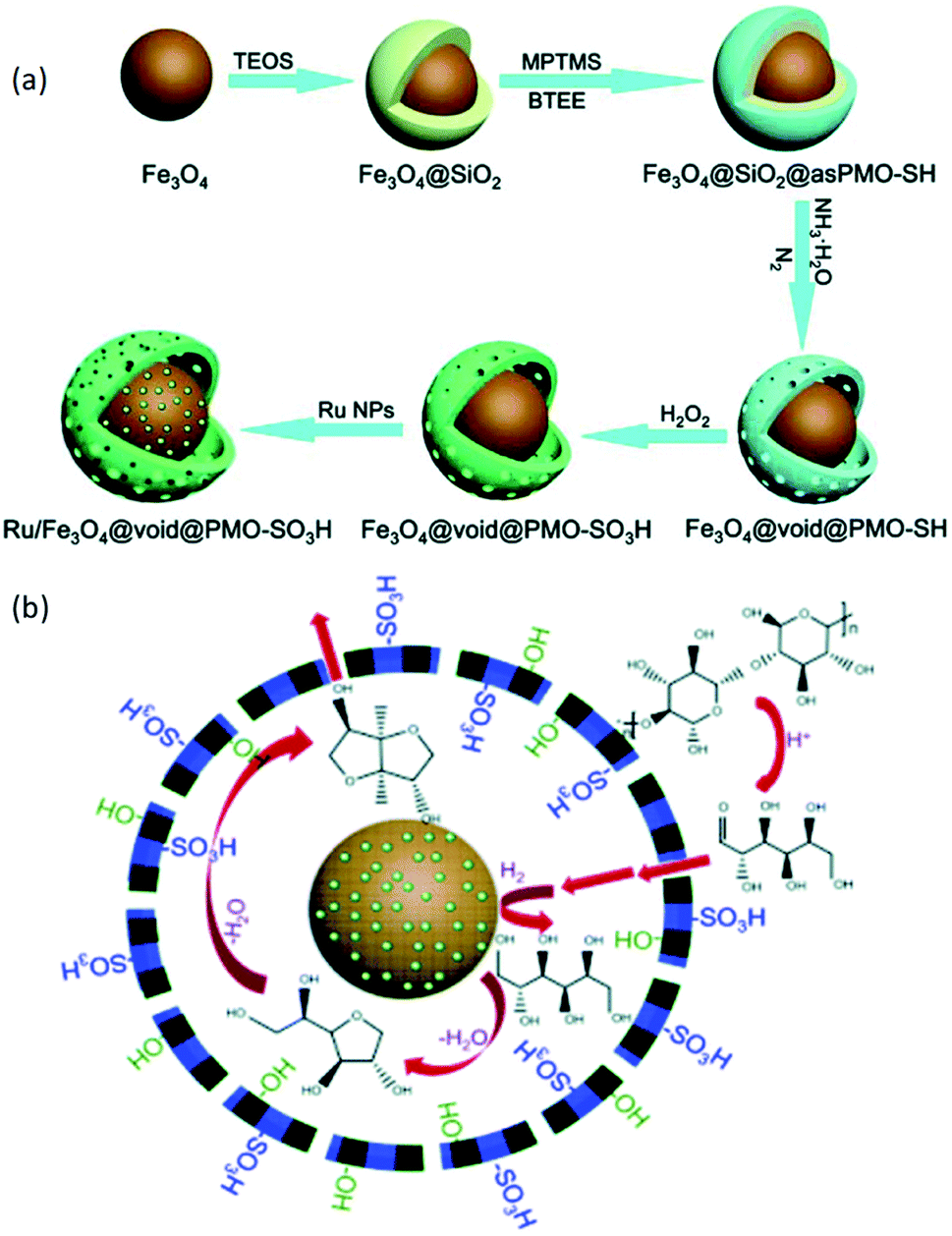 | ||
| Scheme 10 (a) Illustration of the synthesis of Ru–SO3H nanoreactors. (b) Schematic illustration of the cascade reactions in an Ru–SO3H nanoreactor.149 Reproduced from ref. 149 with permission from the Royal Society of Chemistry. | ||
2.6. Group 9 (Co, Rh, Ir)-based PMO nanoreactors and catalytic applications
The study of Co nanoparticle-, Co complex-immobilized or impregnated or anchored on PMO was almost neglected. Only a few explorations were reported. Lu and co-workers grafted an ion liquid organic compound triethoxysilylpropyl-3-butanol-benzotriazolium chloride onto PMO to form benzotriazolium-butanol-based PMO (PMO-IL).150 Different IL contents (x) were used to obtain PMO-IL(x) with various IL contents. Afterwards, cobalt acetate was impregnated into PMO-IL(x) to produce Co-PMO-IL(x) materials, which were used as heterogeneous catalysts in the catalytic transformation of carbon dioxide to cyclic carbonates. The Co-PMO-IL(x)-catalyzed cycloaddition reaction of CO2 with various epoxides was performed in a stainless-steel autoclave at the desired temperature under a fixed pressure. The cycloaddition of propylene oxide with CO2 to propylene carbonate was chosen as a model reaction to evaluate the catalytic performance of Co-PMO-IL(x). By investigating the influence of the catalyst loading, IL content, reaction temperature and pressure on the catalytic activity, optimized reaction conditions were obtained and applied in the cycloaddition of CO2 with various epoxides. The results showed catalytic activity with good to excellent yield (88–97%) and very high selectivity (98.5–99.8%), while Co-PMO-IL could be recycled and reused in the cycloaddition of propylene oxide with CO2 for at least five consecutive runs without significant losses in its catalytic activity, showing an excellent stability and recyclability. Due to the influences of IL loadings and Co active sites, the high catalytic activity could be attributed to the synergistic effects from the cobalt active sites and active hydroxyl group of the IL. Moreover, CoCl2 salt was impregnated into the cyclohexanediamine-urea derivative-bridged-functionalized PMO to form a Co2+-loaded hybrid organosilica material, which was further pyrolyzed at the desired temperature under N2 protection to afford metallic Co-supported ordered mesoporous silica containing metallic Co and carbonyl oxygen-coordinated Co2+ species.151 Note that carbonyl group contents increased with increasing pyrolysis temperature. The hydrogenation of 4-nitrophenol to 4-aminophenol was chosen as a model reaction to evaluate the catalytic activity of such Co-integrated mesoporous silica with the carbonyl group. The results showed a high catalytic activity with a high TOF, high stability and high reusability, compared to those of Co-loaded pure siliceous materials. In addition, catalytic activity increased with increasing carbonyl group content, clearly confirming that the oxygen-coordinated cobalt played a crucial role in enhancing catalytic performance in the hydrogenation of 4-nitrophenol.As the above-mentioned Ru complex-immobilized chiral TsDPEN-functionalized PMO (TsDPEN = 4-methylphenylsulfonyl-1,2-diphenylethylenediamine),135 [Cp*RhCl2]2 was also anchored to a similar support with different organo-bridge groups (ethylene –CH2CH2–, ethyne –CHCH– and phenylene –C6H4–) to afford Cp*RhTsDPEN-PMO.152 Owing to the superior hydrophobicity, Cp*RhTsDPEN-PMO was used as a catalyst in the ATH of aromatic ketones in aqueous solution to the corresponding chiral alcohols. By comparing their catalytic results, the ethylene-bridged chiral Cp*RhTsDPEN-PMO was found to be the best chiral promoter among the three PMOs, thereby showing an excellent catalytic activity in conversion and comparable enantioselectivity to its homogeneous molecular precursor, and meanwhile exhibited an excellent recoverability and reusability in the ATH of acetophenone, which was confirmed by the retained 99.9% conversion and 92.2% ee value after at least 12 recycles (1st run with 95.0% ee). Similarly, the co-condensation of chiral molecular functional organosilane, ion liquid imidazolium-bridged organosilane and BTME with a SDA under acidic conditions formed Imidazolium@ArDPEN@PMO after removal of the SDA (Fig. 23).153 [Cp*RhCl2]2 was then coordinated with the chiral functional group in Imidazolium@ArDPEN@PMO to afford Imidazolium@Cp*RhArDPEN@PMO. This chiral catalyst was used in the ATH of aryl-substituted α-haloketones or benzils with sodium formate in water to the corresponding chiral alcohols (Fig. 23). The results exhibited catalytic activity with good to excellent yields (83–96% and enantioselectivities (93–99% ee), as well as a good recoverability and reusability in the transfer hydrogenation of 2-bromo-phenylethanone for at least 8 recycles (from 99% to 85% yields and 99% to 91% ee during first run to 9 runs).153
 | ||
| Fig. 23 Preparation of Imidazolium@Cp*RhArDPEN@PMO and asymmetric transfer hydrogenation of aryl-substituted α-haloketones.153 Reproduced from ref. 153 with permission from the Royal Society of Chemistry. | ||
As shown in Fig. 24, [Cp*RhCl2]2 was immobilized on BPy-PMO to generate Rh@BPy-PMO with Rh active species [Cp*Rh(bpy)Cl]Cl.154 Such a single-site solid heterogeneous catalyst was used in the transfer hydrogenation of unsaturated nitrogen heterocycles to the corresponding products. The case of transfer hydrogenation of 3-methylquinoxalinone was used to optimize catalytic conditions (Fig. 24). Under the optimized conditions, the transfer hydrogenation of all kinds of unsaturated heterocycles to desirable products was performed. The results gave catalytic activity with good to excellent yields (70–99%). In addition, to confirm Rh species as active sites, a hot reactant suspension was filtrated to remove the solid catalyst and obtain a clear filtrate, and then the substrate was added into the filtrate to further perform a catalytic reaction for a desirable time under identical conditions. The analytic results showed that no noticeable increase in the product yield was detected, clearly corroborating the role of the immobilized Rh complex species on the pore surface of Rh@BPy-PMOT. In addition, for such a solid molecule-like heterogeneous catalyst, a low degree of leaching of the Rh species was detected, reflecting the decline in the yield from 99% in the first run to 71% in the 5th.154 Furthermore, when the Rh@BPy-PMO catalyst was used in the transfer hydrogenation of 2-cyclohexen-1-one to cyclohexanone, a good catalytic activity was obtained, but the reaction rate was lower than that of the molecular catalyst [RhCp*(bpy)Cl]Cl.155 However, when such a heterogeneous catalytic reaction was performed in the presence of bovine serum albumin (BSA), the catalytic activity of Rh-PBy-PMO still remained the same as that in the case without BSA. Oppositely, the [RhCp*(bpy)Cl]Cl-catalyzed reaction of 2-cyclohexen-1-one to form cyclohexanone in the presence of BSA drastically declined due to the quick deactivation of the molecular catalyst. Hence, the preservation of the catalytic activity of Rh-BPy-PMO was attributed to the pore size-sieving effect (the Rh catalyst in the pore channel was protected by the pore structure, which prevented it from directly interacting with proteins), due to the fact that the size (14 × 4 × 4 nm, 66 kDa) of the BSA is bigger than the pore size (3.7 nm) of the catalyst. When small proteins (myoglobin with a molecular size of 2 × 3 × 4 nm, an amino acid histidine) were used, the heterogeneous and homogeneous reactions of 2-cyclohexen-1-one to cyclohexanone were partly or completely inhibited, due to the fact that the access of proteins into pore channels made Rh active species deactivate. Hence, Rh-immobilized BPy-PMO could be applied to a metal complex-catalyzed and enzyme-presented reaction system; for example, Rh-immobilized BPy-PMO combined with an enzyme (horse liver alcohol dehydrogenase; HLADH) promoted sequential reactions involving the transfer hydrogenation of NAD+ to NADH followed by the asymmetric hydrogenation of 4-phenyl-2-butanone to (S)-4-phenyl-2-butanol using DADH.155 This reaction gave high conversion and enantioselectivity, thanks to the compatibility of the two materials. In addition, the Rh complex could be directly integrated into the PMO framework by a co-condensation approach to produce Rh-PPh2-PMO with the RhCl(PPh2CH2CH2)3 unit.139 This catalyst Rh-PPh2-PMO catalyzed a Heck-type reaction between phenylboronic acid and butyl acrylate in water and exhibited a considerable catalytic activity with a yield of 84%, comparable to the homogeneous catalyst Rh(PPh3)3Cl, but higher than that of the grafted Rh-PPh2-PMO (71%) by direct grafting of Rh(PPh3)3Cl onto PPh2-PMO.
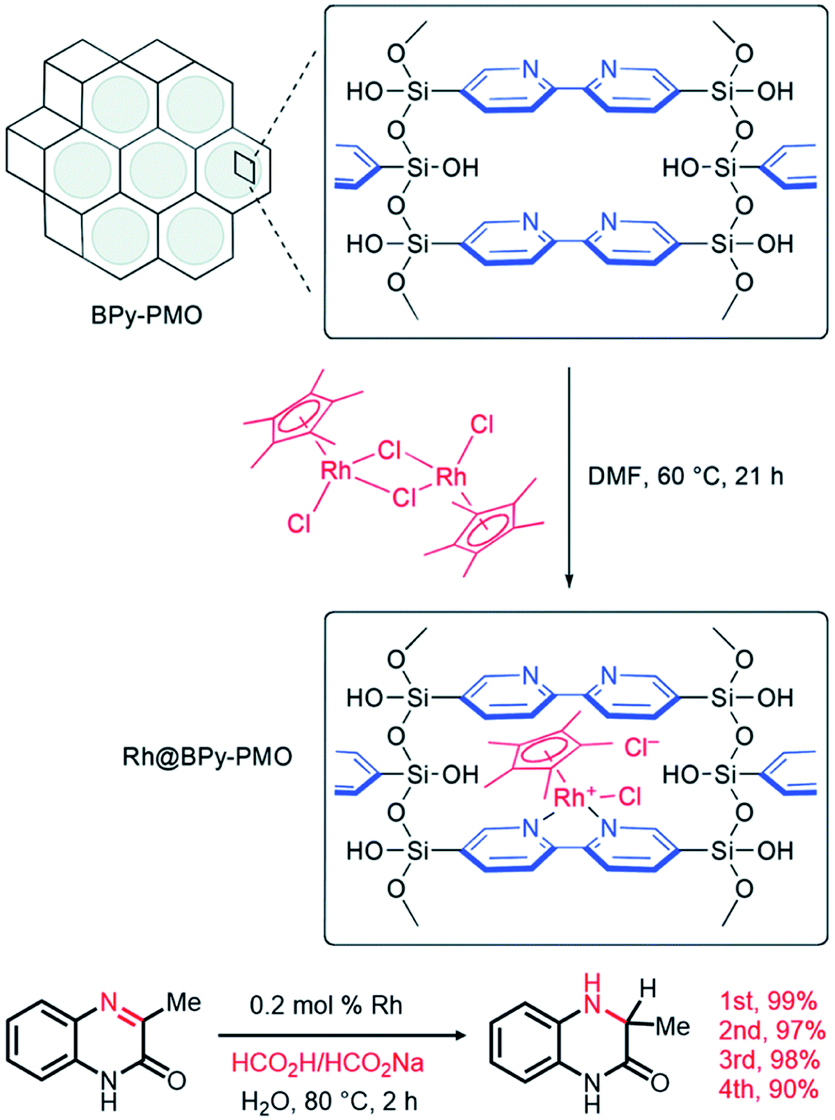 | ||
| Fig. 24 Immobilization of [Cp*RhCl2]2 on the pore surface of BPy-PMO and transfer hydrogenation of 3-methylquinoxalinone.154 Reproduced from Ref. 154 with permission from the Royal Society of Chemistry. | ||
In addition, Rh/Pd@PMO-Ph bifunctional catalysts with an adjustable molar ratio of Rh to Pd were successfully synthesized by surfactant-directing co-condensation of BTEB and organometal silanes RhCl[PPh2(CH2)2Si(EtO)3]3 and PdCl2[PPh2(CH2)2Si(EtO)3]2, where PMO-Ph refers to the phenylene (Ph)-bridged PMO.156 Such a heterobimetal catalyst could be used in one-pot cascade reactions including Rh(I)-catalyzed cinnamaldehyde methylenation and Pd(II)-catalyzed Heck reactions (Scheme 11). The results showed that Rh/Pd@PMO-Ph gave a high catalytic activity with 91% conversion and 71% selectivity for 1,4-diphenyl-1,3-butadiene, which was comparable with those of homogeneous molecular catalysts, RhCl(PPh3)3 and PdCl2(PPh3)2, but the heterogeneous catalyst Rh/Pd@PMO-Ph could be easily recycled and reused. As a result, Rh/Pd@PMO-Ph could greatly reduce the cost and diminish the environmental pollution from heavy metal ions.
 | ||
| Scheme 11 Rh/Pd@PMO-Ph-cocatalyzed one-pot cascade reaction of cinnamaldehyde, trimethylsilyldiazomethane, PPh3, NEt3 and iodobenzene to 1,4-diphenyl-1,3-butadiene at 60 °C in a mixture of isopropanol and tetrahydrofuran. | ||
As above-mentioned Re-BPy-PMO and Re-BPy-PMO-NT, molecular complexes, Ru(bpy)2Cl2·2H2O, [Ir(ppy)2Cl]2 and [Ir(OMe)(cod)]2, have been successfully immobilized on BPy-PMO to form heterogeneous catalysts, Ru(bpy)2(BPy-PMO) (Ru-BPy-PMO), Ir(ppy)2(BPy-PMO) and Ir(OMe)(cod)(BPy-PMO) (Ir-BPy-PMo), respectively.98 As a heterogeneous catalyst, Ir-BPy-PMO showed the highest catalytic performance (94% yield) in C–H borylation of benzene with B2pin2 compared to those (80%, 33%, 63%, and 0) of the homogeneous molecular catalyst Ir(OMe)(cod)(bpy) and heterogeneous catalysts [Ir(OMe)(cod)]2@silica gel, [Ir(OMe)(cod)]2@mesoporous silica and [Ir(OMe)-(cod)]2@polystyrene, respectively, due to the fact that the isolates binding of metals on the well-defined surface in Ir-BPy-PMO suppressed the aggregation and undesired interactions of the metal center. For various types of substrates (arenes), Ir-BPy-PMO catalyzed the C–H borylation of arenes and showed different catalytic activities, due to the steric hindrance of the substituents or pore limitation effect of the catalyst itself. Similarly, Ir-BPy-PMO can also effectively catalyze the C–H borylation of arenes and heteroarenes with HBpin or B2pin2 and multiple borylation of heteroarenes with HBpin. The results gave a high activity with up to 99% yield.157 When dimeric [IrCp*Cl(μ-Cl)]2 (Cp* = η5-pentamethylcyclopentadienyl) reacted with BPy-PMO, a recyclable single-site solid catalyst [IrCp*Cl(BPy-PMO)]Cl was prepared and used as a heterogeneous catalyst for water oxidation.158 By changing the initial content of the Ir complex, a series of heterogeneous catalysts [IrCp*Cl(BPy-PMO)]Cl (Irx-BPy-PMO, x = Ir/bpy molar ratio of Ir/bpy, x = 0.03, 0.07, 0.16) were obtained with various Ir contents (0.10, 0.22, and 0.51 mmol g−1). [IrCp*Cl(BPy-PMO)]Cl-catalyzed water oxidation was performed with cerium(IV) ammonium nitrate (CAN) as an oxidant at room temperature. Based on the amounts of O2 produced per unit of iridium in the first 15 min, the initial turnover frequency values (TOFs) were 2.8, 2.5, and 2.1 min−1 for Irx-BPy-PMOs with x = 0.03, 0.07, and 0.16, respectively,158 showing that the initial TOF for Ir0.03-BPy-PMO was comparable to 3.0 min−1 of the homogeneous catalyst [IrCp*Cl(bpy)]Cl, and was one order of magnitude higher than those of conventional heterogeneous iridium catalysts. The final O2 yield reached 85%. With increasing Ir contents in the mesoporous channel of BPy-PMO, TOFs decreased, implying that water oxidation mainly occurred in mesoporous channels and the limited diffusion had a dominant influence when active sites were located in mesoporous channels. In comparison to other support mesoporous silicas with the same or similar iridium loading, Irx-BPy-PMO exhibited a superior advantage, further corroborating that the uniform distribution of bpy in BPy-PMO ensured the stable existence of single-site metal centers and effectively suppressed the aggregation of active species and the occurrence of undesirable interactions between the iridium active center and support. These results directly verified that BPy-PMO was a unique solid support for the efficient preparation of heterogeneous catalysts Irx-BPy-PMO applied to iridium-catalyzed water oxidation. Additionally, Irx-BPy-PMO also indicated a high stability and reusability compared to homogeneous catalysts. Similarly, [IrCp*Cl(μ-Cl)]2 was immobilized on bpy-/phenylene-bridged PMO nanotubes (BPy-PMO-NT) to afford Ir-BPy-PMO. This catalyst can also be used in water oxidation.159 The results showed that the enhanced durability and high reaction activity were attributed to the stable nanotube structures and the isolated metal active sites as well as the unique one-dimensional nanotubes with large pore diameters. In fact, IrOx-loaded and [Ru(bpy)3]2+-attached acridone-bridged PMO used as a photocatalyst has been explored for water oxidation in the presence of Na2S2O8 in 2014.160
In 2014, Copéret and co-workers used a mixture of bis(triethoxy-silyl)-2,2′-bipyridine (1.0 equiv.) and 5,5′-4,4′-bis(tri-ethoxysilyl)-1,1′-biphenyl (10 equiv.) as the organosilica precursor in the presence of the SDA trimethylstearylammonium chloride under basic conditions to successfully prepare bipyridylene-/biphenylene-bridged PMO (BPy0.1-BP-PMO) after removal of the template. After BPy0.1-BP-PMO was treated with chlorotrimethylsilane in a solvent mixture of N,N-diisopropylethylamine and tetrahydrofuran to afford TMS-capped BPy0.1-BP-PMO(TMS-BPy0.1-BP-PMO), dimeric [{IrCl(COD)}2] was immobilized on TMS-BPy0.1-BP-PMO to generate Ir-BPy0.1-BP-PMO.161 This catalyst was applied to C–H activation and borylation. The results exhibited that Ir-BPy0.1-BP-PMO catalyzed the direct C–H borylation of arenes to the corresponding boronic esters with various conversions and yields, which depended upon the types of substrates. In addition, Ir-BPy0.1-BP-PMO also displayed a high reusability without significant loss of activity.
If the bridged organosilane BTEE replaced BTEB during BPy-PMO synthesis, a mixed ethylene-/bipyridylene-bridged PMO (Et-BPy-PMO) was obtained and showed a 2D hexagonal mesoporous structure with an amorphous pore wall.162 An Ir complex was immobilized on Et-BPy-PMO or BPy-PMO to form Ir-Et-BPy-PMO or Ir-BPy-PMO. Such solid molecular materials could be applied to the disproportionation of formic acid to methanol. The results showed a high selectivity for methanol and high reusability compared to homogeneous Ir-BPy complexes having a similarly effective structure to Ir-Et-BPy-PMO, due to the unique pore confinement effect and high aspect ratio for the retention of generated H2/CO2.162 A detailed reaction mechanism of disproportionation of formic acid catalyzed by Ir-BPy-PMO was elucidated based on kinetics analysis.163
Recently, a new dihydroxybipyridine(BPyOH)-/BP-bridged PMO (BPyOH-BP-PMO) was prepared (Fig. 25). Complexes [Cp*Ir(H2O)3]X [X = 2OTf, SO4] reacted with BPyOH-BP-PMO in water to respectively afford Ir@BPyOH-BP-PMO-OTf and Ir@BPyOH-BP-PMO-SO4.164 Ir@BPyOH-BP-PMO-X can effectively catalyze the dehydrogenative oxidation of 1-phenylethanol to acetophenone in different solvents. The results exhibited that the catalytic activities of Ir@BPyOH-BP-PMO-OTf and Ir@BPyOH-BP-PMO-SO4 (in toluene) were much superior to that of the homogeneous molecular catalyst Cp*Ir(BPyOH)(H2O)(OTf)2, due to the hydrophobicity of the BPyOH-BP-PMO framework and cooperativity between the iridium atom and the BPyOH-BP-PMO support. The cooperativity was confirmed by almost no catalytic activity of Ir@BPy-BP-PMO-SO4 for the dehydrogenative oxidation of 1-phenylethanol to acetophenone. In addition, Ir@BPyOH-BP-PMO-OTf-catalyzed dehydrogenative oxidation could also be extended to primary and second alcohols to the corresponding aldehyde and ketones in good to excellent yields. Note that the solid catalyst Ir@BPyOH-BP-PMO-OTf indicated an excellent reusability for at least 10 cycles without any significant decrease in the product yield. Very recently, [Cp*Ir(H2O)3](OTf)2 was immobilized on more complicate organo-bridged PMO to form a heterogeneous catalyst, which was applied to methanol steam reforming.165 Apart from the aforementioned, an Ir complex-anchored diphenylethylene–diamine-functionalized PMO could also be used for enantioselective reduction of α-cyano and α-nitroacetophenones.166,167
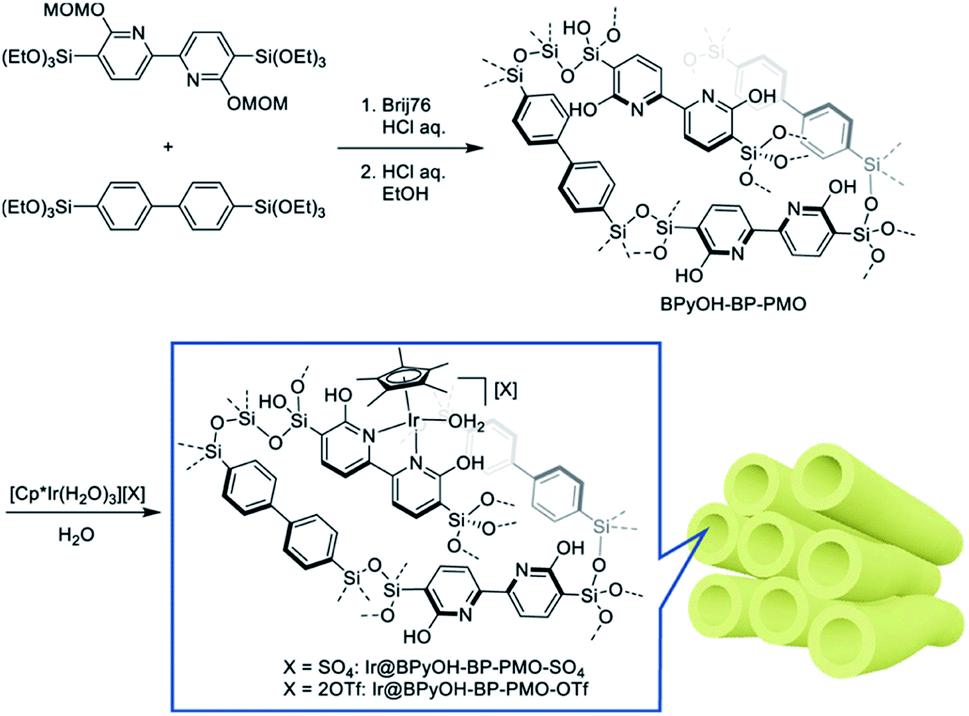 | ||
| Fig. 25 Synthesis of Ir@BPyOH-BP-PMO-SO4 and Ir@BPyOH-BP-PMO-OTf.164 Reproduced with permission.164 Copyright 2020, American Chemical Society. | ||
In 2017, phenylene/bipyridylene-based mesoporous organosilica nanotubes (BPyx-NT, x represents the molar ratio of phenylene-bridged and bipyridylene-bridged organosilica precursors) were synthesized by a sol–gel method and the bipyridylene unit was hung at the external surface of the pore channel/wall (Fig. 26).168 Such a functional group could coordinate with metal atoms to fabricate molecular heterogeneous catalysts. When [IrCp*Cl(μ-Cl)]2 was directly immobilized on BPyx-NT to form a heterogeneous catalyst IrCp*-BPyx-NT. In this case, the formed surface Ir species [IrCp*Cl(bpy)]+ on BPyx-NT were confined inside the pore channel or located on the external surface of the pore wall (Fig. 27a). The pore confinement effect is very conducive to the stability of [IrCp*Cl(bpy)]+ species. [Ir(cod)(OMe)]2 (cod = 1,5-cyclooctadiene) was immobilized on BPy0.3-NT to afford Ir(cod)-BPy0.3-NT with Ir(cod)(OMe) surface species (Fig. 27b). Both catalysts IrCp*-BPyx-NT and IrCp*-BPyx-NT showed high catalytic activities in C–H bond activation, such as IrCp*-BPyx-NT-catalyzed C–H oxidation of different substrates (tetrahydrofuran, cyclohexane, ethylbenzene etc.) and Ir(cod)-BPy0.3-NT-catalyzed C–H borylation of arenes. Catalytic performance strongly depended upon the length of tubes and substrate molecules. IrCp*-BPyx-NT with shorter tubes was beneficial to the diffusion and transport of the reactant and products and thereby showed high catalytic activity. Note that the catalytic activity, stability and reusability of both heterogeneous catalysts IrCp*-BPyx-NT and Ir(cod)-BPy0.3-NT were superior to those of the corresponding homogeneous catalysts and [IrCp*Cl(μ-Cl)]2– or [Ir(cod)(OMe)]2-immobilized bipyridine-grafted phenylene-bridged NT catalysts, revealing the pore confinement effect and the importance of uniform distribution of active surface species. Moreover, a molecular iridium complex [Cp*Ir(H2O)3]SO4 was immobilized on BPy-NT to form a heterogeneous catalyst Cp*Ir-BPy-NT. Such a catalyst can be applied to the hydrogenation of formic acid to methanol.169 The addition of a strong acid (such as triflic acid) at low temperature was conducive to methanol selectivity in a heterogeneous catalytic system, while weakened the conversion of formic acid. Note that selectivity for methanol and conversion of formic acid markedly depended upon acidity, reaction time, H2 pressure and temperature in the reaction system. Under suitable conditions with good HCOOH conversion and methanol selectivity, such a heterogeneous catalyst showed an excellent reusability over four consecutive cycles without any significant loss in activity and still maintained its heterogeneous nature in an extremely high acidic environment.169
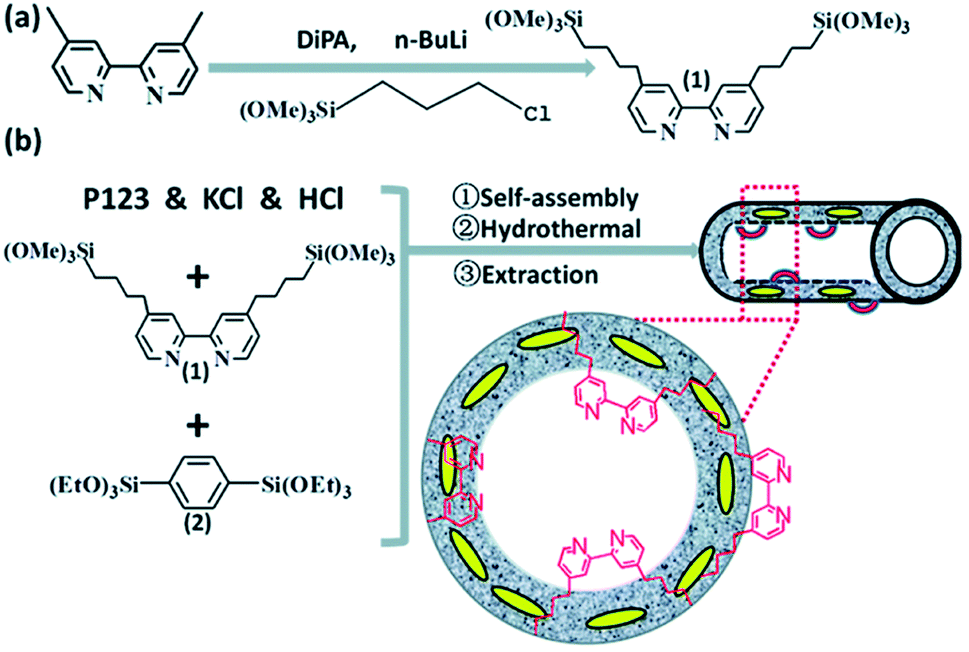 | ||
| Fig. 26 Synthetic routes for the bipyridylene-based organosilica nanotubes (BPy-NT).168 Reproduced from ref. 168 with permission from the Royal Society of Chemistry. | ||
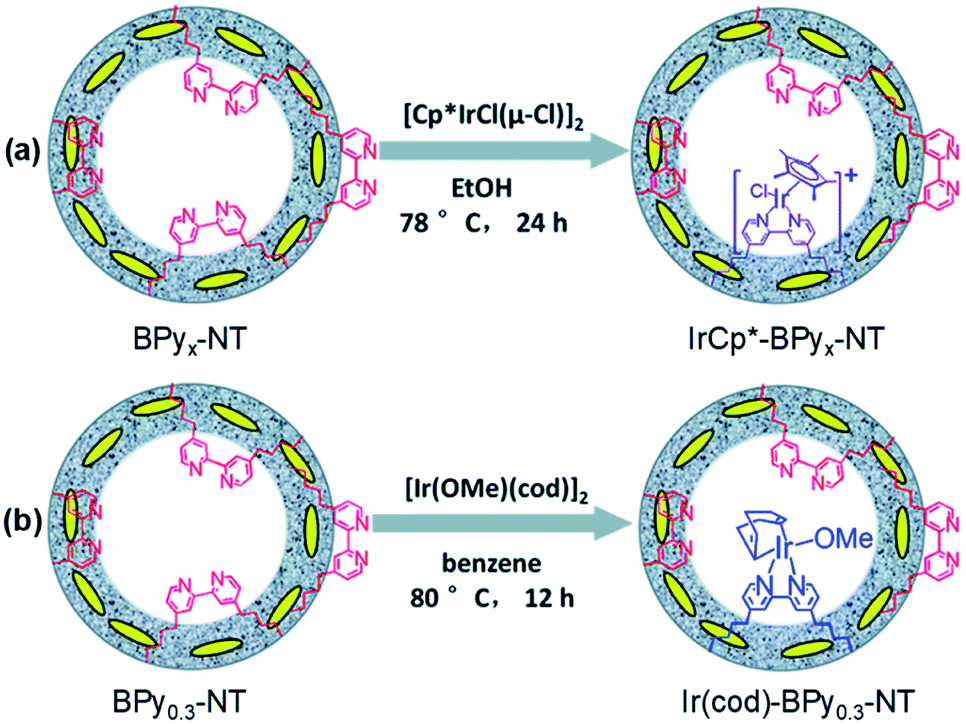 | ||
| Fig. 27 Synthetic route for the molecular heterogeneous solid catalysts (a) IrCp*-BPyx-NT and (b) Ir(cod)-BPy0.3-NT.168 Reproduced from ref. 168 with permission from the Royal Society of Chemistry. | ||
Similarly, a molecular complex [IrCp*Cl(μ-Cl)]2 was anchored on bipyridylene-/ethylene-bridged organosilica nanotubes (Bpy-ENT) to generate an Ir-BPy-ENT heterogeneous catalyst, which was first used as a photocatalyst in hydrogen evolution from formate and aldehyde, showing an enhanced activity and stability compared to homogeneous catalysts.170
2.7. Groups 10 (Ni, Pd, Pt)-based PMO nanoreactors and catalytic applications
 | ||
| Fig. 28 Preparation of the DACH-functionalized PMO and heterogeneous Ni catalyst DACH-Ni-PMO.174 Reproduced with permission.174 Copyright 2012, John Wiley & Sons. | ||
 | ||
| Scheme 12 DACH-Ni-PMO-catalyzed asymmetric Michael addition of malonates to nitroalkenes, where R′ = R′′ = Et and Ar = Ph.174 Reproduced with permission.174 Copyright 2012, John Wiley & Sons. | ||
An alkyl-imidazolium ionic liquid-bridged PMO (PMO-IL) has been used as the support to load metal species, and the role of the IL itself wasn't addressed except for the benzotriazolium ionic liquid containing the substitute group n-butanol.150 NiCl2 was impregnated into PMO-IL to afford a Ni@IL-PMO nanocatalyst with IL+ cation and NiCl3− anion coupling units (Fig. 29).175 A Ni@IL-PMO-catalyzed four-component Hantzsch reaction of ethyl/methylacetoacetate, dimedone, aldehyde and ammonium acetate was performed at 70 °C for the desired time under solvent-free conditions. Under the optimized reaction conditions, polyhydroquinolines were obtained in high to excellent yields and selectivities in the presence of a low loading of Ni@IL-OMO with short reaction times, while Ni@IL-PMO still retained efficient catalytic activity after the 9th run.
 | ||
| Fig. 29 Preparation of Ni@IL-PMO and Hantzsch reaction of ethyl/methylacetoacetate, dimedone, aldehyde and ammonium acetate.175 Reproduced from ref. 175 with permission from the Royal Society of Chemistry. | ||
In addition, as the aforementioned Co-PMO-IL(x),150 NiCl2(DME) (DME = 1,2-dimethoxyethane) reacted with PMO-IL to form NiCl3(DME)− anionic benzotriazolium ionic liquid-functionalized periodic mesoporous organosilicas PMO@ILC4H10O2NiCl3 with various IL contents.176 The catalytic activity was evaluated by the cycloaddition of CO2 with epoxides (Scheme 13). The results showed that catalysts PMO@ILC4H10O2NiCl3 with various IL contents were effective and practical heterogeneous catalysts in the synthesis of cyclic carbonates with high yields (89.5–97.6%) and selectivities (>99%) under solvent- and cocatalyst-free conditions. The hydroxyl group of the IL and silanol groups of PMO significantly influenced the catalytic activity, demonstrating an intensification of the intramolecular synergistic effect. The catalyst also showed a good recoverability and reusability for at least five times without significant loss of the catalytic activity.
 | ||
| Scheme 13 PMO@ILC4H10O2NiCl3-catalyzed cycloaddition of CO2 with epoxides to cyclic carbonates. | ||
In fact, in 2009, a Ni(II) α-diimine complex has been successfully tethered to the pore wall of PMO by one-pot synthesis to get a spherical PMO-supported Ni(II) α-diimine catalyst for ethylene polymerization.177 Moreover, a Ni NP-loaded TiO2-integrated ethylene-bridged PMO catalyst (Ni/TiO2-PMO-Et) was prepared by the impregnation of nickel nitrate into TiO2-PMO-Et, and then by H2 reduction.178 The average size of Ni NPs was about 15 nm in Ni/TiO2-PMO-Et. The hydrogenation of nitrobenzene with H2 was selected to test the catalytic activity of Ni/TiO2-PMO-Et. The results showed that Ni/TiO2-PMO-Et had a high activity in the hydrogenation of nitrobenzene in water; meanwhile, it was a highly recoverable and recyclable catalyst for several cycles with a constant activity and selectivity, due to the fact that the high hydrophobicity of the pore wall composed of ethylene-bridged units greatly protected Ni nanoparticles inside the pore channel from deactivation contacted with water.
2.7.2.1. Pd-catalyzed Suzuki and Suzuki–Miyaura coupling reactions. Due to the functional difference of organo-bridge groups in PMO, Pd salt will interact with these functional groups by forming a Pd(II) coordination complex or random physical adsorption. After being reduced with a reductant, the formed Pd NPs will be stabilized by the functional group to preserve the small size or randomly deposit and aggregate together to form large particles. As a result, their catalytic activity will be strongly influenced. In general, alkyl- and phenylene-bridged groups in the PMO framework did not show any coordination ability. In this case, metal ions are randomly adsorbed on the surface. Only the organo-bridge group containing N, O, and S heteroatoms displays a strong coordination advantage with the metal center. All the above-mentioned organo-bridge groups in PMO frameworks, such as bipyridine, benzotriazolium, imidazolium, thiourea, urea, pyridine Schiff base and so on, can coordinate with metal ions or ensure that metal NPs become more stable in the pore channel. In 2008, Jin and co-workers grafted Pd(OAc)2 on a silica-supported imidazolium-based ionic liquid material (SiO2-IL) to form active N-heterocyclic carbene-Pd species, which could be used in the Suzuki–Miyaura coupling reaction, showing a high catalytic performance.180 Similarly, Pd(OAc)2 was immobilized on an imidazolium-based PMO (PMO-IL) to fabricate a Pd(II)@PMO-IL catalyst with active carbene-Pd species (Fig. 30).181 This catalyst promoted the Suzuki–Miyaura coupling of aryl halides with arylboronic acids in water and showed catalytic activity with moderate to excellent yields (55 ∼ >99%). The soluble Pd species were reserved in the PMO-IL nanostructure. During the catalytic C–C coupling reaction, the nanochannels of PMO-IL as a nanoscaffold recaptured the Pd NPs into the nanoreservoir, thus preventing extensive agglomeration of Pd NPs. As a result, Pd(II)@PMO-IL showed a high catalytic activity, high stability and recyclability.
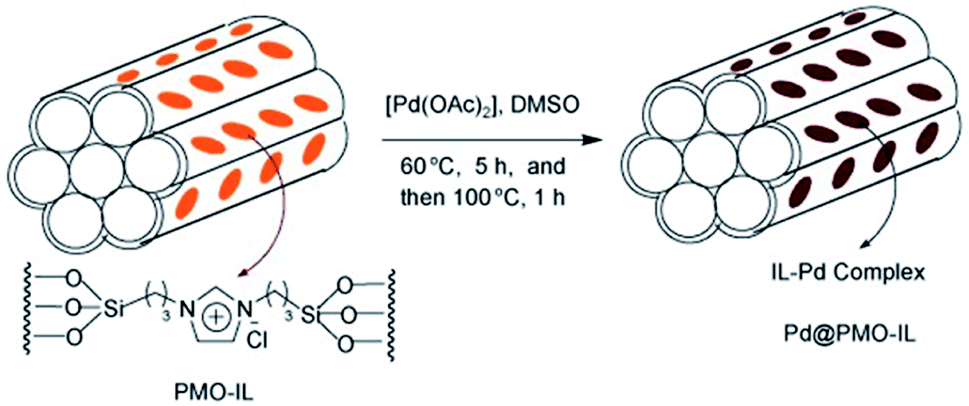 | ||
| Fig. 30 The preparation of the Pd@PMO-IL catalyst.181 Reproduced with permission.181 Copyright 2010, John Wiley & Sons. | ||
Na2PdCl4 reacted with 4,4′-bipyridinium-bridged PMO (Bipy-PMO) in MeOH to form Pd(II)@Bipy-PMO with the (Bipy)2+-PdCl42− cation–anion coupling unit in the PMO framework.182 This solid ion-type heterogeneous catalyst exhibited an efficient catalytic activity (98% yield) in a model C–C cross-coupling reaction of 4-bromoaceto-phenone with phenylboronic acid in water. When various aryl halides and arylboronic acids were used, the catalytic performance strongly depended upon the various substitute groups of both substrates.
Moreover, as the above-mentioned contents, many functional organo-bridged PMOs have been used as supports for immobilizing metal complexes to fabricate catalysts. A carbapalladacycle complex-bridged PMO (PdL∝PMO) in 2005 has been prepared and used as a solid molecular catalyst in the Suzuki–Miyaura cross-coupling reaction of p-bromoacetophenone and phenylboronic acid in the presence of K2CO3.183 The catalytic activity altered with the differences of substitute groups of both substrates.
The complex Na2PdCl4 was immobilized on thiourea-bridged PMO (TU-PMO), followed by reduction to form PdNP@TU-PMO (Fig. 31),184 in which Pd NPs with an average size of 1.8 nm were uniformly distributed inside the pore wall. Such small-sized Pd NPs on TU-PMO catalyzed the Suzuki coupling reaction of aryl halides and arylboronic acids in the presence of K2CO3 and showed an excellent catalytic activity with up to 100% yield and high reusability over several consecutive runs with minimal loss of catalytic activity.
 | ||
| Fig. 31 General pathway for the synthesis of Pd@TU-PMO and Heck reaction of aryl halides and arylboronic acids in the presence of K2CO3.184 Reproduced from ref. 184 with permission from the Royal Society of Chemistry. | ||
Pd(OAc)2 was coordinated with a functional group from melamine (M, 1,3,5-triazine)-functionalized PMO (M-PMO) prepared by the self-assembly co-condensation of N2,N4,N6-tris(3-(triethoxy-silyl)propyl)-1,3,5-triazine-2,4,6-triamine (TTET) with TEOS, to form a Pd-coordination complex-integrated melamine-functionalized PMO (Pd-M-PMO) (Fig. 32), which was used in Suzuki-coupling reactions of bromobenzene and aryl boronic acids in water.185
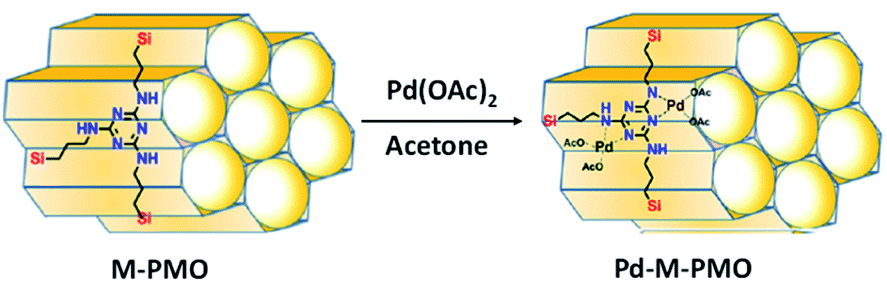 | ||
| Fig. 32 The synthesis of the catalyst Pd-M-PMO for the Suzuki–Miyaura coupling reaction.185 Reproduced with permission.185 Copyright 2019, Elsevier. | ||
2.7.2.2. Pd-catalyzed Heck coupling reactions. Palladium acetate was grafted on bis(propyliminomethyl)phloroglucinol-bridged PMO (LHMS-3) to form Pd-LHMS-3 (Fig. 33) with a proposed Schiff based-chelated dimeric Pd2(μ-O2CCH3)2 unit,186,187 which meant that every Pd(II) atom was chelated with the N atom of the imine, and the O atom of OH of the phenolic unit from the pore wall of LHMS-3 to form a stable six-membered rings,186 while every Pd was coordinated to two O atoms from two acetates, ensuring intact incorporation of the Pd unit into PMO frameworks with uniform distribution. Pd-LHMS-3 catalyzed the Heck reactions of aryl halides with aromatic (styrene) or aliphatic olefins (acrylic acid) and showed a good catalytic activity with yields from 75 to 85% (Fig. 33). Catalyst Pd-LHMS-3 was recycled four times in the Heck reaction of iodobenzene and styrene, and remained 82.1% yield, verifying good reusability.186
 | ||
| Fig. 33 The preparation of the heterogeneous catalyst Pd-LHMS-3 and the Heck C–C bond formation reaction catalyzed by Pd-LHMS-3.186,187 Reproduced from refs 186 and 187 with permission from the Royal Society of Chemistry. | ||
Despite the high activity and stability of Pd-carbene in the imidazolium-based PMO framework, the catalytic activity of Pd@PMO-IL was influenced not only by the Pd loading, reaction temperature, base and solvent, but also by the pore size of the support. In general, a large pore size of the support is conducive to the access and the diffusion of the substrate, while also accompanied by the possibility of leakage of active species without strong interaction between the active species and functional ligand inside the pore channel. Pd@PMO-IL with a large pore (11 nm) catalyzed the Heck coupling reaction of bromobenzene and ethyl acrylate in N-methylpyrolidone under basic conditions. The results showed an excellent activity with >99% yield.188 Meanwhile for various substrates (aryl halides with olefins), under the optimized conditions, the yields ranged from 85% to 96%, mainly depending upon the substitute groups on substrates. Note that Pd@PMO-IL showed an excellent recoverability and reusability for at least 10 runs with >99% yield, but longer reaction times were needed.
2.7.2.3. Pd-catalyzed Hiyama and Sonogashira couplings and cyanation reactions. The above-mentioned Pd-LHMS-3 could also be used in different C–C coupling reactions,187 including (i) the fluoride-free Hiyama coupling reaction of aryl halides with aryl trimethoxysilane in basic conditions (Scheme 14a), Pd-LHMS-3 showed catalytic activity with good to excellent yields (70–95%), (ii) the Sonogashira cross-coupling reactions of aryl halides with terminal alkynes in water or DMF and with hexamine as a base under copper-free conditions, Pd-LHMS-3 exhibited catalytic activity with moderate to good yields (72–90%, Scheme 14b and c), (iii) KCN free cyanation of aryl iodides in good to excellent yields (84–95%, Scheme 14d), and (iv) KCN free cyanation of aryl bromides in moderate to good yields (68–85%, Scheme 14e).
 | ||
| Scheme 14 (a) Hiyama cross couplings of aryl halides with aryl trimethoxysilane promoted by Pd-LHMS-3 in basic aqueous media. (b) Cu-free Sonogashira cross-coupling reactions of aryl-iodides over the Pd-LHMS-3 catalyst in water. (c) Cu-free Sonogashira cross-coupling reaction of aryl bromides over the Pd-LHMS-3 catalyst in DMF. (d) KCN free cyanation of aryl iodides with Pd-LHMS-3. (e) Cyanation of aryl bromides with Pd-LHMS-3.187 Reproduced from ref. 187 with permission from the Royal Society of Chemistry. | ||
Pd@PMO-IL with a Pd-carbene unit could also be used as a catalyst for the Sonogashira cross-coupling reaction of iodobenzene and phenylacetylene in the presence of piperidine as a base in DMF at 70 °C, and the yield reached 94%. When various aryl halides as substrates were used under the optimized conditions, Pd@PMO-IL still exhibited catalytic activity with considerable to excellent yields (54–95%).189 Pd(OAc)2 reacted with BPy-PMO containing 2,2′-BPy bridging groups in the framework to form Pd(OAc)2(BPy-PMO), which was used as an efficient heterogeneous catalyst for the Sonogashira–Hagihara coupling reaction of aryl iodides and acetylene in the presence of CuI, diisopropylamine and tetrahydrofuran.190 The results indicated that the catalytic activity strongly depended upon the substitute of substrates under the optimized conditions, and the catalyst could be easily separated and reused.
2.7.2.4. Pd-catalyzed oxidation reactions. Palladium complexes, [trans-PdCl2L2] (L = 4-C5H4N-(CH2)2Si(OEt)3 (PETS) and PPh2(CH2)2Si(OEt)3 (PPETS)), were directly integrated into ethylene-bridged PMO via the co-condensation of palladium(II) complexes with BTEE in the presence of a SDA and NH4F to form a Pd complex-incorporated PMO-surfactant composite. After removal of the surfactant, Pd complex-integrated PMOs were obtained with various Pd loadings, which depended upon the utilization of the Pd precursor.191 The obtained pyridine–Pd complex (which stemmed from [trans-PdCl2(PETS)2 precursor])-integrated PMO catalyzed the aerobic oxidation of styrene in acetonitrile and showed a low conversion and high selectivity for benzaldehyde. Without solvent, a high conversion could be detected, but the selectivity for the desired product was very low. Diphenylphosphine-based Pd complex-integrated PMO also showed a low conversion and considerable yields for benzaldehyde and benzoic acid in the aerobic oxidation of styrene. Without solvent, high conversion accompanied by very low selectivity for desired benzaldehyde and benzoic acid was found.191 Selective aerobic oxidation of alcohols to the corresponding carbonyl compounds is a very attractive, important and challenging transformation in both academic research and the chemical industry. Different catalysts can effectively control the development of the reaction stage, especially, for primary alcohols. In 2012, Qiao and co-workers successfully synthesized yolk–shell structured silica-PMO spheres with silica as the core and PMO as the shell (YS-PMO), and subtly made Au, Pt and Pd nanoparticles deposit inside the yolk–shell structure to form M-YS-PMO (M = Au, Pt, Pd) (Fig. 34).192 Such a state-of-the-art nanostructured hybrid composite could be used as a catalyst for selectively controlling the development of the reaction stage. For example, Pd-YS-PMO efficiently catalyzed and controlled the selective aerobic oxidation of primary alcohols to aldehydes, showing catalytic activity with moderate to excellent conversions of 53–100% and an excellent selectivity of >99% for aldehydes.
 | ||
| Fig. 34 TEM images (a, d, and g), HRTEM images (b, e, and h) and HAADF-STEM images (c, f, and i) of Au–YS-PMO (a–c), Pt–YS-PMO (d–f) and Pd–YS-PMO (g–i).192 Reproduced with permission.192 Copyright 2012, John Wiley & Sons. | ||
Pd NPs from the reduction of Pd(NO3)2 with NaBH4 were in situ immobilized on imine-bridged PMO with a SBA-15-like structure (PMO-SBA-15) to form Pd/PMO-SBA-15 with a mean size of Pd NPs (inside the pore channel) of 1.8 nm. This catalyst was used in the selective oxidation of benzyl alcohol with H2O2 as the oxidant in water. The results exhibited a high conversion of 97.1% and selectivity of 100% for benzaldehyde, as well as a conveniently recoverable and recyclable feature.193
In 2015, Karimi and co-workers successfully prepared novel heterogeneous catalyst systems, which were comprised of Pd NPs immobilized into the nanospaces of imidazolium-based bifunctional plugged and unplugged periodic mesoporous organosilicas (BFPMO) (Fig. 35).194 In the unplugged Pd catalyst, the corresponding phenylene-/ethylene-/IL-bridged framework regions were a highly hydrophobic microenvironment (Fig. 36a). In the plugged catalyst, plugs mainly consisted of amorphous silica nanocapsules likely originating from the hydrolysis and condensation of TEOS, and contained more silanol groups, and thereby showing strongly hydrophilic nanoregions inside the hydrophobic mesochannels of the BFPMO-IL (Fig. 36b).
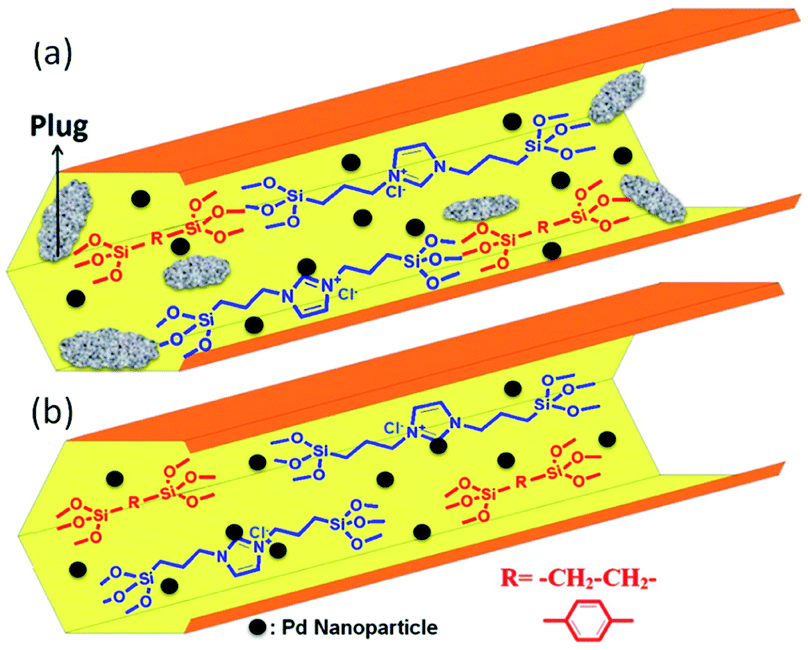 | ||
| Fig. 35 Schematic of plugged (a) and unplugged (b) Pd catalysts.194 Reproduced with permission.194 Copyright 2015, American Chemical Society. | ||
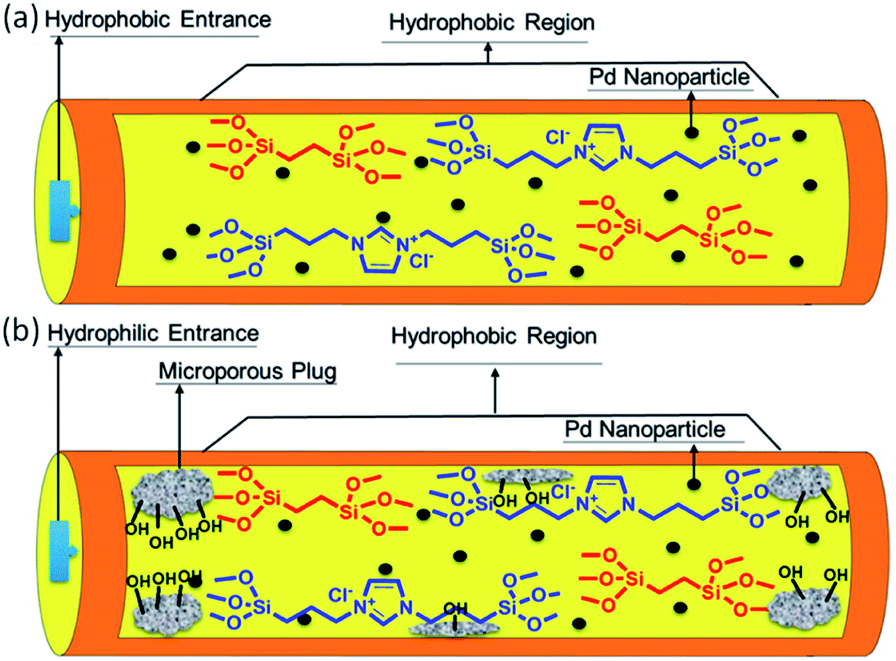 | ||
| Fig. 36 Hydrophilic and hydrophobic regions of IL/ethylene bridge-unplugged (a) and plugged (b) BFPMO-IL catalysts.194 Reproduced with permission.194 Copyright 2015, American Chemical Society. | ||
This kind of adjustable hydrophobic–hydrophilic balance with concomitant control of plugs in the interior of the mesochannels enabled such a type of catalyst to show different catalytic activities and selectivities and therefore enabled the controllable development of different reaction stages. As a result, the unplugged catalysts exhibited much better activity for the selective oxidation of benzyl alcohol to benzaldehyde, while the plugged catalyst showed a high yield and high selectivity for benzoic acid under the exact same reaction conditions (Fig. 37). Both catalysts efficiently controlled the development of oxidation of benzyl alcohol to aldehyde or benzoic acid.
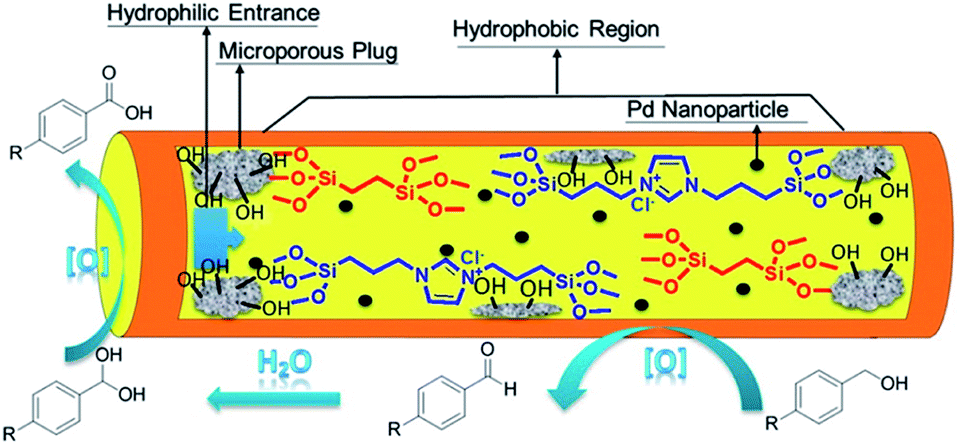 | ||
| Fig. 37 Illustration of selective oxidation of benzyl alcohol catalyzed by the Pd-loaded plugged and unplugged catalysts.194 Reproduced with permission.194 Copyright 2015, American Chemical Society. | ||
According to a reported method,181 Na2PdCl4 was immobilized on PMO-IL to form an efficient and recyclable Pd@PMO-IL heterogeneous catalyst. Such Pd@PMO-IL can effectively apply to the selective aerobic oxidation of a wide variety of different primary and secondary alcohols in α,α,α-trifluorotoluene under normal pressure of oxygen and air atmospheres to the corresponding aldehydes or ketones. The results showed a high catalytic activity with good to excellent yields (65–99%).195 Furthermore, Pd-anchored IL-modified magnetic methylene-bridged PMO (Fe3O4@Me-PMO-IL/Pd) also showed an efficient catalytic activity in the oxidative coupling of various phenols to 2-naphthols under aerobic conditions.196
2.7.2.5. Pd-catalyzed hydrogenation reactions. Pd(OAc)2 was immobilized on ionic liquid imidazolium-bridged PMO (PMO-IL), followed by reduction with NaBH4 to afford Pd NP-loaded PMO-IL (Pd-PMO-IL) with IL contents of 1.0 and 1.7 mmol g−1.64 All Pd NPs were located inside pore channels or well dispersed over the surface of the support. Mesoporous structured Pd-PMO-IL with 1.0 mmol g−1 IL showed an excellent catalytic performance with >99% yield and >99% selectivity in a model hydrogenation reaction of cyclohex-2-enone to cyclohexanone under ambient conditions within 40 min. After the catalyst was separated by filtration or centrifugation, the recoverable catalyst was recycled for at least five runs without any significant loss of activity, verifying an excellent recoverability and reusability. However, microstructured Pd-PMO-IL containing 1.7 mmol g−1 IL gave a similar yield and selectivity, but it must be taken 3 h, clearly demonstrating the very importance of the synergistic effect of the well-defined mesoporous structure and IL sites as a nanoreactor. Pd NPs in the size range of 1.8–2.1 nm deposited by the sputtering technique onto mesoporous silica containing 1-n-butyl-3-(3-trimethoxysilyl-propyl)-imidazolium cations with anions (Cl, NO3, PF6, and NTf2) showed a high selectivity in the selective hydrogenation of dienes and the α, β-unsaturated carbonyl complex to the corresponding monoenes or aldehydes or ketones. The excellent conversion was up to 100% and selectivity was up to 99% for monoenes, aldehydes and ketones, attributed to the intrinsically electron deficient Pd-metallic surfaces and the distinct affinity/diffusion of the diene and monoenes in this IL hybrid organosilica environment.197,198 Pd-PMO-IL could efficiently catalyze the selective hydrogenation of the C
C double bonds adjacent to an aromatic ring or a heteroatom and no byproducts were detected under identical conditions. Na2PdCl4 was loaded on the thiazoline functionalized PMO (TPMO), followed by reduction with NaBH4 to obtain Pd-TPMO (Fig. 38) and Pd NPs inside the pore channel were stabilized by the functional group and pore confinement effect.199 Such a heterogeneous catalyst indicated an efficient catalytic activity in the selective hydrogenation of styrene oxide and styrene & Csp2–Csp2 bond formation reactions for the synthesis of unsymmetrically substituted biaryl ketones and unsymmetrical biaryls as well as the cross-coupling reaction between aryl bromide and phenylboronic acids, while exhibiting high reusability.
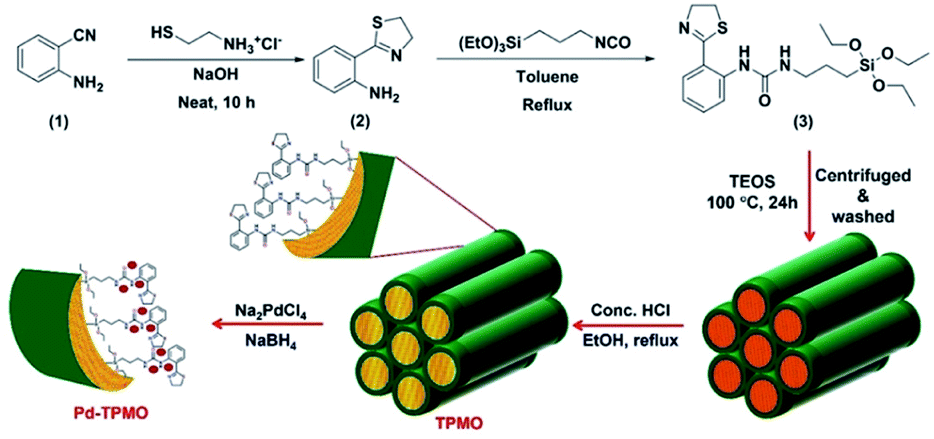 | ||
| Fig. 38 Synthesis of the thiazoline-silyl precursor, thiazoline bridged periodic mesoporous organosilica (TPMO) and Pd NP entrapped TPMO (Pd-TPMO).199 Reproduced with permission.199 Copyright 2012, John Wiley & Sons. | ||
Moreover, except from that the aforementioned the difference of IL functional group from oragnosilica as the support influenced Pd loading, Pd particle size and catalytic activity of catalyst, the inactive organic composite in the support also affected the immobilization of Pd NPs and the resulting catalytic activity. For example, one-pot synthesized Pd-loaded ethylene-bridged hybrid mesoporous silica yolk–shell NPs (Pd@mHSiO2 YSNPs) displayed a markedly higher catalytic activity and hydrothermal stability than Pd-loaded mesoporous silica core–shell NPs (Pd@mSiO2 CSNPs) or Pd-loaded ethylene-bridged hybrid mesoporous silica nanospheres (Pd/mHSiO2 NSs) in the conversion of levulinic acid (LA) into γ-valerolactone (GVL),200 while Pd@mHSiO2 YSNPs also showed highly recoverable and reusable properties without a significant decrease in the catalytic activity for at least 5 runs. The high catalytic activity was attributed to the fact that the hollow cavity greatly decreased mass-transfer resistance and remarkably enhanced the accessibility of the catalytic sites. In addition, the incorporation of organic groups enhanced the hydrophobicity of the pore structure of the support, thereby resulting in a high hydrothermal stability of the outer shell. Pd NPs were immobilized on phenylene-bridged PMO nanotubes (B-PMO-NT) with an inner diameter of ∼7 nm and wall thickness of ∼3 nm to fabricate the Pd@B-PMO-NT catalyst, in which the mean size of Pd NPs located inside pore channels was 1.8 nm. Such a Pd@B-PMO-NT catalyst was used in cyclohexene hydrogenation to cyclohexane. The results showed a 100% conversion.201 However, Pd NPs were immobilized on ethylene-bridged PMO nanotubes (E-PMO-NT) with an inner diameter of ∼6 nm and wall thickness of ∼3 nm to afford Pd@E-PMO-NT, in which the mean size of Pd NPs located inside the pore channel was 4.8 nm. This catalyst exhibited a conversion of 92.3% for the same catalytic reaction. These two results clearly revealed that the well-dispersed small particle size and high hydrophobicity of the catalyst are conducive to the improvement of catalytic activity, such as Pd@B-PMO-NT with high hydrophobicity and small sized Pd NPs compared to that of Pd@E-PMO-NT with relatively low hydrophobicity and large sized Pd NPs. Moreover, both Pd@B-PMO-NT and Pd@E-PMO-NT were used in the catalytic enantioselective hydrogenation of α, β-unsaturated carboxylic acid. The results showed a conversion of >99%, but with different enantioselectivities (48% ee for Pd@B-PMO-NT and 40% ee for Pd@E-PMO-NT).202 Meanwhile, due to one-dimensional nanotube structure and characteristic hydrophobicity, Pd@B-PMO-NT- and Pd@E-PMO-NT-catalyzed oxidation of benzyl alcohol showed a high conversion up to 98% with >85% selectivity for benzaldehyde, which is higher than those of Pd/SBA-15 and Pd/CMK-3 (conversion: 92% for Pd/SBA-15; 55% for Pd/CMK-3; selectivity for benzaldehyde: 70% for Pd/SBA-15; 44% for Pd/CMK-3).202
2.7.2.6. Pd-catalyzed CO2 hydrogenation to formate. CO2 is a main contributor to the greenhouse effect. Transforming CO2 into a storable and sustainable liquid or solid material for energy is still an enormous challenge. At present, CO2 hydrogenation to formic acid as a renewable hydrogen storage compound is considered as an efficient and pivotal reaction to realize hydrogen energy cycles. However, finding and preparing an efficient catalyst to perform this transformation represent a key issue. Pd NPs have been discovered as effective catalysts for this reaction.203 Recent studies manifested that Pd NPs or PdAg NPs immobilized on N-containing mesoporous silica204 or hollow mesoporous carbon spheres205,206 were efficient catalysts for the hydrogenation of CO2 to formate. However, it is interesting to find that hollow mesoporous organosilica spheres (HMOSs) encapsulating PdAg nanoparticles and poly(ethyleneimine) (PEI) as reusable catalysts also showed a high catalytic activity in CO2 hydrogenation to formate.207 Such a state-of-the-art heterogeneous catalyst PdAg + PEI@HMOS was prepared via a multistep process (Fig. 39): (i) adsorption of metal ions into the poly(ethyleneimine) network, (ii) reduction of metal ions by NaBH4, (iii) the formation of a template-directed mesoporous organosilica shell and (iv) ageing and removal of the template agent.
 | ||
| Fig. 39 Illustration of the synthetic procedure for hollow mesoporous organosilica spheres encapsulating PdAg NPs and PEI (PdAg + PEI@HMOS).207 Reproduced with permission.207 Copyright 2020, American Chemical Society. | ||
The structure and morphology as well as elemental distribution of PdAg + PEI@HMOS were well characterized by S/TEM and elemental mapping (Fig. 40), clearly verifying the formation of a hollow mesoporous organosilica structure, the uniform and homogeneous distribution of elements Si, O, N, C, Pd and Ag, and PEI and PdAg NPs located in both shell and hollow cavity regions. PdAg + PEI@HMOS promoted the CO2 hydrogenation to formate and showed a very high formate yield with a turnover number (TON) of over 2754 and high reusability under mild reaction conditions, thanks to the cooperative action of PEI and PdAg NPs confined in a nanospace from direct capture, activation and hydrogenation of the CO2 molecule. In addition, an understandable mechanism of PdAg + PEI@HMOS-catalyzed CO2 hydrogenation to formate was proposed (Fig. 41).
 | ||
| Fig. 40 (a) FE-SEM image, (b and c) TEM images (inset in (c) shows the HAADF-SETM image of an aggregated metal particle), (d) STEM image, (e) HAADF-STEM image, and (f–k) the corresponding STEM elemental maps of (f) Si, (g) O, (h) C, (i) N, (j) Pd, and (k) Ag of PdAg + PEI@HMOS (once used in the CO2 hydrogenation for 22 h). (l) Illustration of PdAg + PEI@HMOS particle and (m) EDX line scanning profile for different elements across the PdAg + PEI@HMOS particle in the direction shown in (e).207 Reproduced with permission.207 Copyright 2020, American Chemical Society. | ||
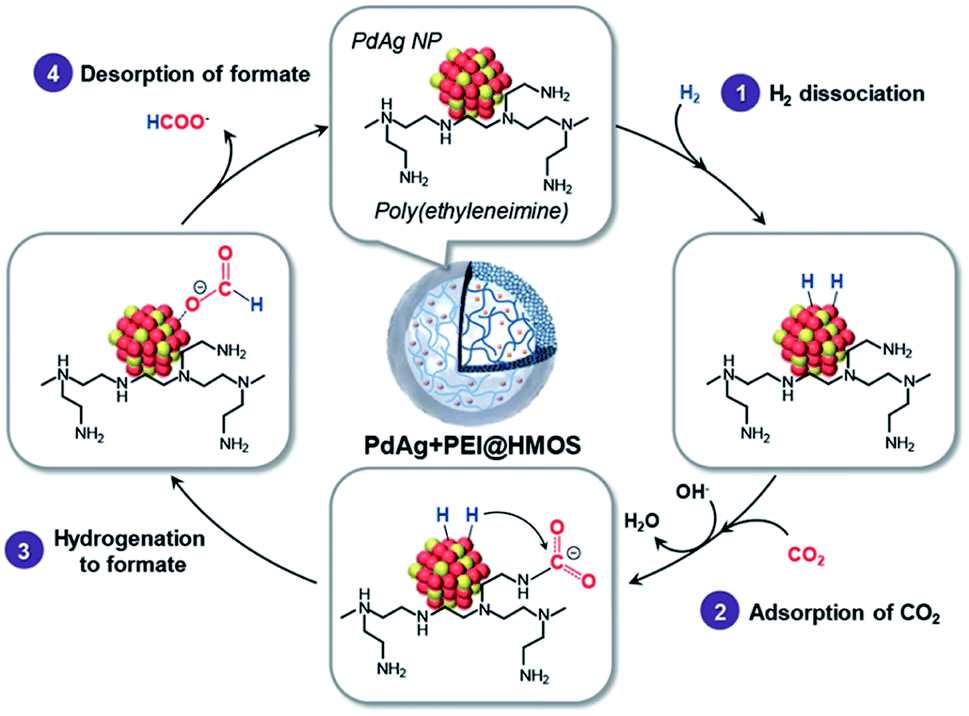 | ||
| Fig. 41 Plausible reaction mechanism for the CO2 hydrogenation to produce formate over the PdAg + PEI@HMOS catalyst.207 Reproduced with permission.207 Copyright 2020, American Chemical Society. | ||
Moreover, Na2PdCl4 immobilized on ketone-Schiff base-bridged N-containing PMO spheres (NPMO) was reduced by NaBH4 to afford Pd@NPMO (Fig. 42).208 Uniform sized NPMO spheres were synthesized by (i) the hydrolysis and self-assembly co-condensation of ketone-Schiff base-based organosilane and TEOS in the presence of CTAB under basic conditions and (ii) removal of CTAB. After Pd particles were loaded, it was clearly observed that Pd NPs with an average size range of 3–5 nm were well dispersed inside the pore channel or on the surface of NPMO spheres. Due to the fact that the role of the N-containing functional group as a stabilizer and pore confinement effect could efficiently stabilize Pd NPs, Pd@NPMO-catalyzed CO2 hydrogenation showed a superior catalytic activity with a TOF of 108 h−1 compared to 9.4 h−1 of Pd-SBA-15, clearly corroborating the vital role of nitrogen sites on the NPMO support in boosting the CO2 reduction with complete selectivity to formate under mild reaction conditions.
 | ||
| Fig. 42 (a) Ketone-Schiff base-based organosilane. (b) TEM image of NPMO. (c) TEM image of Pd@NPMO.208 Reproduced with permission.208 Copyright 2020, American Chemical Society. | ||
2.7.2.7. Pd-catalyzed H2 production. H2 is a very clean energy for the environment and mankind. Pd-based PMO catalysts can be used for H2 generation. The above-mentioned PdNP@TU-PMO with well-dispersed small Pd NPs inside the pore channel was an efficient catalyst in the Suzuki coupling reaction.184 Furthermore, PdNP@TU-PMO could also be applied to H2 generation from the decomposition of formic acid in water. The results showed a superior catalytic activity and revealed that the H2 production rate increased with increasing temperature and metallic Pd NPs were more active than Pd2+ ions.209 The PdNP@TU-PMO catalyst was recyclable while maintaining a good catalytic performance. Furthermore, the Pd NP-immobilized urea-bridged PMO catalyst was also used in the dehydrogenation of formic acid. The results showed that the temperature-dependent effect, catalyst dose effect and solvent effect remarkably influenced the H2 production rate.210 Moreover, it is noted that the formation of Pd NPs on PMO was often reduced by H2, NaBH4 or ammonia borane or a mixture of NaBH4 and ammonia borane. It is interesting to find that the Pd-immobilized carboxylic acid-functionalized PMO could efficiently catalyze hydrogen generation from ammonia borane. The high catalytic activity strongly depended upon the textural properties, intrinsic hydrophobicity of the pore cavity of the PMO and well-dispersed small sized Pd NPs stabilized by the strong electrostatic interactions between the negatively charged carboxylate groups on the support and the positively charged Pd2+.211
2.7.2.8. Pd-catalyzed reduction reactions. Pd-immobilized support catalysts were also used for the reduction reaction. Pd NP-immobilized urea-bridged PMO could be used in the reduction of 4-nitrophenol with NaBH4 to 4-aminophenol. The results indicated a high catalytic activity compared with that of Pd@SAB-15.210 In addition, Pd@PMO-ISO containing the 1,3,5-triazinane-2,4,6-trione-based organo-bridge group was also an efficient catalyst in the reduction of 4-nitrophenol with NaBH4 to 4-aminophenol.212
2.7.2.9. Pd-catalyzed chemoenzymatic asymmetric synthesis of chiral amines and alcohols. The morphology and structure of PMO as a support probably influence the immobilization of metals or metal complexes. Dendritic organosilica nanoparticles (DON) with a large specific surface area and pore volume should be conducive to the subsequent formation of monodisperse, well-separated Pd NPs. Pd NPs by in situ reducing Na2PdCl4 with NaBH4 were successfully immobilized onto the hydrophobic pore channels of DON to afford Pd-loaded DON (DON@Pd). Enzymes were then loaded into the pore channel of DON@Pd via physical adsorption to fabricate chemoenzymatic catalysts with hydrophobic nanopores (DON@Pd-enzymes). Finally, polydopamine (PDA) was coated onto the surface of DON@Pd-enzymes to afford DON@Pd-enzymes@PDA (Fig. 43a).213 When Candida Antarctica lipase B (CALB) as an enzyme was used, a chemoenzymatic catalyst with a polymer shell layer DON@Pd-CALB@PDA was obtained, where DON was prepared by using a mixture of BTEE and TEOS as the silica source, and surfactant CTAB as a SDA under basic conditions via a continuous phase microemulsion method in a mixed n-butanol-cyclohexane-water solvent system. As a result, DON@Pd-CALB@PDA catalyzed one-pot asymmetric synthesis of chiral amines from various amines and ethylmethoxyacetate in the presence of H2 and Na2CO3 and achieved complete racemization within 4 h as well as performed dynamic kinetic resolution of chiral amines, corroborating that the catalytic activity of hydrophobic DON@Pd was much better than that of hydrophilic dendritic silica nanoparticle-supported Pd NPs (DSN@Pd), further verifying the enhanced effect of the hydrophobic microenvironment on catalytic activities (yields of 85–99%) and selectivities (enantioselectivities of 93–99% ee) (Fig. 43b).
 | ||
| Fig. 43 (a) Formation mechanism and process of amphiphilic chemoenzymatic nanocatalysts. (b) Chemoenzymatic asymmetric synthesis of chiral amines by combining Pd-catalyzed racemization and CALB-catalyzed KR. (c) Chemoenzymatic synthesis of chiral alcohols by combining Pd-catalyzed L–S coupling and ADH-catalyzed asymmetric reduction.213 Reproduced from ref. 213 with permission from the Royal Society of Chemistry. | ||
In addition, it is noted that the synthesis of enantiomerically pure chiral benzyl alcohols was performed through two steps by coupling a Pd/Cu-catalyzed Liebeskind–Srogl (L–S) reaction with asymmetric reduction catalyzed by alcohol dehydrogenase (ADH) from Rhodococcus ruber (Fig. 43c). During the preparation of DON@Pd-CALB@PDA, if CALB was replaced by ADH, DON@Pd-ADH@PDA was obtained, which could briefy above-mentioned two-step reactions as one-pot synthesis to prepare chiral benzyl alcohols in Fig. 43c. DON@Pd-ADH@PDA effectively catalyzed the reaction of S-(tert-butyl) ethanethioate and phenylboronic acid with various substitute groups to the corresponding chiral alcohols. The results showed catalytic activity with moderate to high yields (61–86%) and excellent enantioselectivities with 97–99% ee values, clearly confirming that DON@Pd-ADH@PDA was markedly superior to DSN@Pd-ADH@PDA, and revealing that the hydrophobic microenvironment was very critical for the enhancement of stability, activity and cascade efficiency. Moreover, both DON@Pd-CALB@PDA and DON@Pd-ADH@PDA could be easily recovered by centrifugation and reused, and the high catalytic performance was retained without a significant decrease and leaching of the Pd NPs and enzyme.
2.7.2.10. Pd-catalyzed S-arylation coupling reaction of thiols or benzylthiols with arylhalides. It is interesting that Pd@PMO-IL can also be applied to different reactions. A Pd@PMO-IL catalyzed C–S coupling reaction of thiols with arylhalides exhibited excellent activity with the yields of 88–95%. For the reaction of benzylthiol with arylhalides, the yields reached 86–98%. In addition, the recoverable catalyst by filtration could be further used at least 10 times without a significant decline in the yield. The high activity and reusability were mainly attributed to highly active Pd-carbene species and the vital role of the nanopore structure of PMO.214
 | ||
| Scheme 15 Structures of model complexes 1–5 and immobilized complexes 1–5⊂PMO.215 Reproduced with permission.215 Copyright 2018, Elsevier. | ||
The [PtMe2(μ-SMe2)]2 complex could also be immobilized on BPy-PMO without or with surface silylation by using hexamethyldisilazane to form catalysts PtMe2(BPy-PMO) or PtMe2(BPy-PMO-TMS).216 Both heterogeneous catalysts catalyzed the hydrosilylation of phenylacetylene with trimethoxysilane. The results exhibited that PtMe2(BPy-PMO) showed a moderate catalytic activity, due to the partial formation of an undesired Pt complex coordinated with free silanol moieties. PtMe2(BPy-PMO-TMS) showed an improved catalytic behavior due to the high hydrophobicity of TMS-capped BPy-PMO and the passivation of siloxane with Pt species. In addition, owing to the hydrophobic structure of PtMe2(BPy-PMO-TMS) and the dense arrays of the bpy unit on the pore surface/wall, PtMe2(BPy-PMO-TMS) showed good reusability for at least five recycles without the decline of catalytic activity in hydrosilylation. Most recently, the Pt(bpy)Cl2 complex was immobilized on BPy-PMO-NT to afford PtCl2@BPy-PMO-NTs with a homogeneous distribution of Pt species on the pore wall.217 Such PtCl2@BPy-PMO-NTs indicated a highly efficient photocatalytic behavior for H2 evolution under visible light irradiation compared with the pristine molecular Pt complex. The photocatalytic activity increased with increasing loadings of the Pt complex on the BPy-PMO-NTs. Meanwhile, investigations found that the formation of a small number of metal Pt particles on the BPy-NTs was conducive to the enhancement of the H2 evolution reaction during the photoreaction.
Apart from the aforementioned Pt complex-supported PMO materials, Pt NP-immobilized PMOs have also attracted much attention. In 2014, Chen and co-workers reported that platinum(II) salt Pt(NH3)4(NO3)2 and AgNO3 were attached readily to the surface of carboxylic acid-functionalized ethylene-bridged PMO (HOOC-PMO) by electrostatic interaction, and then these metallic ions could be reduced by H2 in Ar to afford Pt- or Ag-loaded HOOC-PMO with a relatively uniform particle distribution on the surface or inside the pore channel.218 Note that the sizes of Pt and Ag NPs reduced with increasing carboxylic acid groups, from 3.6 to 2.5 nm for Pt NPs and from 5.3 to 3.4 nm for Ag NPs. But no catalytic activity was explored for these NPs.
It is noted that small pore space on the PMO is probably beneficial to the stability of metal NPs. The imidazolium-bridged PMO (PMO-IL) prepared under basic conditions often showed a small pore diameter, which probably provided an excellent environment for the stabilization of ultrasmall metal NPs. In 2015, Karimi and co-workers used three different methods to immobilize Na2PtCl4 on PMO-IL to generate Pt salt or Pt NP-loaded PMO-IL catalysts, Pt(II)@PMO-IL, Ptnp@PMO-IL-1 and Ptnp@PMO-IL-2 (Fig. 44).219
 | ||
| Fig. 44 Synthetic routes to various types of Pt-supported PMO-IL.219 Reproduced with permission.219 Copyright 2018, John Wiley & Sons. | ||
N2 physisorption analysis indicated that Pt(II)@PMO-IL, Ptnp@PMO-IL-1 and Ptnp@PMO-IL-2 had different pore diameters, implying different pore structures of materials and the existence of Pt ions or metal Pt particles in different forms. Pt(II)@PMO-IL prepared by simple impregnation of PMO-IL with Na2PtCl4 in water showed a poor activity and selectivity with a very low yield in the aerobic oxidation of benzyl alcohol to benzaldehyde and benzoic acid. PtNP@PMO-IL-1, prepared by impregnation, followed by the reduction of Pt species with NaBH4, only exhibited a significantly improved catalytic activity for benzaldehyde and benzoic acid, demonstrating the catalytic role of Pt NPs located on/inside pore channels. PtNP@PMO-IL-2, synthesized by refluxing an aqueous solution of Na2PtCl4 in the presence of benzyl alcohol (BnOH), gave a high yield for benzoic acid, revealing a strong electrostatic interaction between the IL cation in the PMO-IL framework and [PtCl4]2− before reduction. During reflux, BnOH slowly reduced Pt(II) ions to ultra-small sized metallic Pt NPs and made Pt NPs homogeneously distribute in the entire nanospace of PMO-IL, while the bridged ionic liquids would most likely prevent the agglomeration of the Pt NPs. As a result, ultrasmall Pt particles with 1–1.4 nm were obtained under the reaction conditions. Hence, a heterogeneous catalyst PtNP@PMO-IL-2 with an ultra-small size of Pt NPs was widely applied to the aerobic oxidation of primary and secondary alcohols as well as 5-hydroxymethylfurfural with a high yield.219
Furthermore, Na2PtCl4-bound magnetic PMO-IL via IL+-PtCl42−-IL+ electrostatic interaction was reduced with NaBH4 to form magnetic Pt-PMO-IL, which was used as a heterogeneous catalyst for the carbon sequestration reaction.220 Pt NP-loaded imidazolyl Schiff base (IMIS)-containing PMO (Pt0-IMIS-PMO), which stemmed from the reduction of H2PtCl6·H2O-immobilized IMISPMO with NaBH4, could be used as a heterogeneous catalyst for a hydrosilylation reaction between 1-octene and triethoxylsilane.221 Pt0-loaded thiourea-bridged PMO was used for the reduction of 4-nitrophenol to 4-aminophenol.222 In addition, the highly dispersed bimetallic Pt–Pd NPs were captured and stabilized by imine-bridged PMO with a SBA-15-like structure to form Pt–Pd-PMO-SBA-15. This catalyst was used in the hydrogenation of nitrobenzene. The results showed an excellent catalytic activity compared with monometallic Pt-PMO-SBA-15 and Pd-PMO-SBA-15, thanks to the synergetic effect and the segregation behavior between Pt and Pd interaction.223
2.8. Groups 11, 12 (Cu, Ag, Au, Zn)-based PMO nanoreactors and catalytic applications
Copper atoms are readily coordinated with N and O atoms from the solid surface to form Cu complexes containing Cu–N or Cu–O bonds and thereby anchored on the solid surface. Copper acetate reacted with furfural-imine functionalized mesoporous silica MCM-41/SBA-15 (F-MCM-41/SBA-15) with different pore sizes to form heterogeneous Cu–F-MCM-41/SBA-15 containing a dimeric copper-furfural-imine complex unit. This process enabled the heterogenization of the molecular Cu complex on MCM-41/SBA-15.224 In this case, the pore wall was composed of hydrophilic Si–O–Si frameworks and dinuclear copper species. It is noted that dinuclear copper units were only hung inside the pore channel. Substrates could readily access and diffuse into nanopores and enabled a multicomponent catalytic reaction in the presence of dimeric Cu active units; for example, Cu-F-MCM-41 catalyzed the coupling of aryl bromides and thiophenol under aerobic conditions to afford different thioethers and showed very good catalytic efficiency. Cu-F-SBA-15 catalyzed a one-pot three component coupling reaction of different aryl halides with thiourea and benzyl bromide in aqueous medium to produce different aryl alkyl thioethers (Scheme 16) in very good yields. Note that both catalysts could be reused several times after being collected by filtration.224 | ||
| Scheme 16 Nanocatalyst Cu-F-SBA15-catalyzed three component C–S coupling reactions (a) and (b). | ||
Copper(II) trifluoromethanosulfonate [Cu(OTf)2] reacted with the functional organic group from 2,2′-methylene bis[(4S)-4-phenyl-2-oxazoline](MB4SPO)-functionalized phenylene-bridged PMO, which was prepared by (i) co-condensation of 2,2′-bispropyltrimethoxysilane-2,2′-bis[(4S)-4-phenyl-2-oxazoline]methane and BTEB in the presence of a SDA under basic conditions and (ii) removal of the SDA, to produce Cu-MB4SPO-Ph-PMO containing Cu(MB4SPO)-(OTf)2 units.225 Cu-MB4SPO-Ph-PMO catalyzed the asymmetric benzoylation of hydrobenzoin and exhibited an extremely high efficiency on the kinetic resolution of 1,2-diphenylethane-1,2-diol with persistent high enantioselectivities (91 - > 99%) and yields (46–43% in maximum 50% resolution) for at least five consecutive cycles, thanks to the high bis(oxazoline) density-ensured high metal density and highly stable chemical and structural integrity of the mesoporous material.225
Similarly, chiral Cu complexes, CuMe2PhBox(OTf)2, CuPhBoxCl2 and CuPhBox(OTf)2 (Scheme 17), were directly grafted onto amine-functionalized phenylene- or biphenylene-bridged PMO (NH2PhPMO or NH2BphPMO) to produce CuMe2PhBox(OTf)2@NH2PhPMO, CuPhBoxCl2@NH2PhPMO and CuPhBox(OTf)2@NH2Bph-PMO, respectively.226 Their catalytic activities were evaluated in the kinetic resolution of hydrobenzoin.226 The results exhibited that all the grafted catalysts were active, selective and enantioselective, but different ligands had a significant influence on the yield and enantioselectivity.
 | ||
| Scheme 17 Copper(II) complexes with bis(oxazoline) ligands; OTf = trifluoromethylsulfonyl. | ||
As the aforementioned PMO@ILC4H10O2NiCl3,176 copper oxychloride (Cu2(OH)3Cl) was linked to benzotriazolium ionic liquid-modified PMO (PMO-IL) to afford PMO@ILCu2(OH)3Cl2(x) (x represents the amount of IL used in preparation) with ion-type unit –IL+[Cu2(OH)3Cl2]−. These catalysts were used for the cycloaddition of CO2 with epoxides. The results showed excellent yields and selectivities under solvent- and cocatalyst-free conditions.227 Copper acetate was linked to imidazolium ionic liquid-modified phenylene-bridged PMO to form copper-loaded bifunctional PMO (Cu@BPMO-Ph-IL). As Ni@IL-PMO,175 catalyst Cu@BPMO-Ph-IL promoted the four-component Hantzsch reaction of ethyl/methylacetoacetate, dimedone, aldehyde and ammonium acetate to polyhydroquinolines denoted as Ni@IL-PMO,175 and gave a good catalytic activity, easy separation and reusability, due to the high stability of the mesoporous structure.228 Furthermore, copper nitrate was anchored to alkyl imidazolium-bridged PMO (PMO-IL) to afford a copper-bonded PMO-IL nanocatalyst (Cu@PMO-IL). Due to its highly stable mesoporous structure, Cu@PMO-IL catalyzed solvent-free one-pot three-component Biginelli condensation of aldehydes, urea/thiourea, and alkyl acetoacetate to 3,4-dihydropyrimidinones/thiones under the optimized reaction conditions, showing catalytic activity with good to excellent yields (80–97%), high recoverability and reusability (1st run with 97% yield; 16th run with 87% yield).229 Similarly, a simple Cu salt, such as CuCl2, could also be bound to the bridged IL unit to form a Cu@PMO-IL nanocatalyst with the possible IL+(CuCl3)− unit located in PMO frameworks and the pore wall surface. Such a heterogeneous catalyst was used for the three-component coupling reaction of aldehydes, alkynes and amines to propargylamines under the optimized conditions. The results showed high catalytic performance with a high yield and reusability for at least 7 recycles.230 In fact, CuCl2-bound bisimidazolium ionic liquid-bridged PMO with CuCl42− species for catalytic decomposition of cyclohexyl hydroperoxide has been investigated in 2012.231 A conversion of 99% could be reached for cyclohexanol with a selectivity of 55% and for cyclohexanone with a selectivity of 29%. In addition, copper-imprinted functionalized mesoporous organosilica nanocomposites (Cu@PMO NCs) could also be used as a heterogeneous catalyst for the synthesis of β-hydroxy-1,2,3-triazoles from styrene oxide, NaN3 and alkyne, and the preparation of β-hydroxytriazoles from epoxides, phenylacetylene and NaN3 in water.232,233 Recently, CuCl2 was immobilized on highly hydrophobic TMS-caped BPy-PMO (BPy-PMO-TMS) to form Cu-BPy-PMO-TMS with single-site CuCl2(bpy) species.234 Cu-BPy-PMO-TMS catalyzed Mukaiyama epoxidation at a low substrate concentration, but free radical auto-oxidation also occurred at a high substrate concentration. Both reactions simultaneously occurred inside and outside pore channels, but the addition of a solid scavenger only quenched the epoxidation outside the pore channel not inside the pore channel, due to the pore size effect. Hence, the combination of a mesoporous catalyst with a solid radical scavenger would offer a new promising strategy to make the free radical reaction only occur inside the pore channel and probably lead to high selectivity for products. Most recently, Cu-coordinated tris(triazolylmethyl)amine (TTA)-bridged organosilica could be used as a small-molecule-selective organosilica nanoreactor for copper-catalyzed azide–alkyne cycloaddition reactions in cellular and living systems (Fig. 45), further extending the application of Cu-PMO catalysts.235
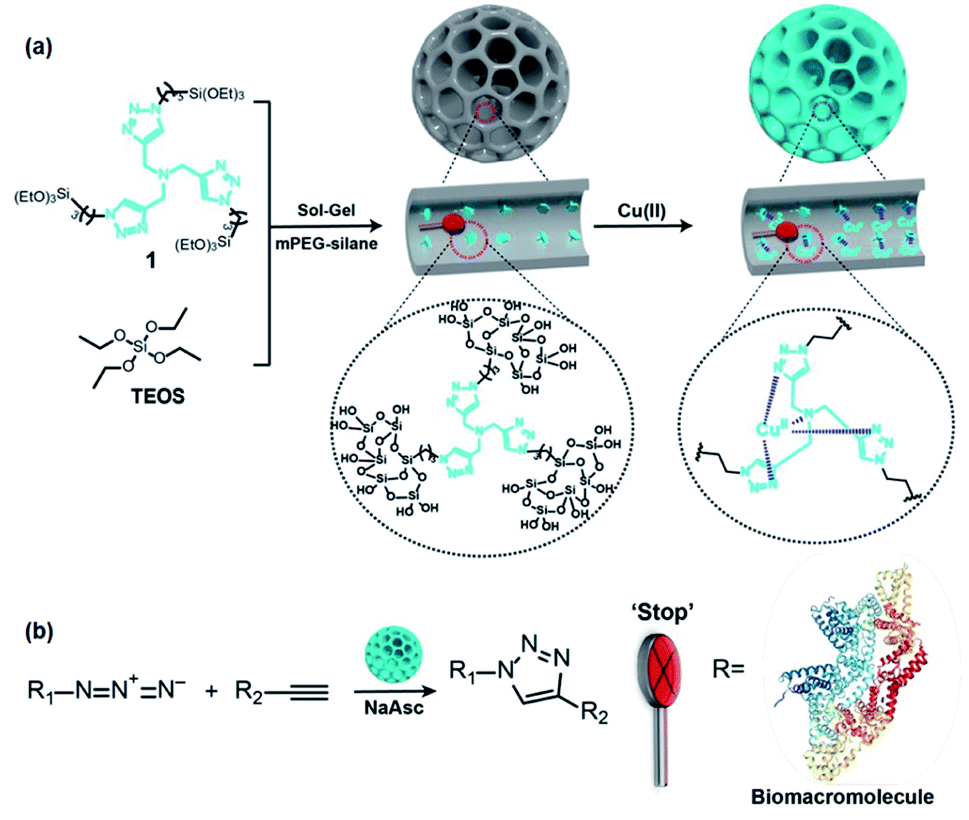 | ||
| Fig. 45 (a) Cu-TTASi nanoreactors and the detailed coordination relations of CuII and N(C3N3)3 ligands and (b) CuII-TTASi-catalyzed CuAAC reactions in the presence of sodium ascorbate. mPEG-silane: PEGylation triethoxysilane; NaAsc: sodium ascorbate.235 Reproduced with permission.235 Copyright 2021, American Chemical Society. | ||
Noble metal-based nanostructured catalysts have attracted much attention. As a member of noble metal elements, the immobilization of Ag on PMO often adopts different approaches, such as in situ photo-reduced deposition, chemical reduction, ultrasound dispersion, polymer-assisted Ag salt adsorption and reduction and so on. For example, a silver mirror reaction was used as a versatile approach to immobilize Ag NPs on hierarchically mesoporous/macroporous organosilica (HMMO) microspheres. Fresh Ag(NH3)2OH solution was poured into a mixture of HMMO microspheres and glucose for a desired time to obtain an Ag-loaded hierarchically structured hybrid material Ag/HMMO.236 Ag particles were uniformly dispersed on HMMO. Such a Ag/HMMO effectively catalyzed the reduction of 4-nitrophenol or 4-nitrophenolate with NaBH4 to 4-aminophenol or 4-aminophenolate, respectively, and showed a high catalytic activity and high reusability. Ag was immobilized on polyacrylic acid-coated hollow mesoporous organosilica spheres (PAA@HMOS) to form Ag@PAA@HMOS. Ag NPs with a size of about ∼3 nm were located in the cavity or on the shell. Such a composite was used in 4-nitrophenol reduction with NaBH4 to 4-aminophenol, and Congo red (CR) and Rhodamine B (RhB) reduction with NaBH4 in water.237 Furthermore, Ag NPs were immobilized onto hydrophobic and highly interconnected macro-mesopore structured multi-compartment (HIMC) vinyl organosilica microspheres to fabricate Ag/HIMC vinyl organosilica microspheres. Such microspheres could be used as an efficient catalytic microreactor to perform the complete degradation of methylene blue to leucomethylene blue by using NaBH4 in a short time.238 In addition, Ag-loaded PMO could effectively catalyze the photodegradation of methylene blue and reached an efficient degradation of about 81% within 1 h.239
Au as a vital noble metal can be immobilized on PMO with or with surface modification. Au can exist in PMO as an Au complex or Au particle form. But examples of anchored gold(I) complexes on PMO for catalytic applications are rather very scarce. In 2010, Li and coworkers synthesized a novel organosilazane AuCl[PPh2CH2–CH2Si(OEt)3]2 containing Au complex-based bridged unit.139 The co-condensation of BTEB and AuCl[PPh2CH2CH2Si(OEt)3]2 in the presence of Pluronic P123 under acidic conditions generated organogold complex-/phenylene-bridged PMO after removal of P123 (Au-PPh2-PMO). As a heterogeneous catalyst with considerably uniform homogeneous Au(I) distribution, Au-PPh2-PMO catalyzed the hydration of phenylacetylene and showed a high yield of 91%, much higher than those of the molecular catalyst AuCl(PPh3) (26%) and grafted Au(I)-PPh2-PMO (24%).139 In 2018, gold(I)–NHC complexes, [Au(OH)(IPr)] (IPr = N,N′-bis[2,6-(di-isopropyl)phenyl] imidazole-2-ylidene) and [Au(OH)(IPr*)] (IPr* = N,N′-1,3-bis[2,6-bis(diphenylmethyl)-4-methylphenyl]imidazole-2-ylidene), were immobilized on sulfonic acid-based ethylene-bridged PMO (PMO-SO3H) and TMS-capped PMO-SO3H (cPMO-SO3H) via an acid–base reaction to afford PMO-SO3-Au(IPr), cPMO-SO3-Au(IPr) and cPMO-SO3-Au(IPr*), respectively(Fig. 46).240 The catalytic activities of PMO-SO3-Au(IPr), cPMO-SO3-Au(IPr) and cPMO-SO3-Au(IPr*) were evaluated by the hydration of diphenylacetylene in a mixture of dioxane and water. 10 mol% of PMO-SO3-Au(IPr) was used in the hydration of diphenylacetylene, and the yield reached 91% after reaction for 5 h. When PMO-SO3H was used, no catalytic activity was observed. When 7.5 mol% of cPMO-SO3-Au(IPr) and cPMO-SO3-Au(IPr*) were used, the yields of the product reached >99% after reaction for 3 h. In addition, for the reusability of catalysts, an acid work-up was necessary after each cycle in order to reactivate the catalyst and obtain high yields in the subsequent cycles.240
 | ||
| Fig. 46 Synthesis of the PMO-based gold(I)–NHC catalysts PMO-SO3-Au(IPr), cPMO-SO3-Au(IPr) and cPMO-SO3-Au(IPr*).240 Reproduced with permission.240 Copyright 2018, John Wiley & Sons. | ||
Note that NaAuCl4 was directly impregnated into ionic liquid-bridged PMO (PMO-IL) to form Au(III)@PMO-IL with the electrostatic interaction between IL+ and Au(III)Cl4−. Without further reduction, Au(III)@PMO-IL catalyzed the aerobic oxidation of benzyl alcohol to benzaldehyde in the presence of Cs2CO3 and showed a wonderful catalytic activity with 99% conversion and >99% selectivity for benzaldehyde.241 The high catalytic activity was attributed to the fact that alkyl imidazolium groups in the interior of mesochannels of Au@PMO-IL not only served as a handle for in situ generation and stabilization of gold nanoparticles from AuCl4− but also effectively prevented the leaching of Au species into solution during the reaction process. Hence, during catalysis, in situ formed Au NPs as the active species played a vital role in driving the reaction. More interestingly, Au(III)@PMO-IL could catalyze various types of alcohols including activated benzylic, primary and secondary aliphatic, heterocyclic, and challenging cyclic aliphatic alcohols to the expected carbonyl compounds in good to excellent yields and selectivities. The catalyst was also recovered and reused for at least seven runs. However, when Au(III)@PMO-IL was reduced by NaBH4 to produce Au(0)@PMO-IL, its catalytic activity was lower than that of Au(III)@PMO-IL and took a long reaction time for the aerobic oxidation of benzylalcohol under identical conditions. But Au(0)@PMO-IL could effectively catalyze the three-component coupling reaction of aldehydes, alkynes and amines to the corresponding propargylamine in good to excellent yields.242 However, NaAuCl4 was directly adsorbed onto the mixed ethylene-/phenylene-bridged PMO and was reduced by NaBH4 to afford the Au@PMO catalyst. This catalyst could catalyze not only Ullmann coupling of iodobenzene under basic conditions, showing catalytic activity with up to 95% yield,243 but also room-temperature aerobic oxidation of benzyl alcohol and various alcohols to the corresponding aldehydes or ketones in good to excellent yields depending upon the substitute group.244.
Thiol(HS)-functionalized ethylene-bridged PMO (HS-PMO(Et)) was synthesized and used as a support for the immobilization of HAuCl4 (Scheme 18). HS-PMO(Et) was briefly suspended in HAuCl4 ethanol solution at room temperature for 24 h to prepare Au-HS/SO3H-PMO(Et) with 0.1 mmol g−1 Au.245 During this period, three processes including in situ reduction of Au3+ to Au NPs, the oxidation of HS to SO3H, and coordination of the Au NPs with the unreacted HS ligand occurred simultaneously, leading to covalent bonding of Au NPs to HS/SO3H-functionalized PMO to form a metal–organic group bifunctional catalyst Au-HS/SO3H-PMO(Et). The catalytic activity of Au-HS/SO3H-PMO(Et) was evaluated in water-medium and solvent-free phenylacetylene hydration, alkyne hydration, intramolecular hydroamination, styrene oxidation reactions and three-component coupling reactions (Scheme 18).245 The catalytic results showed that Au-HS/SO3H-PMO(Et) had a high catalytic activity for these organic reactions compared with homogeneous catalysts and heterogeneous catalysts Au/HS-PMO and Au/SO3H-PMO, which was mainly attributable to (i) the uniform and narrow size distribution of Au NPs with a size of about 1–2 nm located inside the pore surface not on the external surface, ensuring maximal exposure to reaction substrates and minimization of Au NP leaching, (ii) the unique amphiphilic compartment of Au-HS/SO3H-PMO(Et), conducive to the access of the substrate, and (iii) the synergistic effect of bifunctional roles of Au and HS/SO3H groups.
 | ||
| Scheme 18 Illustration of the preparation of Au-SH/SO3H-PMO(Et) and the corresponding catalytic organic reactions.245 Reproduced with permission.245 Copyright 2011, American Chemical Society. | ||
Moreover, selective alkaline etching of core–shell structure Au@SiO2@PMO led to the formation of Au@Void@PMO with empty cavities and Au NPs were anchored on the surface of the shell, which ensured the maximal exposure of active sites to substrates. Au@Void@PMO catalyzed 4-nitrophenol reduction with NaBH4 to 4-aminophenol and showed high catalytic activity.246
By the reduction of Au(III) ions with NaBH4 or an in situ hydrothermal reaction, Au NPs were immobilized onto different structured and morphological PMOs to afford Au-PMOs for the reduction of 4-nitrophenol to 4-aminophenol with NaBH4, such as, highly hydrophobic yolk–shell Au@Ph-PMO,247 Au&PMO Junas NPs, Au@PMO yolk–shell NPs, Au@PMO@mSiO2 yolk–shell double shell NPs (Fig. 47),248 and Au NPs confined into the pore walls of morphology-controlled PMO, and so on.249 Polyvinylpyrrolidone (PVP)-protected ultrasmall Au NPs confined in mesoporous silica-ethylene-bridged PMO yolk–shell nanospheres through a ‘‘one-pot’’ method were also addressed and used as a nanocatalytic reactor for the aerobic oxidation of styrene.250 HAuCl4 was immobilized on BPy-PMO to form solid molecular single-site AuCl3-BPy-PMO, followed by reduction with H2, to afford Au NP-supported BPy-PMO (AuNPs/BPy-PMO). Au NPs with an average size of 3.8 nm were homogeneously distributed inside and outside the mesopore channels of BPy-PMO, while some large Au NPs with sizes of 6–8 nm were located on the external surface.251 As a heterogeneous catalyst, AuNPs/BPy-PMO showed a high catalytic activity with 91% conversion and 87% yield in the aerobic oxidation of benzaldehyde to benzoic acid, compared with 39% conversion and 22% yield of Au/MCM-41, due to the highly hydrophobic mesopore structure and ultrasmall Au nanoparticle. Additionally, Au was immobilized on (1-hydroxyl-2-ethylenediamine)-ethylene-bridged PMO to form a bifunctional Au-loaded PMO heterogeneous catalyst. Such a catalyst was used in Knoevenagel condensation and one-pot tandem alcohol oxidation. The results showed catalytic activity with considerable to excellent conversions and yields.252
 | ||
| Fig. 47 The synthesis illustration of Au&PMO Janus, Au@PMO yolk–shell, and Au@PMO/mSiO2 yolk-double shell nanostructures.248 Reproduced from ref. 248 with permission from the Royal Society of Chemistry. | ||
More recently, gold-loaded mesoporous organosilica-silica core–shell NPs as catalytic nanoreactors were rationally designed and successfully synthesized (Scheme 19).253 A thiol-functionalized mesoporous organosilica nanosphere (MON) as the core and as the capturing center for Au(III) ions and pure mesoporous silica (MS) as the shell formed core–shell structured SH-MON@MS. The distinct core/shell composition enabled selective loading of Au NPs into the core domain affording hierarchically structured materials Au/SH-MON-n@MS for the reduction of 4-nitrophenol to 4-aminophenol with NaBH4.253 The larger mesopore sizes of the shell layer facilitate easy access and diffusion of reactants and products, while the inner organosilica pores provide a hydrophobic microenvironment. Due to the superior structural properties, the materials Au/SH-MON-n@MS (n = 1, 2, 3, 4) exhibited an excellent catalytic performance in the reduction of 4-nitrophenol to 4-aminophenol and considerable conversions and excellent selectivities for benzaldehyde in the aerobic oxidation of benzyl alcohol to benzaldehyde.
 | ||
| Scheme 19 Schematic illustration of the preparation process of core-shell-structured Au/SH-MON@MS by controlling the functional group content.253 Reproduced with permission.253 Copyright 2020, John Wiley & Sons. | ||
Except from studies on Au-based PMO catalysis, in fact, the catalytic activity of Au alloy-based PMO was also investigated. In 2018, Liu and co-workers prepared short aminopropyl-functionalized phenylene-bridged PMO nanotubes (AMx-PMO) with various aminopropyl group contents (x = 0.1, 0.2, 0.4, corresponding to a 1 mol total initial silica precursor). HAuCl4 (y) and H2PdCl4 (z) with different molar ratios (molar ratio of y:z = 1:0, 4:1, 1:1, 1:4, 0:1) were co-reduced with NaBH4 and co-immobilized on AMx-PMO to afford AuyPdz@AMx-PMO (Fig. 48).254 The resulting AuPd@AM0.4-PMO was used as a catalyst to investigate its catalytic activity in visible light-driven hydrogen evolution from formaldehyde. The experimental results exhibited a superior hydrogen evolution performance from formaldehyde aqueous solution under visible light illumination, due to the strong interactions and the alloying synergistic effect between ultrasmall Au and Pd nanoparticle in the ultrafine AuPd alloy nanostructures and the efficient electron transfer from Au with a localized surface plasmon resonance effect to active Pd sites.
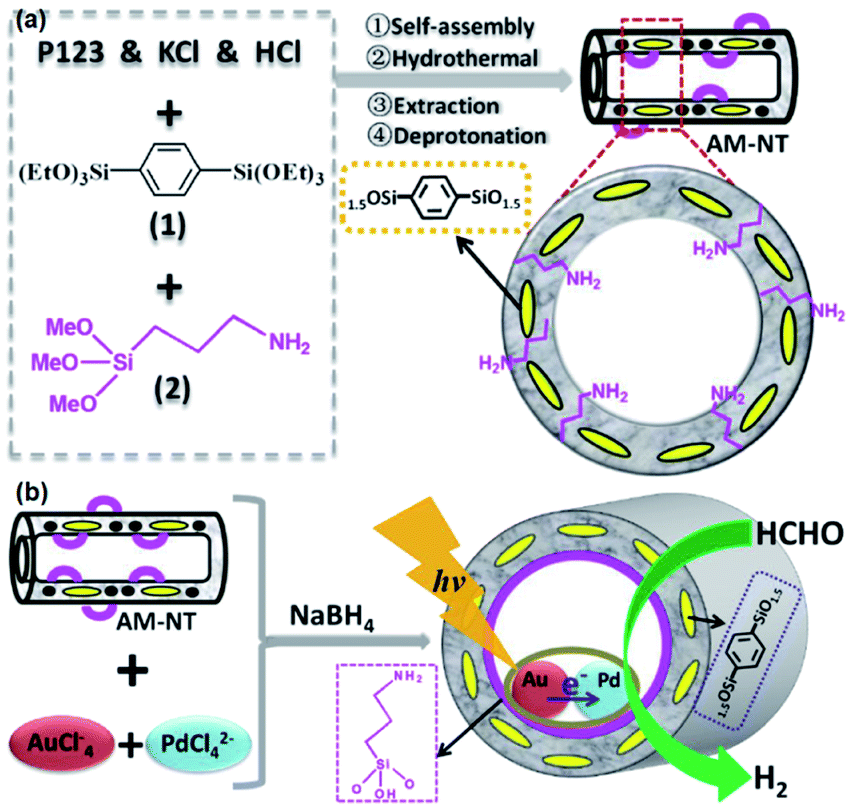 | ||
| Fig. 48 (a) Synthetic routes of amine-incorporated organosilica nanotubes. (b) Synthetic routes and photocatalytic hydrogen evolution of amino-functionalized organosilica nanotube supported AuPd alloy nanoparticles.254 Reproduced with permission.254 Copyright 2018, Elsevier. | ||
Furthermore, more interestingly, a very sophisticated composite material, ultrasmall Au-loaded in situ polymerized functionalized polymer-confined hollow mesoporous organosilica nanoparticle (HMON)-combined collagenase (Col) enzyme-supported PVP-encapsulated Cu-tannic acid (TA) composite (HMON-Au-Col@Cu-TA-PVP), was used as a biocatalytic nanoreactor for enhancing reactive oxygen species (ROS)-mediated anticancer therapy.255 In this complicated composite, the hollow cavity of HMONs was exploited as a nanoreactor for in situ polymerization of a polymer containing thiol groups, which was further used to immobilize glucose oxidase (GOx)-like ultrasmall Au NPs to obtain HMON-Au. Col and Cu-TA complexes were then deposited onto the surface of HMONs-Au, followed by coating of PVP to fabricate HMON-Au-Col@Cu-TA-PVP (Fig. 49). The ultrasmall Au NPs, which behave like glucose oxidase-like nanozym, could catalyze glucose into H2O2 and gluconic acid with high efficiency. H2O2 was further converted into ·OH by a Cu-TA-catalyzed Fenton-like reaction. The enhanced ROS generation after the combination of chemodynamic therapy (CDT) and photodynamic therapy (PDT) led to an effective antitumor therapeutic efficacy, while Col in the composite enhanced tumor accumulation of nanoparticles and alleviated the hypoxia of the tumor tissues, thereby further enhancing the antitumor efficacy.
 | ||
| Fig. 49 Schematic showing the process of preparing an in situ polymerized hollow mesoporous organosilica biocatalytic nanoreactor for synergistic photodynamic (PDT)/chemo-dynamic (CDT) therapy.255 Reproduced with permission.255 Copyright 2020, John Wiley & Sons. | ||
Moreover, the highly dispersed gold NPs were deposited on Ti-incorporated mesoporous organosilica (Ti-MO) to afford Au/Ti-MO. This material was used for hydrophobicity-induced vapor-phase propene epoxidation to propene oxide using O2 and H2. The results showed an excellent catalytic performance.256
For zinc-immobilized PMO materials, only a few investigations are shown. Zinc acetate Zn(OAc)2 could be immobilized on IL-bridged PMO (PMO-IL) to afford Zn(II)@PMO-IL, which was used in a three-component coupling reaction of various aldehydes, amines and arylacetylene to the corresponding propargylamines. The results exhibited a high efficiency and reusability.257 Furthermore, zinc acetate could also be successfully coordinated with TMS-capped bipyridylene-bridged PMO (BPy-PMO-TMS) to form an immobilized Zn complex, Zn(OAc)2(BPy-PMO-TMS), which was used as a heterogeneous catalyst for the N-formylations of amines and amides with CO2 and PhSiH3 to produce the corresponding formamides. The result showed that Zn(OAc)2(BPy-PMO-TMS) with a lower Zn loading exhibited a high catalytic activity.258 Moreover, the active zinc-substituted polyoxotungstate [PW11Zn(H2O)39]5− (PW11Zn) was immobilized into amine-functionalized ethylene (E)- or phenylene (B)-bridged PMO to form PW11Zn@NH2-PMO-E or B.259 Their catalytic activities were investigated under an oxidative desulfurization system (CODS) and a biphasic (diesel/extraction solvent) oxidative desulfurization system (ECODS). The results confirmed the high desulfurization efficiency of both PW11Zn-based NH2-PMO-E and NH2-PMO-B, and more stable PW11Zn@NH2-PMO-E. In addition, PW11Zn@NH2-PMO-E could efficiently catalyze the desulfurization of real diesel, and the yield reached 75.9% after 2 h, while this catalyst could be easily recovered by centrifugation and reused for the treatment of real diesel, implying that it is a promising candidate for practical applications.
2.9. Group 3 and lanthanide-based PMO nanoreactors and catalyses
Sodium benzenesulfonate group-modified phenylene-bridged PMO (NaSO3Ph-PMO) was used as a support to graft scandium triflate and generate a mesoporous Lewis acid catalyst (Sc(OTf)2-SO3Ph-PMO). This catalyst promoted the Barbier–Grignard and Mukaiyama–Aldol reactions and exhibited superior catalytic reactivity compared to those of homogeneous catalysts scandium triflate and Sc(OTf)2-SO3Ph-SBA-15, thanks to the fact that the ordered mesoporous channel and highly hydrophobic microenvironment stabilized and concentrated the substrates, and weakened intrinsic mass transfer resistance inside nanochannels.260 In addition, for lanthanide-based PMO materials, the main applications were focused on optic properties, which are beyond the scope of this review.3. Main influence factors on the catalytic performance of metal-loaded PMO heterogeneous catalysts
Heterogeneous catalysis is a relatively complex process and the heterocatalytic reaction often takes place at the liquid–solid interface. Once the heterocatalytic reaction only occurs at the liquid–liquid interface, active species can quickly leach into solution and thereby lead to the deactivation of the catalyst. In fact, for a heterocatalytic reaction, many factors strongly influence the catalytic performance of catalysts, such as the composite and structure of the heterogeneous catalyst, catalyst dose, solvent, reaction temperature and time. For metal-loaded PMO heterogeneous catalysts as nanoreactors, the catalytic reaction is becoming more complicated, due to the intricate structure of the catalyst itself including the pore size, amount and size of active metal particles, hydrophobic or hydrophilic environment of the support surface, nature of the substrate and so on. On the basis of the above-mentioned investigations of metal-loaded PMO heterogeneous catalysts, the results showed that the catalytic performance of the heterogeneous catalyst in different chemical reactions was strongly influenced by the loading of metal species,66,71,86,149,191,217 particle size of metal NPs,64,149,201 hydrophobic microenvironment of PMO,46,99,124 and size of the available microenvironment such as the pore size or cavity size.106,134,192 In terms of the preparation of the heterogeneous catalyst itself, it is obvious that the amount of loaded metal species of metal complex-grafted PMO heterogeneous catalysts mainly depended upon the reaction of the metal complex with silanol/hydroxyl groups and the populations of surface silanol/hydroxyl groups on PMOs.71,72 A previous review by Anwander has outlined the advanced synthesis concepts of organometallic complex-modified inorganic–organic hybrid PMS to optimize local-, micro- and mesoenvironments for catalytic performance.261 For metal-grafted PMO heterogeneous catalysts, the grafting reaction relies on (i) whether the ligands of the metal complex precursor can be easily displaced, e.g., by protonolysis with the hydroxyl group on PMO to form alkanes, (silyl)amines or alcohols as a byproduct after grafting alkyl, (silyl)amide, and alkoxide reagents, and (ii) the molecular size of the ligands in the metal complex or actual molecular size of the metal complex itself. In the case of (i), metal-loaded PMOs probably contain a high metal loading. In the case of (ii), ligands or metal complex precursors of small molecular size can readily access the pore surface and external surface of PMO to react with the silanol groups and form metal species by –Si–O–M– bonds. For ligands or metal complexes of large molecular size, the metal loading on PMO mainly depends not only upon the pore size and regular shape of the pore channels of the PMO support, but also upon the efficacy of the ligand to act as a leaving group. Hence, for heterogeneous catalysts prepared by the grafting strategy, an optimal support with a high surface area, suitable pore size, regular pore shape and structure, as well as an appropriate metal complex precursor are pivotal for the fabrication and improvement of the catalytic performance of heterogeneous catalysts.For the preparation of metal complex-bearing PMO heterogeneous catalysts, metal loading mainly relies on the number of functional groups on PMO.146,175,192,253 The distribution of metal species on the PMO is governed by the distribution of functional groups on PMO. Only functional groups uniformly distributed over the mesopore surface or the external surface are available to coordinate with metal complexes and to form active metal sites for catalysis. All catalytic performances will strongly rely on the available metal active sites loaded on PMO.
For metal-loaded PMO catalysts prepared by the incipient wetness impregnation method or one-pot co-assembly synthetic approach, metal loadings on PMO are up to the initial metal amount used. In this case, the aggregation of metal species is unavoidable. The amount of initial metal precursor needs to be controlled and a uniform dispersion of active metal species with a small particle size on the support is necessary in order to improve the catalytic activity. Low loading or overloading of metal species cannot sufficiently exert maximal catalytic efficiency of heterogeneous catalysts.
In addition, for metal-loaded PMO heterogeneous catalysts, pore confinement effects also strongly affect the activity of heterogeneous catalysts in organic synthesis.106,134,162,168,199,208
Finally, it is necessary to emphasize that the hydrophobic microenvironment of PMOs also has an important influence on the catalytic performance. For example, the Ti-PMO-catalyzed oxidation of 1-octene with TBHP indicated a remarkably different catalytic performance.46 With increasing initial BTEE contents in the mixture of TEOS and BTEE (from 0 to 33%, to 100%) in Ti-PMO, the catalytic activity correspondingly increased (TOF changed from 65.5 to 83.9, to 104.4 h−1). Especially, upon silylation of the PMO with hexamethyldisilazane, the TOF markedly enhanced from 83.9 to 143.2 h−1, although the active Ti content was lower in this case, clearly confirming the key role of the hydrophobic microenvironment. Similarly, 10%Mo-BPy-PMO-TMS (Mo loading of 0.30 g) showed an improved catalytic activity compared to 11% Mo-BPy-PMO (Mo loading of 0.35 g) for the epoxidation of cis-cyclooctene with TBHP, verifying that the hydrophobic environment is conducive to the improvement of catalytic performance.99 Surface silylated Re-BPy0.3-NT-Me exhibited a high catalytic activity in the photochemical reduction of CO2 in comparison with Re-BPy0.3-NT.124 Overall, many metal-loaded PMO heterogeneous catalysts surveyed in this article showed a strong hydrophobicity effect on catalytic performance.164,178,194,202,211,216,234,256,260
4. Conclusions and prospects
In this review, the rational design, successful fabrication and catalytic activity of transition metal-based complexes or particle-supported PMO materials were summarized. At the beginning of this review, in describing the development of PMO materials, it was pointed out that the integration of any non-functional organic bridge group couldn't intrinsically alter the physicochemical properties except for their adsorption ability. However, PMO materials indicated some remarkable advantages regarding the hydrophobicity and uniform distribution of bridging groups in the whole framework compared to PMS. Hence, the rational design and successful integration of a functional organic group (carboxylic, thio, amine, chelating, pyridine etc.) into a bridged organosilane and the introduction of active metal species are the key and extraordinarily pivotal issues to modify the physical and chemical properties of PMOs. In terms of catalytic applications, perfect combinations of a high surface area, uniform pore structure, high hydrophobicity, uniformly distributed and intact integrated functional group and catalytically active species in composite catalysts have to be found. The preparation of some important functionalized bridging groups and the corresponding integrated PMO was elucidated. The immobilization of total 24 transition metal elements on PMOs to afford the corresponding metal NP or metal complex-loaded PMO materials as nanoreactors has been reviewed. Consideration has been given to synthesis approaches, surface active species, catalytic activities in selective oxidation reactions, reduction reactions, hydrogenation, chiral organic compound synthesis, coupling reactions, CO2 reduction, addition reactions, desulfurization, catalytic reactions in biosystems, H2 production and so on. Such metal-loaded PMO-catalyzed organic reactions revealed that the catalytic activity of the nanocatalyst was strongly influenced by pore confinement effects, the hydrophobicity of the pore channels, particle size, uniform or inhomogeneous distribution of active species, the stability of active species, the role of functional groups, catalyst dose, the synthetic method of the nanocatalyst and reaction conditions. Especially, the key role of functional groups, the unique structure of PMOs and synergistic effects of the PMO and active species, and between active species, was elucidated.Although functionalized PMOs showed some important advantages compared with PMSs, the functionalized bridging organic groups still can be an issue regarding their uniform distribution in the respective PMO. On the one hand, a bulky functionalized bridged organosilane as a single organosilica source will hardly produce highly ordered PMOs. On the other hand, the presence/co-condensation of TEOS or TMOS leads to an inhomogeneous distribution of the bridging organic group in the PMO frameworks. Also, the stability of the functional group itself needs to be considered. At present, bipyridine- and IL-bridged functional groups are considered very stable. In the future, an advanced PMO support must possess a highly stable functional group and mesopore structure, and a readily adjustable pore size. Moreover, how to perform the heterogenization of molecular catalysts on PMO to draw near the level of single-atom molecular catalysis? At present, single-atom catalysis is an optimal state for catalytic reactions due to an outstanding activity and selectivity in chemical catalysis caused by a unique electronic structure and unsaturated coordination environment. If every dispersed metal species immobilized on the support can serve as a catalytic center to achieve maximum atomic utilization efficiency, heterogeneous catalysts can reach the level of single-atom catalysis. At present, lots of studies are focusing on this aspect, including the applications of noble-metal single atoms in thermocatalysis, electrocatalysis, and photocatalysis,262 cooperativity in supported metal single atom catalysis,263 synthesis of oxygen defect-stabilized heterogeneous single atom catalysts,264 and transformation of heterogeneous catalysts from the nanoscale to single atoms for CO2 hydrogenation to formic acid/formate.265 All these investigations have clearly indicated that single-atom catalysis is becoming a sustainable pathway for advanced catalytic applications.266 Hence in order to richly utilize the catalytic efficiency of metal species on PMO, the preparation of a metal-based PMO nanocatalyst must consider the transformation of a metal loaded catalyst from the nanoscale to single atoms, especially, metal nanoparticles on PMO. In fact, bipyridylene-bridged PMO can probably show a certain potential in the fabrication of a single atom catalyst. Many advanced synthesis strategies need to be developed for the preparation of a great number of atom-controllable precise heterogeneous catalysts including the design of novel PMO supports.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dr Yucang Liang is grateful to Prof. Dr Reiner Anwander for his continued support for this research.References
- T. Asefa, M. J. MacLachlan, N. Coombs and G. A. Ozin, Nature, 1999, 402, 867–871 Search PubMed.
- B. J. Melde, B. T. Holland, C. F. Blanford and A. Stein, Chem. Mater., 1999, 11, 3302–3308 Search PubMed.
- S. Inagaki, S. Guan, Y. Fukushima, T. Ohsuna and O. Terasaki, J. Am. Chem. Soc., 1999, 121, 9611–9614 Search PubMed.
- J. G. Croissant, X. Cattoën, J.-O. Durand, M. W. C. Man and N. M. Khashab, Nanoscale, 2016, 8, 19945–19972 Search PubMed.
- M. Kuroki, T. Asefa, W. Whitnal, M. Kruk, C. Yoshina-Ishii, M. Jaroniec and G. A. Ozin, J. Am. Chem. Soc., 2002, 124, 13886–13895 Search PubMed.
- K. Landskron, B. D. Hatton, D. D. Perovic and G. A. Ozin, Science, 2003, 302, 266–269 Search PubMed.
- H. Zhu, D. J. Jones, J. Zajac, R. Dutartre, M. Rhomari and J. Rozière, Chem. Mater., 2002, 14, 4886–4894 Search PubMed.
- Ö. Dag, C. Yoshina-Ishii, T. Asefa, M. J. MacLachlan, H. Grondey, N. Coombs and G. A. Ozin, Adv. Funct. Mater., 2001, 11, 213–217 Search PubMed.
- T. Asefa, M. Kruk, M. J. MacLachlan, N. Coombs, H. Grondey, M. Jaroniec and G. A. Ozin, J. Am. Chem. Soc., 2001, 123, 8520–8530 Search PubMed.
- Y. Lu, H. Fan, N. Doke, D. A. Loy, R. A. Assink, D. A. LaVan and C. J. Brinker, J. Am. Chem. Soc., 2000, 122, 5258–5261 Search PubMed.
- H. Zhu, D. J. Jones, J. Zajac, J. Rozière and R. Dutartre, Chem. Commun., 2001, 2568–2569 Search PubMed.
- O. Muth, C. Schellbach and M. Fröba, Chem. Commun., 2001, 2032–2033 Search PubMed.
- M. C. Burleigh, M. A. Markowitz, E. M. Wong, J.-S. Lin and B. P. Gaber, Chem. Mater., 2001, 13, 4411–4412 Search PubMed.
- Y. Liang, M. Hanzlik and R. Anwander, Chem. Commun., 2005, 525–527 Search PubMed.
- L. Zhao, G. Zhu, D. Zhang, Y. Di, Y. Chen, O. Terasaki and S. Qiu, J. Phys. Chem. B, 2005, 109, 764–768 Search PubMed.
- W. Guo, I. Kim and C.-S. Ha, Chem. Commun., 2003, 2692–2693 Search PubMed.
- Z. Zhang, X. Yan, B. Tian, S. Shen, D. Chen, G. Zhu, S. Qiu and D. Zhao, Chem. Lett., 2005, 34, 182–183 Search PubMed.
- S. Inagaki, S. Guan, T. Ohsuna and O. Terasaki, Nature, 2002, 416, 304–307 Search PubMed.
- B. Hatton, K. Landskron, W. Whitnall, D. Perovic and G. A. Ozin, Acc. Chem. Res., 2005, 38, 305–312 Search PubMed.
- F. Hoffmann, M. Cornelius, J. Morell and M. Fröba, Angew. Chem., Int. Ed., 2006, 45, 3216–3251 Search PubMed.
- S. Guan, S. Inagaki, T. Ohsuna and O. Terasaki, J. Am. Chem. Soc., 2000, 122, 5660–5661 Search PubMed.
- A. Sayari, S. Hamoudi, Y. Yang, I. L. Moudrakovski and J. R. Ripmeester, Chem. Mater., 2000, 12, 3857–3863 Search PubMed.
- S. S. Park, C. H. Lee, J. H. Cheon and D. H. Park, J. Mater. Chem., 2001, 11, 3397–3403 Search PubMed.
- M. P. Kapoor and S. Inagaki, Chem. Lett., 2004, 33, 88–89 Search PubMed.
- D.-J. Kim, J.-S. Chung, W.-S. Ahn, G.-W. Kang and W.-J. Cheong, Chem. Lett., 2004, 33, 422–423 Search PubMed.
- V. Rebbin, R. Schmidt and M. Fröba, Angew. Chem., Int. Ed., 2006, 45, 5210–5214 Search PubMed.
- N. Muramoto, T. Sugiyama, T. Matsuno, H. Wada, K. Kuroda and A. Shimojima, Nanoscale, 2020, 12, 21155–21164 Search PubMed.
- T. Suteewong, H. Sai, R. Hovden, D. Muller, M. S. Bradbury, S. M. Gruner and U. Wiesner, Science, 2013, 340, 337–341 Search PubMed.
- X. Li, L. Zhou, Y. Wei, A. M. El-Toni, F. Zhang and D. Zhao, J. Am. Chem. Soc., 2014, 136, 15086–15092 Search PubMed.
- L. Luo, Y. Liang, E. S. Erichsen and R. Anwander, Chem.–Eur. J., 2018, 24, 7200–7209 Search PubMed.
- J. Croissant, X. Cattoën, M. W. C. Man, P. Dieudonné, C. Charnay, L. Raehm and J.-O. Durand, Adv. Mater., 2015, 27, 145–149 Search PubMed.
- T. Zhao, A. Elzatahry, X. Li and D. Zhao, Nat. Rev. Mater., 2019, 4, 775–791 Search PubMed.
- X. Su, Y. Tang, Y. Li, Z. Wang, J. Tao, K. Chen, Y. Liu, J. Wu, D. Wang and Z. Teng, ACS Appl. Mater. Interfaces, 2019, 11, 12063–12069 Search PubMed.
- Z. Teng, W. Li, Y. Tang, A. Elzatahry, G. Lu and D. Zhao, Adv. Mater., 2019, 31, 1707612 Search PubMed.
- Z. Fu, L. Li, F. Li, R. A. Bhutto, X. Niu, D. Liu and X. Guo, Ind. Eng. Chem. Res., 2020, 59, 14797–14805 Search PubMed.
- J. G. Croissant, Y. Fatieiev and N. M. Khashab, Adv. Mater., 2017, 29, 1604634 Search PubMed.
- A. Noureddine and C. J. Brinker, Chem. Eng. J., 2018, 340, 125–147 Search PubMed.
- Z. K. Lyles, M. Tarannum, C. Mena, N. M. Inada, V. S. Bagnato and J. L. Vivero-Escoto, Adv. Ther., 2020, 3, 2000022 Search PubMed.
- Z. Lin, L. Xu, J. Zhang, Z. Li and J. Zhao, Nano, 2019, 14, 1950141 Search PubMed.
- B. Yang, Y. Chen and J. Shi, Mater. Sci. Eng., R, 2019, 137, 66–105 Search PubMed.
- B. Yang, Y. Chen and J. Shi, Adv. Mater., 2019, 31, 1901778 Search PubMed.
- D. Shao, F. Zhang, F. Chen, X. Zheng, H. Hu, C. Yang, Z. Tu, Z. Wang, Z. Chang, J. Lu, T. Li, Y. Zhang, L. Chen, K. W. Leong and W. Dong, Adv. Mater., 2020, 32, 2004385 Search PubMed.
- N. Lu, W. Fan, X. Yi, S. Wang, Z. Wang, R. Tian, O. Jacobson, Y. Liu, B. C. Yung, G. Zhang, Z. Teng, K. Yang, M. Zhang, G. Niu, G. Lu and X. Chen, ACS Nano, 2018, 12, 1580–1591 Search PubMed.
- F. Hoffmann and M. Fröba, Chem. Soc. Rev., 2011, 40, 608–620 Search PubMed.
- U. Díaz, D. Brunel and A. Corma, Chem. Soc. Rev., 2013, 42, 4083–4097 Search PubMed.
- J. A. Melero, J. Iglesias, J. M. Arsuaga, J. Sainz-Pardo, P. de Frutos and S. Blazquez, J. Mater. Chem., 2007, 17, 377–385 Search PubMed.
- A. Kuschel, M. Luka, M. Wessig, M. Drescher, M. Fonin, G. Kiliani and S. Polarz, Adv. Funct. Mater., 2010, 20, 1133–1143 Search PubMed.
- J. G. Croissant, X. Cattoën, M. W. C. Man, J.-O. Durand and N. M. Khashab, Nanoscale, 2015, 7, 20318–20334 Search PubMed.
- S. S. Park, M. S. Moorthy and C.-S. Ha, NPG Asia Mater., 2014, 6, e96, DOI:10.1038/am.2014.13.
- S. S. Park, M. S. Moorthy and C.-S. Ha, Korean J. Chem. Eng., 2014, 31, 1707–1719 Search PubMed.
- N. Mizoshita, T. Tani and S. Inagaki, Chem. Soc. Rev., 2011, 40, 789–800 Search PubMed.
- X. Liu, S. Inagaki and J. Gong, Angew. Chem., Int. Ed., 2016, 55, 14924–14950 Search PubMed.
- P. Van Der Voort, D. Esquivel, E. De Canck, F. Goethals, I. Van Driessche and F. J. Romero-Salguero, Chem. Soc. Rev., 2013, 42, 3913–3955 Search PubMed.
- Y. Liang and R. Anwander, Dalton Trans., 2013, 42, 12521–12545 Search PubMed.
- C. Copéret, A. Comas-Vives, M. P. Conley, D. P. Estes, A. Fedorov, V. Mougel, H. Nagae, F. Núñez-Zarur and P. A. Zhizhko, Chem. Rev., 2016, 116, 323–421 Search PubMed.
- S. H. Petrosko, R. Johnson, H. White and C. A. Mirkin, J. Am. Chem. Soc., 2016, 138, 7443–7445 Search PubMed.
- A. Modak, M. Nandi and A. Bhaumik, Catal. Today, 2012, 198, 45–51 Search PubMed.
- Y. Huang, P. Yuan, Z. Wu and X. Yuan, J. Porous Mater., 2016, 23, 895–903 Search PubMed.
- P. He, Y. Huang, P. Yuan and X. Yuan, J. Porous Mater., 2018, 25, 1625–1632 Search PubMed.
- P. Yuan, Y. Huang, X. Yuan and H. Luo, J. Mol. Catal., 2015, 29, 135–142 Search PubMed.
- Z. Wang and K. J. Balkus Jr., Microporous Mesoporous Mater., 2017, 243, 76–84 Search PubMed.
- M. Rico-Santacruz, E. Serrano, G. Marcì, E. I. García-López and J. García-Martínez, Chem.–Eur. J., 2015, 21, 18338–18344 Search PubMed.
- C. S. J. Cazin, M. Veith, P. Braunstein and R. B. Bedford, Synthesis, 2005, 622–626 Search PubMed.
- L. Wang, S. Shylesh, D. Dehe, T. Philippi, G. Dörr, A. Seifert, Z. Zhou, M. Hartmann, R. N. K. Taylor, M. Jia, S. Ernst and W. R. Thiel, ChemCatChem, 2012, 4, 395–400 Search PubMed.
- D. Elhamifar, O. Yari and S. Hajati, Appl. Organomet. Chem., 2018, 32, e4471 Search PubMed.
- D. Song, P. Zhang, Y. Sun, Q. Zhang and Y. Guo, Microporous Mesoporous Mater., 2019, 279, 352–363 Search PubMed.
- M. Shaker and D. Elhamifar, Colloids Surf., A, 2021, 608, 125603 Search PubMed.
- Y. Wei, G. Li, C. Wang and H. Guo, J. Colloid Interface Sci., 2021, 586, 233–242 Search PubMed.
- O. Yari, D. Elhamifar and M. Shaker, J. Organomet. Chem., 2021, 940, 121787 Search PubMed.
- Y. Maki, Y. Ide and T. Okada, Chem. Eng. J., 2016, 299, 367–372 Search PubMed.
- S. Abedi, B. Karimi, F. Kazemi, M. Bostina and H. Vali, Org. Biomol. Chem., 2013, 11, 416–419 Search PubMed.
- W. Yan, S. M. Mahurin, S. H. Overbury and S. Dai, Chem. Mater., 2005, 17, 1923–1925 Search PubMed.
- A. Ahadi, H. Alamgholiloo, S. Rostamnia, X. Liu, M. Shokouhimehr, D. A. Alonso and R. Luque, ChemCatChem, 2019, 11, 4803–4809 Search PubMed.
- D. Elhamifar, O. Yari and B. Karimi, J. Colloid Interface Sci., 2017, 500, 212–219 Search PubMed.
- D. Song, Q. Zhang, Y. Sun, P. Zhang, Y.-H. Guo and J.-L. Hu, ChemCatChem, 2018, 10, 4953–4965 Search PubMed.
- F. Su, S. An, D. Song, X. Zhang, B. Lu and Y. Guo, J. Mater. Chem. A, 2014, 2, 14127–14138 Search PubMed.
- D. Song, S. An, Y. Sun and Y. Guo, J. Catal., 2016, 333, 184–199 Search PubMed.
- C. Baleizão, B. Gigante, D. Das, M. Álvaro, H. Garcia and A. Corma, J. Catal., 2004, 223, 106–113 Search PubMed.
- S. Verma, M. Nandi, A. Modak, S. L. Jain and A. Bhaumik, Adv. Synth. Catal., 2011, 353, 1897–1902 Search PubMed.
- P. Borah, X. Ma, K. T. Nguyen and Y. Zhao, Angew. Chem., Int. Ed., 2012, 51, 7756–7761 Search PubMed.
- S. Farahmand, M. Ghiaci, M. Vatanparast and J. S. Razavizadeh, New J. Chem., 2020, 44, 7517–7527 Search PubMed.
- S. Zhou, B. Wang, S. Gao, Y. Ding and Y. Kong, Appl. Surf. Sci., 2017, 397, 183–191 Search PubMed.
- A. Feliczak, K. Walczak, A. Wawrzyńczak and I. Nowak, Catal. Today, 2009, 140, 23–29 Search PubMed.
- S. An, D. Song, Y. Sun, Y. Guo and Q. Shang, Microporous Mesoporous Mater., 2016, 226, 396–405 Search PubMed.
- L. Xu, Y. Wang, X. Yang, X. Yu, Y. Guo and J. H. Clark, Green Chem., 2008, 10, 746–755 Search PubMed.
- L. Xu, W. Li, J. Hu, K. Li, X. Yang, F. Ma, Y. Guo, X. Yu and Y. Guo, J. Mater. Chem., 2009, 19, 8571–8579 Search PubMed.
- A. P. Singh, N. Torita, S. Shylesh, N. Iwasa and M. Arai, Catal. Lett., 2009, 132, 492–499 Search PubMed.
- S. Shylesh, C. Srilakshmi, A. P. Singh and B. G. Anderson, Microporous Mesoporous Mater., 2007, 99, 334–344 Search PubMed.
- Y. Shiraishi, H. Ohara and T. Hirai, J. Catal., 2008, 254, 365–373 Search PubMed.
- M. Amini, M. M. Haghdoost and M. Bagherzadeh, Coord. Chem. Rev., 2013, 257, 1093–1121 Search PubMed.
- M. Jia and W. R. Thiel, Chem. Commun., 2002, 2, 2392–2393 Search PubMed.
- A. G. Moaser, A. Ahadi, S. Rouhani, B. B. Mamba, T. A.-M. Msagati, S. Rostamnia, T. Kavetskyy, S. Dugheri, S. Khaksar, A. Hasanzadeh and M. Shokouhimehr, J. Mol. Liq., 2020, 312, 113388 Search PubMed.
- M. Vasconcellos-Dias, C. D. Nunes, P. D. Vaz, P. Ferreira, P. Brandão, V. Félix and M. J. Calhorda, J. Catal., 2008, 256, 301–311 Search PubMed.
- A. Castro, J. C. Alonso, A. A. Valente, P. Neves, P. Brandão, V. Félix and P. Ferreira, Eur. J. Inorg. Chem., 2010, 1405–1412 Search PubMed.
- E. D. Koutsouroubi, A. K. Xylouri and G. S. Armatas, Chem. Commun., 2015, 51, 4481–4484 Search PubMed.
- E. D. Koutsouroubi, I. T. Papadas and G. S. Armatas, ChemPlusChem, 2016, 81, 947–954 Search PubMed.
- M. Waki, N. Mizoshita, T. Tani and S. Inagaki, Angew. Chem., Int. Ed., 2011, 50, 11667–11671 Search PubMed.
- M. Waki, Y. Maegawa, K. Hara, Y. Goto, S. Shirai, Y. Yamada, N. Mizoshita, T. Tani, W. J. Chun, S. Muratsugu, M. Tada, A. Fukuoka and S. Inagaki, J. Am. Chem. Soc., 2014, 136, 4003–4011 Search PubMed.
- S. Ishikawa, Y. Maegawa, M. Waki and S. Inagaki, ACS Catal., 2018, 8, 4160–4169 Search PubMed.
- S. Kataoka and S. Inagaki, Eur. J. Inorg. Chem., 2020, 4083–4087 Search PubMed.
- B. Karimi, M. Ghoreishi-Nezhad and J. H. Clark, Org. Lett., 2005, 7, 625–628 Search PubMed.
- B. Karimi and M. Khorasani, ACS Catal., 2013, 3, 1657–1664 Search PubMed.
- R. Zhang, W. Ding, B. Tu and D. Zhao, Chem. Mater., 2007, 19, 4379–4381 Search PubMed.
- B. Karimi, F. B. Rostami, M. Khorasani, D. Elhamifar and H. Vali, Tetrahydron, 2014, 70, 6114–6119 Search PubMed.
- B. Karimi, M. Khorasani, F. B. Rostami, D. Elhamifar and H. Vali, ChemPlusChem, 2015, 80, 990–999 Search PubMed.
- Q. Yang, D. Han, H. Yang and C. Li, Chem.–Asian J., 2008, 3, 1214–1229 Search PubMed.
- K. Kamata, K. Yonehara, Y. Sumida, K. Yamaguchi, S. Hikichi and N. Mizuno, Science, 2003, 300, 964–966 Search PubMed.
- C. Baleizão and H. Garcia, Chem. Rev., 2006, 106, 3987–4043 Search PubMed.
- A. Nodzewska, A. Wadolowska and M. Watkinson, Coord. Chem. Rev., 2019, 382, 181–216 Search PubMed.
- J. Hu, Q. Wu, W. Li, L. Ma, F. Su, Y. Guo and Y. Qiu, ChemSusChem, 2011, 4, 1813–1822 Search PubMed.
- L. Ma, F. Su, W. Guo, S. Zhang, Y. Guo and J. Hu, Microporous Mesoporous Mater., 2013, 169, 16–24 Search PubMed.
- D. Elhamifar and A. Shábani, Chem.–Eur. J., 2014, 20, 3212–3217 Search PubMed.
- M. Nasr-Esfahani, D. Elhamifar, T. Amadeh and B. Karimi, RSC Adv., 2015, 5, 13087–13094 Search PubMed.
- M. Neysi, D. Elhamifar and M. Norouzi, Mater. Chem. Phys., 2020, 243, 122589 Search PubMed.
- M. Norouzi and D. Elhamifar, Catal. Lett., 2019, 149, 619–628 Search PubMed.
- X. Wang, I. Thiel, A. Fedorov, C. Copéret, V. Mougel and M. Fontecave, Chem. Sci., 2017, 8, 8204–8213 Search PubMed.
- M. D. Sampson, A. D. Nguyen, K. A. Grice, C. E. Moore, A. L. Rheingold and C. P. Kubiak, J. Am. Chem. Soc., 2014, 136, 5460–5471 Search PubMed.
- M. Bourrez, F. Molton, S. Chardon-Noblat and A. Deronzier, Angew. Chem., Int. Ed., 2011, 50, 9903–9906 Search PubMed.
- J. Hawecker, J. M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1983, 536–538 Search PubMed.
- J. Hawecker, J. M. Lehn and R. Ziessel, Helv. Chim. Acta, 1986, 69, 1990–2012 Search PubMed.
- M. Waki, K.-i. Yamanaka, S. Shirai, Y. Maegawa, Y. Goto, Y. Yamada and S. Inagaki, Chem.–Eur. J., 2018, 24, 3846–3853 Search PubMed.
- S. Shirai, M. Waki, Y. Maegawa, Y. Yamada and S. Inagaki, New J. Chem., 2019, 43, 2471–2478 Search PubMed.
- X. Liu, Y. Goto, Y. Maegawa, T. Ohsuna and S. Inagaki, APL Mater., 2014, 2, 113308 Search PubMed.
- S. Zhang, M. Li, W. Qiu, J. Han, H. Wang and X. Liu, Appl. Catal., B, 2019, 259, 118113 Search PubMed.
- M. Waki, M. Ikai, Y. Goto, Y. Maegawa and S. Inagaki, Eur. J. Inorg. Chem., 2021, 1624–1631 Search PubMed.
- Z. Sun, X. Zhou, W. Luo, Q. Yue, Y. Zhang, X. Cheng, W. Li, B. Kong, Y. Deng and D. Zhao, Nano Today, 2016, 11, 464–482 Search PubMed.
- T. Cheng, D. Zhang, H. Li and G. Liu, Green Chem., 2014, 16, 3401–3427 Search PubMed.
- A. C. Gomes, M. J. Ferreira, S. M. Bruno, N. Bion, P. Ferreira, A. A. Valente, M. Pillinger, J. Rocha and I. S. Gonçalves, Dalton Trans., 2013, 42, 14612–14620 Search PubMed.
- A. C. Gomes, M. A. O. Lourenço, S. M. Bruno, P. Ferreira, A. A. Valente, M. Pillinger and I. S. Gonçalves, J. Organomet. Chem., 2014, 751, 501–507 Search PubMed.
- D. Elhamifar and E. Nazari, ChemPlusChem, 2015, 80, 820–826 Search PubMed.
- E. Gu, W. Zhong and X. Liu, RSC Adv., 2016, 6, 98406–98412 Search PubMed.
- A. Akbari, M. G. Dekamin, A. Yaghoubi and M. R. Naimi-Jamal, Sci. Rep., 2020, 10, 10646 Search PubMed.
- T. Seki, K. McEleney and C. M. Crudden, Chem. Commun., 2012, 48, 6369–6371 Search PubMed.
- S. Sisodiya, A. Lazar, S. Shylesh, L. Wang, W. R. Thiel and A. P. Singh, Catal. Commun., 2012, 25, 22–27 Search PubMed.
- D. Zhang, J. Xu, Q. Zhao, T. Cheng and G. Liu, ChemCatChem, 2014, 6, 2998–3003 Search PubMed.
- Y. Zhao, R. Jin, Y. Chou, Y. Li, J. Lin and G. Liu, RSC Adv., 2017, 7, 22592–22598 Search PubMed.
- A. Lazar, S. C. George, P. R. Jithesh, C. P. Vinod and A. P. Singh, Appl. Catal., A, 2016, 513, 138–146 Search PubMed.
- A. Lazar, S. Silpa, C. P. Vinod and A. P. Singh, Mol. Catal., 2017, 440, 66–74 Search PubMed.
- J. Huang, F. Zhu, W. He, F. Zhang, W. Wang and H. Li, J. Am. Chem. Soc., 2010, 132, 1492–1493 Search PubMed.
- V. Balzani and A. Juris, Coord. Chem. Rev., 2001, 211, 97–115 Search PubMed.
- A. Jana, J. Mondal, P. Borah, S. Mondal, A. Bhaumik and Y. Zhao, Chem. Commun., 2015, 51, 10746–10749 Search PubMed.
- H. Takeda, M. Ohashi, Y. Goto, T. Ohsuna, T. Tani and S. Inagaki, Adv. Funct. Mater., 2016, 26, 5068–5077 Search PubMed.
- N. Ishito, H. Kobayashi, K. Nakajima, Y. Maegawa, S. Inagaki, K. Hara and A. Fukuoka, Chem.–Eur. J., 2015, 21, 15564–15569 Search PubMed.
- Y. Kuramochi, M. Sekine, K. Kitamura, Y. Maegawa, Y. Goto, S. Shirai, S. Inagaki and H. Ishida, Chem.–Eur. J., 2017, 23, 10301–10309 Search PubMed.
- M. Waki, S. Shirai, K.-i. Yamanaka, Y. Maegawa and S. Inagaki, RSC Adv., 2020, 10, 13960–13967 Search PubMed.
- B. Karimi, D. Elhamifar, O. Yari, M. Khorasani, H. Vali, J. H. Clark and A. J. Hunt, Chem.–Eur. J., 2012, 18, 13520–13530 Search PubMed.
- B. Karimi, O. Yari, M. Khorasani, H. Vali and F. Mansouri, ChemCatChem, 2018, 10, 1783–1787 Search PubMed.
- S. Omar and R. Abu-Reziq, Appl. Sci., 2020, 10, 5769 Search PubMed.
- Y. Yang, W. Zhang, F. Yang, B. Zhou, D. Zeng, N. Zhang, G. Zhao, S. Hao and X. Zhang, Nanoscale, 2018, 10, 2199–2206 Search PubMed.
- N. Yao, C. Chen, D. J. Li and Y. L. Hu, J. Environ. Chem. Eng., 2020, 8, 103953 Search PubMed.
- H. Huo, Y. Jiang, Z. Wang, Y. Hu, T. Zhao, X. Liu, X. Xu and K. Lin, J. Mater. Sci., 2021, 56, 364–379 Search PubMed.
- R. Liu, R. Jin, L. Kong, J. Wang, C. Chen, T. Cheng and G. Liu, Chem.–Asian J., 2013, 8, 3108–3115 Search PubMed.
- F. Zhou, X. Hu, M. Gao, T. Cheng and G. Liu, Green Chem., 2016, 18, 5651–5657 Search PubMed.
- K. Matsui, Y. Maegawa, M. Waki, S. Inagaki and Y. Yamamoto, Catal. Sci. Technol., 2018, 8, 534–539 Search PubMed.
- T. Himiyama, M. Waki, Y. Maegawa and S. Inagaki, Angew. Chem., Int. Ed., 2019, 58, 9150–9154 Search PubMed.
- J. Huang, F. Zhang and H. Li, Appl. Catal., A, 2012, 431–432, 95–103 Search PubMed.
- Y. Maegawa and S. Inagaki, Dalton Trans., 2015, 44, 13007–13016 Search PubMed.
- X. Liu, Y. Maegawa, Y. Goto, K. Hara and S. Inagaki, Angew. Chem., Int. Ed., 2016, 55, 7943–7947 Search PubMed.
- S. Zhang, H. Wang, M. Li, J. Han, S. Inagaki and X. Liu, Dalton Trans., 2017, 46, 9369–9374 Search PubMed.
- H. Takeda, M. Ohashi, Y. Goto, T. Ohsuna, T. Tani and S. Inagaki, Chem.–Eur. J., 2014, 20, 9130–9136 Search PubMed.
- W. R. Grüning, G. Siddiqi, O. V. Safonova and C. Copéret, Adv. Synth. Catal., 2014, 356, 673–679 Search PubMed.
- S. Yamaguchi, Y. Maegawa, N. Onishi, R. Kanega, M. Waki, Y. Himeda and S. Inagaki, ChemCatChem, 2019, 11, 4797–4802 Search PubMed.
- S. Yamaguchi and S. Hashimoto, Asian J. Org. Chem., 2020, 9, 99–104 Search PubMed.
- M. Shimizu, K. Michikawa, Y. Maegawa, S. Inagaki and K.-i. Fujita, ACS Appl. Nano Mater., 2020, 3, 2527–2535 Search PubMed.
- S. Yamaguchi, Y. Maegawa, K.-i. Fujita and S. Inagaki, ChemSusChem, 2021, 14, 1074–1081 Search PubMed.
- C. Chen, L. Kong, T. Cheng, R. Jin and G. Liu, Chem. Commun., 2014, 50, 10891–10893 Search PubMed.
- B. Deng, W. Xiao, C. Li, F. Zhou, X. Xia, T. Cheng and G. Liu, J. Catal., 2014, 320, 70–76 Search PubMed.
- S. Zhang, H. Wang, M. Li, J. Han, X. Liu and J. Gong, Chem. Sci., 2017, 8, 4489–4496 Search PubMed.
- S. De, L. Gevers, A.-H. Emwas and J. Gascon, ACS Sustainable Chem. Eng., 2019, 7, 3933–3939 Search PubMed.
- S. Zhang, M. Li, Q. Wu, H. Yang, J. Han, H. Wang and X. Liu, Appl. Catal., B, 2018, 236, 466–474 Search PubMed.
- A. D. Marchese, T. Adrianov and M. Lautens, Angew. Chem., Int. Ed., 2021, 60, 16750–16762 Search PubMed.
- E. D. Boyes, A. P. LaGrow, M. R. Ward, R. W. Mitchell and P. L. Gai, Acc. Chem. Res., 2020, 53, 390–399 Search PubMed.
- S. L. Poe, M. Kobašlija and D. T. McQuade, J. Am. Chem. Soc., 2007, 129, 9216–9221 Search PubMed.
- R. Jin, K. Liu, D. Xia, Q. Qian, G. Liu and H. Li, Adv. Synth. Catal., 2012, 354, 3265–3274 Search PubMed.
- D. Elhamifar, H. Khanmohammadi and D. Elhamifar, RSC Adv., 2017, 7, 54789–54796 Search PubMed.
- J. R. Li and Y. L. Hu, Croat. Chem. Acta, 2020, 93, 105–110 Search PubMed.
- B. K. Bahuleyan, B. R. Jermy, I. Y. Ahn, H. Suh, D.-W. Park, C. S. Ha and I. Kim, Catal. Commun., 2009, 11, 252–256 Search PubMed.
- W. Li, H. Cheng, W. Lin, G. Liang, C. Zhang and F. Zhao, J. Mol. Catal. A: Chem., 2016, 411, 214–221 Search PubMed.
- K. Hong, M. Sajjadi, J. M. Suh, K. Zhang, M. Nasrollahzadeh, H. W. Jang, R. S. Varma and M. Shokouhimehr, ACS Appl. Nano Mater., 2020, 3, 2070–2103 Search PubMed.
- H. Qiu, S. M. Sarkar, D.-H. Lee and M.-J. Jin, Green Chem., 2008, 10, 37–40 Search PubMed.
- B. Karimi, D. Elhamifar, J. H. Clark and A. J. Hunt, Chem.–Eur. J., 2010, 16, 8047–8053 Search PubMed.
- A. Ahadi, S. Rostamnia, P. Panahi, L. D. Wilson, Q. Kong, Z. An and M. Shokouhimehr, Catalysts, 2019, 9, 140 Search PubMed.
- A. Corma, D. Das, H. García and A. Leyva, J. Catal., 2005, 229, 322–331 Search PubMed.
- E. Doustkhah, S. Rostamnia, M. Imura, Y. Ide, S. Mohammadi, C. J. T. Hyland, J. You, N. Tsunoji, B. Zeynizadeh and Y. Yamauchi, RSC Adv., 2017, 7, 56306–56310 Search PubMed.
- S. Elavarasan, K. Kala, I. Muhammad, A. Bhaumik and M. Sasidharan, Mol. Catal., 2019, 476, 110521 Search PubMed.
- A. Modak, J. Mondal, V. K. Aswal and A. Bhaumik, J. Mater. Chem., 2010, 20, 8099–8106 Search PubMed.
- A. Modak, J. Mondal and A. Bhaumik, Green Chem., 2012, 14, 2840–2855 Search PubMed.
- D. Elhamifar, B. Karimi, J. Rastegar and M. H. Banakar, ChemCatChem, 2013, 5, 2418–2424 Search PubMed.
- M. Shaker and D. Elhamifar, Tetrahedron Lett., 2020, 61, 152481 Search PubMed.
- M. Waki and S. Inagaki, Appl. Organomet. Chem., 2021, 35, e6341 Search PubMed.
- N. Linares, A. E. Sepúlveda, J. R. Berenguer, E. Lalinde and J. Garcia-Martinez, Microporous Mesoporous Mater., 2012, 158, 300–308 Search PubMed.
- J. Liu, H. Q. Yang, F. Kleitz, Z. G. Chen, T. Yang, E. Strounina, G. Q. Lu and S. Z. Qiao, Adv. Funct. Mater., 2012, 22, 591–599 Search PubMed.
- C. Liu, R. Tan, D. Yin, N. Yu and Y. Zhou, Chin. J. Catal., 2010, 31, 1369–1373 Search PubMed.
- B. Karimi, M. Khorasani, H. Vali, C. Vargas and R. Luque, ACS Catal., 2015, 5, 4189–4200 Search PubMed.
- B. Karimi, D. Elhamifar, J. H. Clark and A. J. Hunt, Org. Biomol. Chem., 2011, 9, 7420–7426 Search PubMed.
- M. Shaker and D. Elhamifar, Mater. Today Chem., 2020, 18, 100377 Search PubMed.
- L. Luza, A. Gual, C. P. Rambor, D. Eberhardt, S. R. Teixeira, F. Bernardi, D. L. Baptista and J. Dupont, Phys. Chem. Chem. Phys., 2014, 16, 18088–18091 Search PubMed.
- L. Luza, C. P. Rambor, A. Gual, F. Bernardi, J. B. Domingos, T. Grehl, P. Brüner and J. Dupont, ACS Catal., 2016, 6, 6478–6486 Search PubMed.
- M. Sudharsan, S. Subramanian, A. J. Amali and D. Suresh, ChemistrySelect, 2020, 5, 6131–6140 Search PubMed.
- M. Lv, Q. Xin, B. Bian, S. Yu, S. Liu, L. Li, C. Xie and Y. Liu, Dalton Trans., 2020, 49, 418–430 Search PubMed.
- X. Liu, X. Li, Z. Guan, J. Liu, J. Zhao, Y. Yang and Q. Yang, Chem. Commun., 2011, 47, 8073–8075 Search PubMed.
- J. Sun, H. Wang, X. Gao, X. Zhu, Q. Ge, X. Liu and J. Han, Microporous Mesoporous Mater., 2017, 247, 1–8 Search PubMed.
- K. Mori, T. Sano, H. Kobayashi and H. Yamashita, J. Am. Chem. Soc., 2018, 140, 8902–8909 Search PubMed.
- K. Mori, S. Masuda, H. Tanaka, K. Yoshizawa, M. Che and H. Yamashita, Chem. Commun., 2017, 53, 4677–4680 Search PubMed.
- S. Masuda, K. Mori, Y. Futamura and H. Yamashita, ACS Catal., 2018, 8, 2277–2285 Search PubMed.
- G. Yang, Y. Kuwahara, S. Masuda, K. Mori, C. Louis and H. Yamashita, J. Mater. Chem. A, 2020, 8, 4437–4446 Search PubMed.
- Y. Kuwahara, Y. Fujie, T. Mihogi and H. Yamashita, ACS Catal., 2020, 10, 6356–6366 Search PubMed.
- K. J. Betsy, A. Lazar, A. Pavithran and C. P. Vinod, ACS Sustainable Chem. Eng., 2020, 8, 14765–14774 Search PubMed.
- E. Doustkhah, S. Rostamnia, B. Zeynizadeh, J. Kim, Y. Yamauchi and Y. Ide, Chem. Lett., 2018, 47, 1243–1245 Search PubMed.
- E. Doustkhah, H. Mohtasham, M. Farajzadeh, S. Rostamnia, Y. Wang, H. Arandiyan and M. H. N. Assadi, Microporous Mesoporous Mater., 2020, 293, 109832 Search PubMed.
- J. R. Deka, D. Saikia, P.-H. Chen, K.-T. Chen, H.-M. Kao and Y.-C. Yang, Mater. Res. Bull., 2020, 125, 110786 Search PubMed.
- A. Zebardasti, M. G. Dekamin and E. Doustkhah, Catalysts, 2021, 11, 621 Search PubMed.
- L. Gao, Z. Wang, Y. Liu, P. Liu, S. Gao, J. Gao and Y. Jiang, Chem. Commun., 2020, 56, 13547–13550 Search PubMed.
- S. Rostamnia, E. Doustkhah, R. Bulgar and B. Zeynizadeh, Microporous Mesoporous Mater., 2016, 225, 272–279 Search PubMed.
- M. Yoshida, K. Saito, H. Matsukawa, S. Yanagida, M. Ebina, Y. Maegawa, S. Inagaki, A. Kobayashi and M. Kato, J. Photochem. Photobiol., A, 2018, 358, 334–344 Search PubMed.
- Y. Naganawa, Y. Maegawa, H. Guo, S. S. Gholap, S. Tanaka, K. Sato, S. Inagaki and Y. Nakajima, Dalton Trans., 2019, 48, 5534–5540 Search PubMed.
- Y. Goto, Y. Maegawa, M. Horii and S. Inagaki, Microporous Mesoporous Mater., 2021, 313, 110854 Search PubMed.
- J. R. Deka, H.-M. Kao, S.-Y. Huang, W.-C. Chang, C.-C. Ting, P. C. Rath and C.-S. Chen, Chem.–Eur. J., 2014, 20, 894–903 Search PubMed.
- B. Karimi, Z. Naderi, M. Khorasani, H. M. Mirzaei and H. Vali, ChemCatChem, 2016, 8, 906–910 Search PubMed.
- P. R. Upadhyay, P. Gautam and V. Srivastava, Chem. Pap., 2019, 73, 2241–2253 Search PubMed.
- Y. Huo, J. Hu, Y. Tu, Z. Huang, S. Lin, Y. Hu and C. Feng, Appl. Organomet. Chem., 2020, 34, e5697 Search PubMed.
- M. Esmat, H. Mohtasham, Y. GadelHak, R. T. Mehrebani, R. Tahawy, S. Rostamnia, N. Fukata, S. Khaksar and E. Doustkhah, Catalysts, 2020, 10, 167, DOI:10.3390/catal10020167.
- C. Liu, R. Tan, N. Yu and D. Yin, Microporous Mesoporous Mater., 2010, 131, 162–169 Search PubMed.
- J. Mondal, N. Salam and A. Bhaumik, J. Nanosci. Nanotechnol., 2013, 13, 4883–4895 Search PubMed.
- M. A. O. Lourenço, L. Carneiro, A. Mayoral, I. Diaz, A. R. Silva and P. Ferreira, J. Catal., 2014, 320, 63–69 Search PubMed.
- L. Carneiro, A. R. Silva, M. A. O. Lourenço, A. Mayoral, I. Diaz and P. Ferreira, Eur. J. Inorg. Chem., 2016, 413–421 Search PubMed.
- X. B. Liu and Y. L. Hu, Silicon, 2021, 13, 4013–4024 Search PubMed.
- D. Elhamifar and H. Ardeshirfard, J. Colloid Interface Sci., 2017, 505, 1177–1184 Search PubMed.
- D. Elhamifar, F. Hosseinpoor, B. Karimi and S. Hajati, Microporous Mesoporous Mater., 2015, 204, 269–275 Search PubMed.
- M. Gholinejad, B. Karimi, A. Aminianfar and M. Khorasani, ChemPlusChem, 2015, 80, 1573–1579 Search PubMed.
- X. Zheng, M. Wang, Z. Sun, C. Chen, J. Ma and J. Xu, Catal. Commun., 2012, 29, 149–152 Search PubMed.
- H. Naeimi, V. Nejadshafiee and S. Masoum, RSC Adv., 2015, 5, 15006–15016 Search PubMed.
- H. Naeimi, V. Nejadshafiee and M. R. Islami, Bull. Chem. Soc. Jpn., 2016, 89, 212–219 Search PubMed.
- S. Ishikawa, Y. Maegawa, M. Waki and S. Inagaki, Appl. Catal., A, 2019, 575, 87–92 Search PubMed.
- Z. Gao, Y. Li, Z. Liu, Y. Zhang, F. Chen, P. An, W. Lu, J. Hu, C. You, J. Xu, X. Zhang and B. Sun, Nano Lett., 2021, 21, 3401–3409 Search PubMed.
- Y. Gao, S. Zhao, G. Zhang, L. Deng, J. Li, R. Sun, L. Li and C.-P. Wong, J. Mater. Sci., 2015, 50, 3399–3408 Search PubMed.
- P. Xu, Z. Wu, W. Dai, Y. Wang, M. Zheng, X. Su and Z. Teng, J. Taiwan Inst. Chem. Eng., 2020, 117, 287–293 Search PubMed.
- G. Du, J. Peng, Y. Zhang, H. Zhang, J. Lü and Y. Fang, Langmuir, 2017, 33, 5223–5235 Search PubMed.
- K. Shahzad, T. Najam, M. S. Bashir, M. A. Nazir, A. ur Rehman, M. A. Bashir and S. S. A. Shah, Inorg. Chem. Commun., 2021, 123, 108357 Search PubMed.
- E. De Canck, F. Nahra, K. Bevernaege, S. V. Broeck, J. Ouwehand, D. Maes, S. P. Nolan and P. Van Der Voort, ChemPhysChem, 2018, 19, 430–436 Search PubMed.
- B. Karimi, A. Bigdeli, A. A. Safari, M. Khorasani, H. Vali and S. K. Karimvand, ACS Comb. Sci., 2020, 22, 70–79 Search PubMed.
- B. Karimi, M. Gholinejad and M. Khorasani, Chem. Commun., 2012, 48, 8961–8963 Search PubMed.
- B. Karimi and F. K. Esfahani, Chem. Commun., 2011, 47, 10452–10454 Search PubMed.
- B. Karimi and F. K. Esfahani, Adv. Synth. Catal., 2012, 354, 1319–1326 Search PubMed.
- F.-X. Zhu, W. Wang and H.-X. Li, J. Am. Chem. Soc., 2011, 133, 11632–11640 Search PubMed.
- Y. Yang, W. Zhang, Y. Zhang, A. Zheng, H. Sun, X. Li, S. Liu, P. Zhang and X. Zhang, Nano Res., 2015, 8, 3404–3411 Search PubMed.
- H. Zou, R. Wang, X. Li, X. Wang, S. Zeng, S. Ding, L. Li, Z. Zhang and S. Qiu, J. Mater. Chem. A, 2014, 2, 12403–12412 Search PubMed.
- X. Wang, Y. He, C. Liu, Y. Liu, Z.-A. Qiao and Q. Huo, Nanoscale, 2016, 8, 13581–13588 Search PubMed.
- X. Huang, M. Zhang, M. Wang, W. Li, C. Wang, X. Hou, S. Luan and Q. Wang, Langmuir, 2018, 34, 3642–3653 Search PubMed.
- Y. Yao, X. Zhang, J. Peng and Q. Yang, Chem. Commun., 2015, 51, 3750–3753 Search PubMed.
- N. Ishito, K. Nakajima, Y. Maegawa, S. Inagaki and A. Fukuoka, Catal. Today, 2017, 298, 258–262 Search PubMed.
- Y. Horiuchi, D. D. Van, Y. Yonezawa, M. Saito, S. Dohshi, T.-H. Kim and M. Matsuoka, RSC Adv., 2015, 5, 72653–72658 Search PubMed.
- L. Luo, L. Bock, Y. Liang and R. Anwander, Eur. J. Inorg. Chem., 2020, 3967–3976 Search PubMed.
- S. Zhang, H. Wang, L. Tang, M. Li, J. Tian, Y. Cui, J. Han, X. Zhu and X. Liu, Appl. Catal., B, 2018, 220, 303–313 Search PubMed.
- L. Li, Z. Yang, W. Fan, L. He, C. Cui, J. Zou, W. Tang, O. Jacobson, Z. Wang, G. Niu, S. Hu and X. Chen, Adv. Funct. Mater., 2020, 30, 1907716 Search PubMed.
- M. P. Kapoor, A. K. Sinha, S. Seelan, S. Inagaki, S. Tsubota, H. Yoshida and M. Haruta, Chem. Commun., 2002, 2902–2903 Search PubMed.
- S. L. Allahgholipour and R. Baharfar, Monatsh. Chem., 2020, 151, 991–997 Search PubMed.
- X.-T. Lin, K. Matsumoto, Y. Maegawa, K. Takeuchi, N. Fukaya, K. Sato, S. Inagaki and J.-C. Choi, New J. Chem., 2021, 45, 9501–9505 Search PubMed.
- S. O. Ribeiro, P. L. Almeida, J. Pires, B. de Castro and S. S. Balula, Microporous Mesoporous Mater., 2020, 302, 110193 Search PubMed.
- M. Chen, C. Liang, F. Zhang and H. Li, ACS Sustainable Chem. Eng., 2014, 2, 486–492 Search PubMed.
- R. Anwander, Chem. Mater., 2001, 13, 4419–4438 Search PubMed.
- F. Zhang, Y. Zhu, Q. Lin, L. Zhang, X. Zhang and H. Wang, Energy Environ. Sci., 2021, 14, 2954–3009 Search PubMed.
- P. Serp, Nanoscale, 2021, 13, 5985–6004 Search PubMed.
- L. Zhang, X. Zhao, Z. Yuan, M. Wu and H. Zhou, J. Mater. Chem. A, 2021, 9, 3855–3879 Search PubMed.
- R. Sun, Y. Liao, S.-T. Bai, M. Zheng, C. Zhou, T. Zhang and B. F. Sels, Energy Environ. Sci., 2021, 14, 1247–1285 Search PubMed.
- B. Singh, V. Sharma, R. P. Gaikwad, P. Fornasiero, R. Zbořil and M. B. Gawande, Small, 2021, 17, 2006473 Search PubMed.
Footnote |
| † Dedicated to Professor Reiner Anwander on the occasion of his 60th birthday. |
| This journal is © The Royal Society of Chemistry 2021 |