
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign principles of noble metal-free electrocatalysts for hydrogen production in alkaline media: combining theory and experiment
Hyeonjung
Jung†
,
Seokhyun
Choung†
and
Jeong Woo
Han
 *
*
Department of Chemical Engineering, Pohang University of Science and Technology (POSTECH), Pohang, Gyeongbuk 37673, Republic of Korea. E-mail: jwhan@postech.ac.kr
First published on 19th October 2021
Abstract
Water electrolysis is a promising solution to convert renewable energy sources to hydrogen as a high-energy-density energy carrier. Although alkaline conditions extend the scope of electrocatalysts beyond precious metal-based materials to earth-abundant materials, the sluggish kinetics of cathodic and anodic reactions (hydrogen and oxygen evolution reactions, respectively) impede the development of practical electrocatalysts that do not use precious metals. This review discusses the rational design of efficient electrocatalysts by exploiting the understanding of alkaline hydrogen evolution reaction and oxygen evolution reaction mechanisms and of the electron structure–activity relationship, as achieved by combining experimental and computational approaches. The enhancement of water splitting not only deals with intrinsic catalytic activity but also includes the aspect of electrical conductivity and stability. Future perspectives to increase the synergy between theory and experiment are also proposed.
1. Introduction
Clean energy transitions to replace traditional carbon sources are being sought to mitigate fossil-fuel depletion, carbon emissions and environmental pollution.1,2 Conversion and storage of sustainable energy such as wind or solar energy to hydrogen as an energy-dense carrier has been advocated as a promising solution.3 Electrochemical water splitting (2H2O → 2H2 + O2) has been accepted as a potential hydrogen-production technology, due to the availability of water as a reactant, and stable output efficiency.4–8 It avoids emission of huge amounts of carbon dioxide and does not exacerbate fossil-fuel depletion, which are two major objections to conventional steam reforming of natural gas to yield hydrogen.9Water electrolysis consists of the hydrogen evolution reaction (HER; water reduction to hydrogen gas) as a cathodic half-reaction and the oxygen evolution reaction (OER; water oxidation to oxygen gas) as an anodic half-reaction. The thermodynamic potential requirement for splitting water into hydrogen and oxygen is theoretically 1.23 V vs. standard hydrogen electrode (SHE) under the standard conditions, but it always takes more energy (overpotential) than this in practice. To minimize the overpotential requirement and achieve efficient water electrolysis, it should be conducted in either a highly acidic medium or a highly alkaline medium.10–12 Acid electrolysis is usually performed using a proton exchange membrane (PEM), and has the advantages of high energy efficiency and fast hydrogen production rate, but uses expensive noble-metal electrocatalysts. Promising and relatively inexpensive transition metal oxides (TMOs) for OER catalysts mostly suffer from serious instability such as decomposition and metal dissolution in acidic and strongly oxidative environments at high operation voltages.13 Alkaline electrolysis overcomes some of these instability problems and extends the scope of electrocatalysts to earth-abundant materials.14,15 However, the alkaline medium of water splitting causes slow HER kinetics due to the low proton concentration.16–19 Therefore, increase of both activity and stability in alkaline media for the OER and HER has been huge research topics in electrochemistry.
To obtain an economically viable alkaline electrolyzer, the first choice should be to replace noble metal-based catalysts (Pt/C, IrO2) with inexpensive, earth-abundant metal-based catalysts, given that catalysts account for a large portion of the stack price.20 Although water electrolysis is considered a promising hydrogen-production method, the sluggish OER kinetics at the anode and sluggish HER kinetics at the cathode under alkaline conditions remain primary impediments to the development of low-cost electrocatalysts.21,22 Emerging materials based on earth-abundant transition metal (TM) elements have been evaluated for the alkaline HER and OER, and if applicable can solve the long-standing problem of low price efficiency. TMs and TM alloys have been widely studied theoretically and experimentally as HER catalysts.23–25 Emerging TM materials with ligands such as sulfides,26–32 phosphides,33–37,43 carbides,38–42 and hydroxides44–47 have been extensively investigated owing to their high activity comparable to noble metal-based catalysts.
For the OER, substantial research efforts have been devoted to investigating the activity of TMO catalysts that use perovskites,48–51 spinels,52–56 layered oxides including hydr(oxy)oxides44,57–60 and other types of oxides61–65 owing to their low cost, easy synthesizability, rich selection of structural combinations, and environmentally benign nature. Furthermore, they are stable in alkaline solution and have moderate conductivities, which make them good candidates for the electrocatalytic OER. Non-oxide catalysts like metal chalcogenides that include sulfides,66–69 selenides,70–73 and carbides74–77 have also been reported as promising electrocatalytic materials for the alkaline OER.
For these emerging materials to be widely accepted and further developed, the atomic-level mechanisms of catalysis must be understood and exploited. These mechanisms can be identified using interdisciplinary theoretical methods and high-resolution characterization and imaging techniques. Computational simulations that use density functional theory (DFT) have illuminated the intrinsic properties of catalysts by observing the properties of transition states and reaction intermediates, and have provided insights that enable rational design of catalysts.78 The simulations provide the interaction energies of atoms or molecules on a catalytic surface with sufficient accuracy, and thereby provide thermodynamic data for the targeted reaction. The nudged elastic band (NEB) method enables identification of the minimum-energy path (MEP) for a given chemical process if both the initial and final states are known, and provides kinetic data.79 DFT calculations can also investigate the charge distribution and electronic structure of catalyst systems, whereas such information is not easily obtained experimentally.80 Finding the relationship between the thermodynamic properties of catalysts and their electronic structural properties is an important step in increasing the performance of catalysts. By exploiting a deep understanding of material properties at the atomic scale, a combination of theoretical approaches with experimental methods has been successfully implemented to study catalytic activity, search for new material combinations, and eliminate inefficient trial-and-error searches. In addition, computer speed and memory are being increased rapidly, so high-throughput screening of material search spaces is being performed extensively by considering the accumulated knowledge about activity descriptors.
The blooming of the massive ab initio calculation database has opened up a new possibility of exploring the limitless material search space by means of machine learning (ML) techniques. Recently, ML-based high-throughput screening studies have successfully discovered a large number of new catalysts for the HER114 and OER.146 Furthermore, ML approaches have expanded our understanding on the physical nature of catalytic materials for water splitting, igniting the huge research interest of data-driven ML studies in the materials design field.
Several great review articles have covered the HER, OER, and overall water splitting catalysis, but a comprehensive theory-specialized perspective on the topic is missing. Two reviews18,81 have considered only one side of the water-splitting electrode. Many other researchers focused on specific materials such as homogeneous Co catalysts,82,83 heterogeneous Ni catalysts,84 borides and sulfides,85 specific morphology (low dimensional (1D/2D) and single/double atomic coordination),86,87 precious metals,88 photocatalysis,89 or bifunctional catalysts.90 Some other reviews have focused on specific design strategies such as support, interface and strain engineering.91,92 Here, we will consider the specificity of alkaline media while addressing a universal strategy to develop cost-efficient catalysts.
This review looks at recent progress towards overcoming the slow kinetics of the HER and OER under alkaline conditions, by combining computation and experimentation. First, several powerful descriptors for the HER and OER are introduced, focusing on the strategies to which the descriptors are applied in rational design to increase intrinsic catalytic activity. For the HER, hydrogen-binding strength has been a widely applied descriptor that can be tuned by electronic structure engineering, various nano-structuring concepts, and new active materials identified using machine learning. Several influential papers regarding the water dissociation barrier descriptor are highlighted, as a result of their efforts to increase the understanding and ability to manipulate the descriptor. The function of promoter species in the electrolyte is highlighted with important theoretical and experimental findings. For the OER, the descriptors for determining the two major mechanisms, i.e., the adsorbate-evolution mechanism (AEM) and lattice-oxygen oxidation mechanism (LOM), are introduced; oxygen vacancy formation energy, electron donating ability of B cations in the ABO3 perovskites, oxygen 2p-band center (Op) and metal 3d-band center (Md). In the AEM, several activity determining descriptors such as the Gibbs free energy difference of adsorbed O and OH intermediates (ΔGO − ΔGOH) and electronic structural descriptors including eg orbital occupancy, metal–oxygen covalency, and number of excess electrons (NEE) are explored. In the case of LOM, metal–oxygen covalency, oxygen anion and vacancy diffusion are discussed experimentally and computationally, with practical examples. Although the alkaline environment generally provides better catalyst stability than the acidic environment, the stability issues still exist under alkaline conditions and have not yet been completely elucidated. The stability and electrical conductivity of the materials are summarized comprehensively. The relationships among stability, conductivity and activity are reviewed, considering several strategies to increase the stability and the electrical conductivity.
We hope our review broadens the design principles of inexpensive catalysts for alkaline water splitting reactions by combining theory and experiment, and that it extends insight into the hydrogen oxidation and oxygen reduction reactions (HOR/ORR), which share similar intermediates with the HER and OER, and CO2 and NOx reduction that involves water dissociation reactions similar to those in the alkaline HER. We will also cover important experimental and computational considerations of alkaline media, so our design principles may also be applicable to other alkaline environmental reactions.
2. Alkaline hydrogen evolution reaction
2.1. HER mechanism under alkaline conditions
The HER under alkaline conditions involves water dissociation and coupling of hydrogen atoms. The general consensus on the mechanism has been well established to occur in two sequential steps; first, the water dissociation step occurs:| H2O + e− → H* + OH− (Volmer step, 120 mV dec−1), |
| H2O + H* + e− → H2(g) + OH− (Heyrovsky step, 40 mV dec−1), |
| H* + H* → H2(g) (Tafel step, 30 mV dec−1) |
The protons under the alkaline conditions are obtained by the Volmer reaction, which dissociates water molecules. These hydrogen atoms then recombine by either the Heyrovsky recombination reaction or the Tafel recombination reaction. For those two steps, hydrogen binding energy (HBE) is a determinant in predicting the reaction rate of the HER. Under acidic conditions, the H atom is the sole intermediate of the HER, so the HBE determines the HER activity, following the Sabatier principle that the key intermediate should have modest binding strength, neither strong nor weak. Strong binding of the catalyst to the H hinders the hydrogen desorption in the subsequent Tafel or Heyrovsky steps; weak binding hinders the Volmer step.
Despite the successful implementation of the HBE descriptor in acidic media, HBE could not solely explain the HER under alkaline conditions due to the complexity of the reaction environments. Long-standing debates have been conducted on the much lower HER activity in alkaline than in acidic media for the state-of-the-art Pt and Au catalysts (Fig. 1a).93 Although the HER mechanism under alkaline conditions is still elusive, many researchers consider that the Volmer step is the activity-limiting reaction, in which excess energy is required to overcome the high water dissociation barrier. The Markovic group have reported a series of results which implied that the water dissociation is a key step which actually limits HER kinetics.46 Oxophilic sites such as Ni(OH)2 can accelerate the water dissociation, increasing the HER activity three to five times under alkaline conditions, so it may serve as the active site for the water dissociation step (Fig. 1b). Therefore, along with HBE, other important descriptors have been highlighted including hydroxyl binding energy (OHBE), water dissociation barrier, solvated electrolyte cations, chemical dynamics in the double layer and co-adsorbed spectator species.
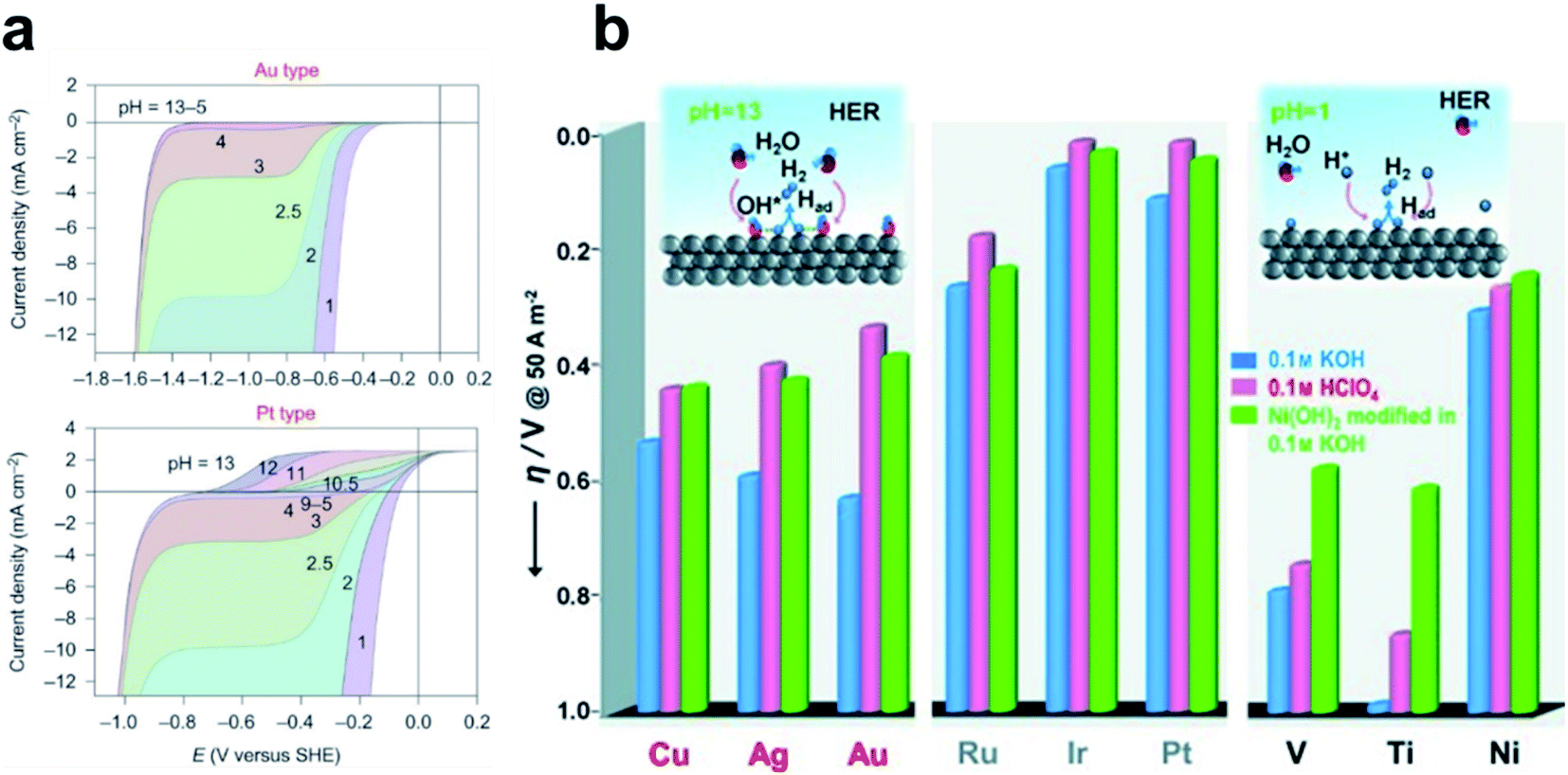 | ||
| Fig. 1 (a) pH-dependent current–potential polarization curves at rotation rates of 1600 rpm and sweep rate of 50 mV s−1 for Au(111) and Pt(111). Reproduced from ref. 93 Copyright 2013, Springer Nature. (b) Overpotential η required for a 5 mA cm−2 current density, in 0.1 M HClO4 and 0.1 M KOH for bare metal surfaces (Cu, Ag, Au, Ru, Ir, Pt, V, Ti and Ni) and Ni(OH)2-modified surfaces. Reproduced from ref. 46. Copyright 2012, Wiley. | ||
First, HBE as a practical descriptor under alkaline conditions will be discussed in Section 2.2, focusing on four aspects: (1) the volcano relationship between HBE and alkaline HER activity, (2) the linear relationship between HBE and the electronic structure, (3) nano-structuring of materials towards the optimal HBE, and (4) machine learning approaches towards the optimal HBE. Then, the importance of the water dissociation barrier, OHBE, and ‘dual active site’ strategy in the alkaline HER will be covered in Section 2.3. Finally, the role of electrolyte species will be discussed in Section 2.4, and conductivity issues will be covered in Section 2.5.
2.2. Hydrogen binding
Although many other influential factors such as OHBE, water dissociation barrier, and electrolyte dynamics should be considered to obtain a complete picture of HER kinetics under alkaline conditions, the HBE is still widely used as a HER activity descriptor under alkaline conditions due to its practicality. Sheng et al. provided important experimental evidence that the HER under alkaline conditions follows the HBE trend by performing the fundamental study of combining theoretical HBE calculation and the exchange current density measurement (Fig. 2a).95 Their results also demonstrated that HBE and current density have the volcano relationship under both acidic and alkaline conditions. A follow-up study that also combined theory and experiment96 showed that HBE was correlated well with current density, and overpotential in universal pH. Durst et al. reached a similar conclusion that the HBE serves as a powerful descriptor of the HER under alkaline conditions by conducting control experiments using platinum-group metals.97 In addition, several reports have successfully described the high alkaline HER activity of carbides and selenides using HBE as a single descriptor. Vrubel et al. found that Mo2C shows excellent activity and stability under both alkaline and acidic conditions comparable to that of the Pt/C catalyst, which was confirmed by the optimal HBE.104 The HBEs of monometallic carbide surfaces have been evaluated by DFT calculations in low-coverage and high-coverage carbide systems, which showed a clear correlation between HBE and exchange current density under the alkaline conditions.107 Zheng et al. synthesized a phase controlled CoSe2 by modifying the d-electron states of the Co atoms via P doping, hence optimizing the HBE in alkaline solution.110
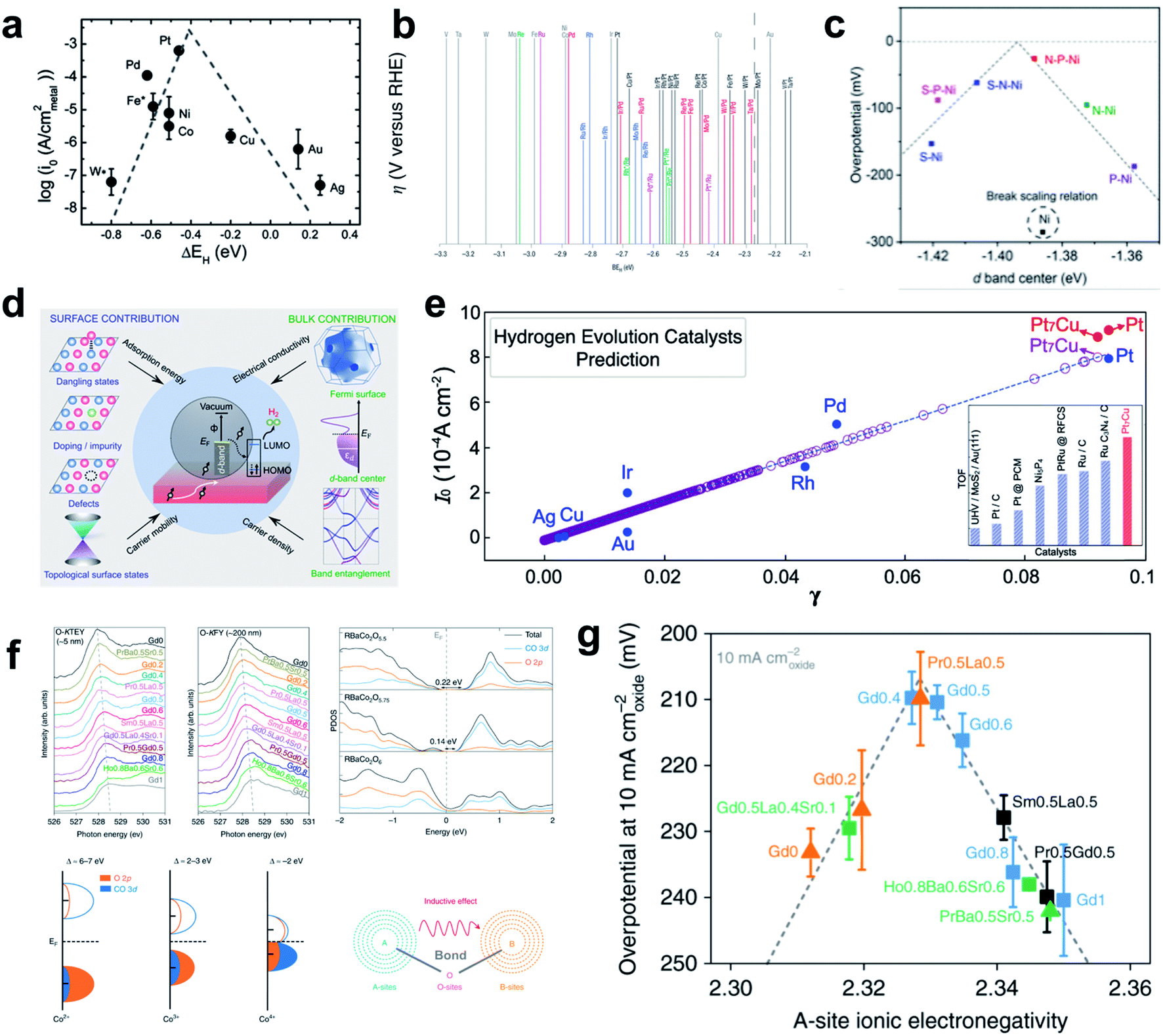 | ||
| Fig. 2 (a) Exchange current density as a function of HBE. Reproduced from ref. 95. Copyright 2013, Royal Society of Chemistry. (b) Overpotential as a function of HBE. Reproduced from ref. 100. Copyright 2004, Springer Nature. (c) Overpotential as a function of the d-band center of the heteroatom doped Ni catalyst. Reproduced from ref. 101. Copyright 2019, American Chemical Society. (d) Schematic of the electronic properties considered; electronic conductivity, carrier density, carrier mobility and adsorption energy. (e) Exchange-current density as a function of PBP. Reproduced from ref. 102. Copyright 2020, American Chemical Society. (f) Soft XAS spectra at the O–K edge collected in TEY mode, FY modes, PDOS calculations of Co 3d and O 2p orbitals for three perovskites, charge-transfer models of Co2+, Co3+, and Co4+ in covalent systems. (g) Inductive effects and electron exchange interactions between A-sites and B-sites in perovskite overpotential at 10 mA cm−2 as a function of A-site ionic electronegativity. Reproduced from ref. 103. Copyright 2019, Springer Nature. | ||
Xu et al. found that the ‘projected berry phase’ (PBP) value, which depends solely on the topological structure and electronic structure, has a good linear relationship with HER exchange current density (Fig. 2d and e).102 They found that the PBP descriptor is linearly correlated with the HBE, electronic structure, work function, Fermi level and d-band center model. To test the idea, and to apply the strategy in practical search for an HER material, they considered the PBP to select Pt7Cu model catalyst, and synthesized it. It showed superior HER activity, and thereby confirmed the relationship between the electronic structure and HBE.
Tuning the A site ionic electronegativity (AIE) of perovskite materials103 is a good example of a rational strategy suggested by Shao and coworkers to design novel materials by optimizing the electronic structure. Comparison of cobalt-containing single perovskites ABO3−δ and a double perovskite family AA′B2O6−δ. (A′ ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) = lanthanide) showed a solid volcano relationship between AIE and the HER activity (Fig. 2f and g). They found that a material with AIE = ∼2.33 would have better catalytic activity than Pt/C; measurements using (Gd0.5La0.5)BaCo2O5.5+δ confirmed this prediction.
= lanthanide) showed a solid volcano relationship between AIE and the HER activity (Fig. 2f and g). They found that a material with AIE = ∼2.33 would have better catalytic activity than Pt/C; measurements using (Gd0.5La0.5)BaCo2O5.5+δ confirmed this prediction.
Despite some studies that questioned the ability of the electronic structure to serve as a universal descriptor,44 this approach has been effective in providing an important physical background for materials design and enabling a direct correlation between catalytic activity and surface electronic properties.
Monometallic carbide surfaces have been evaluated by using DFT to investigate the HBE in low-coverage and high-coverage carbide systems.107 Analysis results identified carbides that maximize the HER; when synthesized, they showed a clear correlation between the adsorption free energy of H (ΔGH) and exchange current density concluding that HBE is a useful guideline for HER activity in predicting carbide activity under the alkaline conditions. In addition, the coverage dependent ΔGH implied that the surface coverage effects are three times more sensitive on the carbide system than on the pure-metal system. This result may suggest a viable design strategy to optimize the HBE on carbide and similar systems (Fig. 3a and b).
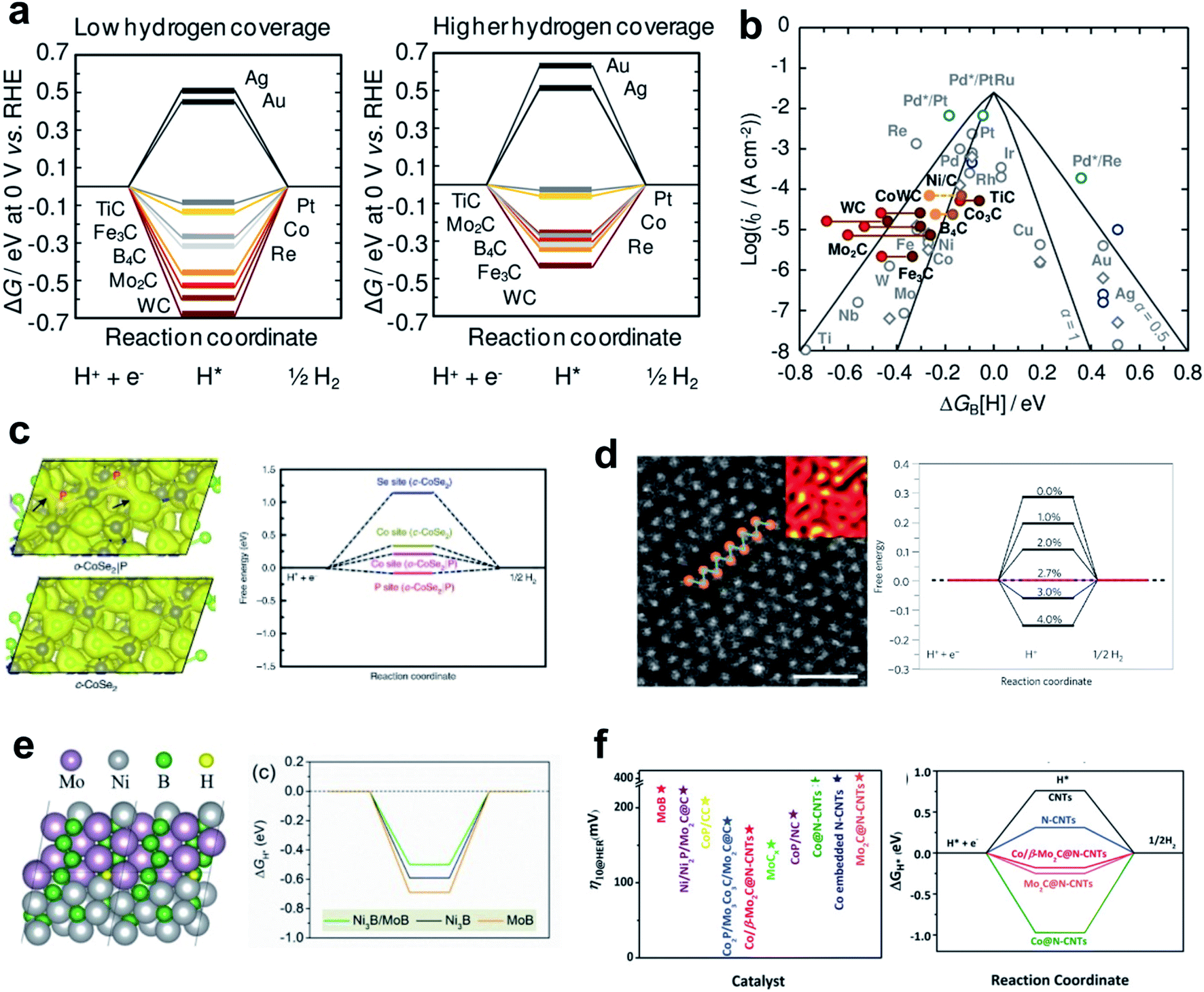 | ||
| Fig. 3 (a) Free-energy diagrams for the electrochemical reduction of H+ in 1/4 ML coverage and 1/2 ML coverage. (b) Volcano plot for the HER on polycrystalline (gray circles) or single-crystal (dark blue circles) TMs (1/4 ML H*). Reproduced from ref. 107. Copyright 2014, American Chemical Society. (c). Calculated charge density distribution for o-CoSe2|P and free energy diagrams for hydrogen adsorption. Reproduced from ref. 110. Copyright 2018, Springer Nature. (d) High-resolution STEM images of an as-exfoliated WS2 monolayer 1T superlattice and the free energy diagram of H+ adsorption. Reproduced from ref. 111. Copyright 2013, Springer Nature. (e) H* adsorbed on the Ni3B/MoB heterosturcture; free-energy diagram (at U = 0 V) for the HER on the Ni3B/MoB, Ni3B and MoB structure. Reproduced from ref. 113. Copyright 2020, Elsevier. (f) η10 of recently reported catalysts and calculated Gibbs free energy for H* adsorption on different catalysts. Reproduced from ref. 106. Copyright 2019, Wiley. | ||
Nørskov and his coworkers conducted several pioneering studies that have broadened the material space of sulfides for HER materials.108 They opened up the potential application of MoS2 in the HER by showing the theoretical HBE of the material followed by in-depth characterizations. They found that the edge sites of the MoS2 have similar adsorption behavior to Pt, whereas the MoS2(0001) facet was inactive to the H binding. Subsequent studies maximized the fraction of active edge sites by nanostructuring.27 Although MoS2 showed poor activity under alkaline conditions, breakthrough results were obtained by introducing conductive elements such as Ni and Co, which can foster the water dissociation steps.109
Active and stable phases of dichalcogenides also have been evaluated as HER catalysts. Zheng et al. developed o-CoSe2 by phase transforming c-CoSe2 with controllable P doping that provided weakened electronegative sites and thereby tuned the d-electron states of the Co atoms.110 Their nano-structuring strategy, phase transformation by hetero-atom doping, has achieved high HER activity and stability in alkaline solution. The active sites of o-CoSe2 are always closer to the optimal HER activity than c-CoSe2 (Fig. 3c).
WS2 also shows phase-dependent reactivity. Voiry et al. found that the abundant 1T phase of WS2 has high HER activity.111 In the 1T WS2 phase, the exfoliated nanosheet structure of WS2 has a certain amount of tensile and compressive strain, which contribute to HER activity. DFT calculation revealed that the 1T phase is stable enough owing to a high phase transformation barrier. In addition, an ideally introduced tensile strain controlled the optimal free energy of atomic hydrogen adsorption (Fig. 3d). The applied tensile strain induces shift of the density of states near the Fermi level. Similar activity enhancement strategies by strain engineering can be applied generally under the alkaline conditions.112
Heterostructuring is another nano-structuring strategy that can tune catalyst activity. Huang et al. designed a grain boundary structure between Ni3B and MoB which serves as the active center with induced strain.113 The grain boundary enriched with bimetallic borides created additional defects and large voids, which served as hydrogen-binding sites and yielded near-optimal HBE (Fig. 3e). Ouyang et al. developed an efficient heterostructure HER catalyst comprising Co/β-Mo2C@N-CNTs. The heterointerface site enriched the active sites, resulting in much lower overpotential, which was confirmed by overpotential measurement and theoretical analysis (Fig. 3f).106
Tran et al. provided an automated machine learning scheme to seek materials, which can automatically identify the best candidates for the HER from millions of possible structures by using only the HBE descriptor.114 In this study, they obtained 1499 intermetallic bulk structures from the Materials Project database, and possible surfaces were cleaved from the bulk structures. The HBE on each surface was calculated using DFT to make a fingerprint-like input database combined with three atomic features. The surrogate ML model was then trained by the input database using the Tree-based Pipeline Optimization Tool (TPOT), and the trained ML model was used to predict the HBE of unknown candidate materials. They identified all possible surface structures and suggested that even for the same materials the HBE can vary depending on the coordination structure. The model identified 102 candidate HER alloys, and all have optimal HBE (Fig. 4a).
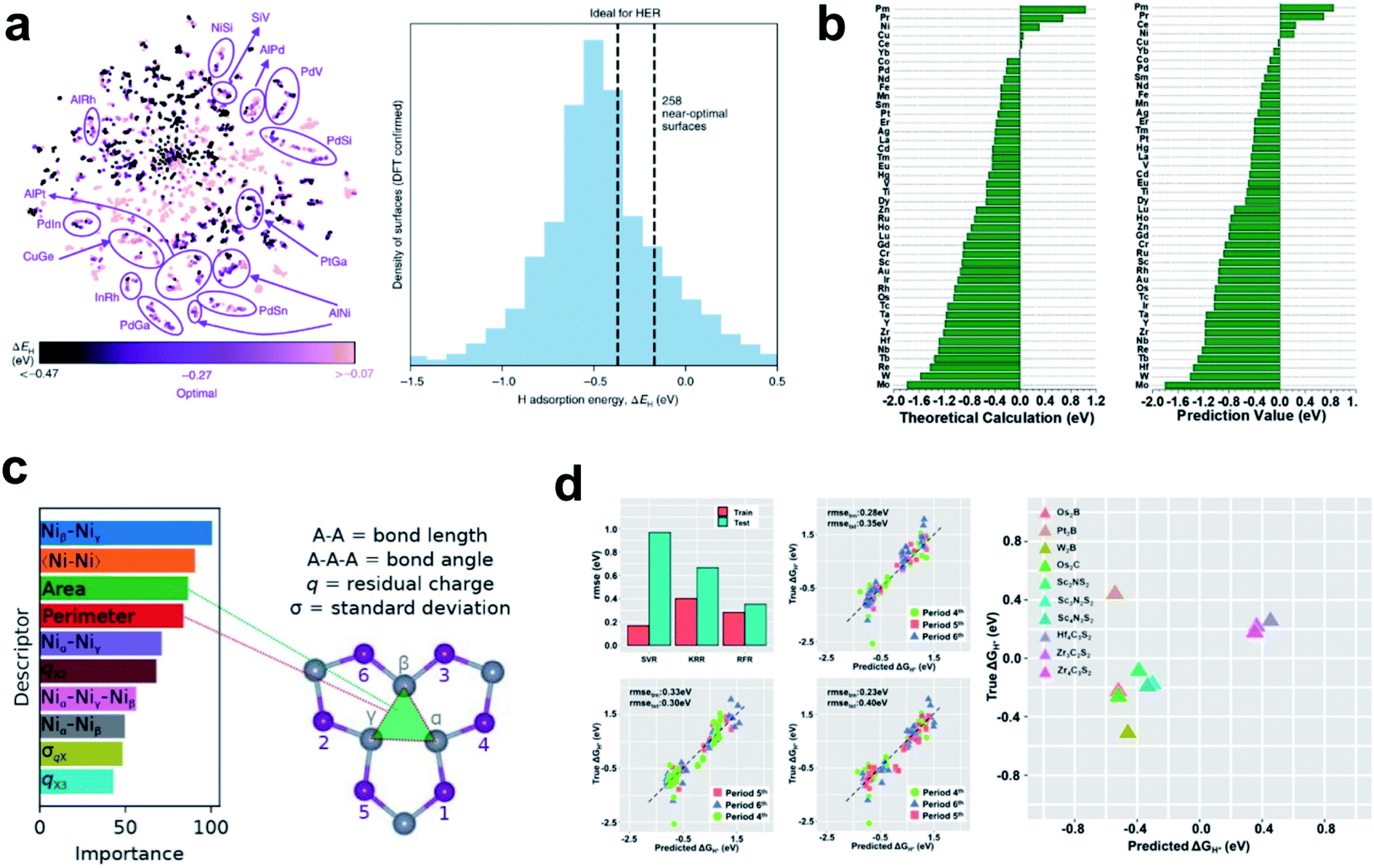 | ||
| Fig. 4 (a) t-SNE40 visualization of all the adsorption sites simulated using DFT, and normalized distribution of low-coverage ΔEH values calculated using DFT. Reproduced from ref. 114. Copyright 2018, Wiley. (b) Comparison of DFT calculation and (b) machine-learning predictions of 2H adsorption energies. Reproduced from ref. 116. Copyright 2019, Wiley. (c) Relative importance of descriptors calculated from the RRF model. Reproduced from ref. 118. Copyright 2018, American Chemical Society. (d) Average root mean square error of SVR, KRR, and RFR obtained using the LOCO CV method. Predicted vs. DFT-calculated ΔGH* of MXenes by using RFR. Reproduced from ref. 117. Copyright 2020, American Chemical Society. | ||
Back et al. conducted a machine learning aided screening process of HER materials using available databases such as Materials Project and GASpy databases.115 A convolutional neural network called CGCNN was used to predict the HBE and CO poisoning tolerance. They discovered 18 promising alloy combinations and 4 cost-effective materials by considering the HBE descriptor, and confirmed that data-driven search for materials can be easily performed using a single powerful descriptor. Sun et al. performed DFT combined with ML for the high-throughput screening study of atomic transition and lanthanide metals anchored on graphdiyne.116 They first calculated the adsorption strengths of all possible HER intermediates on the catalyst surfaces, and then together with electronic structure features as the input data, they trained an ML model using bag-tree algorithm. The trained machine learning model predicted almost the same HBE as the result calculated by DFT, and the results were passed to experimentalists for further investigation (Fig. 4b). Zheng et al. combined DFT and machine learning models to identify 8 MXenes as promising HER candidates from 299 MXenes.117 In addition, they provided data-driven physical insights into the material that S groups are HER-regulating species due to the antibonding states (Fig. 4d).117
The emerging machine learning techniques provide new tools to help understand the most important factors in designing HER catalysts. Wexler et al. studied the theoretical activity of dopant-substituted Ni2P using DFT and RRF ML model.118 By performing relative importance analysis, they found that structure-based descriptors such as Ni–Ni bond length in Ni2P materials and existence of Ni3-hollow structural motifs are the most influential factors (Fig. 4c).118
2.3. Water dissociation
While HBE has been widely accepted as the HER activity descriptor under a wide range of pH conditions due to its practicality in the rational design of electrocatalysts, it is increasingly accepted that the HBE Sabatier principle does not fully explain the significantly slower reaction kinetics under alkaline conditions than under acidic conditions. The sluggish kinetics of the HER in an alkaline environment is generally attributed to the huge potential required for the water dissociation step, known as the Volmer step (H2O + e− → OH− + H*). Thus, enhancing the alkaline HER can be accomplished by overcoming the requirement for a high rate-limiting potential. The general consensus for improving the Volmer step is to introduce active sites suitable for easily cleaving the H–OH bond and moderately binding the resultant hydroxyl ion. Thus, OHBE as a second thermodynamic descriptor and the water dissociation energy barrier as an intrinsic kinetic parameter have been widely used in the rational design of next-generation alkaline HER catalysts. This section will explore the significance of the water dissociation barrier and OHBE in the design of alkaline HER catalysts, followed by ‘dual-active site’ strategies for accelerating the Volmer step.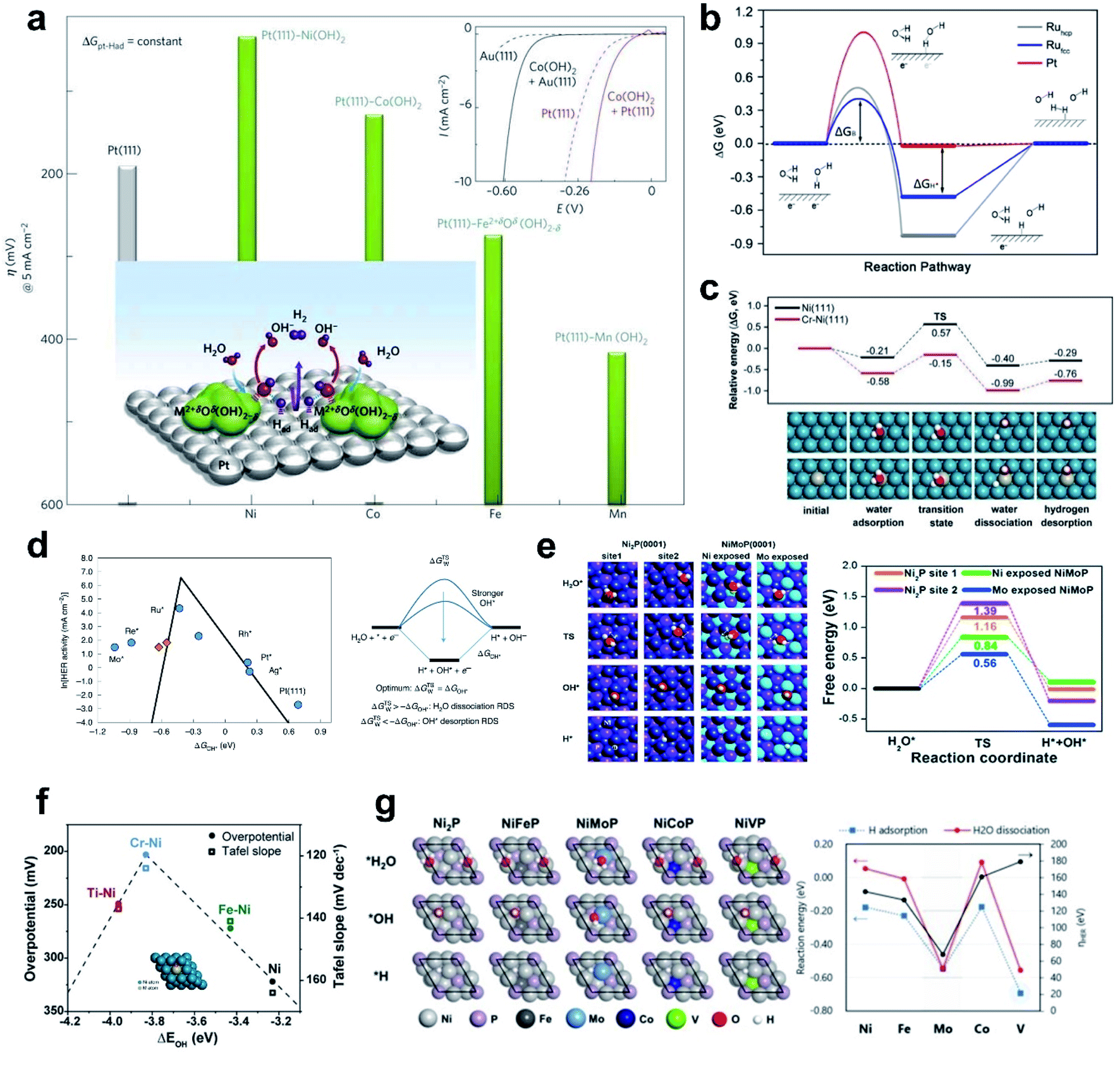 | ||
| Fig. 5 (a) Trend in overpotential for the HER as a function of 3d transition elements. Reproduced from ref. 44. Copyright 2012, Springer Nature. (b) Gibbs free energy diagram of the HER on different surfaces including reactant initial state, intermediate state, and final state. Reproduced from ref. 119. Copyright 2016, American Chemical Society. (c) Theoretical calculation of HER energy profile over Ni and Cr–Ni catalysts. (d) Simulated and experimentally measured HER activity and HER reaction mechanism. Reproduced from ref. 124. Copyright 2020, Springer Nature. (e) Free energy diagram for H2O activation. Reproduced from ref. 43. Copyright 2018, Elsevier. (f) Volcano plot of overpotentials at 10 mA cm−2, and Tafel slopes as a function of the ΔEOH for M–Ni and Ni catalysts. (g) DFT calculation results of NiP and NiMPs (M = Fe, Mo, Co, V) for electrocatalytic activity analysis in the HER. Reproduced from ref. 120. Copyright 2020, American Chemical Society. | ||
Zheng et al. recently provided a comprehensive study that revealed the importance of water dissociation in determining the HER kinetics in an alkaline environment by performing characterization and in-depth theoretical analysis using a well-defined model system.119 The Ru catalyst showed better H2O dissociation ability than the Pt catalyst; theoretical analysis showed that the difference is a result of the high barrier to water dissociation on Pt, so the Ru catalyst has higher activity than the Pt catalyst, even though the Ru catalyst does not have an optimal HBE. This work showed that the kinetic barrier to H2O dissociation is more important in the HER than hydrogen binding is (Fig. 5b).
Kim et al. developed Cr-incorporated nickel nano-helices which can accelerate the dissociative adsorption of H2O. Oxophilic dopants such as Fe, Ti and Cr can provide active sites. Theoretical analysis of the reaction barrier by using the NEB method suggested that the Cr–Ni bimetallic system had a significantly lower reaction barrier (0.42 eV) than the bare Ni surface. The nano-helical morphology of the catalyst may also have enriched the active stepped structure of Ni and Cr (Fig. 5c).120
Wu et al. elucidated that the diminished kinetic barrier of water dissociation on N-doped NiCo2S4 over NiCo2S4 is the key to the enhanced HER kinetics. DFT calculation revealed that the N incorporation on NiCo2S4 decreased the water dissociation barrier from 1.32 eV to 1.06 eV in the Volmer step, and also accelerated the H–H coupling (Heyrovsky) step.121 DFT analysis indicated that the barrier of the Heyrovsky step was higher on N-doped NiCo2S4 than on NiCo2S4; this result emphasizes the importance of considering the kinetic water dissociation descriptor.
Lee et al. developed a Co-doped NiFe phosphide which outperformed most of the alkaline HER catalysts. The HBE of NiFe–Co phosphide was the same as that of NiFe phosphide and the water dissociation thermodynamics were even worse on NiFe–Co phosphide (−0.10 eV) than on NiFe phosphide (−0.13 eV). However, the water dissociation kinetic barrier was effectively reduced, so HER kinetics was increased; these results imply that to understand the full reaction, kinetic analysis is necessary.122
Several reports suggested that the hydroxide binding energy (OHBE) can be useful as an HER descriptor. The Koper group extensively studied to reveal the correlation between HER activity and OHBE.124 A well-defined catalyst surface is modeled to capture the atomic scale picture of the HER using DFT. Similar to the HBE descriptor, the OHBE formed a volcano activity relationship; if the OHBE is too strong, it hinders hydroxide desorption, but if it is too weak, it slows the water dissociation step (Fig. 5d). An overall volcano plot between HER activity and OHBE, considering all possible HER reaction paths, suggested that Ru incorporation will place Pt at the top of the volcano, owing to optimal OHBE and HBE; this conclusion is consistent with previous reports.93 They concluded that both HBE and OHBE should be optimized simultaneously.
The Ni nano-helix system also shows a clear volcano-shaped relationship between OHBE and HER activity.120 Oxophilic dopants like Fe, Ti and Cr can provide OH binding sites that have higher OHBE than pure Ni (Fig. 5f). Ni and Fe–Ni do not have strong enough OHBE, so water dissociation is not favored. However, Ti–Ni has a too-strong OHBE, which implies that adsorbed OH can block the active site limiting the water adsorption. Cr–Ni had an optimal OHBE, and therefore was likely to have an optimal water dissociation barrier; this suggestion was confirmed by theoretical calculation and experimental measurement.
The adsorption energy of a water molecule can explain the enhanced HER activity, given that water adsorption is the first step of the Volmer reaction. Yu et al. compared the water adsorption property of Ni2P and NiMoP structures as well as HBE and water dissociation (Fig. 5e).43 The results showed that the water dissociation barrier decreases as the strength of water binding increases. Water adsorption alone cannot explain HER activity, but can at least provide thermodynamic insights into the reaction and trend.
Water dissociation and hydrogen binding occur together in the complex alkaline environment, so incorporating a water dissociation active site together with a hydrogen-binding active site to form a dual active site should synergistically increase the catalytic efficiency. The Marković group is one of the pioneers in designing a dual active site HER catalyst. By introducing water dissociation active materials such as Ni(OH)2, CoO(OH), FeO(OH) and MnO(OH), they significantly increased the HER activity under alkaline conditions. This result implies that the water dissociation reaction occurs near the metal hydroxide species, and that the hydrogen binds to the optimal Pt site.44 This finding stimulated many other studies of dual active sites to exploit their synergistic effect.
The idea of destabilizing the water molecule to facilitate the Volmer step was realized by the Sargent group.126 They designed a Cu-based catalyst that had a surface that was doped with Ni atoms, and decorated with a CrOx complex (Fig. 6a). They achieved an exceptional performance of 48 mV overpotential at a current density of 10 mA cm−2 under neutral conditions. DFT calculation of the energies of water dissociation, and H and OH binding energy showed that the water dissociation was much lower when the CrOx complex was present, and that the OHBE was significantly increased on the doped Ni atom. The conclusion was that dual active sites created by Ni atom and CrOx dissociate H2O much faster with smaller barriers than other model systems.
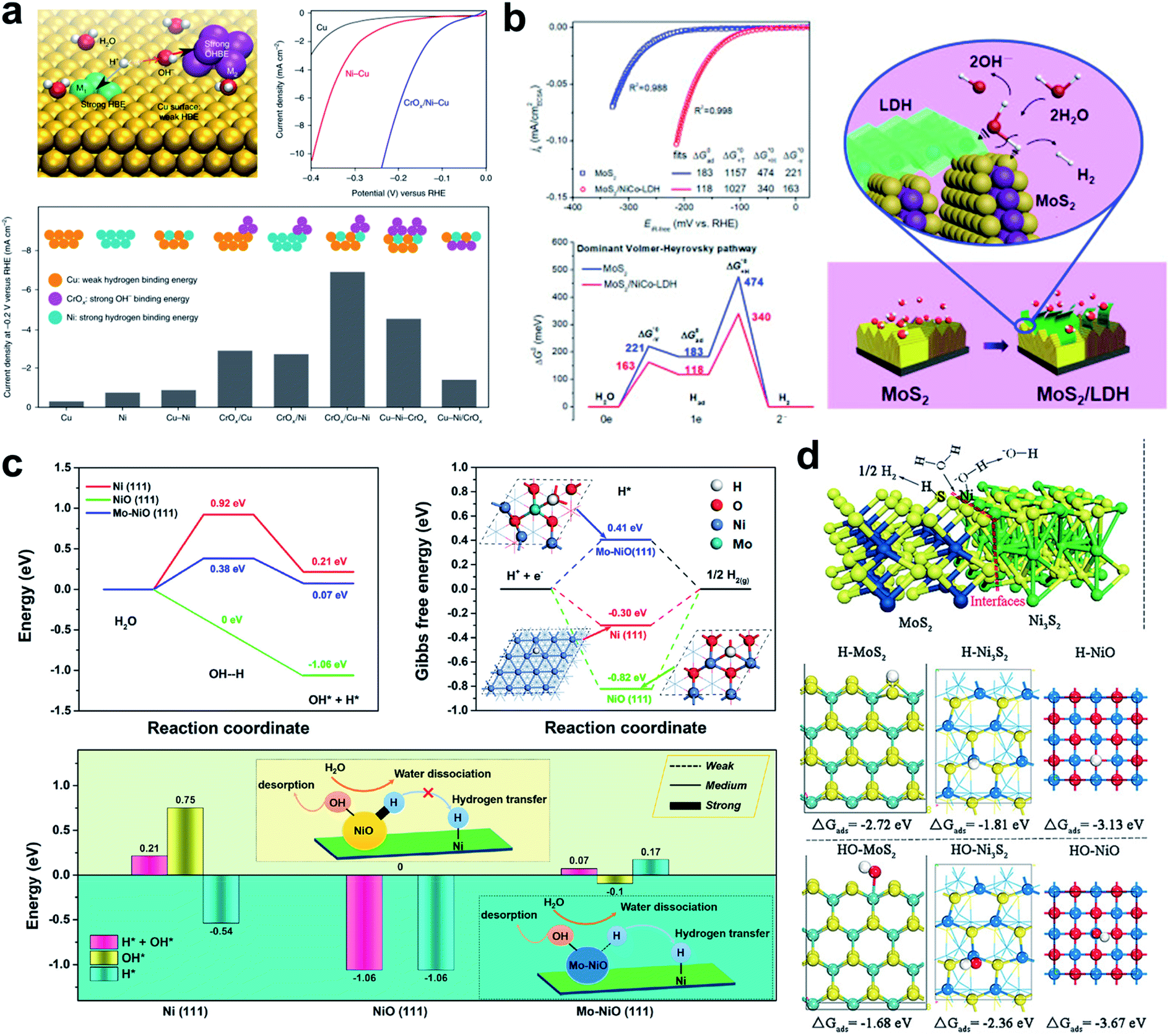 | ||
| Fig. 6 (a) Design principle of HER catalysts in neutral water by anisotropic surface doping to destabilize the water molecule on the catalyst surface; representative current density–voltage (J–V) curves before iR correction; comparison of HER activities on various Cu–Ni–CrOx catalysts. Reproduced from ref. 126. Copyright 2019, Springer Nature. (b) CSA normalized polarization curves; free energy diagram of the dominant Volmer–Heyrovsky pathway for the HER in an alkaline electrolyte; schematic illustration of the HER in the MoS2/LDH interface. Reproduced from ref. 32. Copyright 2017, Cell Press. (c) Calculated H2O-dissociation barriers; calculated Gibbs free energies of H adsorption on Ni(111), NiO(111), and Mo–NiO(111); summary of adsorption energy of H* + OH*, OH*, and H* on the three surfaces. Reproduced from ref. 127. Copyright 2019, American Chemical Society. (d) Chemisorption models of H and OH intermediates on the surfaces of MoS2, Ni3S2, NiO, and MoS2/Ni3S2 heterostructures. Reproduced from ref. 128. Copyright 2016, Wiley. | ||
Hu et al. developed a synergistic hybrid HER catalyst composed of MoS2 and LDH which have a low HER overpotential of 78 mV at 10 mA cm2; the Heyrovsky step was rate limiting for both the MoS2 and the MoS2/NiCo-LDH system (Fig. 6b).32 However, every water dissociation step was facilitated by the NiCo-LDH, and the H species that formed then transfer to MoS2 which has an optimal ΔGH ∼ 0.08 eV.
Huang et al. developed a Mo–NiO and Ni dual active site catalyst which can dissociate the water molecule with ease on the Mo–NiO while transferring the hydrogen atom dissociated from H2O on the Mo–NiO site to the Ni site easily for the next Tafel or Heyrovsky reaction (Fig. 6c).127 A heterostructured MoS2/Ni3S2 presented high activity toward both the HER and OER under alkaline conditions.128 DFT analysis revealed that the outstanding HER activity is a result of dual active properties of the material. The H atom is preferentially adsorbed on the MoS2, and the hydroxyl binds preferentially to the Ni3S2. The created interfacial sites therefore act as a water dissociation center and the formed H and OH species are partitioned among the materials (Fig. 6d).
2.4. The role of water and cations
In this section, the importance of water and solvated cation species in the HER process as a reactant and a carrier will be discussed. As pointed out by a recent perspective, “beyond adsorption” such as the solvation strength of electrolyte cations, chemical dynamics in the double layer and co-adsorbed spectator species can play important roles in the HER, and thus emphasis should be made on this.246 While the underlying role of these species is still unclear, pioneering studies have shed light on the role of cations and water molecules in the alkaline HER. The Marković group found that Li+ cation in Ni(OH)2/Pt-islands/Pt(111) can contribute to HER reaction kinetics by controlling water molecules.45 The metal hydroxide species provides an anchoring site that stabilizes an HO–Li–H2O complex. The hydrated Li+ may act as a destabilizer of HO–H bonding and thereby increase the HER under alkaline conditions. Removing hydroxyl ions from the surface to prevent poisoning of the active site was highlighted since it would then facilitate further dissociative water adsorption.44 In the following work, they proposed that Sadδ−–Cn+–H2O clusters may promote HER kinetics by activating water molecules in the double layer regions.129 In-depth experiment was performed to reveal the elusive role of the cation in the electrolyte. Liu et al. proposed the role of a cation complex of hydroxyl and water which can promote the HER.130 They found that increasing the amount of Li+ and Na+ caused increase in HER activity (Fig. 7a). Li+ alone did not increase the HER rate of Pt, so other species like Ni(OH)2 must interact with Li+. A series of these studies indicated the importance of water and solvated cation species in the HER process as a reactant and a carrier.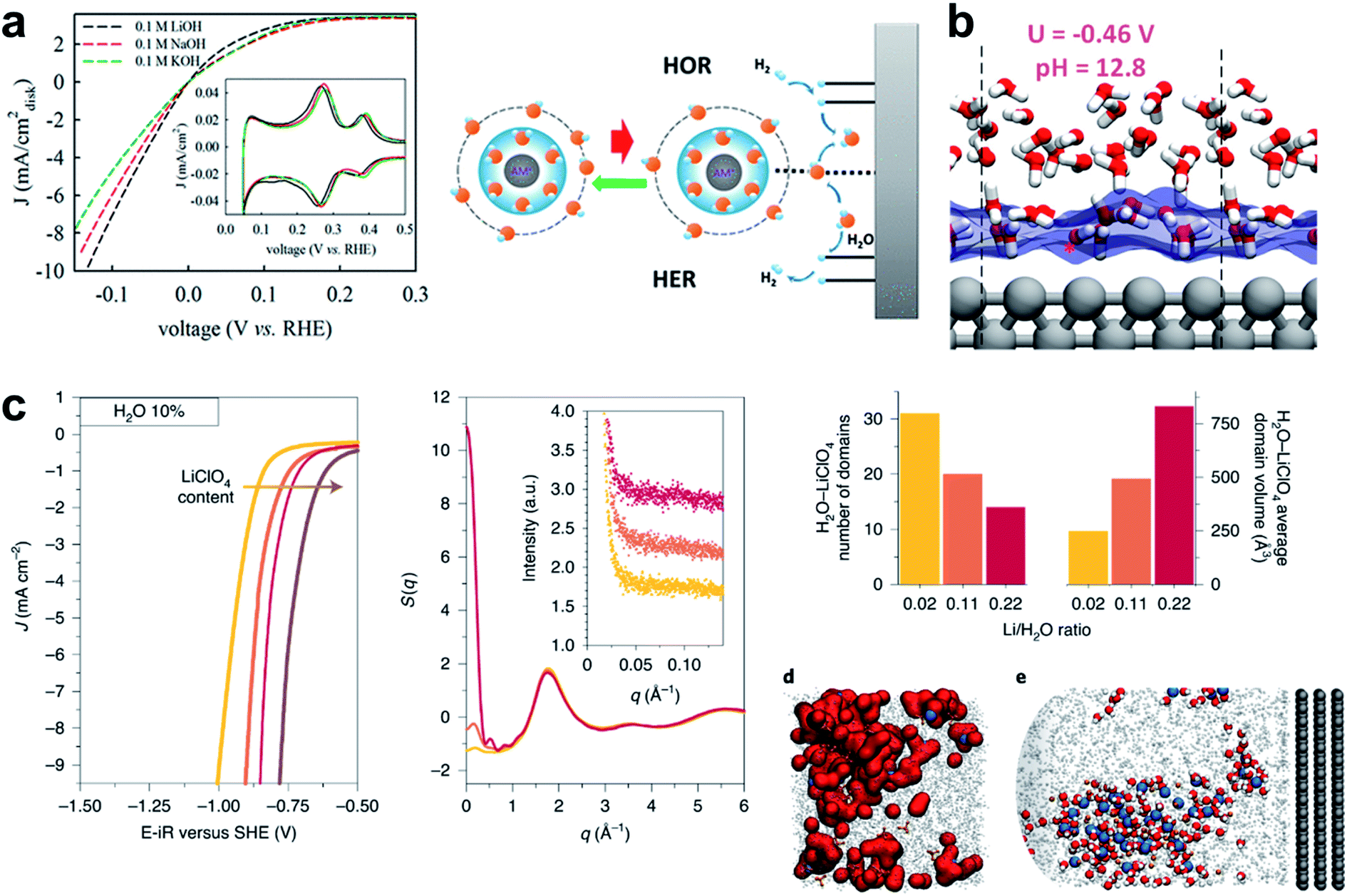 | ||
| Fig. 7 (a) Cyclic voltammetry on Pt/C with various concentrations of Ni2+, and on the Pt1Ni1/C alloy. Reproduced from ref. 130. Copyright 2019, American Chemical Society. (b) Snapshots with atomic details of the interfaces at U = −0.46 V. Reproduced from ref. 131. Copyright 2018, American Chemical Society. (c) Linear sweep voltammograms recorded in ACN with 10% in mass of added H2O in the presence of 100 mM (yellow), 500 mM (orange), 1 M (red), or 2 M (brown) LiClO4 on a rotating disc (1600rpm) Pt electrode with a 50 mV s−1 sweeping rate and E–iR being the potential corrected for ohmic drop; total X-ray-weighted structure factor of the electrolytes containing 10% H2O, 100 mM (yellow), 500 mM (orange), 1 M (red) LiClO4 as computed from the MD simulations; number of LiClO4−–H2O domains and their average volume depending on the LiClO4/H2O ratio at 10% in mass of water calculated from the MD simulations; (d) and (e) MD snapshots of the concentrated electrolyte (1 M LiClO4 and 10% H2O) showing the nanodomains in the bulk. Reproduced from ref. 133. Copyright 2020, Springer Nature. | ||
To reveal the role of the cations in the solution, a study combining theory and experiment was conducted by the Grimaud group. They utilized organic solvents such as acetonitrile to decouple the H2O–H2O effect, and left H2O as the only reactant. They also performed molecular dynamics simulation of bulk liquid and electric interface to investigate the long- and short-range cation–H2O interaction. The aqueous-rich nano-domains were captured; their size and reactivity can be controlled by the electrolyte. The water molecules engaged in both short-range and long-range interaction (Fig. 7c),131 which is consistent with an earlier theoretical report for the function of the cation in the electrolyte.132
Another theoretical study to identify the role of water in HBE was conducted using a Quantum Mechanics Molecular Dynamics (QMMD) study performed by the Goddard group. They concluded that the water adsorption preference depends largely on the pH, so the HBE can be manipulated by adjusting the pH.133 They also proposed that the electrode tends to repel water when the applied voltage is negative, and thereby increases the HBE. They raised the possibility that the change of water adsorption is a key to the pH dependence (Fig. 7b).
Many enlightening theoretical studies have been performed to describe the realistic adsorption/desorption behaviors of the reaction intermediate at the electrolyte–electrode interface, which is one of the major issues in the DFT modeling of electrocatalysis. Including solvent molecules explicitly with excess protons has been used for an atomistic level picture of electrode potential and the solvation effect on the HER. Rossmeisl et al. constructed a realistic atomic model for the solid–liquid interface at electrode surfaces.247 They established a model with protons solvated in water layers to naturally distribute the electrons and control the electrode potential by protons. Their model accurately described interface capacitance, and the work function was related directly to the electrode potential. In addition, they suggested that a change in the configurational entropy of the proton upon approaching the electrode surface largely drives the pH-dependence of the electrochemical proton transmission.248 When a proton is carried from the bulk solution to the electrode surface, entropy is converted to enthalpy, and this conversion is greater in an alkaline environment. This work emphasized the critical role of a restructured interfacial water solvation environment in affecting the HBE in relation to pH. Chan and Nørskov proposed a capacitor-like model to precisely determine the HER energetics at a constant potential for the charge transfer reactions by explicitly including solvent molecules.249 Using a single water layer, they could develop a simple but realistic model to depict the HER.
2.5. Electronic conductivity
Sufficient electronic conductivity is highly demanding for an electrode material to effectively carry out an electrochemical reaction by transferring electrons from the electrode to the surface. However, the development of emerging HER materials such as TM oxides and TM chalcogenides has been largely hampered due to the semiconductive nature of those materials. Substantial efforts have been devoted to promote the intrinsic electronic conductivity using a variety of strategies such as chemical doping, strain control and phase transition. These strategies have been effectively implemented by the combination of the theoretical predictions and experimental validations.The electronic structure of a material can be theoretically predicted by analyzing DFT calculated density of states where the band gap between the conduction band and the valence band of the electronic structure represents the conductive nature. By analyzing the electronic band structure of a material upon the modification such as doping and structural transformation, the conductivity of the material can be predicted in the atomic scale. For the experimental validations, the charge transfer resistance (Rct) of the material can be evaluated by electrochemical impedance spectroscopy (EIS) and the resulting Nyquist plot. The following series of reports will provide a general guideline for the improvement of the electronic conductivity of semiconductive materials such as MoS2. The Nørskov group found that MoS2 nanoparticle supported on graphite is a promising HER catalyst, which was inspired by nitrogenase enzymes in nature that can effectively evolve hydrogen.108 MoS2 has a hexagonal layered crystal structure and the HER activity of MoS2 is considered to originate from the edges rather than basal planes. However, intrinsically low conductivity through the in-plane S–Mo–S layers and deficient carrier as a prototype Mott-insulator still greatly impede wide applications. Therefore, various attempts to dope heteroatoms into MoS2 have been made to improve the conductivity of the materials. The 2D graphene-like structure of MoS2 transferred electrons between in-plane nanosheets, and avoided an inferior 3D transfer process between S–Mo–S layers in bulk MoS2. The dopants introduced some new states in the band gap and narrowed it, which leads to improved electron transfer kinetics. Sun et al. doped vanadium in a MoS2 interlayer and engineered its 2D structure to increase its electronic conductivity.134 V element in the MoS2 interlayer formed new states in the conduction band that were much closer to the valence band than in pure Mo2S, so the V-doped MoS2 behaved as a semimetal, in which valence electrons could be easily activated to the defect states (Fig. 8a and b).
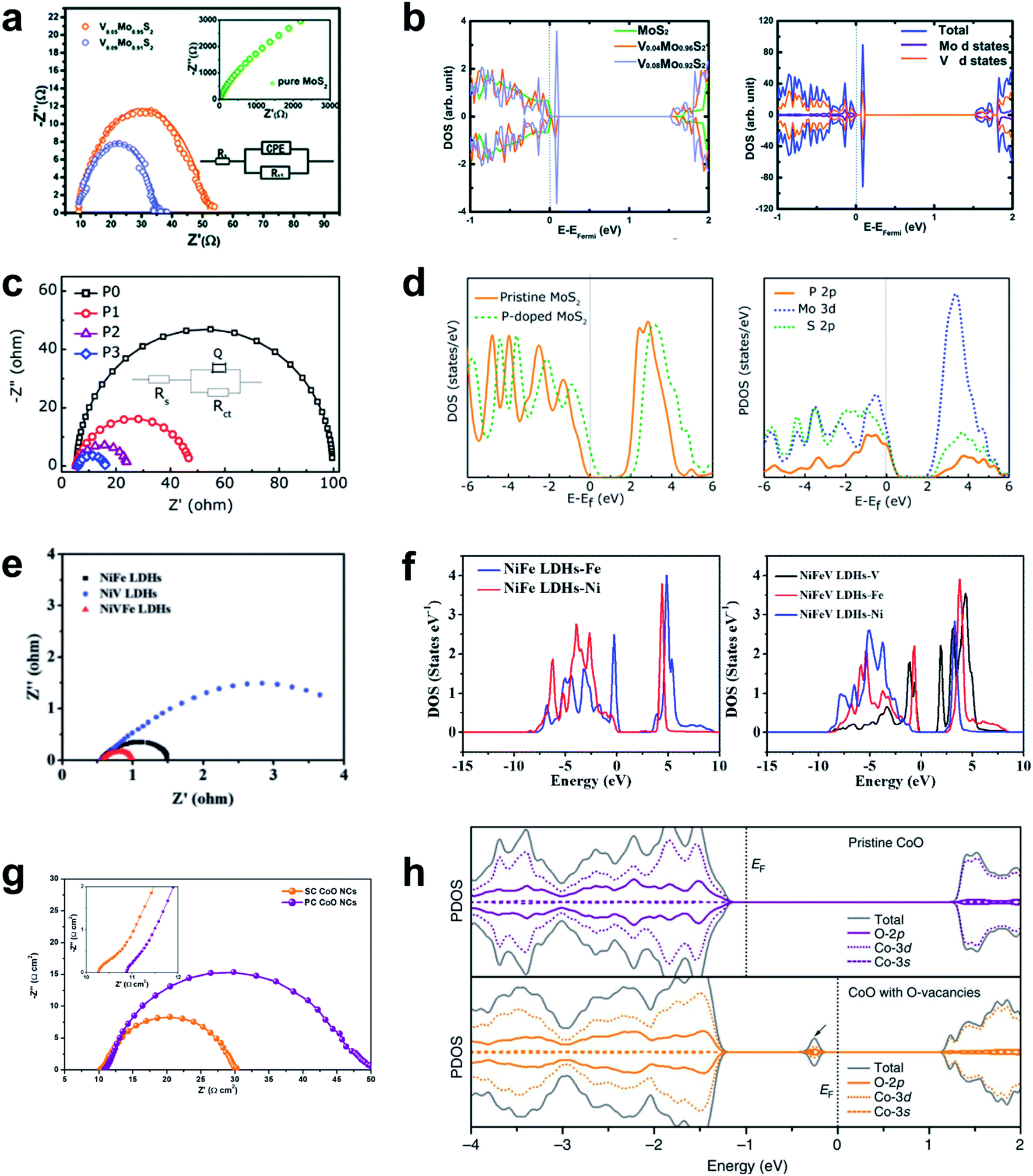 | ||
| Fig. 8 (a) Nyquist plot and (b) PDOS of pristine and V-doped MoS2 nanosheets. Reproduced from ref. 134. Copyright 2014, Royal Society of Chemistry. (c) Nyquist plot and (d) PDOS of pristine and P-doped MoS2 nanosheets. Reproduced from ref. 135. Copyright 2017, American Chemical Society. (e) Nyquist plot and (f) PDOS of NiFe LDHs and NiFeV LDHs. Reproduced from ref. 182. Copyright 2018, Wiley. (g) Nyquist plot and (h) PDOS of pristine and oxygen-defective CoO. Reproduced from ref. 183. Copyright 2016, Springer Nature. | ||
P doping also increases the electronic conductivity and stability of MoS2 nanosheet, and increases its alkaline HER activity compared to pure MoS2.135 The electrical resistivity decreased as the P doping ratio was increased to 5%; this trend indicates that the P is responsible for improving the electrical conductivity of the MoS2 nanosheet (Fig. 8c). The P-doped MoS2 monolayer has 0.3 eV narrower bandgap than the pristine MoS2 monolayer, and the 2p-band of P atom strongly hybridized with the 3d orbital of its neighboring Mo atoms and to the 2p orbital of S atoms, and thereby promoted fast charge transfer in the basal plane (Fig. 8d). Other heteroatoms such as Se,136 Nb,137 N,138 and TMs (Fe, Co, Ni, Cu, and Zn)139,140 can also be doped into MoS2 to increase its electronic conductivity. A different system consists of single-atomic Co supported on g-C3N4 (Co1/CN). Doping the system with P increased its electrical conductivity.141 The P atom was well incorporated on the C3N4, and promoted HER kinetics by increasing its electronic conductivity.
3. Alkaline oxygen evolution reaction
3.1. OER mechanism
The adsorbate evolution mechanism (AEM) and the lattice oxygen oxidation mechanism (LOM) are widely accepted as possible OER mechanisms. In the AEM, the two oxygen atoms that constitute one oxygen molecule are supplied by an aqueous solution, whereas in LOM one oxygen comes from the lattice and leaves an oxygen vacancy. Lattice oxygen can be oxidized when the O2/H2O redox potential is aligned with the lattice oxygen 2p-band, so the LOM can show pH-dependent kinetics.142–144To predict whether AEM or LOM dominates in OER catalysts, the oxygen vacancy formation energy (ΔEVof) and the N–V descriptor have been applied to ABO3 perovskites (A = La, La0.5Sr0.5, Sr and B = Mn, Fe, Co, Ni),145 where N is the number of unpaired electrons on the isolated B atom, and V is the nominal charge of B in the stoichiometric bulk ABO3. According to the reaction energy calculation of AEM and LOM, the OER mechanism of this system is determined by the free energy difference between the A0-like intermediate (*O) of the AEM and the A1-like intermediate (*OO with oxygen vacancy) of the LOM. The free energy difference decreases linearly with decreasing oxygen vacancy formation energy; this trend occurs because the B cation's electron-donating ability weakens, which is well explained by the N–V parameter (Fig. 9a).
 | ||
| Fig. 9 (a) Linear relationship of free-energy difference between A1-like and A0-like intermediates with ΔEVof and N–V. Reproduced from ref. 145. Copyright 2016, American Chemical Society. (b) Co-regulation of the OER mechanism on spinel oxides to Op and the relative Op to Md. Reproduced from ref. 146. Copyright 2020, Springer Nature. (c) Schematic of the cationic d-band entering the ligand p-band, transferring electrons to the metal forming ligand holes. Reproduced from ref. 148. Copyright 2016, Springer Nature. (d) AEM and LOM on ZnxCo1−xO2 slabs. Reproduced from ref. 147. Copyright 2019, Springer Nature. | ||
The oxygen 2p-band center (Op) and its relative value to the metal 3d-band center (Md) have been proposed to determine the OER mechanism of spinels after analyzing the relationship between thirty experimental references and DFT-calculated band centers (Fig. 9b).146 For the LOM, the lattice oxygen of the spinel should meet both the activity and priority criteria. The activity criterion is met when Op is high enough (greater than −1.75 eV) to ensure escape from the lattice. The priority criterion is met when Op is higher than Md for intramolecular charge transfer from the oxygen to the cation. Therefore, to define the OER mechanism in spinels, both absolute Op and Op relative to Md must be evaluated.
In transition metal oxyhydroxides (MOOH), the LOM occurs only if two neighboring oxidized oxygens can hybridize their oxygen holes without significantly sacrificing M–O hybridization.147 Incorporation of catalytically inactive Zn2+ (d10) ion into CoOOH increased the Co–O covalency and formed an accessible oxygen non-bonding state, which was confirmed by Bader charge distribution and projected density of state (PDOS) analysis. As the zinc content of ZnxCo1−xO2 increased (x = 0, 0.1, 0.2), the relative stability of A2 (AEM intermediate) to L2 (LOM intermediate) (Fig. 9d) and the activation energy from A2 to L2 all decreased. As a result, the Zn0.2Co0.8O2 slab proceeded through the preferential LOM path, and the potential barrier was 0.60 V lower than that of the CoO2 slab. However, the excess Zn component (Zn0.5Co0.5O2 slab) impedes recharging of the remaining oxygen vacancies after O2 gas formation (energy barrier ∼0.96 eV), so structural instability develops. This study showed that the OER of MOOH can undergo LOM with sufficiently high M–O covalency and smooth recovery of lattice oxygen. M–O covalency can be strengthened by the metal's high oxidation state, which has low Md close to Op, and thereby lattice oxygen can be activated in the presence of highly oxidized metal species (Fig. 9c).148 For example, Ni4+ species derived by the Fe component of the (FeCoCrNi)OOH surface accompanied holes into the related oxygen 2p-band and facilitated lattice oxygen coupling, and thereby triggered lattice oxygen activation at the Fe–Ni dual-site center, which have high intrinsic OER activity.144
3.2. Adsorbate evolution mechanism
This section presents the factors that determine the catalytic activity of TM alkaline OER catalysts, and strategies to control the factors. The four-electron pathway for AEM on metal surfaces was established first,149 and the LOM was demonstrated in next section.142,150,151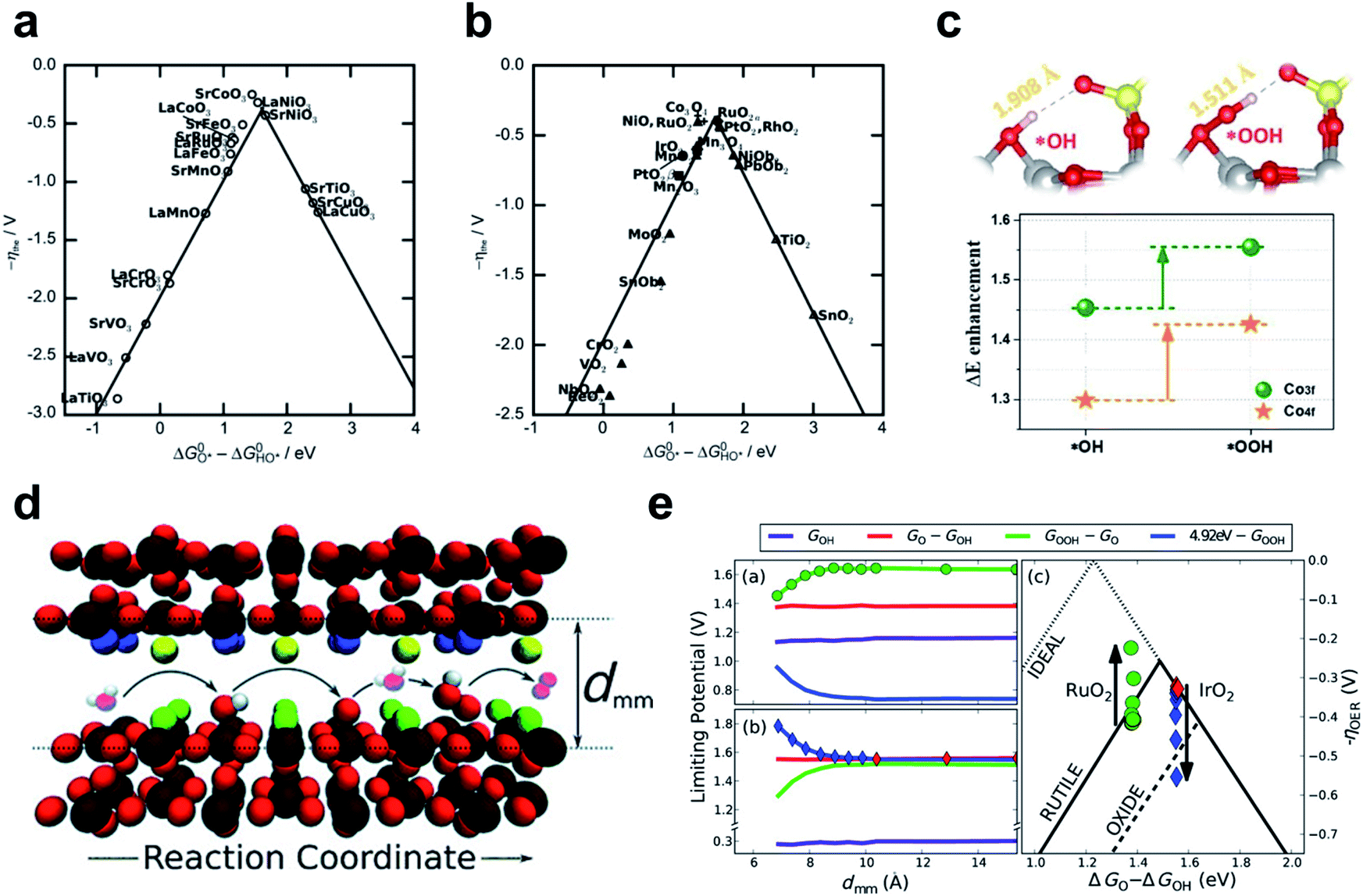 | ||
| Fig. 10 The negative theoretical overpotential plotted against ΔGO − ΔGOH (a) for perovskites and (b) for rutile, anatase, Co3O4, MnxOy oxides. Reproduced from ref. 154. Copyright 2011, Wiley. (c) The optimized *OH and *OOH structures on Co3O4–SO4 surface and the stabilization of intermediate binding at the Co3O4–SO4 surface compared to Co3O4. Reproduced from ref. 155. Copyright 2020, Elsevier. (d) Schematic of OER elementary steps in the microscopic channel with dmm width. (e) Limiting potential change of each OER elementary step and corresponding overpotential change as dmm decreases on RuO2 (up) and IrO2 (down). Reproduced from ref. 156. Copyright 2015, Wiley. | ||
One attempt to circumvent this limitation is to use hydrogen bonding to separate the *OOH stability from the scaling relationship. SO42− modification of the Co3O4 surface strengthens *OOH adsorption relatively more than *OH adsorption, so the OER overpotential decreases by 40 mV.155 *OOH is longer and more flexible than *OH, so *OOH could form closer hydrogen bonds to the upper O atom of the adjacent SO42− than *OH could (Fig. 10c).
Another way to design additional hydrogen bonding sites for *OOH is to use three-dimensional nanoscopic channels. Doyle and coworkers investigated the OCI stability with nanoscopic channel spacing by means of DFT calculation (Fig. 10e).156 In this system, the second surface of the opposite site interacts with the *OOH intermediate, whereas *O and *OH remain unaffected (Fig. 10d). A channel width of about 7 Å increased the theoretical catalyst efficiency of RuO2 (111) by 10%, and width less than this caused proton transfer from *OOH to the opposite surface. This strategy may be applicable to planar catalysts, such as γ-NiOOH, in which interlayer spacing can be controlled if sufficient catalytic durability is ensured. However, methods to stabilize *OOH are limited to improving the catalysts on the “left leg” (ΔGO − ΔGOH < 1.6 eV) of the activity volcano, which are hindered by the *OOH formation step.
The simplicity of the intermediate adsorption energy calculation and the universality of ΔGO − ΔGOH as a descriptor have made it applicable to many OER catalyst screening studies.157–161 However, the ΔGO − ΔGOH descriptor does not much decrease calculation cost from the overall OER pathway investigation, and is consequential rather than fundamental. Rational design of catalysts with optimal ΔGO − ΔGOH is a difficult task, because we do not know which electronic properties directly determine ΔGO − ΔGOH. This difficulty raised the need for parameters related to electronic structure as essential OER activity descriptors.
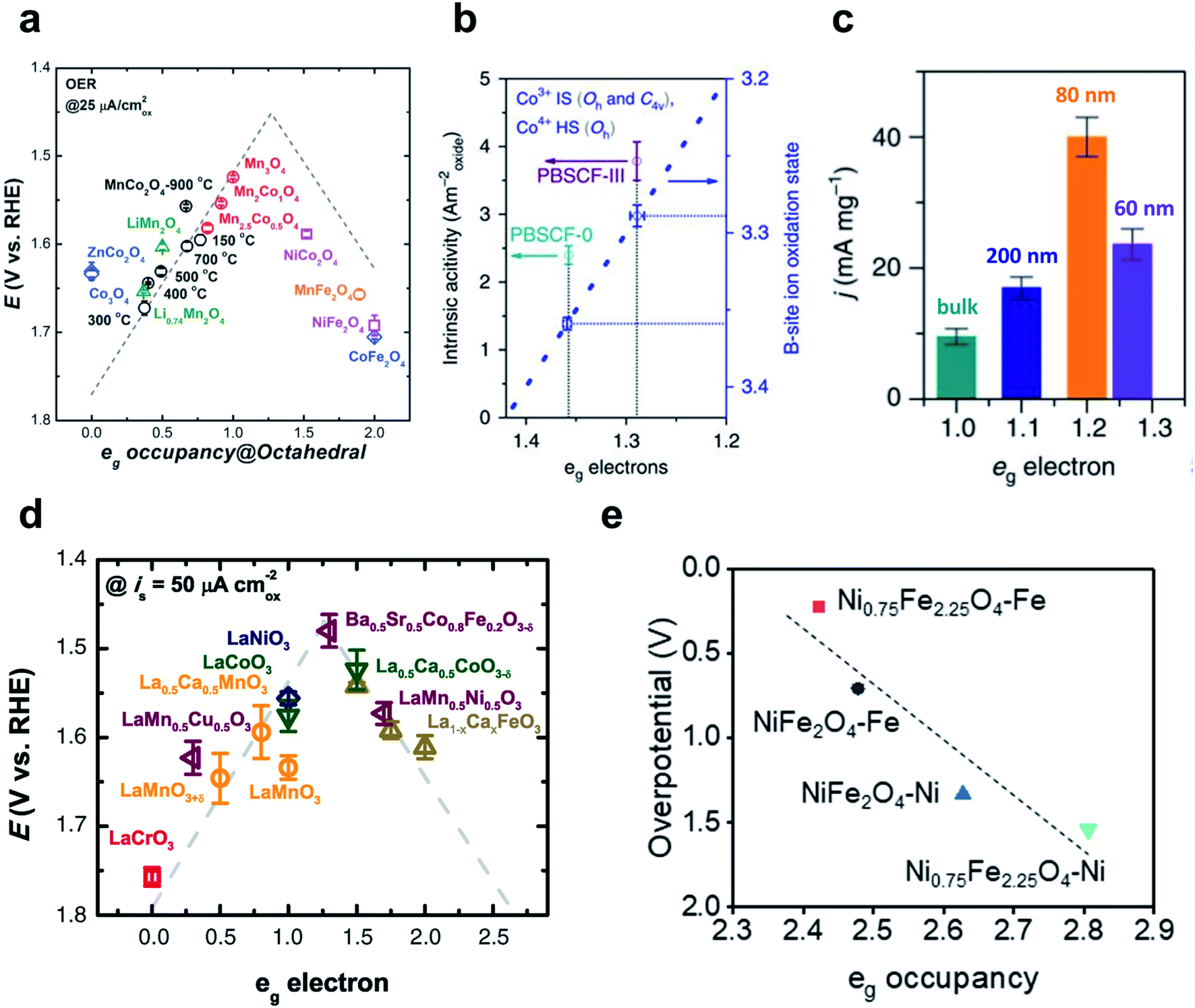 | ||
| Fig. 11 (a) Relationship between OER overpotentials and eg occupancy of TMs in perovskites. Reproduced from ref. 163. Copyright 2017, Wiley. (b) Intrinsic activity versus eg occupancy of PBSCF-0 and PBSCF-III. Reproduced from ref. 165. Copyright 2017, Springer Nature. (c) Mass activities for the bulk and nanosized LCO. Reproduced from ref. 164. Copyright 2016, Springer Nature. (d) Relationship between OER overpotentials and eg occupancy of octahedral metals in spinels. Reproduced from ref. 162. Copyright 2011, AAAS. (e) Relationship between theoretical OER overpotentials and eg occupancy of the octahedral TM in NiFe2O4 and Ni0.75Fe2.25O4. Reproduced from ref. 167. Copyright 2021, Elsevier. | ||
Zhao et al. have reported a perovskite nanostructure designing method to engineer eg occupancy. Reducing the particle size of a single perovskite LaCoO3 (LCO) to ∼80 nm increased the eg occupancy of cobalt ions from 1 to ∼1.2 with optimal OER activity.164 The eg occupancy of the LCO nanoparticle was estimated assuming a core–shell model in which the spin states of Co3+ ions in the core were maintained at 50% high spin (HS: t42ge2g) and 50% low spin (LS: t52ge0g), while the spin states of the shell were in the 100% HS state. Starting from bulk LCO with only LS state Co3+ ions (eg occupancy ∼1), decreasing LCO particle sizes to 200, 80, and 60 nm resulted in 1.09, 1.21, and 1.27 eg occupancy, respectively. The OER activity, expressed as the current density of 10mAcm−2 per unit mass, showed a volcano curve as a function of eg occupancy with an optimal point at ∼80 nm particle size (Fig. 11c). In summary, particle size reduction increased the ratio of core-to-shell composed of HS state Co3+ ions and eg occupancy was successfully tuned to increase OER activity.
Their subsequent study tuned the eg occupancy of B metals in the double perovskite PrBa0.5Sr0.5Co1.5Fe0.5O5+δ (PBSCF) by synthesizing nanofiber morphology and controlling the diameter.165 Reducing the diameter of PBSCF nanofibers from 196 to 20 nm increased the mass activity, and the thinnest (PBSCF-III) showed 1.6 times higher intrinsic activity than PBSCF powder (PBSCF-0), and was far superior to commercial IrO2 catalyst and reported advanced perovskite catalysts. The eg occupancy of PBSCF-0 and PBSCF-III was estimated using a first-order approximation to the B-metal oxidation state proposed by Grimaud and their coworkers.166 Compared to PBSCF-0, PBSCF-III had a lower B-site cobalt ion oxidation state; this difference indicates a lower and closer eg occupancy (∼1.29) to the optimal value than PBSCF-0 (∼1.36) (Fig. 11b).
The metal composition can also be adjusted to optimize eg occupancy. Lee et al. have altered the Ni/Fe composition in NiFe2O4 through conventional corrosion of the Ni foam in the presence of exotic Fe3+ cations.167 Ni0.75Fe2.25O4 nanoparticles required 82 mV lower overpotential than NiFe2O4 to reach a current density of 10 mA cm−2, and the Tafel slope showed a 14 mV dec−1 faster OER rate than NiFe2O4. DFT calculation confirmed the increased intrinsic OER activity of Ni0.75Fe2.25O4 than NiFe2O4, and the correlation of eg occupancy and theoretical overpotential also revealed that Ni is the major active site for these nickel ferrites (Fig. 11e).
However, metal oxide may become amorphous under OER conditions, and the presence of two crystal fields, i.e., octahedral Oh and square pyramidal C4v, causes ambiguity in eg occupancy estimates.166 In addition, eg occupancy cannot represent electron sharing in M–O bonds, which specifically affects the LOM process. LaMnO3, LaCoO3, and LaNiO3 have the same ∼1 eg occupancy but contrasting OER activity; these are representative examples of the limitations of eg occupancy as a descriptor.168 Descriptors that consider M–O covalency avoid these limitations.
 | ||
| Fig. 12 (a) Overpotential and ΔEd–p values of the OER depending on the oxygen-vacancy position in Sm0.5Sr0.5CoO3−δ. Reproduced from ref. 169. Copyright 2020, American Chemical Society. (b) Schematics of charge-transfer energies in bulk perovskites. Reproduced from ref. 171. Copyright 2018, American Chemical Society. (c) The experimentally measured OER overpotential plotted as a function of the calculated max (DT, DO) (eV). Reproduced from ref. 146. Copyright 2020, Springer Nature. (d) OER overpotential plotted as a function of the N–V parameter. (e) TM valence states (left) and OER operating potentials (right) as a function of composition x in ZnFexCo2−xO4 oxides. Reproduced from ref. 173. Copyright 2018, Wiley. (f) OCI binding energies and (g) OER overpotentials as a function of NEE. (h) Volcano plot of OER activities of V, Nb, Ta, Mo, and W atom doped TiO2(110). Reproduced from ref. 174. Copyright 2019, Royal Society of Chemistry. | ||
For spinels, the ΔEd–p descriptor used for perovskites should be modified due to the covalency competition between octahedral and tetrahedral sites.146 M–O breakage occurs from the weaker covalent bond in the MO–O–MT backbone, where MO denotes an octahedral metal and MT denotes a tetrahedral metal; thus one with a higher 3d-band center with lower bond durability should be the OER descriptor in spinels (Fig. 12c).
As mentioned in Section 3.1, N–V could predict the M–O covalency and dominant OER mechanism of perovskites. Zhou et al. used the same descriptor to explain the trend in AEM activity vs. B-metal composition in spinel ZnFexCo2−xO4 oxides (x = 0–2.0).173 In this system the highly crystalline surface is well preserved over OER cycling, and Op is lower than the Fermi level, so the LOM is not a favorable route. Among the series of ZnFexCo2−xO4 oxides, ZnFe0.4Co1.6O4 catalyst showed the lowest overpotential (35 mV at 25 μA cm−2) and the highest valence state of cobalt (Fig. 12e). A higher valence state of Co cation (Co4+) compared with Co3+ indicates a lower energy 3d state and would produce higher hybridization with oxygens. Consequently, the experimentally measured OER overpotential was linearly related to N–V; this trend indicates M–O covalency (Fig. 12d).
Replacing some Ti atoms with hetero-metal dopants (V, Nb, Ta, Mo, W) could tune the NEE and OER activity of the TiO2(110) surface (Fig. 12h). This strategy can be extended to other reducible metal oxides such as SnO2, NiO, WO3, and ZnO. Quantitative NEE measurements are difficult both experimentally and computationally, but indirect analysis of NEE can be attempted using Bader charge analysis or calculation of valence electron configuration.
3.3. Lattice oxygen oxidation mechanism
Yoo et al. demonstrated the importance of considering lattice oxygen participation in understanding the trends in the OER on ABO3 perovskites.151 Using DFT, they showed that LOM can lead to higher OER activity than the conventional AEM by bypassing the scaling relationships dictated in the AEM-based reaction process (Fig. 13a). They also showed that the OER activity volcano for AEM is universal for all perovskites, whereas that for LOM depends on the identity of the A cation in ABO3. This observation explains experimental observations that perovskites such as Pr0.5Ba0.5CoO3−δ and SrCoO3−δ show higher OER activities than the conventionally predicted optimal compounds such as LaNiO3 and SrCoO3, and consequently emphasized the need for detailed evaluation of LOM activity. | ||
| Fig. 13 (a) Overall OER activity volcano that considers AEM for all perovskites, LOM for lanthanum-bearing perovskites, and LOM for strontium-bearing perovskites. Reproduced from ref. 151. Copyright 2018, American Chemical Society. (b) OER potential versus the DFT computed Op of (Ln0.5Ba0.5)CoO3−δ with Ln = Pr, Sm, Gd or Ho. Reproduced from ref. 166. Copyright 2013, Springer Nature. (c) Correlation of OER current density with the vacancy parameter δ of La1−xSrxCoO3−δ. Reproduced from ref. 150. Copyright 2016, Springer Nature. (d) Schematic rigid band diagrams of LaCoO3 and SrCoO3. Reproduced from ref. 142. Copyright 2017, Springer Nature. (e) Correlation of intrinsic OER activity with the hybridization factor of P-LSCF, RP-LSCF, and RP/P-LSCF. Reproduced from ref. 176. Copyright 2020, Wiley. (f) Structural characterization of hex-BSCF (left) and the electronic structure of Co ion at the tetrahedral and octahedral sites in hex-BSCF (right). Reproduced from ref. 175. Copyright 2020, Wiley. | ||
The correlation between LOM activity and M–O covalency occurs because oxygen vacancies form during the LOM process.150 The degree of oxygen vacancy formation is affected by the relative positions of the TM 3d-band compared with the oxygen 2p-band of the crystal; a more covalent system exhibits a higher vacancy concentration. A high oxidation state of the TM increases the overlap of the metal 3d-band with the oxygen 2p-band, and downshifts the Fermi level. When the oxygen 2p-band states at the Fermi level lie above the redox energy of the O2/H2O couple, ligand holes are created in the oxygen 2p-band; this process indicates that the oxidation of lattice oxygen becomes thermodynamically favorable (Fig. 13d).142 Therefore, the charge transfer energy and oxidation state that represent M–O covalency in TMs can represent LOM activity and be tuned to increase OER activity.
Transforming the structure of perovskites is one way to use the highly oxidized TM for higher LOM activity. Mefford et al. adjusted A-site metal composition in the La1−xSrxCoO3−δ perovskite to increase both the bulk oxidation state of Co and the concentration of oxygen vacancies; as a result the vacancy parameter increased and current density at 1.63 V vs. RHE also increased (Fig. 13c).150 Zhu et al. have reported the unique hexagonal structure composed of one honeycomb-like network, Ba4Sr4(Co0.8Fe0.2)4O15 (hex-BSCF), as a highly active and durable OER electrocatalyst in alkaline solution.175 Its unique structure ensures that both the high spin Co3+/Co4+ ions in the tetrahedral site and intermediate spin Co4+ ions in the octahedral site are active, which give rise to AEM and LOM, respectively. Increase in the oxidation state of Co ions increased the Co–O covalency, and the high-lying unoccupied eg orbitals of Co4+ ions that have Oh symmetry have stronger covalency than the t2g orbitals of Co4+ ions that have Td symmetry (Fig. 13f). Therefore, the strong M–O covalency between the Co4+Oh and the 2p ligand hole can trigger oxidation of lattice oxygen. The distinct OER mechanisms of the two active sites were further confirmed by combined XAS analysis and DFT calculations.
They also hybridized Ruddlesden–Popper phase LaSr3Co1.5Fe1.5O10−δ with perovskite phase La0.25Sr0.75Co0.5Fe0.5O3−δ (RP/P-LCSF) which increased M–O covalency and oxygen ion diffusion (Fig. 13e).176 RP/P-LSCF delivers a much lower OER overpotential and smaller Tafel slope than RP-LSCF, P-LSCF, and RuO2. The strengthened M–O covalence of RP/P-LCSF was confirmed by O K-edge XAS spectra, considering the pre-edge peak below ∼530 eV in the O–K XAS spectra which correspond to the unoccupied O 2p orbitals hybridized with TM 3d orbitals.166,177,178
 | ||
| Fig. 14 (a) Correlation of OER current density with the oxygen ion diffusion rate of La1−xSrxCoO3−δ. Reproduced from ref. 150. Copyright 2016, Springer Nature. (b) Correlation of intrinsic OER activity with the oxygen-ion diffusion coefficient of P-LSCF, RP-LSCF, and RP/P-LSCF. Reproduced from ref. 176. Copyright 2020, Wiley. Correlation of intrinsic OER activity with (c) the oxygen anion diffusion rate and (d) the oxygen vacancy diffusion rate. Reproduced from ref. 181. Copyright 2020, Springer Nature. | ||
3.4. Electronic conductivity
Attempts to accelerate the Faraday process have also been made in OER kinetics. V doping in NiFe LDHs reduces the onset potential and achieved efficient electrocatalysis for water oxidation.182 This doping significantly lowered the electron-transfer resistance (Fig. 8e) and the sheet resistance from (2.4 ± 0.3) × 103 Ω sq−1 to (1.3 ± 0.2) × 103 Ω sq−1; these changes are direct evidence of increased conductivity of NiFeV LDHs. The PDOS plot showed that V narrowed the bandgap, and thereby changed the conduction band state of NiFe LDHs (Fig. 8f).Creation of oxygen vacancies on pyramidal facets can tune the atomic structure of one-dimensional single crystal (SC) CoO nanorods and increased their OER activity.183 The SC nature and the oxygen vacancies increased the carrier concentration in SC CoO compared to polycrystalline (PS) CoO, and thereby lowered the electronic resistance of SC CoO (Fig. 8g). The PDOS analysis showed that oxygen vacancies created new electronic states in the bandgap by hybridization of O 2p, Co 3d and Co 3s bands for increased charge transfer (Fig. 8h).
4. Stability
The electrode material should be chosen by considering its stability for long operation and practical usages. Undesired oxidation and subsequent dissolution of the catalyst surface impede the development of TM based electrocatalysts, given that the surface properties of the materials often dictate the activity. The susceptibility of TMs to oxidation and corrosion can greatly reduce the catalyst efficiency by limiting the duration for which they are effective. Cheap TM elements have been introduced, but their poor stability gives them inferior catalytic activity compared to noble metal catalysts. Alkaline conditions generally increase the stability of TMs by forming passivation structures such as oxides and hydroxides, but the catalyst can corrode regardless of the environment (Fig. 15a).184 | ||
| Fig. 15 (a) Corrosion of the MxPy in acidic and alkaline environments. Chronoamperometry at 0.8 V (solid line) and 1.2 V (dashed line) vs. RHE for 1000 s; corrosion percentages (wt%) calculated on the basis of the metal phosphides. Reproduced from ref. 184. Copyright 2019, American Chemical Society. (b) Activity–stability relationship of TMSx for the HER in acid and alkaline environments. Reproduced from ref. 129. Copyright 2016, Springer Nature. (c) Two-dimensional histograms of ΔGH* and ΔGCO*; electrochemical stability ΔGPourbaix under reduction (−0.2 VRHE) and oxidation conditions (+0.2 VRHE) at pH = 0. Reproduced from ref. 115. Copyright 2020, Royal Society of Chemistry. (d) Fe segregation energies for pristine M(OH)2 and the sample at the OER potential (K1/3MO2). Reproduced from ref. 199. Copyright 2020, Springer Nature. (e) Computational model for CoFe0.25Al1.75O4 after Al3+ leaching and schematic band diagrams with and without Al3+ vacancies. Reproduced from ref. 188. Copyright 2019, Springer Nature. | ||
4.1. Stability measurement
Several measurement techniques have been used to evaluate the stability of the catalyst. The long-term stability of the HER catalyst is often studied by monitoring the variation of current density or overpotential over time. For example, the electrochemical long-term stability of Cu/NiCu was assessed by monitoring the current density, overpotential, and charge transfer resistance over a 120 hour period.185 Zadick et al. studied the large instability of Pt/C under alkaline conditions, and found that the degradation rate is three times worse than in acidic medium.186 Accelerated stress tests (ASTs) were used to determine the deterioration of Pt/C samples under three different pH conditions. The degradation of the catalyst surface was then clearly identified by analyzing the electrochemical surface area (ECSA) measurements and identical location transmission electron microscopy (ILTEM) images. They suggested that the extreme carbon surface modification under alkaline operating conditions can dissolve up to 63% of the Pt nanoparticles, as demonstrated by Raman spectroscopy and X-ray photoelectron spectroscopy (XPS) measurements.Another way to measure the stability of the materials is to analyze the dissolution concentration of the element. Hofmann et al. quantified the amount of cobalt and phosphorus dissolved in Co2P both under acidic and alkaline conditions.187 They determined the pH-dependent stability using linear sweep voltammetry (LSV). Using Inductively Coupled Plasma Optical Emission Spectrometry (ICP-OES) analysis, the amount of dissolved elements in the catalytic materials was then compared before and after the repeated LSV measurement. They found that Co2P preferentially dissolves P atoms over Co atoms under alkaline conditions, which largely degrades the activity. In contrast, in an acidic environment, the dissolution occurred stoichiometrically (two Co per P), so the activity and ECSA were retained uniformly. The degraded Co2P formed Co(OH)2, then deactivated.
The direct detection of stability can be achieved before, after and even in real time, using state-of-the-art advanced operando spectroscopy and microscopic high-resolution imaging techniques. XAS,141,219–222 surface X-ray scattering,223 mass spectrometry,224 attenuated total reflectance Fourier transform infrared spectroscopy (ATR FT-IR),225 and Raman spectroscopy226 have been used for in-depth stability analysis and mechanistic studies, as we have discussed in Section 5.
In summary, electrochemical stability can be assessed by several stability evaluation criteria. To determine long-term stability, conventional electrochemical measurements such as linear/cyclic sweep voltammetry and amperometry have been extensively used. To detect degradation and dissolution of catalytic materials, high resolution spectroscopy and in situ techniques have been frequently employed in conjunction with ECSA. These state-of-the-art measurement techniques accelerate our understanding of the catalyst stability and aid in the development of stable materials.
4.2. Stability–activity relationship
Corrosion of a component in a multicomponent system often creates additional defect sites, which can act as active sites for the HER and OER. The Marković group demonstrated the stability and activity relationship in TMS that the dissolution of cations from Co–Mo–Sx chalcogens in strong alkaline media induces formation of active defects which serve as an active site. They confirmed that the activity–stability has strong correlation in that a less stable catalyst is more likely to have better activity.129 They compared the amount of the dissolved cations (Mo and Co) to find that a moderately defective structure has the smallest overpotential after 500 hours of operation and decent stability (Fig. 15b).Moderate instability of the catalyst surface promotes LOM in metal oxides, and increases their overall OER activity. Some studies have shown that descriptors for predicting the OER mechanism or activity can also be used to predict the stability of the catalyst.145,147,166,188 However, excessive destruction such as metal dissolution, structural reconstruction and oxidative decomposition decreases OER operation and remains a problem to be solved.
Recently, it has been found that many OER active materials suffer severe dissolution during the reaction;189,190 this observation implies that high activity in the OER is always accompanied by elevated metal dissolution rates (thermodynamic material instability).191,192 The traditional view that all components are ‘frozen’ in space (static active sites) and that only the reactants and products are mobile has been revised as dynamic phenomena have been observed during the OER on metal oxide193,194 and perovskite142,150,195 surfaces. These changes suggest that activity and stability can be increased simultaneously. To consider both activity and stability, the activity–stability factor (ASF) has been suggested as a metric; it is the ratio of the rate of O2 production (activity) to the rate of metal dissolution (stability), measured simultaneously using in situ inductively coupled plasma mass spectrometry.196
4.3. Predicting the stability
DFT can determine the stability of the electrochemical catalyst by calculating the thermodynamic energy of reactions that destroy it, such as metal dissolution,197–199 lattice oxygen escape,188,200 surface segregation201,202 and thermodynamic phase stability.203 For example, Kuai et al. revealed that Fe segregation in mixed Ni–Fe hydroxide is energetically favorable at edge sites and at high potential in the alkaline OER (Fig. 15d).199 Wu et al. theoretically showed that the surface reconstruction of CoFe0.25Al1.75O4 can be self-terminated by Al3+ leaching (Fig. 15e).188 Introduction of Al vacancies into the CoFe0.25Al1.75O4 lattice downshifted Op and blocked further formation of oxygen vacancies; the modulated surface reconstruction provided a highly active oxyhydroxide surface without disruption of bulk structures.A data driven screening study conducted by Back et al. has performed phase stability analysis to filter out unstable materials.115 They constructed Pourbaix diagrams, which evaluate thermodynamic phase stability with respect to applied potential and pH. The Pourbaix Gibbs free energy (ΔGPourbaix) was calculated at between −0.2 and +0.2 VRHE, at pH = 0, 7 and 14. ΔGPourbaix alone cannot capture the kinetic stability such as dissolution and segregation, but does provide a thermodynamic picture which serves as the least requirement for the materials. Structural stability represented by convex Hull energy was also used to filter out unstable materials. From a dataset of 23050 DFT calculations, 573 candidates were identified by referring with respect to activity criteria (ηHER/HOR > 0.2 V), and the set was then reduced to 134 by considering electrochemical stability criteria (ΔGPourbaix < 0.1 eV) (Fig. 15c).
4.4. Strategies to enhance stability
To prevent the dissolution of metals in hydr(oxy)oxides, methods of adding metal cations to the electrolyte have been proposed. Chung et al. added Fe cations to the electrolyte for the increased stability of Fe–MOxHy hydr(oxy)oxide clusters (M = Ni, Co, Fe) in the alkaline OER.204 In pure KOH solutions, the activities of the Fe–MOxHy samples were deactivated and Fe was depleted after just 1 h of potential hold at 1.7 V (Fig. 16a). However, addition of 0.1 ppm Fen+(aq.) to the electrolyte effectively prevented activity loss under the same test conditions, without decrease in the initial OER activity (Fig. 16c). Isotope experiment with 56Fe in the initial electrode and 57Fe in the initial electrolyte showed that two isotope Fe were switched during OER operation so the electrode maintained its total Fe content (Fig. 16b and d). The results indicate that Fe–MOxHy itself was not absolutely stable in 0.1 ppm Fen+(aq.) electrolyte, but that OER activity could be sustained over time by enabling continuous Fe dynamic exchange at the interface. In the case of Ni–Co3O4, it was also reported that Fe impurities in KOH electrolyte suppressed the lattice-metal corrosion under OER conditions.250 Fe impurities replacing the lattice metal of the Ni (oxy)hydroxide catalyst not only improved the dynamic stability, but also provided the highly active site by themselves.160,251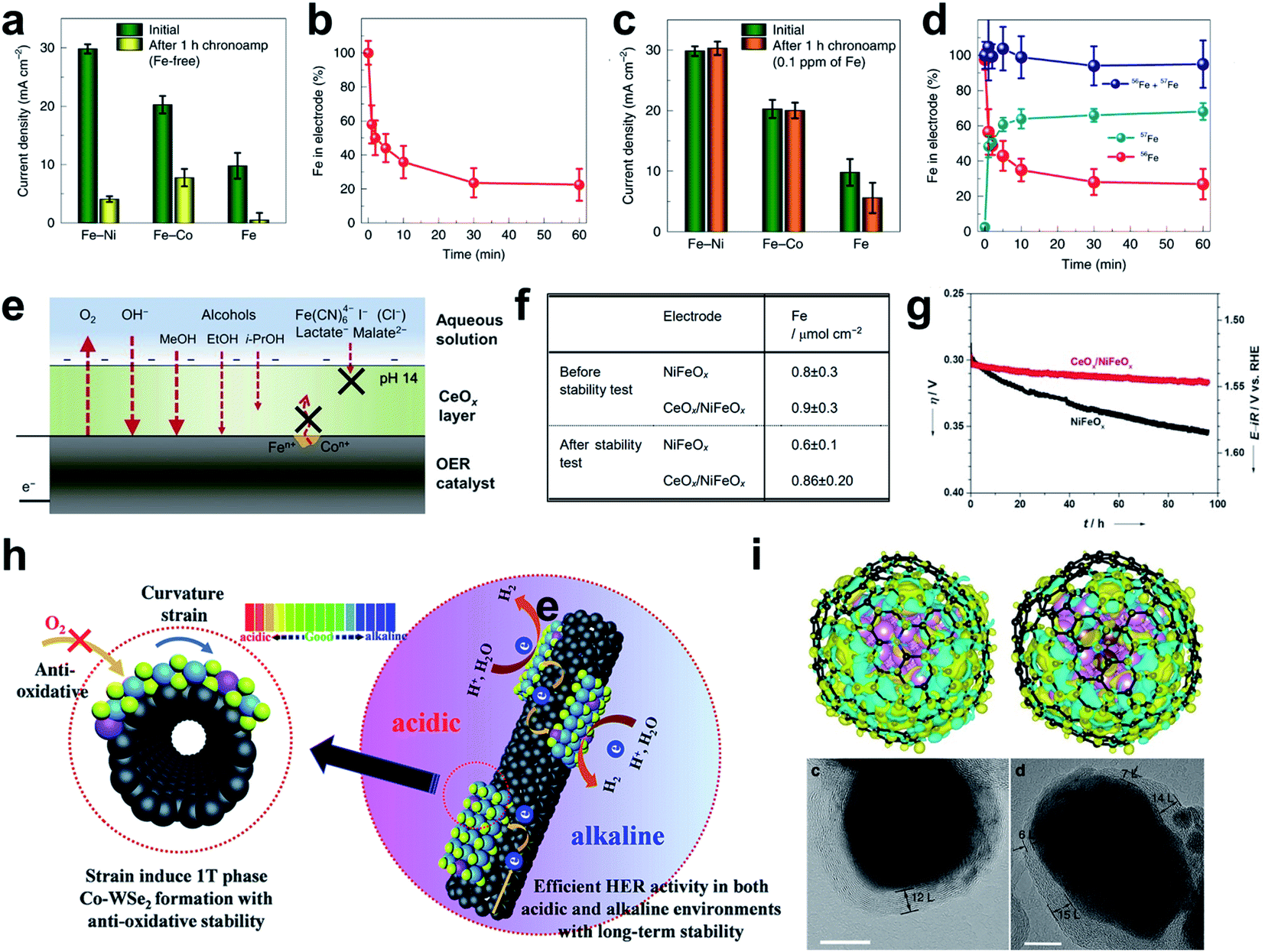 | ||
| Fig. 16 Stability test of the Fe-NiOxHy electrode for 1 h (a) in Fe-free purified KOH and (b) in KOH solution containing 0.1 ppm Fe. Change of total Fe amount in the Fe-NiOxHy electrode over time (c) in Fe-free purified KOH and (d) in a KOH solution containing 0.1 ppm Fe. Reproduced from ref. 204. Copyright 2020, Springer Nature. (e) Schematic of the deposition of a permselective CeOx layer on the OER catalyst. (f) Compositions of bare and CeOx-coated NiFeOx measured by ICP before and after the stability test for 96 h. (g) Electrocatalytic stability test by controlled current electrolysis. Reproduced from ref. 205. Copyright 2018, Wiley. (h) HER scheme for the Co-WSe2/MWNT heterostructure catalyst. Reproduced from ref. 206. Copyright 2018, Royal Society of Chemistry. (i) HAADF image and calculated charge-density differences of different models. Reproduced from ref. 208. Copyright 2017, Springer Nature. | ||
Fe impurities in alkaline electrolytes also help in the phase maintenance of the Ni- and Co-based OER catalysts. Ni(OH)2 films aged in Fe-containing 1 M KOH exhibited a stable double (oxy)hydroxide (LDH) phase, inhibiting the phase transformation over aging or overcharging. In contrast, Ni(OH)2 films aged in an Fe-containing electrolyte predominantly presented the β-Ni(OH)2 phase, which undergoes phase transformation over aging (into β-Ni(OH)2 and β-NiOOH) and overcharging (from β-NiOOH to γ-NiOOH).252 The Pourbaix diagram of Ni–H2O with/without Fe doping showed that Fe incorporation lowers the free energy of all compounds and expands the phase region of Ni(OH)2, NiOOH, and NiO2 under OER conditions (near 1.5 V vs. SHE). In the same study, ab initio molecular dynamics (AIMD) simulation showed that the intercalation and adsorption of the hydrated Fe cations are thermodynamically and kinetically stable in the layered γ-NiOOH system, and those adsorbed on the catalyst surface lower the OER overpotential of Ni active sites.253 On the other hand, Fe cations adsorbed on the oxygen-defective Co oxide catalyst (CoOx) serve as main active sites of the alkaline OER.254
Leakage of metal cations from the OER electrode can also be prevented by coating the active surface with a layer that is permselective to reactants. Highly active NiFeOx electrocatalysts for the OER gradually deactivate over time, owing to loss of Fe species from the active sites into solution during catalysis. Anodic deposition of a CeOx layer prevented the loss of such Fe species from the OER catalysts (Fig. 16f), and yielded a highly stable activity (Fig. 16g).205 The CeOx layer did not affect the OER activity or electronic conductivity of the catalyst underneath, but allowed the selective permeation of OH− and O2 while preventing diffusion of redox ions (Fig. 16e). The deposition of the CeOx layer even retained Co species in CoOx nanoparticles; this result suggests that CeOx layer addition could increase the stability, without significant loss of the OER activity.
To prevent oxidation and dissolution of a material, strain engineering to induce phase transformation to a more stable phase is a feasible strategy. Jun et al. developed a synthetic technique for cobalt-doped tungsten selenide (Co-WSe2) using multi-walled carbon nanotubes (MWNTs) as a topological template. The nucleation and growth occur on the carbon template, which induces curvature strain to the Co-WSe2 and exposes the 1 T phase (Fig. 16h). The 1 T phase Co-WSe2/MWNT heterostructure showed better HER activity and stability than other Co-WSe2 which were degraded by oxidation of the surface. The improved stability is attributed to the stable curved 1T phase decreasing the oxidation degree.206
Encapsulation is widely applied to increase stability by increasing dissolution resistance and conductivity.207 Chen et al. have synthesized a cobalt based bimetallic alloy encapsulated by six to fifteen layers of nitrogen-doped graphene layers208 (Fig. 16i). They achieved perfect stability, with corrosion resistance even after 10000 cycles of HER operation under alkaline conditions, and had superior activity to commercial Pt/C. DFT calculations reveal that a small number of Ru atoms helped to optimize the HBE, and charges were transferred well to the carbon sheets, so the HBE was optimized.
In summary, the oxidation and dissolution of metal components in a high potential environment can significantly degrade a material's electrochemical activity even under alkaline conditions. Many experimental and theoretical developments in recent years have led to significant stability improvements through various strategies. One potential strategy is to dynamically recover the dissolved metals by adding metal cations to the electrolyte. Impurity metal elements can be added to increase the inherent dissolution resistance by minimizing unwanted phase transformation. Furthermore, the phase of a material can be altered intentionally by strain engineering to increase its resistance to the oxidation and dissolution. Coating the active materials with a permselective CeOx layer or graphene layers can also be applied to enhance the corrosion resistance.
5. Scope and future perspectives
DFT calculations that are used with suitable activity descriptors enable rational design of highly active catalysts by executing a theoretical screening process; they also explain the reason for activity, stability and conductivity increase observed in experiments. Despite the undeniable success of the theoretical approaches in the field, researchers should pay attention to the accuracy of the simulation and consistency with the experiments. Although descriptors based on the adsorption strength of the intermediates of the reaction, such as ΔGH for the HER and ΔGO − ΔGOH for the OER, have guided the development of novel catalysts, these only give a thermodynamic picture of the reaction, and thus are used to rapidly capture the trends of electrochemical reactions. Although the activation barriers can be examined by various optimization methods, the results rely largely on the transition state pathways. To ensure reliability and reproducibility, the transition state should be carefully characterized by providing configurations and vibrational frequencies, and the key simulation details should be open to other researchers.The solvent effect is also required for the adsorption energy calculation given that the reaction takes place in a solvation environment.209,210 Inclusion of the solvent effect increases the computational complexity, but some compromises have been proposed, such as implicit solvation211 or microsolvation212 with just three water molecules. Those methods cannot capture the overall contribution of co-adsorbed water to the adsorbates, but can be a good compromise between calculation cost and the need to consider the solvation effect.
The effects of spectator species such as hydroxide anion or aqueous metal cations should also be considered. They can induce electrostatic interaction at the double layer region of the electrode surface. The double layer structures contain substrate and adsorbate interaction, which have important functions.213 The full effect of the system should be considered when elucidating the atomic nature of the catalytic system by simulation. In addition, the electronic structure-based descriptors such as orbital occupancy or band center, must select an appropriate U parameter for TMs in oxides.214–217
Accurate mechanistic studies can increase the understanding of HER and OER catalysis to guide the development of new materials. However, their reaction mechanisms are still in debate. Even from the same perovskite family, different LOM pathways142,145 or further refined steps218 have been proposed. Operando and in situ spectroscopies can be used to reveal the nature of the active sites, metal oxidation states and local coordination structure of the catalyst during the water splitting reaction. XAS,141,219–222 surface X-ray scattering,223 mass spectrometry,224 attenuated total reflectance Fourier transform infrared spectroscopy (ATR FT-IR)225 and Raman spectroscopy226 have been used for mechanistic studies of electrochemistry, coupled with DFT calculation. Thorough operando spectroscopy and computational identification of the active sites and structure-dependent activity origin also can extend the perspectives and considerations of each material.
Most in situ and theoretical efforts so far have been conducted only on well-defined surfaces of polycrystalline catalysts. However, the material search space has expanded to include hydroxides, phosphides, sulfides, and nitrides, so fundamental studies on those surfaces are necessary. The surface structure and reconstruction vary in the electrochemical environment, so these various structures must be considered when designing a surface-reaction simulation.227,228 The Pourbaix diagram could give a simple and powerful description to help identify stable structures.229
Electronic structural characteristics differ among different types of catalysts, so setting a suitable descriptor for each catalyst could be a way to increase the accuracy of the OER activity descriptor. Various OER candidates (e.g., hydroxides, oxyhydroxides, phosphides, sulfides, carbides) do not have OER activity descriptors; their identification requires analysis of the electronic structure and water oxidation mechanism of these materials, although previous research mainly focused on perovskites and simple oxides. In oxyhydroxides, OER cycling of the low valence of Co can reduce the overpotential,22,230 and the increased OER activity with V dopant in NiFe(OH)2 could be explained by an increased overlap between Md and Op.231 In nickel phosphides, the OER overpotential shows a volcano plot with change in the Ni 3d-band center, which could be decreased by Fe substitution.125,232 Further analysis of OER mechanisms and activity descriptors must be expanded to diverse material groups.
Finally, recent research on water electrolysis has focused on applications on a real commercial scale. Such applications require reduction of prices, use of abundant water resources such as seawater, and must be validated experimentally on a commercial scale. For compatible integration of the catalyst to increase the cost efficiency of electrolyzers, the need to develop an HER-and-OER bifunctional catalyst has been emphasized. Several important findings have shown great opportunity in the field of bifunctional catalysts, such as NiFe-layered double hydroxides, CoMnO/CN, and N, S co-doped carbon nanotubes.233–235 Compatibility with different operating conditions is also important to reduce the overall cost, so a pH-universal catalyst that can be effective both in acidic and alkaline environments has been sought.37,129,236,237 Abundant seawater is also being evaluated as a resource for water electrolysis,238,239 and the alkaline media alleviates the OER selectivity problem, which is in competition with an accompanying chlorine evolution reaction (CER).240 Therefore, mechanistic study on the CER in alkaline seawater splitting is required, although recent CER research has been focused on noble metal oxide catalysts in acidic environments.241–245 Consequently, commercial-scale activity and durability tests are also expected for both alkaline OER and HER.233–235
6. Conclusion
Fundamental understanding on the nature of the catalytic reaction has advanced the development of the TM-based HER and OER catalysts in recent decades. In the case of the HER, substantial effort has been devoted both to find a catalyst that has optimal hydrogen binding thermodynamics, and to minimize the water dissociation energy barrier kinetics. These two important criteria have been proven to determine the HER activity of the materials. We have first explored from classical HBE dependent volcano relationships to the state-of-the-art design strategies such as electronic structure tuning, various nano-structuring concepts and machine learning-based expansion of search space. Thereafter, we discussed the Volmer reaction, with main focus on the water dissociation barrier, from the perspectives of kinetics and thermodynamics. Several studies have determined that fine tuning the adsorption strength of OH, H and H2O provides huge room for increase in catalytic activity. From the kinetics perspective, introducing a water dissociation center to make a dual active site is the most powerful design strategy, and has proven to be viable. The functions of promoter species in the electrolyte have also been highlighted.Two major mechanisms of the OER, i.e., AEM and LOM, and parameters for determining favorable paths, such as oxygen vacancy formation energy, N–V, Op and Md were introduced. In AEM, ΔGO − ΔGOH has been proposed as a conventional OER activity descriptor according to the scaling relationship between the adsorption strength of intermediates, and electronic structural descriptors including eg occupancy, M–O covalency, and NEE could predict catalytic activity. In the LOM, in which lattice oxygen participates, M–O covalency, oxygen anion and vacancy diffusion are significantly involved in catalytic activity. The pros and cons of each descriptor and their suitability for specific material families should be considered when choosing an appropriate descriptor. We also discussed ways to tune that property to increase OER activity or investigate it experimentally and computationally.
High conductivity of the electrode material is an essential requirement when developing semiconductor materials as an electrode. Given that the electrical conductivity of a material is determined by the band gap of the electronic structure, the computationally obtained density of states is widely used to predict the conductivity. Several strategies are reviewed, including doping with conductive heteroatoms such as Co, Ni, V and P atoms, and introducing oxygen vacancies. The broadening of the material search space to TM-based materials is inevitably accompanied by deactivation of the catalyst, and this problem should be considered when evaluating catalysts for practical uses. The detection of the activity criteria by time dependent high-resolution spectroscopy and advanced operando spectroscopy revealed the origin of stability and activity trends. Several computational descriptors have been implemented to gain mechanistic insight towards the stability, and to filter out unstable materials. Successful synthesis methods were reviewed.
Focusing on the combination of theory and experiment, this review has described the design principles of inexpensive catalysts for alkaline water splitting reactions, by increasing the understanding of alkaline HER and OER mechanisms and the relationship between the electronic structure and catalytic activity. Electrical conductivity and instability problems were also mentioned, and future perspectives for increased synergy between theory and experiment were proposed. We hope that this review broadens the perspective of a design principle of inexpensive catalyst for alkaline water splitting reactions, and can provide insight into the field of electrochemistry such as hydrogen oxidation and oxygen reduction reactions (HOR/ORR), which share similar intermediates.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (MSIT) (NRF-2018M3A7B4062825, NRF-2021M3H7A1026173, and NRF-2021M3H4A1A01079300) and the Korea Institute of Energy Technology Evaluation and Planning (20193410100050) in the Ministry of Trade, Industry and Energy.References
- M. Dresselhaus and I. Thomas, Nature, 2001, 414, 332–337 CrossRef CAS PubMed.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- J. A. Turner, Science, 2004, 305, 972–974 CrossRef CAS PubMed.
- S. K. Sahoo, Y. Ye, S. Lee, J. Park, H. Lee, J. Lee and J. W. Han, ACS Energy Lett., 2018, 4, 126–132 CrossRef.
- J. Park, S. Lee, H. E. Kim, A. Cho, S. Kim, Y. Ye, J. W. Han, H. Lee, J. H. Jang and J. Lee, Angew. Chem., Int. Ed., 2019, 58, 16038–16042 CrossRef CAS PubMed.
- J. Kim, Y. Yang, A. Seong, H.-J. Noh, C. Kim, S. Joo, A. Cho, L. Zhang, J. Zhou, J. Q. Wang, J. W. Han, J. Mahmood, J. B. Baek and G. Kim, J. Mater. Chem. A, 2020, 8, 14927–14934 RSC.
- A. Cho, J. Ko, B.-K. Kim and J. W. Han, ACS Catal., 2018, 9, 967–976 CrossRef.
- H. Zhao, Y. P. Zhu and Z. Y. Yuan, Eur. J. Inorg. Chem., 2016, 2016, 1916–1923 CrossRef CAS.
- A. Le Goff, V. Artero, B. Jousselme, P. D. Tran, N. Guillet, R. Métayé, A. Fihri, S. Palacin and M. Fontecave, Science, 2009, 326, 1384–1387 CrossRef CAS PubMed.
- J. Wang, W. Cui, Q. Liu, Z. Xing, A. M. Asiri and X. Sun, Adv. Mater., 2016, 28, 215–230 CrossRef CAS PubMed.
- J. Rossmeisl, K. Dimitrievski, P. Siegbahn and J. K. Nørskov, J. Phys. Chem. C, 2007, 111, 18821–18823 CrossRef CAS.
- S. Anantharaj, S. Ede, K. Karthick, S. S. Sankar, K. Sangeetha, P. Karthik and S. Kundu, Energy Environ. Sci., 2018, 11, 744–771 RSC.
- H. Jin, J. Joo, N. K. Chaudhari, S. I. Choi and K. Lee, ChemElectroChem, 2019, 6, 3244–3253 CrossRef CAS.
- L. Chen, X. Dong, Y. Wang and Y. Xia, Nat. Commun., 2016, 7, 11741 CrossRef CAS PubMed.
- L. Han, S. Dong and E. Wang, Adv. Mater., 2016, 28, 9266–9291 CrossRef CAS PubMed.
- F. Safizadeh, E. Ghali and G. Houlachi, Int. J. Hydrogen Energy, 2015, 40, 256–274 CrossRef CAS.
- F. Lu, M. Zhou, Y. Zhou and X. Zeng, Small, 2017, 13, 1701931 CrossRef PubMed.
- N. Mahmood, Y. Yao, J. W. Zhang, L. Pan, X. Zhang and J. J. Zou, Adv. Sci., 2018, 5, 1700464 CrossRef PubMed.
- Y. Zheng, Y. Jiao, A. Vasileff and S. Z. Qiao, Angew. Chem., Int. Ed., 2018, 57, 7568–7579 CrossRef CAS PubMed.
- M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, Int. J. Hydrogen Energy, 2013, 38, 4901–4934 CrossRef CAS.
- G. Glenk and S. Reichelstein, Nat. Energy, 2019, 4, 216–222 CrossRef CAS.
- B. Zhang, L. Wang, Z. Cao, S. M. Kozlov, F. P. G. de Arquer, C. T. Dinh, J. Li, Z. Wang, X. Zheng, L. Zhang, Y. Wen, O. Voznyy, R. Comin, P. De Luna, T. Regier, W. Bi, E. E. Alp, C.-W. Pao, L. Zheng, Y. Hu, Y. Ji, Y. Li, Y. Zhang, L. Cavallo, H. Peng and E. H. Sargent, Nat. Catal., 2020, 3, 985–992 CrossRef CAS.
- L. Birry and A. Lasia, J. Appl. Electrochem., 2004, 34, 735–749 CrossRef CAS.
- J. Greeley, T. F. Jaramillo, J. Bonde, I. Chorkendorff and J. K. Nørskov, Nat. Mater., 2006, 5, 909–913 CrossRef CAS PubMed.
- X. Mao, L. Wang, Y. Xu, P. Wang, Y. Li and J. Zhao, npj Comput. Mater., 2021, 7, 46 CrossRef CAS.
- T. F. Jaramillo, K. P. Jørgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100–102 CrossRef CAS PubMed.
- J. Kibsgaard, Z. Chen, B. N. Reinecke and T. F. Jaramillo, Nat. Mater., 2012, 11, 963–969 CrossRef CAS PubMed.
- J. Kibsgaard, T. F. Jaramillo and F. Besenbacher, Nat. Chem., 2014, 6, 248–253 CrossRef CAS PubMed.
- M. S. Faber, R. Dziedzic, M. A. Lukowski, N. S. Kaiser, Q. Ding and S. Jin, J. Am. Chem. Soc., 2014, 136, 10053–10061 CrossRef CAS PubMed.
- H. Li, C. Tsai, A. L. Koh, L. Cai, A. W. Contryman, A. H. Fragapane, J. Zhao, H. S. Han, H. C. Manoharan, F. Abild-Pedersen, J. K. Nørskov and X. Zheng, Nat. Mater., 2016, 15, 48–53 CrossRef CAS PubMed.
- P. Wang, X. Zhang, J. Zhang, S. Wan, S. Guo, G. Lu, J. Yao and X. Huang, Nat. Commun., 2017, 8, 14580 CrossRef CAS PubMed.
- J. Hu, C. Zhang, L. Jiang, H. Lin, Y. An, D. Zhou, M. K. Leung and S. Yang, Joule, 2017, 1, 383–393 CrossRef CAS.
- M. Caban-Acevedo, M. L. Stone, J. R. Schmidt, J. G. Thomas, Q. Ding, H. C. Chang, M. L. Tsai, J. H. He and S. Jin, Nat. Mater., 2015, 14, 1245–1251 CrossRef CAS PubMed.
- Y. Wen, J. Qi, D. Zhao, J. Liu, P. Wei, X. Kang and X. Li, Appl. Catal., B, 2021, 293, 120196 CrossRef CAS.
- K. Xu, Y. Sun, Y. Sun, Y. Zhang, G. Jia, Q. Zhang, L. Gu, S. Li, Y. Li and H. J. Fan, ACS Energy Lett., 2018, 3, 2750–2756 CrossRef CAS.
- X. Yang, A.-Y. Lu, Y. Zhu, M. N. Hedhili, S. Min, K.-W. Huang, Y. Han and L.-J. Li, Nano Energy, 2015, 15, 634–641 CrossRef CAS.
- Z. H. Xue, H. Su, Q. Y. Yu, B. Zhang, H. H. Wang, X. H. Li and J. S. Chen, Adv. Energy Mater., 2017, 7, 1602355 CrossRef.
- Q. Gao, W. Zhang, Z. Shi, L. Yang and Y. Tang, Adv. Mater., 2019, 31, 1802880 CrossRef PubMed.
- Y.-Y. Ma, Z.-L. Lang, L.-K. Yan, Y.-H. Wang, H.-Q. Tan, K. Feng, Y.-J. Xia, J. Zhong, Y. Liu, Z.-H. Kang and Y.-G. Li, Energy Environ. Sci., 2018, 11, 2114–2123 RSC.
- Q. Hu, X. Liu, B. Zhu, L. Fan, X. Chai, Q. Zhang, J. Liu, C. He and Z. Lin, Nano Energy, 2018, 50, 212–219 CrossRef CAS.
- W. Xiong, Q. Guo, Z. Guo, H. Li, R. Zhao, Q. Chen, Z. Liu and X. Wang, J. Mater. Chem. A, 2018, 6, 4297–4304 RSC.
- Y. Huang, Q. Gong, X. Song, K. Feng, K. Nie, F. Zhao, Y. Wang, M. Zeng, J. Zhong and Y. Li, ACS Nano, 2016, 10, 11337–11343 CrossRef CAS PubMed.
- L. Yu, I. K. Mishra, Y. Xie, H. Zhou, J. Sun, J. Zhou, Y. Ni, D. Luo, F. Yu and Y. Yu, Nano Energy, 2018, 53, 492–500 CrossRef CAS.
- R. Subbaraman, D. Tripkovic, K.-C. Chang, D. Strmcnik, A. P. Paulikas, P. Hirunsit, M. Chan, J. Greeley, V. Stamenkovic and N. M. Markovic, Nat. Mater., 2012, 11, 550–557 CrossRef CAS PubMed.
- R. Subbaraman, D. Tripkovic, D. Strmcnik, K.-C. Chang, M. Uchimura, A. P. Paulikas, V. Stamenkovic and N. M. Markovic, Science, 2011, 334, 1256–1260 CrossRef CAS PubMed.
- N. Danilovic, R. Subbaraman, D. Strmcnik, K. C. Chang, A. Paulikas, V. Stamenkovic and N. M. Markovic, Angew. Chem., Int. Ed., 2012, 51, 12495–12498 CrossRef CAS PubMed.
- G. Chen, T. Wang, J. Zhang, P. Liu, H. Sun, X. Zhuang, M. Chen and X. Feng, Adv. Mater., 2018, 30, 1706279 CrossRef PubMed.
- Y. Matsumoto, S. Yamada, T. Nishida and E. Sato, J. Electrochem. Soc., 1980, 127, 2360 CrossRef CAS.
- J. O. M. Bockris and T. Otagawa, J. Electrochem. Soc., 1984, 131, 290 CrossRef CAS.
- A. Vojvodic and J. K. Nørskov, Science, 2011, 334, 1355–1356 CrossRef CAS PubMed.
- Y. Zhu, W. Zhou, J. Sunarso, Y. Zhong and Z. Shao, Adv. Funct. Mater., 2016, 26, 5862–5872 CrossRef CAS.
- B. Marsan, N. Fradette and G. Beaudoin, J. Electrochem. Soc., 1992, 139, 1889 CrossRef.
- I. Nikolov, R. Darkaoui, E. Zhecheva, R. Stoyanova, N. Dimitrov and T. Vitanov, J. Electroanal. Chem., 1997, 429, 157–168 CrossRef CAS.
- M. Li, Y. Xiong, X. Liu, X. Bo, Y. Zhang, C. Han and L. Guo, Nanoscale, 2015, 7, 8920–8930 RSC.
- R. Chen, H.-Y. Wang, J. Miao, H. Yang and B. Liu, Nano Energy, 2015, 11, 333–340 CrossRef CAS.
- Y. Wang, T. Zhou, K. Jiang, P. Da, Z. Peng, J. Tang, B. Kong, W. B. Cai, Z. Yang and G. Zheng, Adv. Energy Mater., 2014, 4, 1400696 CrossRef.
- H. Liang, F. Meng, M. Cabán-Acevedo, L. Li, A. Forticaux, L. Xiu, Z. Wang and S. Jin, Nano Lett., 2015, 15, 1421–1427 CrossRef CAS PubMed.
- Z. Lu, H. Wang, D. Kong, K. Yan, P.-C. Hsu, G. Zheng, H. Yao, Z. Liang, X. Sun and Y. Cui, Nat. Commun., 2014, 5, 4345 CrossRef CAS PubMed.
- F. Song and X. Hu, Nat. Commun., 2014, 5, 4477 CrossRef CAS PubMed.
- J. X. Feng, S. H. Ye, H. Xu, Y. X. Tong and G. R. Li, Adv. Mater., 2016, 28, 4698–4703 CrossRef CAS PubMed.
- T. Gao, Z. Jin, M. Liao, J. Xiao, H. Yuan and D. Xiao, J. Mater. Chem. A, 2015, 3, 17763–17770 RSC.
- Y. Liu, H. Wang, D. Lin, C. Liu, P.-C. Hsu, W. Liu, W. Chen and Y. Cui, Energy Environ. Sci., 2015, 8, 1719–1724 RSC.
- M. Xing, L.-B. Kong, M.-C. Liu, L.-Y. Liu, L. Kang and Y.-C. Luo, J. Mater. Chem. A, 2014, 2, 18435–18443 RSC.
- Y. Meng, W. Song, H. Huang, Z. Ren, S.-Y. Chen and S. L. Suib, J. Am. Chem. Soc., 2014, 136, 11452–11464 CrossRef CAS PubMed.
- M. Fekete, R. K. Hocking, S. L. Chang, C. Italiano, A. F. Patti, F. Arena and L. Spiccia, Energy Environ. Sci., 2013, 6, 2222–2232 RSC.
- P. Ganesan, M. Prabu, J. Sanetuntikul and S. Shanmugam, ACS Catal., 2015, 5, 3625–3637 CrossRef CAS.
- W. Zhu, X. Yue, W. Zhang, S. Yu, Y. Zhang, J. Wang and J. Wang, Chem. Commun., 2016, 52, 1486–1489 RSC.
- S. Dou, L. Tao, J. Huo, S. Wang and L. Dai, Energy Environ. Sci., 2016, 9, 1320–1326 RSC.
- B. Dong, X. Zhao, G.-Q. Han, X. Li, X. Shang, Y.-R. Liu, W.-H. Hu, Y.-M. Chai, H. Zhao and C.-G. Liu, J. Mater. Chem. A, 2016, 4, 13499–13508 RSC.
- C. Xia, Q. Jiang, C. Zhao, M. N. Hedhili and H. N. Alshareef, Adv. Mater., 2016, 28, 77–85 CrossRef CAS PubMed.
- Y. Liu, H. Cheng, M. Lyu, S. Fan, Q. Liu, W. Zhang, Y. Zhi, C. Wang, C. Xiao, S. Wei, B. Ye and Y. Xie, J. Am. Chem. Soc., 2014, 136, 15670–15675 CrossRef CAS PubMed.
- X. Xu, F. Song and X. Hu, Nat. Commun., 2016, 7, 12324 CrossRef CAS PubMed.
- A. T. Swesi, J. Masud and M. Nath, Energy Environ. Sci., 2016, 9, 1771–1782 RSC.
- H. Jiang, Y. Yao, Y. Zhu, Y. Liu, Y. Su, X. Yang and C. Li, ACS Appl. Mater. Interfaces, 2015, 7, 21511–21520 CrossRef CAS PubMed.
- Z. Kou, L. Zhang, Y. Ma, X. Liu, W. Zang, J. Zhang, S. Huang, Y. Du, A. K. Cheetham and J. Wang, Appl. Catal., B, 2019, 243, 678–685 CrossRef CAS.
- K. Xu, H. Ding, H. Lv, P. Chen, X. Lu, H. Cheng, T. Zhou, S. Liu, X. Wu, C. Wu and Y. Xie, Adv. Mater., 2016, 28, 3326–3332 CrossRef CAS PubMed.
- Y.-J. Tang, C.-H. Liu, W. Huang, X.-L. Wang, L.-Z. Dong, S.-L. Li and Y.-Q. Lan, ACS Appl. Mater. Interfaces, 2017, 9, 16977–16985 CrossRef CAS PubMed.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133 CrossRef.
- H. Jónsson, G. Mills and K. W. Jacobsen, Classical and Quantum Dynamics in Condensed Phase Simulations, in Proceedings of the International School of Physics, LERICI, Villa Marigola, 1998, pp. 385–404 Search PubMed.
- J. I. Steinfeld, J. S. Francisco and W. L. Hase, Chemical Kinetics and Dynamics, Prentice Hall Upper, Saddle River, NJ, 1999 Search PubMed.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- V. Artero, M. Chavarot-Kerlidou and M. Fontecave, Angew. Chem., Int. Ed., 2011, 50, 7238–7266 CrossRef CAS PubMed.
- A. Badreldin, A. E. Abusrafa and A. Abdel-Wahab, ChemSusChem, 2021, 14, 10 CrossRef CAS PubMed.
- N. K. Chaudhari, H. Jin, B. Kim and K. Lee, Nanoscale, 2017, 9, 12231–12247 RSC.
- Y. Guo, T. Park, J. W. Yi, J. Henzie, J. Kim, Z. Wang, B. Jiang, Y. Bando, Y. Sugahara, J. Tang and Y. Yamauchi, Adv. Mater., 2019, 31, 1807134 CrossRef PubMed.
- L. Li, P. Wang, Q. Shao and X. Huang, Chem. Soc. Rev., 2020, 49, 3072–3106 RSC.
- D. Zhao, Z. Zhuang, X. Cao, C. Zhang, Q. Peng, C. Chen and Y. Li, Chem. Soc. Rev., 2020, 49, 2215–2264 RSC.
- J. Creus, J. De Tovar, N. Romero, J. García-Antón, K. Philippot, R. Bofill and X. Sala, ChemSusChem, 2019, 12, 2493–2514 CrossRef CAS PubMed.
- D. Li, J. Shi and C. Li, Small, 2018, 14, 1704179 CrossRef PubMed.
- X. Wu, C. Tang, Y. Cheng, X. Min, S. P. Jiang and S. Wang, Chem.–Eur. J., 2020, 26, 3906–3929 CAS.
- J. Zhang, Q. Zhang and X. Feng, Adv. Mater., 2019, 31, 1808167 CrossRef PubMed.
- B. You, M. T. Tang, C. Tsai, F. Abild-Pedersen, X. Zheng and H. Li, Adv. Mater., 2019, 31, 1807001 CrossRef PubMed.
- D. Strmcnik, M. Uchimura, C. Wang, R. Subbaraman, N. Danilovic, D. Van Der Vliet, A. P. Paulikas, V. R. Stamenkovic and N. M. Markovic, Nat. Chem., 2013, 5, 300–306 CrossRef CAS PubMed.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, J23 CrossRef.
- W. Sheng, M. Myint, J. G. Chen and Y. Yan, Energy Environ. Sci., 2013, 6, 1509–1512 RSC.
- W. Sheng, Z. Zhuang, M. Gao, J. Zheng, J. G. Chen and Y. Yan, Nat. Commun., 2015, 6, 5848 CrossRef CAS PubMed.
- J. Durst, A. Siebel, C. Simon, F. Hasché, J. Herranz and H. Gasteiger, Energy Environ. Sci., 2014, 7, 2255–2260 RSC.
- A. Nilsson, L. Pettersson, B. Hammer, T. Bligaard, C. Christensen and J. Nørskov, Catal. Lett., 2005, 100, 111–114 CrossRef CAS.
- J. K. Nørskov, F. Abild-Pedersen, F. Studt and T. Bligaard, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 937–943 CrossRef PubMed.
- J. Greeley and M. Mavrikakis, Nat. Mater., 2004, 3, 810–815 CrossRef CAS PubMed.
- H. Jin, X. Liu, S. Chen, A. Vasileff, L. Li, Y. Jiao, L. Song, Y. Zheng and S.-Z. Qiao, ACS Energy Lett., 2019, 4, 805–810 CrossRef CAS.
- Q. Xu, G. Li, Y. Zhang, Q. Yang, Y. Sun and C. Felser, ACS Catal., 2020, 10, 5042–5048 CrossRef CAS PubMed.
- D. Guan, J. Zhou, Y.-C. Huang, C.-L. Dong, J.-Q. Wang, W. Zhou and Z. Shao, Nat. Commun., 2019, 10, 3755 CrossRef PubMed.
- H. Vrubel and X. Hu, Angew. Chem., Int. Ed., 2012, 51, 12703–12706 CrossRef CAS PubMed.
- Y. Liu, G. Yu, G. D. Li, Y. Sun, T. Asefa, W. Chen and X. Zou, Angew. Chem., Int. Ed., 2015, 54, 10752–10757 CrossRef CAS PubMed.
- T. Ouyang, Y. Q. Ye, C. Y. Wu, K. Xiao and Z. Q. Liu, Angew. Chem., Int. Ed., 2019, 58, 4923–4928 CrossRef CAS PubMed.
- R. Michalsky, Y.-J. Zhang and A. A. Peterson, ACS Catal., 2014, 4, 1274–1278 CrossRef CAS.
- B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jørgensen, J. H. Nielsen, S. Horch, I. Chorkendorff and J. K. Nørskov, J. Am. Chem. Soc., 2005, 127, 5308–5309 CrossRef CAS PubMed.
- J. Jiang, M. Gao, W. Sheng and Y. Yan, Angew. Chem., Int. Ed., 2016, 55, 15240–15245 CrossRef CAS PubMed.
- Y. R. Zheng, P. Wu, M. R. Gao, X. L. Zhang, F. Y. Gao, H. X. Ju, R. Wu, Q. Gao, R. You, W. X. Huang, S. J. Liu, S. W. Hu, J. F. Zhu, Z. Y. Li and S. H. Yu, Nat. Commun., 2018, 9, 2533 CrossRef PubMed.
- D. Voiry, H. Yamaguchi, J. Li, R. Silva, D. C. Alves, T. Fujita, M. Chen, T. Asefa, V. B. Shenoy, G. Eda and M. Chhowalla, Nat. Mater., 2013, 12, 850–855 CrossRef CAS PubMed.
- S. Maiti, K. Maiti, M. T. Curnan, K. Kim, K.-J. Noh and J. W. Han, Energy Environ. Sci., 2021, 14, 3717–3756 RSC.
- H. Huang, H. Jung, H. Jun, D. Y. Woo, J. W. Han and J. Lee, Chem. Eng. J., 2021, 405, 126977 CrossRef CAS.
- K. Tran and Z. W. Ulissi, Nat. Catal., 2018, 1, 696–703 CrossRef CAS.
- S. Back, J. Na, K. Tran and Z. W. Ulissi, Phys. Chem. Chem. Phys., 2020, 22, 19454–19458 RSC.
- M. Sun, A. W. Dougherty, B. Huang, Y. Li and C. H. Yan, Adv. Energy Mater., 2020, 10, 1903949 CrossRef CAS.
- J. Zheng, X. Sun, C. Qiu, Y. Yan, Z. Yao, S. Deng, X. Zhong, G. Zhuang, Z. Wei and J. Wang, J. Phys. Chem. C, 2020, 124, 13695–13705 CrossRef CAS.
- R. B. Wexler, J. M. P. Martirez and A. M. Rappe, J. Am. Chem. Soc., 2018, 140, 4678–4683 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, Y. Zhu, L. H. Li, Y. Han, Y. Chen, M. Jaroniec and S.-Z. Qiao, J. Am. Chem. Soc., 2016, 138, 16174–16181 CrossRef CAS PubMed.
- J. Kim, H. Jung, S.-M. Jung, J. Hwang, D. Y. Kim, N. Lee, K.-S. Kim, H. Kwon, Y.-T. Kim, J. W. Han and J. K. Kim, J. Am. Chem. Soc., 2021, 143, 1399–1408 CrossRef CAS PubMed.
- Y. Wu, X. Liu, D. Han, X. Song, L. Shi, Y. Song, S. Niu, Y. Xie, J. Cai, S. Wu, J. Kang, J. Zhou, Z. Chen, X. Zheng, X. Xiao and G. Wang, Nat. Commun., 2018, 9, 1425 CrossRef PubMed.
- J. Lee, H. Jung, Y. S. Park, N. Kwon, S. Woo, N. C. S. Selvam, G. S. Han, H. S. Jung, P. J. Yoo, S. M. Choi, J. W. Han and B. Lim, Appl. Catal., B, 2021, 294, 120246 CrossRef CAS.
- J. L. Fajín, M. N. l. DS Cordeiro and J. R. Gomes, J. Phys. Chem. A, 2014, 118, 5832–5840 CrossRef PubMed.
- I. T. McCrum and M. T. Koper, Nat. Energy, 2020, 5, 891–899 CrossRef CAS.
- H. Roh, H. Jung, H. Choi, J. W. Han, T. Park, S. Kim and K. Yong, Appl. Catal., B, 2021, 120434 CrossRef CAS.
- C.-T. Dinh, A. Jain, F. P. G. de Arquer, P. De Luna, J. Li, N. Wang, X. Zheng, J. Cai, B. Z. Gregory, O. Voznyy, B. Liu, M. Sinton, D. Crumlin and E. J. Sargent, Nat. Energy, 2019, 4, 107–114 CrossRef CAS.
- J. Huang, J. Han, T. Wu, K. Feng, T. Yao, X. Wang, S. Liu, J. Zhong, Z. Zhang, Y. Zhang and B. Song, ACS Energy Lett., 2019, 4, 3002–3010 CrossRef CAS.
- J. Zhang, T. Wang, D. Pohl, B. Rellinghaus, R. Dong, S. Liu, X. Zhuang and X. Feng, Angew. Chem., Int. Ed., 2016, 55, 6702–6707 CrossRef CAS PubMed.
- J. Staszak-Jirkovský, C. D. Malliakas, P. P. Lopes, N. Danilovic, S. S. Kota, K.-C. Chang, B. Genorio, D. Strmcnik, V. R. Stamenkovic, M. G. Kanatzidis and N. M. Markovic, Nat. Mater., 2016, 15, 197–203 CrossRef PubMed.
- E. Liu, J. Li, L. Jiao, H. T. T. Doan, Z. Liu, Z. Zhao, Y. Huang, K. Abraham, S. Mukerjee and Q. Jia, J. Am. Chem. Soc., 2019, 141, 3232–3239 CrossRef CAS PubMed.
- N. Dubouis, A. Serva, R. Berthin, G. Jeanmairet, B. Porcheron, E. Salager, M. Salanne and A. Grimaud, Nat. Catal., 2020, 3, 656–663 CrossRef CAS.
- G. Cassone, F. Creazzo, P. V. Giaquinta, J. Sponer and F. Saija, Phys. Chem. Chem. Phys., 2017, 19, 20420–20429 RSC.
- T. Cheng, L. Wang, B. V. Merinov and W. A. Goddard III, J. Am. Chem. Soc., 2018, 140, 7787–7790 CrossRef CAS PubMed.
- X. Sun, J. Dai, Y. Guo, C. Wu, F. Hu, J. Zhao, X. Zeng and Y. Xie, Nanoscale, 2014, 6, 8359–8367 RSC.
- P. Liu, J. Zhu, J. Zhang, P. Xi, K. Tao, D. Gao and D. Xue, ACS Energy Lett., 2017, 2, 745–752 CrossRef CAS.
- Z. Zheng, L. Yu, M. Gao, X. Chen, W. Zhou, C. Ma, L. Wu, J. Zhu, X. Meng, J. Hu, Y. Tu, S. Wu, J. Mao, Z. Tian and D. Deng, Nat. Commun., 2020, 11, 3315 CrossRef CAS PubMed.
- D. C. Nguyen, T. L. L. Doan, S. Prabhakaran, D. T. Tran, D. H. Kim, J. H. Lee and N. H. Kim, Nano Energy, 2021, 82, 105750 CrossRef CAS.
- S. Bolar, S. Shit, N. C. Murmu, P. Samanta and T. Kuila, ACS Appl. Mater. Interfaces, 2021, 13, 765–780 CrossRef CAS PubMed.
- Q. Xiong, Y. Wang, P. F. Liu, L. R. Zheng, G. Wang, H. G. Yang, P. K. Wong, H. Zhang and H. Zhao, Adv. Mater., 2018, 30, 1801450 CrossRef PubMed.
- Y. Shi, Y. Zhou, D.-R. Yang, W.-X. Xu, C. Wang, F.-B. Wang, J.-J. Xu, X.-H. Xia and H.-Y. Chen, J. Am. Chem. Soc., 2017, 139, 15479–15485 CrossRef CAS PubMed.
- L. Cao, Q. Luo, W. Liu, Y. Lin, X. Liu, Y. Cao, W. Zhang, Y. Wu, J. Yang, T. Yao and S. Wei, Nat. Catal., 2019, 2, 134–141 CrossRef CAS.
- A. Grimaud, O. Diaz-Morales, B. Han, W. T. Hong, Y.-L. Lee, L. Giordano, K. A. Stoerzinger, M. T. Koper and Y. Shao-Horn, Nat. Chem., 2017, 9, 457–465 CrossRef CAS PubMed.
- C. Yang, C. Laberty-Robert, D. Batuk, G. Cibin, A. V. Chadwick, V. Pimenta, W. Yin, L. Zhang, J.-M. Tarascon and A. Grimaud, J. Phys. Chem. Lett., 2017, 8, 3466–3472 CrossRef CAS PubMed.
- N. Zhang, X. Feng, D. Rao, X. Deng, L. Cai, B. Qiu, R. Long, Y. Xiong, Y. Lu and Y. Chai, Nat. Commun., 2020, 11, 4066 CrossRef CAS PubMed.
- X. Rong, J. Parolin and A. M. Kolpak, ACS Catal., 2016, 6, 1153–1158 CrossRef CAS.
- Y. Sun, H. Liao, J. Wang, B. Chen, S. Sun, S. J. H. Ong, S. Xi, C. Diao, Y. Du, J.-O. Wang, M. B. H. Breese, S. Li, H. Zhang and Z. J. Xu, Nat. Catal., 2020, 3, 554–563 CrossRef CAS.
- Z.-F. Huang, J. Song, Y. Du, S. Xi, S. Dou, J. M. V. Nsanzimana, C. Wang, Z. J. Xu and X. Wang, Nat. Energy, 2019, 4, 329–338 CrossRef CAS.
- A. Grimaud, W. T. Hong, Y. Shao-Horn and J.-M. Tarascon, Nat. Mater., 2016, 15, 121–126 CrossRef CAS PubMed.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- J. T. Mefford, X. Rong, A. M. Abakumov, W. G. Hardin, S. Dai, A. M. Kolpak, K. P. Johnston and K. J. Stevenson, Nat. Commun., 2016, 7, 11053 CrossRef CAS PubMed.
- J. S. Yoo, X. Rong, Y. Liu and A. M. Kolpak, ACS Catal., 2018, 8, 4628–4636 CrossRef CAS.
- J. Rossmeisl, A. Logadottir and J. K. Nørskov, Chem. Phys., 2005, 319, 178–184 CrossRef CAS.
- J. Rossmeisl, Z.-W. Qu, H. Zhu, G.-J. Kroes and J. K. Nørskov, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS.
- I. C. Man, H. Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- R. Zhang, L. Wang, L. Pan, Z. Chen, W. Jia, X. Zhang and J.-J. Zou, Appl. Catal., B, 2020, 277, 119237 CrossRef CAS.
- A. D. Doyle, J. H. Montoya and A. Vojvodic, ChemCatChem, 2015, 7, 738–742 CrossRef CAS.
- D. R. Kauffman, X. Deng, D. C. Sorescu, T.-D. Nguyen-Phan, C. Wang, C. M. Marin, E. Stavitski, I. Waluyo and A. Hunt, ACS Catal., 2019, 9, 5375–5382 CrossRef CAS.
- X. Li, D. Jiao, Y. Liang and J.-X. Zhao, Sustainable Energy Fuels, 2021, 5, 3330–3339 RSC.
- X. Xu, H. Xu and D. Cheng, Nanoscale, 2019, 11, 20228–20237 RSC.
- D. Friebel, M. W. Louie, M. Bajdich, K. E. Sanwald, Y. Cai, A. M. Wise, M.-J. Cheng, D. Sokaras, T.-C. Weng, R. Alonso-Mori, R. C. Davis, J. R. Bargar, J. K. Nørskov, A. Nilsson and A. T. Bell, J. Am. Chem. Soc., 2015, 137, 1305–1313 CrossRef CAS PubMed.
- B. Zhang, X. Zheng, O. Vozny, R. Comin, M. Bajdich, M. Garcia-Melchor, J. Xu, M. Liu, F. P. García de Arquer, C. T. Dinh, F. Fan, M. Yuan, E. Yassitepe, A. Janmohamed, N. Chen, T. Regier, L. Han, H. L. Xin, L. Zheng, H. Yang, A. Vojvodic and E. H. Sargent, Science, 2016, 352, 333–337 CrossRef CAS PubMed.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- C. Wei, Z. Feng, G. G. Scherer, J. Barber, Y. Shao-Horn and Z. J. Xu, Adv. Mater., 2017, 29, 1606800 CrossRef PubMed.
- S. Zhou, X. Miao, X. Zhao, C. Ma, Y. Qiu, Z. Hu, J. Zhao, L. Shi and J. Zeng, Nat. Commun., 2016, 7, 11510 CrossRef CAS PubMed.
- B. Zhao, L. Zhang, D. Zhen, S. Yoo, Y. Ding, D. Chen, Y. Chen, Q. Zhang, B. Doyle, X. Xiong and M. Liu, Nat. Commun., 2017, 8, 14586 CrossRef CAS PubMed.
- A. Grimaud, K. J. May, C. E. Carlton, Y.-L. Lee, M. Risch, W. T. Hong, J. Zhou and Y. Shao-Horn, Nat. Commun., 2013, 4, 2439 CrossRef PubMed.
- J. Lee, H. Jung, Y. S. Park, S. Woo, N. Kwon, Y. Xing, S. H. Oh, S. M. Choi, J. W. Han and B. Lim, Chem. Eng. J., 2021, 420, 127670 CrossRef CAS.
- J. Suntivich, W. T. Hong, Y.-L. Lee, J. M. Rondinelli, W. Yang, J. B. Goodenough, B. Dabrowski, J. W. Freeland and Y. Shao-Horn, J. Phys. Chem. C, 2014, 118, 1856–1863 CrossRef CAS.
- H. Lee, O. Gwon, K. Choi, L. Zhang, J. Zhou, J. Park, J.-W. Yoo, J.-Q. Wang, J. H. Lee and G. Kim, ACS Catal., 2020, 10, 4664–4670 CrossRef CAS.
- Y. Duan, S. Sun, S. Xi, X. Ren, Y. Zhou, G. Zhang, H. Yang, Y. Du and Z. J. Xu, Chem. Mater., 2017, 29, 10534–10541 CrossRef CAS.
- I. Yamada, A. Takamatsu, K. Asai, T. Shirakawa, H. Ohzuku, A. Seno, T. Uchimura, H. Fujii, S. Kawaguchi, K. Wada, H. Ikeno and S. Yagi, J. Phys. Chem. C, 2018, 122, 27885–27892 CrossRef CAS.
- W. T. Hong, R. E. Welsch and Y. Shao-Horn, J. Phys. Chem. C, 2016, 120, 78–86 CrossRef CAS.
- Y. Zhou, S. Sun, J. Song, S. Xi, B. Chen, Y. Du, A. C. Fisher, F. Cheng, X. Wang, H. Zhang and Z. J. Xu, Adv. Mater., 2018, 30, 1802912 CrossRef PubMed.
- X. Huang, J. Wang, H. B. Tao, H. Tian and H. Xu, Chem. Sci., 2019, 10, 3340–3345 RSC.
- Y. Zhu, H. A. Tahini, Z. Hu, Z. G. Chen, W. Zhou, A. C. Komarek, Q. Lin, H. J. Lin, C. T. Chen, Y. Zhong, M. T. Fernandez-Diaz, S. C. Smith, H. Wang, M. Liu and Z. Shao, Adv. Mater., 2020, 32, 1905025 CrossRef CAS PubMed.
- Y. Zhu, Q. Lin, Z. Hu, Y. Chen, Y. Yin, H. A. Tahini, H. J. Lin, C. T. Chen, X. Zhang, Z. Shao and H. Wang, Small, 2020, 16, 2001204 CrossRef CAS PubMed.
- X. Wang, Z. Pan, X. Chu, K. Huang, Y. Cong, R. Cao, R. Sarangi, L. Li, G. Li and S. Feng, Angew. Chem., Int. Ed., 2019, 58, 11720–11725 CrossRef CAS PubMed.
- D. Guan, J. Zhou, Z. Hu, W. Zhou, X. Xu, Y. Zhong, B. Liu, Y. Chen, M. Xu, H. J. Lin, C. T. Chen, J. Q. Wang and Z. Shao, Adv. Funct. Mater., 2019, 29, 1900704 CrossRef.
- F. Van Buren, G. Broers, A. Bouman and C. Boesveld, J. Electroanal. Chem. Interfacial Electrochem., 1978, 87, 389–394 CrossRef CAS.
- S. Royer, D. Duprez and S. Kaliaguine, Catal. Today, 2006, 112, 99–102 CrossRef CAS.
- Y. Pan, X. Xu, Y. Zhong, L. Ge, Y. Chen, J.-P. M. Veder, D. Guan, R. O'Hayre, M. Li, G. Wang, H. Wang, W. Zhou and Z. Shao, Nat. Commun., 2020, 11, 2002 CrossRef CAS PubMed.
- P. Li, X. Duan, Y. Kuang, Y. Li, G. Zhang, W. Liu and X. Sun, Adv. Energy Mater., 2018, 8, 1703341 CrossRef.
- T. Ling, D.-Y. Yan, Y. Jiao, H. Wang, Y. Zheng, X. Zheng, J. Mao, X.-W. Du, Z. Hu, M. Jaroniec and S. Z. Qiao, Nat. Commun., 2016, 7, 12876 CrossRef CAS PubMed.
- A. Parra-Puerto, K. L. Ng, K. Fahy, A. E. Goode, M. P. Ryan and A. Kucernak, ACS Catal., 2019, 9, 11515–11529 CrossRef CAS.
- R. Solmaz, A. Döner and G. Kardaş, Int. J. Hydrogen Energy, 2009, 34, 2089–2094 CrossRef CAS.
- A. Zadick, L. Dubau, N. Sergent, G. Berthome and M. Chatenet, ACS Catal., 2015, 5, 4819–4824 CrossRef CAS.
- Y. Zhang, L. Gao, E. J. M. Hensen and J. P. Hofmann, ACS Energy Lett., 2018, 3, 1360–1365 CrossRef CAS PubMed.
- T. Wu, S. Sun, J. Song, S. Xi, Y. Du, B. Chen, W. A. Sasangka, H. Liao, C. L. Gan, G. G. Scherer, L. Zeng, H. Wang, H. Li, A. Grimaud and Z. J. Xu, Nat. Catal., 2019, 2, 763–772 CrossRef CAS.
- A. C. Garcia, T. Touzalin, C. Nieuwland, N. Perini and M. T. Koper, Angew. Chem., Int. Ed., 2019, 58, 12999–13003 CrossRef CAS PubMed.
- S. H. Chang, N. Danilovic, K.-C. Chang, R. Subbaraman, A. P. Paulikas, D. D. Fong, M. J. Highland, P. M. Baldo, V. R. Stamenkovic, J. W. Freeland, J. A. Eastman and N. M. Markovic, Nat. Commun., 2014, 5, 4191 CrossRef CAS PubMed.
- E. Fabbri and T. J. Schmidt, ACS Catal., 2018, 8, 9765–9774 CrossRef CAS.
- T. Binninger, R. Mohamed, K. Waltar, E. Fabbri, P. Levecque, R. Kötz and T. J. Schmidt, Sci. Rep., 2015, 5, 12167 CrossRef CAS PubMed.
- D. A. Lutterman, Y. Surendranath and D. G. Nocera, J. Am. Chem. Soc., 2009, 131, 3838–3839 CrossRef CAS PubMed.
- M. Huynh, D. K. Bediako and D. G. Nocera, J. Am. Chem. Soc., 2014, 136, 6002–6010 CrossRef CAS PubMed.
- E. Fabbri, M. Nachtegaal, T. Binninger, X. Cheng, B.-J. Kim, J. Durst, F. Bozza, T. Graule, R. Schäublin, L. Wiles, M. Pertoso, N. Danilovic, K. E. Ayers and T. J. Schmidt, Nat. Mater., 2017, 16, 925–931 CrossRef CAS PubMed.
- Y. T. Kim, P. P. Lopes, S. A. Park, A. Y. Lee, J. Lim, H. Lee, S. Back, Y. Jung, N. Danilovic, V. Stamenkovic, J. Erlebacher, J. Snyder and N. M. Markovic, Nat. Commun., 2017, 8, 1449 CrossRef PubMed.
- J. W. Bennett, D. Jones, X. Huang, R. J. Hamers and S. E. Mason, Environ. Sci. Technol., 2018, 52, 5792–5802 CrossRef CAS PubMed.
- A. Abbaspour-Tamijani, J. W. Bennett, D. T. Jones, N. Cartagena-Gonzalez, Z. R. Jones, E. D. Laudadio, R. J. Hamers, J. A. Santana and S. E. Mason, Appl. Surf. Sci., 2020, 515, 145865 CrossRef CAS.
- C. Kuai, Z. Xu, C. Xi, A. Hu, Z. Yang, Y. Zhang, C.-J. Sun, L. Li, D. Sokaras, C. Dong, S.-Z. Qiao, X.-W. Du and F. Lin, Nat. Catal., 2020, 3, 743–753 CrossRef CAS.
- A. Bergmann, T. E. Jones, E. M. Moreno, D. Teschner, P. Chernev, M. Gliech, T. Reier, H. Dau and P. Strasser, Nat. Catal., 2018, 1, 711–719 CrossRef CAS.
- H. Kwon, W. Lee and J. W. Han, RSC Adv., 2016, 6, 69782–69789 RSC.
- M. Choi, I. A. Ibrahim, K. Kim, J. Y. Koo, S. J. Kim, J.-W. Son, J. W. Han and W. Lee, ACS Appl. Mater. Interfaces, 2020, 12, 21494–21504 CrossRef CAS PubMed.
- K. A. Persson, B. Waldwick, P. Lazic and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 235438 CrossRef.
- D. Y. Chung, P. P. Lopes, P. F. B. D. Martins, H. He, T. Kawaguchi, P. Zapol, H. You, D. Tripkovic, D. Strmcnik, Y. Zhu, S. Seifert, S. Lee, V. R. Stamenkovic and N. M. Markovic, Nat. Energy, 2020, 5, 222–230 CrossRef.
- K. Obata and K. Takanabe, Angew. Chem., Int. Ed., 2018, 57, 1616–1620 CrossRef CAS PubMed.
- G. Zhang, X. Zheng, Q. Xu, J. Zhang, W. Liu and J. Chen, J. Mater. Chem. A, 2018, 6, 4793–4800 RSC.
- K. Maiti, K. Kim, K.-J. Noh and J. W. Han, Chem. Eng. J., 2021, 423, 130233 CrossRef CAS.
- J. Su, Y. Yang, G. Xia, J. Chen, P. Jiang and Q. Chen, Nat. Commun., 2017, 8, 14969 CrossRef PubMed.
- J. A. Gauthier, C. F. Dickens, L. D. Chen, A. D. Doyle and J. K. Nørskov, J. Phys. Chem. C, 2017, 121, 11455–11463 CrossRef CAS.
- P. Gono, F. Ambrosio and A. Pasquarello, J. Phys. Chem. C, 2019, 123, 18467–18474 CrossRef CAS.
- K. Mathew, R. Sundararaman, K. Letchworth-Weaver, T. Arias and R. G. Hennig, J. Chem. Phys., 2014, 140, 084106 CrossRef PubMed.
- F. Calle-Vallejo, R. F. de Morais, F. Illas, D. Loffreda and P. Sautet, J. Phys. Chem. C, 2019, 123, 5578–5582 CrossRef CAS.
- V. R. Stamenkovic, D. Strmcnik, P. P. Lopes and N. M. Markovic, Nat. Mater., 2017, 16, 57–69 CrossRef CAS PubMed.
- V. I. Anisimov, J. Zaanen and O. K. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 943 CrossRef CAS PubMed.
- L. Wang, T. Maxisch and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 195107 CrossRef.
- S. Lutfalla, V. Shapovalov and A. T. Bell, J. Chem. Theory Comput., 2011, 7, 2218–2223 CrossRef CAS PubMed.
- E. A. Carter, Science, 2008, 321, 800–803 CrossRef CAS PubMed.
- J. S. Yoo, Y. Liu, X. Rong and A. M. Kolpak, J. Phys. Chem. Lett., 2018, 9, 1473–1479 CrossRef CAS PubMed.
- P. Li, M. Wang, X. Duan, L. Zheng, X. Chen, Y. Zhang, Y. Kuang, Y. Li, Q. Ma, Z. Feng, W. Liu and X. Sun, Nat. Commun., 2019, 10, 1711 CrossRef PubMed.
- H. N. Nong, T. Reier, H.-S. Oh, M. Gliech, P. Paciok, T. H. T. Vu, D. Teschner, M. Heggen, V. Petkov, R. Schlögl, T. Jones and P. Strasser, Nat. Catal., 2018, 1, 841–851 CrossRef CAS.
- F. Dionigi and P. Strasser, Adv. Energy Mater., 2016, 6, 1600621 CrossRef.
- M. Kim, J. Park, H. Ju, J. Y. Kim, H.-S. Cho, C.-H. Kim, B.-H. Kim and S. W. Lee, Energy Environ. Sci., 2021, 14, 3053–3063 RSC.
- R. R. Rao, M. J. Kolb, L. Giordano, A. F. Pedersen, Y. Katayama, J. Hwang, A. Mehta, H. You, J. R. Lunger, H. Zhou, N. B. Halck, T. Vegge, I. Chorkendorff, I. E. L. Stephens and Y. Shao-Horn, Nat. Catal., 2020, 3, 516–525 CrossRef CAS.
- C. Roy, B. Sebok, S. Scott, E. Fiordaliso, J. Sørensen, A. Bodin, D. Trimarco, C. Damsgaard, P. Vesborg and O. Hansen, Nat. Catal., 2018, 1, 820–829 CrossRef CAS.
- B. Wang, K. Zhao, Z. Yu, C. Sun, Z. Wang, N. Feng, L. Mai, Y. Wang and Y. Xia, Energy Environ. Sci., 2020, 13, 2200–2208 RSC.
- X. Bo, R. K. Hocking, S. Zhou, Y. Li, X. Chen, J. Zhuang, Y. Du and C. Zhao, Energy Environ. Sci., 2020, 13, 4225–4237 RSC.
- P. Christopher, ACS Energy Lett., 2018, 3, 3015–3016 CrossRef CAS.
- P. Quaino, F. Juarez, E. Santos and W. Schmickler, Beilstein J. Nanotechnol., 2014, 5, 846–854 CrossRef PubMed.
- H. A. Hansen, J. Rossmeisl and J. K. Nørskov, Phys. Chem. Chem. Phys., 2008, 10, 3722–3730 RSC.
- M. Bajdich, M. García-Mota, A. Vojvodic, J. K. Nørskov and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 13521–13530 CrossRef CAS PubMed.
- Y. Jeung, H. Jung, D. Kim, H. Roh, C. Lim, J. W. Han and K. Yong, J. Mater. Chem. A, 2021, 9, 12203–12213 RSC.
- S. Sun, X. Zhou, B. Cong, W. Hong and G. Chen, ACS Catal., 2020, 10, 9086–9097 CrossRef CAS.
- K. Qu, Y. Zheng, Y. Jiao, X. Zhang, S. Dai and S. Z. Qiao, Adv. Energy Mater., 2017, 7, 1602068 CrossRef.
- J. Luo, J.-H. Im, M. T. Mayer, M. Schreier, M. K. Nazeeruddin, N.-G. Park, S. D. Tilley, H. J. Fan and M. Grätzel, Science, 2014, 345, 1593–1596 CrossRef CAS PubMed.
- J. Li, Y. Wang, T. Zhou, H. Zhang, X. Sun, J. Tang, L. Zhang, A. M. Al-Enizi, Z. Yang and G. Zheng, J. Am. Chem. Soc., 2015, 137, 14305–14312 CrossRef CAS PubMed.
- D. Kong, J. J. Cha, H. Wang, H. R. Lee and Y. Cui, Energy Environ. Sci., 2013, 6, 3553–3558 RSC.
- Z. Peng, S. Yang, D. Jia, P. Da, P. He, A. M. Al-Enizi, G. Ding, X. Xie and G. Zheng, J. Mater. Chem. A, 2016, 4, 12878–12883 RSC.
- S. Khatun, H. Hirani and P. Roy, J. Mater. Chem. A, 2021, 9, 74–86 RSC.
- S. r. Dresp, F. Dionigi, M. Klingenhof and P. Strasser, ACS Energy Lett., 2019, 4, 933–942 CrossRef CAS.
- F. Dionigi, T. Reier, Z. Pawolek, M. Gliech and P. Strasser, ChemSusChem, 2016, 9, 962–972 CrossRef CAS PubMed.
- K. S. Exner, I. Sohrabnejad-Eskan and H. Over, ACS Catal., 2018, 8, 1864–1879 CrossRef CAS.
- K. S. Exner, J. Anton, T. Jacob and H. Over, Angew. Chem., Int. Ed., 2016, 55, 7501–7504 CrossRef CAS PubMed.
- K. S. Exner, J. Anton, T. Jacob and H. Over, ChemElectroChem, 2015, 2, 707–713 CrossRef CAS.
- K. S. Exner, J. Anton, T. Jacob and H. Over, Angew. Chem., Int. Ed., 2014, 53, 11032–11035 CrossRef CAS PubMed.
- K. S. Exner, J. Anton, T. Jacob and H. Over, Electrochim. Acta, 2014, 120, 460–466 CrossRef CAS.
- L. Rebollar, S. Intikhab, N. J. Oliveira, Y. Yan, B. Xu, I. T. McCrum, J. D. Snyder and M. H. Tang, ACS Catal., 2020, 10, 14747–14762 CrossRef CAS.
- J. Rossmeisl, E. Skúlason, M. E. Björketun, V. Tripkovic and J. K. Nørskov, Chem. Phys. Lett., 2008, 466, 68–71 CrossRef CAS.
- J. Rossmeisl, K. Chan, E. Skúlason, M. E. Björketun and V. Tripkovic, Catal. Today, 2016, 262, 36–40 CrossRef CAS.
- K. Chan and J. K. Nørskov, J. Phys. Chem. Lett., 2015, 6, 2663–2668 CrossRef CAS PubMed.
- I. Spanos, M. F. Tesch, M. Yu, H. Tüysüz, J. Zhang, X. Feng, K. Müllen, R. Schlögl and A. K. Mechler, ACS Catal., 2019, 9, 8165–8170 CrossRef CAS.
- J. M. P. Martirez and E. A. Carter, ACS Catal., 2020, 10, 2720–2734 CrossRef CAS.
- S. Klaus, Y. Cai, M. W. Louie, L. Trotochaud and A. T. Bell, J. Phys. Chem. C, 2015, 119, 7243–7254 CrossRef CAS.
- Y. Zhou and N. López, ACS Catal., 2020, 10, 6254–6261 CrossRef CAS.
- L. Gong, X. Y. E. Chng, Y. Du, S. Xi and B. S. Yeo, ACS Catal., 2018, 8, 807–814 CrossRef CAS.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |