
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fischer indole synthesis applied to the total synthesis of natural products
Majid M. Heravi
 *,
Sahar Rohani
,
Vahideh Zadsirjan
* and
Nazli Zahedi
*,
Sahar Rohani
,
Vahideh Zadsirjan
* and
Nazli Zahedi
Department of Chemistry, School of Science, Alzahra University, Vanak, Tehran, Iran. E-mail: mmheravi@alzahra.ac.ir; z_zadsirjan@yahoo.com
First published on 15th November 2017
Abstract
One of the oldest and most useful reactions in organic chemistry is the Fischer indole synthesis (FIS). It is known to have a wide variety of applications including the synthesis of indole rings, often present as the framework in the total synthesis of natural products, particularly those found in the realm of alkaloids, which comprise a ring system known as an indole alkaloid. In this review, we are trying to emphasize the applications of FIS as an old reaction, which is currently applied to the total synthesis of biologically active natural products and some other complex targets.
 Majid M. Heravi | Majid M Heravi was born in 1952 in Mashhad, Iran. He received his B. Sc. degree from the National University of Iran in 1975 and his M. Sc. and Ph. D. degrees from Salford University, England in 1977 and 1980, respectively. He completed his doctoral thesis under the supervision of the late Jim Clarck in Salford University, England. He started his career as a research fellow in Daroupakhsh (a pharmaceutical company) in 1981 Tehran, Iran and joined as an assistant professor to Ferdowsi University of Mashhad, Iran in 1983, and was promoted to associate professor in 1993 and full professor in 1997 in the aforementioned university. In 1999 he moved to Alzahra University of Tehran, Iran as professor of chemistry where he still works. He has previously been a visiting professor at UC Riverside, California, USA and Hamburg University, Hamburg, Germany. His research interests focus on heterocyclic chemistry, catalysis, organic methodology and green synthetic organic chemistry. |
 Sahar Rohani | Sahar Rohani was born in 1987 in Shiraz, Iran. She received her B.Sc. degree in pure Chemistry from the Kharazmi University, Tehran, Iran, in 2009 and her MSc. degree in Organic Chemistry at Kashan University, Iran (2012) under the supervision of Dr Abdolhamid Bamoniri. She is currently working towards her PhD in Organic chemistry at Alzahra University under the supervision of Dr Ghodsi Mohammadi Ziarani. Her research field is synthesis of various nanocatalysts and their application in coupling reactions. |
 Vahideh Zadsirjan | Vahideh Zadsirjan was born in Shiraz, Iran in 1979. She received her B. Sc. in Pure Chemistry from Kharazmi University in 2002, and her M. Sc by course in 2007 and Ph. D degree in organic chemistry in 2016 under the supervision of Professor Majid M. Heravi from Alzahra University Tehran, Iran. Her research is focused on heterocyclic chemistry, catalysis, organic methodology and green synthetic organic chemistry. |
 Nazli Zahedi | Nazli Zahedi was born in Tehran, Iran in 1987. She received her M.D. (G.P) from Medical School, Islamic Azad University, Tehran, Iran, in 2014. She is working towards, taking a place as resident in a Medical School. |
1. Introduction
Nowadays, there has been increasing attention on the total synthesis of bioactive natural products and their synthetic analogues in the arena of organic chemistry. Novel synthetic approaches and strategies are available that permit formation of novel complex molecules or already structurally known naturally occurring compounds which can be used as prescribed drugs or medications.1 Remarkably, the name indole is a combination of the words indigo and oleum because initially, indole was prepared and identified from the reaction of the indigo dye with oleum.2 As a matter of fact, one of the most plentiful heterocyclic systems found in nature is indole. It is a vital functional nucleus in the structures of different dyes, fragrances, pharmaceuticals and agricultural chemicals.3,4 Indole ring moieties became important structural components in diverse natural pharmaceutical agents, hence their synthesis and functionalization is a key field in heterocyclic chemistry, which has attracted the attention of synthetic organic chemists.5 Therefore, in the last century, different strategies for producing an indole moiety have been established6 and among them, the FIS is the most well-established and practical strategy.7Pd-promoted reaction is also a perfect approach for the synthesis of indoles. In this approach o-haloaniline is usually used as starting material, which upon treatment with appropriate unsaturated units, generates new carbon–carbon and carbon–nitrogen bonds for the construction of the indole core.8–12
Buchwald et al. presented an approach for the syntheses of FIS precursors. They developed a Pd-catalyzed cross coupling reaction for the synthesis of N-aryl benzophenone hydrazones, which are used as common precursors in the typical FIS.13 The same group also reported a facile, efficient and general Pd-catalyzed approach for the synthesis of a wide range of arylhydrazines or arylhydrazones, which are typical precursors for the FIS.14
FIS which is rarely called Fischer indolization, has been accomplished first by Emil Fischer and Friedrich Jourdan in 1883.15 FIS is significant among the well-established classical methods which efficiently results in the synthesis of the bioactive indole scaffold that is usually found in alkaloids and in different valuable medicament.16,17 The FIS usually gives a facile, effective protocol for the conversion of enolizable N-arylhydrazones into indoles using an acid as catalyst.18 In FIS, the selection of acid catalyst is very decisive. Brønsted acids such as HCl, H2SO4 and PTS were frequently employed effectively in this reaction.19,20 Lewis acids such BF3/etherate, ZnCl2, FeCl3, and AlCl3 are also beneficial catalysts for this reaction.21,22 Several reviews have been reported the selected examples of FIS acid catalysis.23
A few arylhydrazines are commercially available; they are generally synthesized by reduction of aryl diazonium salts, which in turn can be provided from the appropriate aniline derivatives. Alternatively, aryl diazonium salts can directly be transformed to hydrazones via the Japp–Klingemann reaction.24 The Japp–Klingemann reaction involves the reaction of the aryl diazonium salt with an active methylenyl or methinyl compound in the presence of an appropriate either acid or base to afford an azo compound, which under either basic or acidic, or even thermal conditions can be transformed into the corresponding hydrazone.24
Indole is the most powerful pharmacodynamic core known in several naturally occurring compounds.12–25 Indoles are known as privileged structures due to their unique roles in different biochemical procedures.26,27
In the group of molecules having biological properties that were known for a long period of time, melatonin 3 as a common cycle of circadian, tryptophan as a vital amino acid utilized in sleep disorders and depressive states treatment, tryptamine 1 like serotonin 2 realized as growth factors in plants, are important neurotransmitter. Furthermore, strychnine 5 is a potent stimulant of central nervous system (CNS), LSD 6 is a powerful hallucinogen, and also reserpine was applied as antihypertensive (Fig. 1).28
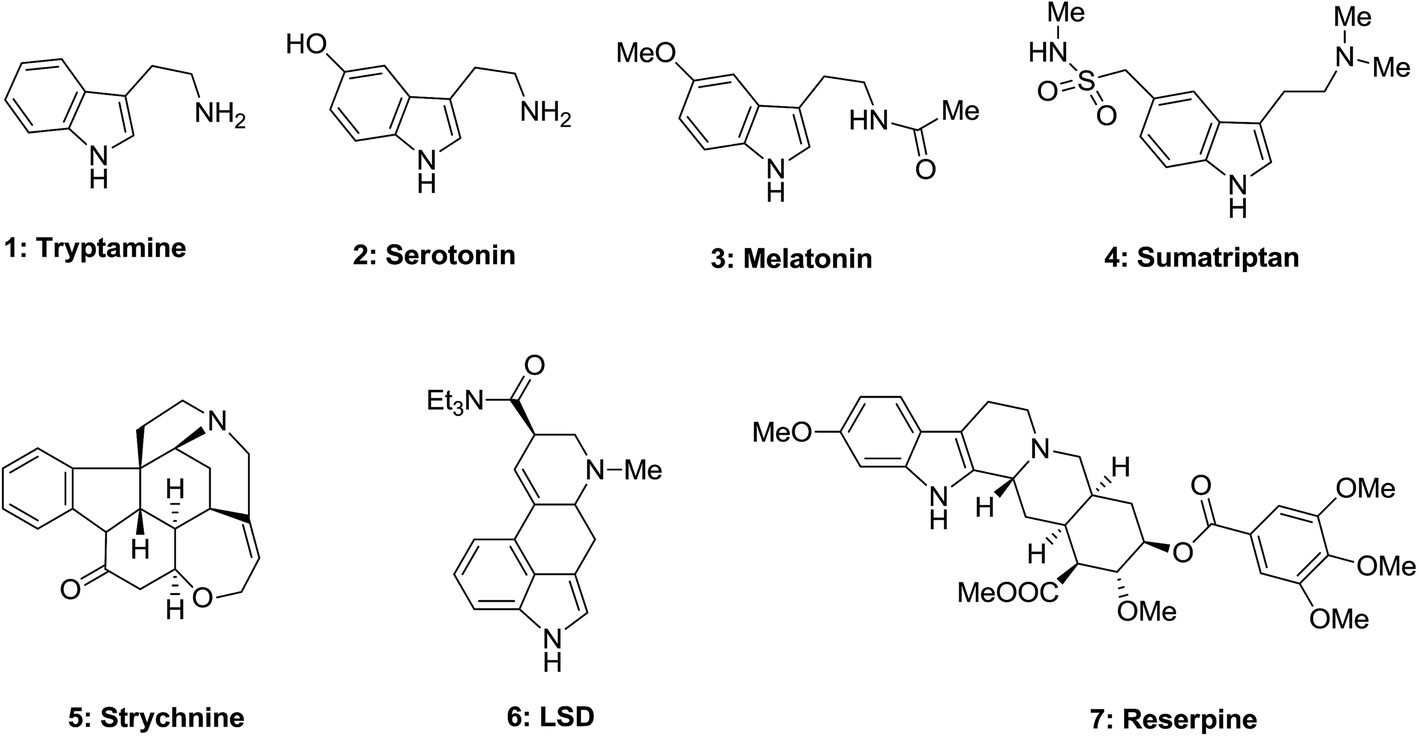 | ||
| Fig. 1 Biologically potent molecules containing indole as a moiety. | ||
Esrine appears in the Physostigma venenosums seeds has been suggested for the curing of Alzheimer's disease. Serotonin, the vasoconstrictor hormone, serves as a neurotransmitter in animals. Dimeric vinca alkaloids including vincristine and vinblastine extracted from Catharanthus roseus, which are utilized in cancer and Hodgkin's diseases treatment.28
Due to the importance of indole derivatives,29 it has been published different reviews in the synthesis of indoles and their applications in the total synthesis of natural product.25,30–33 Because of the large number of biologically fascinating natural products containing poly-substituted indole moieties, FIS has attained significant synthetic attention.34
In continuation of our interest in applications of name reactions in the total synthesis of natural products35–47 and in the synthesis of heterocyclic systems,48–52 in this review, we try to highlight the applications of FIS in total synthesis of biologically active natural products.
2. Applications of Fischer indole synthesis in the total synthesis of natural products using
2.1. Aryl hydrazines
Strychnine 5 is the chief molecule of the strychnos alkaloids group, one of the most crowded groups of indole alkaloids. Initially, strychnine since 1818 was extracted from the Strychnos-nux vomica's bark and seeds by Pelletier also Caventou and its essential form has been recognized by Regnault, about 20 years later.53,54 In 1954 the historic strychnine's total synthesis 5 by Woodward55 signified a milestone in the arena of organic chemistry. Strychnine 5 (C21H22N2O2) has a complex structure. It contains six adjacent stereocenters, which five of them are positioned in the cyclohexane ring's center and includes merely 24 skeletal atoms, closely packed and organized in seven rings. Specified its complicated construction, coupled with its highly toxic activities and pharmacological, strychnine 5 has attracted organic chemists much attention. It is a disreputable poison that ∼50 mg of it can be deadly for an adult human. That obstructs postsynaptic hindrance in the spinal cord where it irritates the transmitter glycine. These effects have made strychnine more valuable and useful in trial pharmacology.56The starting materials for the total synthesis of strychnine 5 was the 2-veratrylindole 10 that was synthesized by FIS from phenylhydrazine 8 and acetoveratrone 9. The first steps in this methodology involves the introduction of the 2-aminoethyl chain to the β-position of 2-veratrylindole 10. After several steps, compound 10 was transformed to strychnine 5 (Scheme 1).57,58
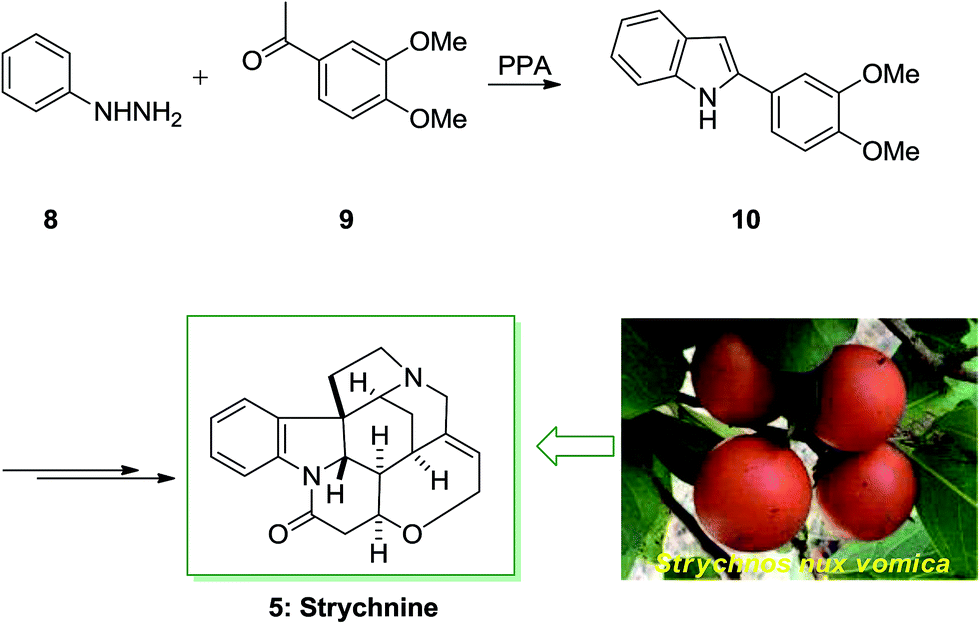 | ||
| Scheme 1 Total synthesis of strychnine 5. | ||
Aspidospermidine is a member of aspidosperma alkaloids including a pentacyclic ring system, which was separated from Aspidosperma (a genus of flowering plant in the family Apocynaceae) quebracho blanco and other aspidosperma species.59,60 The total synthesis of alkaloids aspidospermine 11 was first achieved and reported in 1963 by Stork and Dolfini.61 It was used in the treatment of erectile impotence, and decreasing the benign prostatic hyperplasia (BPH) symptoms, in guinea pig and also in rabbit corpus spongiosum and cavernosum due to its inhibition of smooth muscle contractions.
The parent indole is an achiral molecular unit; the formation of chiral products by using a FIS is by no means unusual. The application of α-branched carbonyl molecules for example can result in the synthesis of indolenine derivatives having a quaternary stereocenter in the 3-position. For the total synthesis of (±)-aspidospermine 11, Stork and co-workers acquired merit of this reactivity. This method was started from butyraldehyde 12, which transformed into complex cyclohexanone 13 upon several steps. The FIS of the cyclohexanone 13 and hydrazine 14 afforded the indolenine 15 that has been transformed into the desired 11 via imine reduction and N-acetylation (Scheme 2).61
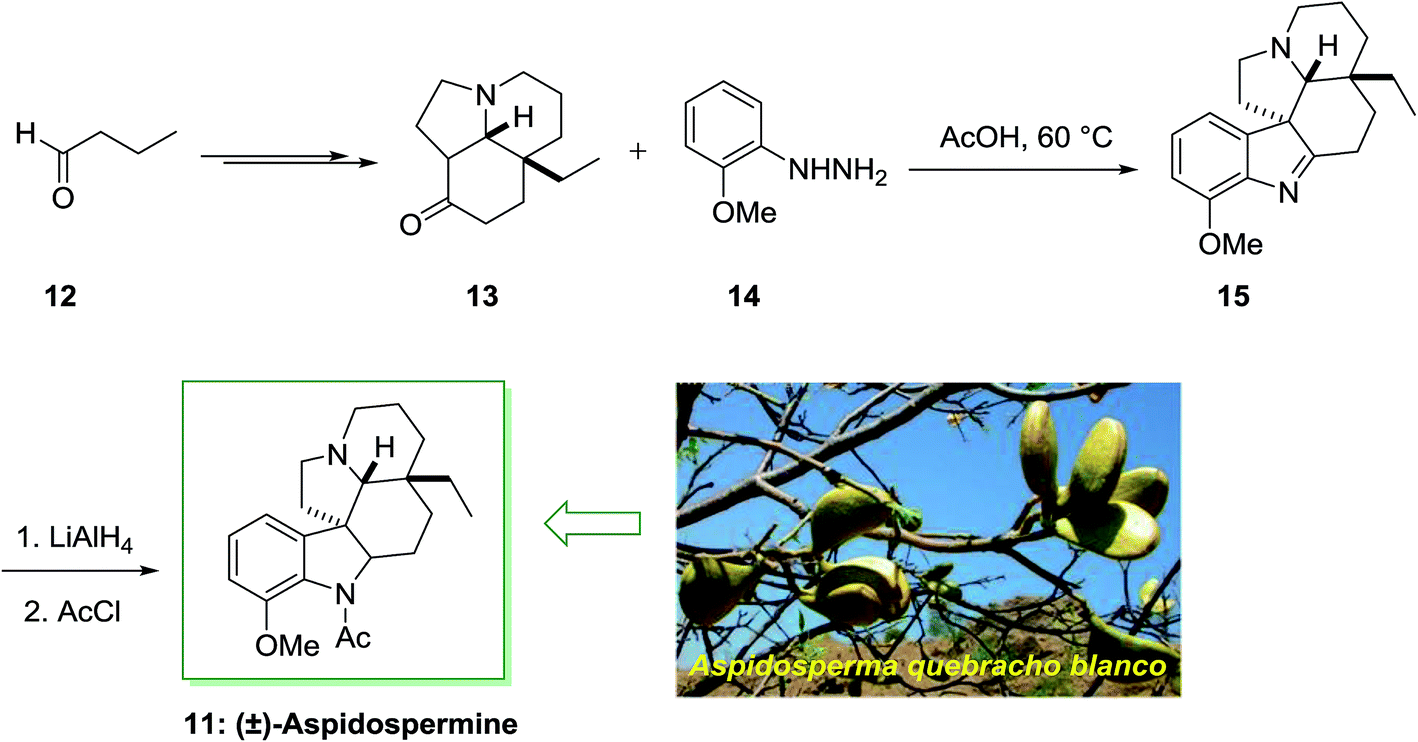 | ||
| Scheme 2 Total synthesis of (±)-aspidospermine 11. | ||
Remarkably, in chemistry of alkaloid, attention in carbazole alkaloids has raised significantly throughout the past years because of the desired ability of novel kinds of pharmacologically active compounds. Therefore, for example various carbazoles, oxotetrahydrocarbazoles, tetrahydrocarbazoles, mukonine and glycozoline isomers, prenylcarbazoles, carbazomycins, amino-and nitrocarbazoles as well as pyrido[b]carbazole derivatives (such as ellipticines 16 and analogues) contain anti-convulsant, anti-tumour, anti-inflammatory, anti-histamine, psychotropic, antibiotic and fungistatic activities.62
The tetracyclic natural product ellipticine (5,11-dimethyl-6H-pyrido[4,3-b]carbazole) 16 has been extracted in 1959 from the Ochrosia elliptica Labill plant material.63 This small tropical evergreen tree goes to the Apocynaceae family and included various other alkaloids, involving 9-methoxyellipticine. As ellipticine 16 was extracted from various other Apocynaceae plants class (Ochrosia acuminate, Ochrosia moorei and Ochrosia vieillardii) and from strychnos dinkagei of the Loganiaceae class. The ellipticine class of complexes use their biological property through various styles of action, the most well-developed of that are insertion with topoisomerase II inhibition and DNA. Recently, other types of action were demonstrated, involving kinase inhibition, communication with bio-oxidation, p53 transcription factor and adduct formation.64
As depicted in Scheme 3, the total synthesis of ellipticine 16 initiated from the allylic alcohol 17. A [3,3]-sigmatropic rearrangement with propionic anhydride 18 gave the carboxylic acid 19. Next, the carboxylic acid 19 treated with phenylhydrazine 8 to provide phenylhydrazone 20 continued by the FIS to provide indole 21. The latter afforded the corresponding natural product ellipticine 16 in several reaction steps.65
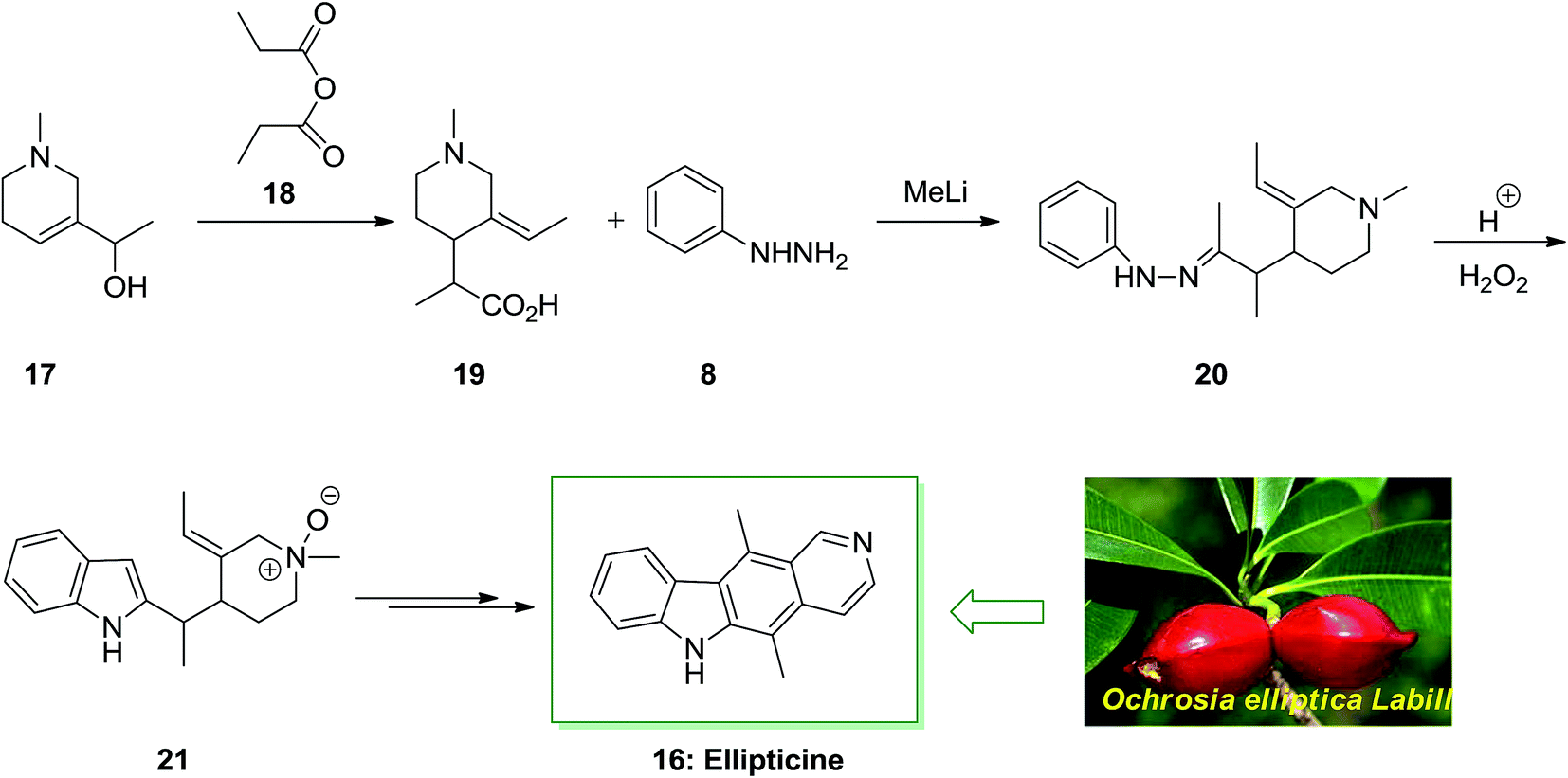 | ||
| Scheme 3 Total synthesis of ellipticine 16. | ||
A series of 11-alkylbenzo[a]carbazole derivatives 22 and their dihydro analogues have been prepared and examined for their binding attraction for the estrogen receptor and their antiestrogenic and estrogenic effects in the immature mouse. They also showed mammary tumor inhibiting property. Furthermore, benzo[a]-carbazole derivatives are flat polycycles, which contain the intercalating potential into the DNA. In 1986 von Angerer and Prekajac described the total synthesis of the 11-alkyl-11H-benzo[a]carbazoles 22. In this approach, the FIS of the arylhydrazine hydrochlorides 23 and the tetralones 24 gave the 5,6-dihydro-11H-benzo[a]carbazoles 25. Next, the latter has been transformed into the corresponding natural product 22b through subjection to different chemical reactions (Scheme 4).66
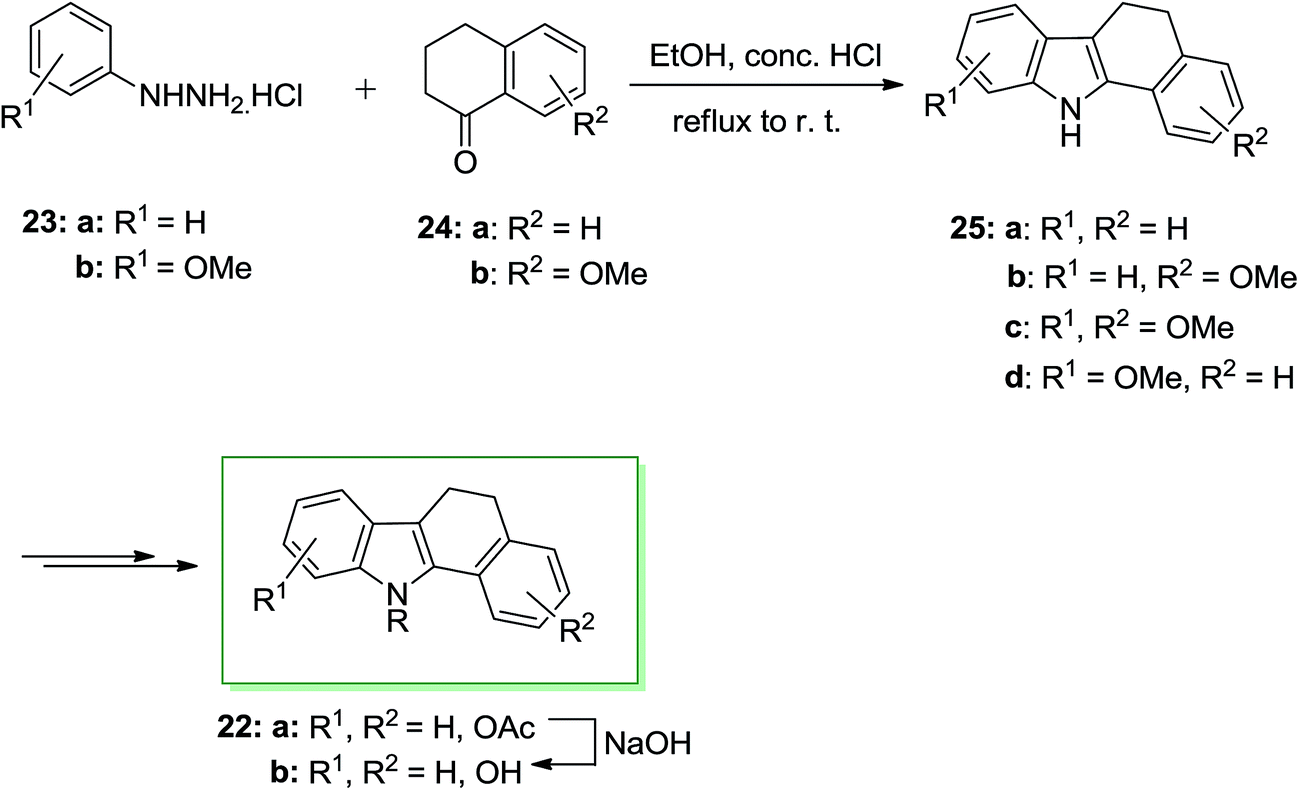 | ||
| Scheme 4 Total synthesis of 11-alkyl-11H-benzo[a]carbazoles 22. | ||
The Strychnos alkaloids are obtained from preakuammicine and appearently generated from secologanin and tryptophan through geissoschizine and strictosidine which is actually a monoterpenoid indole alkaloids biosynthetic pathway. Several mechanisms have been presupposed to interconnect those of the Strychnos type with the Corynanthe alkaloid geissoschizine, but the main features of the rearrangement to preakuammicine and dehydropreakuammicine stay still unknown. The Strychnos alkaloids have obtained less attention from synthetic standpoint than other kinds of indole alkaloids such as Yohimbe, Iboga, Aspidosperma and Corynanthe.67
The indole alkaloids bearing a nonrearranged secologanin scaffold contain different structural varieties. Among these, the alkaloids of the uleine family (dasycarpidan stereoparent) and the Strychnos alkaloids along with the Aspidospermatan biogenetic subtype (condyfolan stereoparent) are identified by the presence of a 1,5-methanoazocino[4,3-b] indole moiety having two-carbon chain, typically an ethyl group, at the bridge carbon.
An asymmetric total synthesis of the alkaloids of the uleine family, nordasycarpidone 26, dasycarpidol 27, dasycarpidone 28 and tubotaiwin 29 were achieved via formation of the tetracyclic intermediate 34, that were synthesized through FI reaction. This approach was initiated from 4-piperidineacetates cis-31 and trans-31, which formed from l-benzyl-3-ethyl-4-piperidone 30 and their transformation to the desired 4-acetonylpiperidines 32. Then, the conversion of piperidine cis-32 into the bridged 2-azabicyclo [3.3.1]-nonane 33 and the FIS of the latter afforded the desired compound 34. The FIS of ketone 33 has been examined by applying different acid catalysts. The best consequence has been provided once the phenylhydrazone from 33 has been refluxed in acetic acid. Based on these reaction conditions the desired tetracycle 34 has been produced as the only isolable product, but in satisfactory yield. The methanoazocinoindole 34 as a usual main intermediate. This tetracyclic compound includes a C-20 ethyl group equatorial with respect to the piperidine ring, namely, with the identical relative stereochemistry as uleine, dasycarpidone, and the Aspidospermatan alkaloids. Therefore, after several steps, tetracycle 34 afforded the alkaloids nordasycarpidone 26 and dasycarpidone 27 in 73% and 76% yields, respectively. Lastly, NaBH4 reduction of dasycarpidone 27 resulted in the alkaloid dasycarpidol 28. On the other hand, methanoazocinoindole 34, afforded (±)-tubotaiwin 29 upon several steps (Scheme 5).68,69
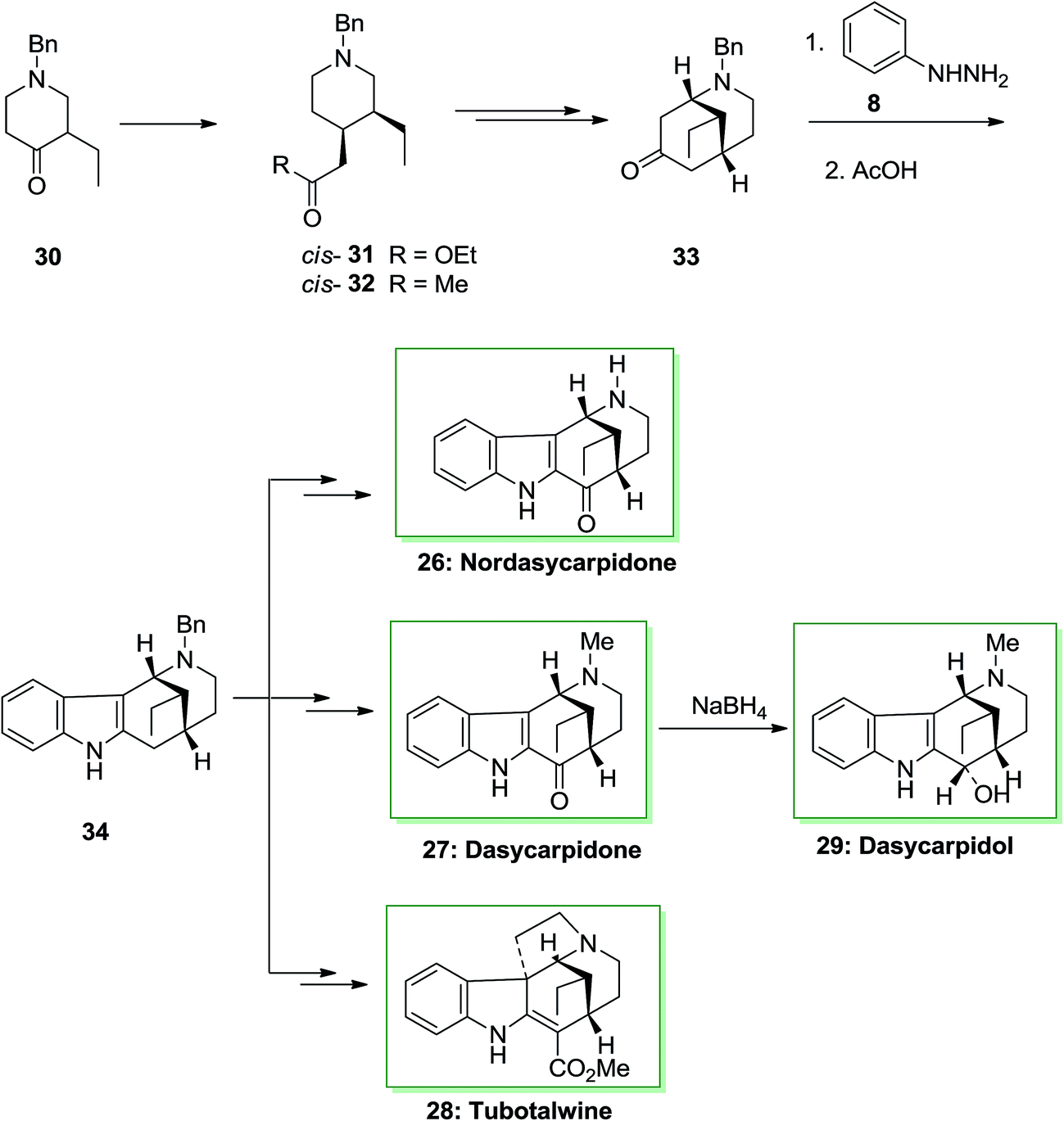 | ||
| Scheme 5 Total synthesis of, nordasycarpidone 26, dasycarpidone 27, dasycarpidol 28 and tubotalwine 29. | ||
Archer and co-workers employed an identical approach for the synthesis of 5,11-demethylellipticines (9-hydroxy-6H-pyrido[4,3-b] carbazole).68 In the current method, initially, the reaction of enamine 36 and methyl vinyl ketone 37 afforded a mixture of trans- and cis-ketones 38 that have individually transformed into indole 40 through FI reaction. Some of the nonlinear pyrido[3,4-c]carbazole (17%) have been synthesized from the cis-ketone. Next, dehydrogenation and demethylation gave the corresponding natural product 9-hydroxy-6H-pyrido[4,3-b]carbazole 35 (Scheme 6).70
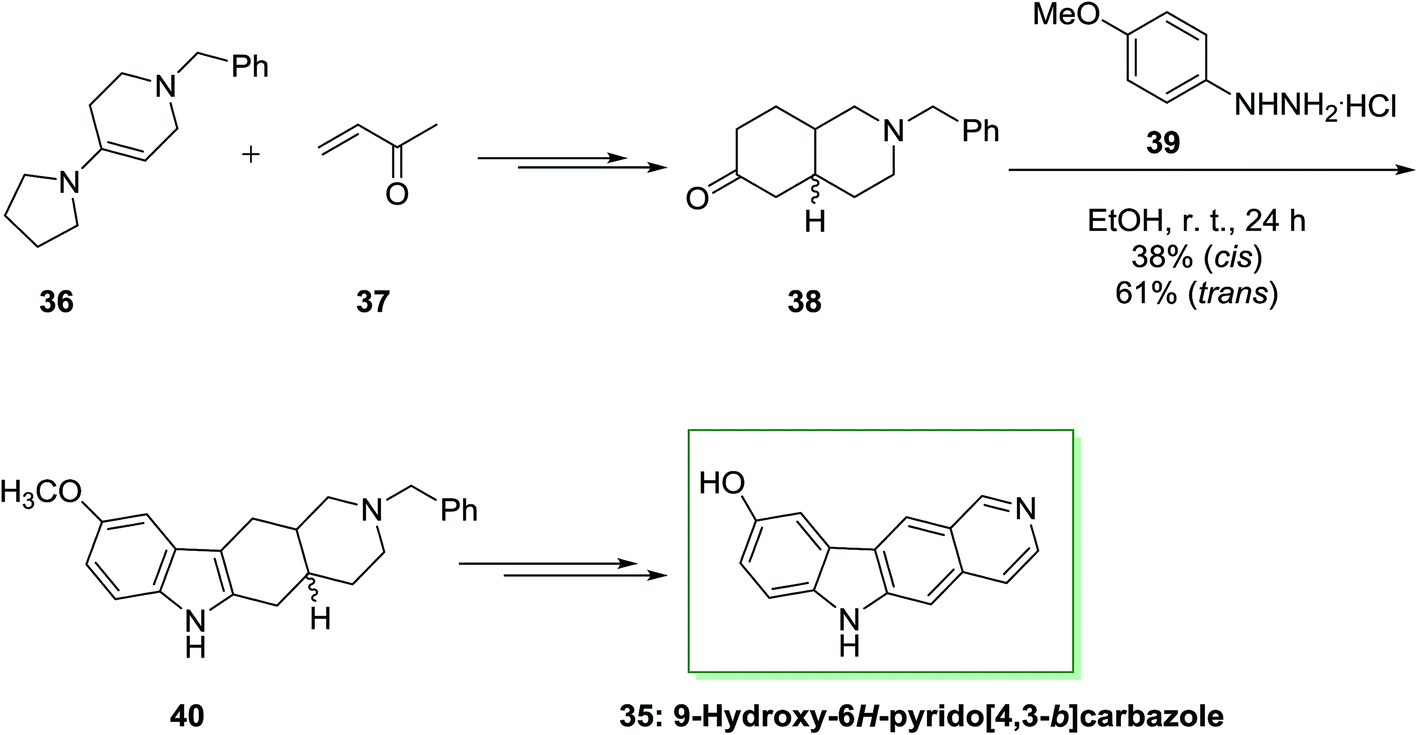 | ||
| Scheme 6 Synthesis of 9-hydroxy-6H-pyrido[4,3-b]carbazole 35. | ||
Indolocarbazole alkaloids exhibit an increasing figure of natural products extracted from slime molds, marine sources and soil organisms. Several of them exhibited significant biological property. For example, staurosporine and rebeccamycin are known as antitumor, protein kinase C and topoisomerase I inhibitors, respectively. Arcyriaflavin A 41 is proven to be an inhibitor of human cytomegalovirus replication. Particularly, a synthetic derivative, NB-506, is now under clinical trials as antitumor agents.71
Similarly, a concise and extremely significant synthesis of indolo[2,3-a]pyrrolo[3,4-c]carbazole derivatives like arcyriaflavin A 41 via a double FIS has been effectively demonstrated by Bergman and Pelcman in 1989.70,71 This approach is based on a double FIS of the bis(phenylhydrazone) 45. The latter was synthesized through Diels–Alder reaction of market purchasable 2,3-bis(trimethylsilyloxy) butadiene 42 with the dienophiles 43, affording the cycloadducts 44. This cycloadduct was treated with phenylhydrazine 8 in acetic acid and MeOH to afford 45. The double FIS of 45 using polyphosphoric acid trimethylsilyl ester (PPSE) as the cyclization agent, led to arcyriaflavin A 41 in 68% yield (Scheme 7). The novel method gave a concise and very significant synthesis of indolo[2,3-a]pyrrolo[3,4-c]carbazole derivatives and appropriate in the synthesis of a large range of functionalized products and does not need expensive or in available starting compounds.72
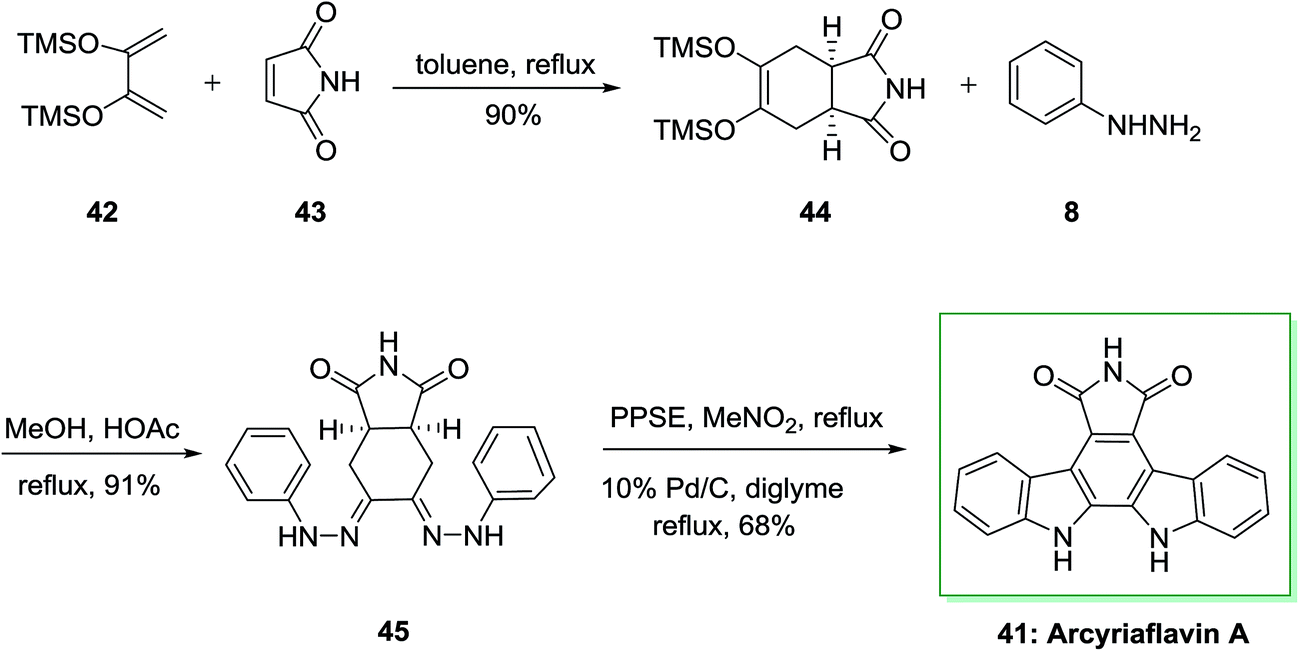 | ||
| Scheme 7 Total synthesis of arcyriaflavin A 41. | ||
A novel synthetic method to the synthesis of natural product arcyriaflavin-A 41 and unsymmetrical analogs was demonstrated by Tomé and co-workers in 2000.73 The method relied on consecutive Diels–Alder cycloaddition, FI and formal nitrene insertion procedures. This total synthesis was initiated from the market purchasable 2-nitrobenzaldehyde 46. Upon various steps, 2-nitrobenzaldehyde 46 transformed into the perhydroisoindole-1,3,5-trione 47a. Then, trione 47a has been exposed to a FIS with p-methoxyphenylhydrazine 48, providing a mixture of compounds 49a and 50a in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. This ratio looks to depend on the stability of the ene-hydrazine intermediate. Upon two steps, the corresponding compound 50a gave the pure arcyriaflavin-A analogue 41a and the parent natural product arcyriaflavin-A 41b in 55% yield (Scheme 8).73
:1 ratio. This ratio looks to depend on the stability of the ene-hydrazine intermediate. Upon two steps, the corresponding compound 50a gave the pure arcyriaflavin-A analogue 41a and the parent natural product arcyriaflavin-A 41b in 55% yield (Scheme 8).73
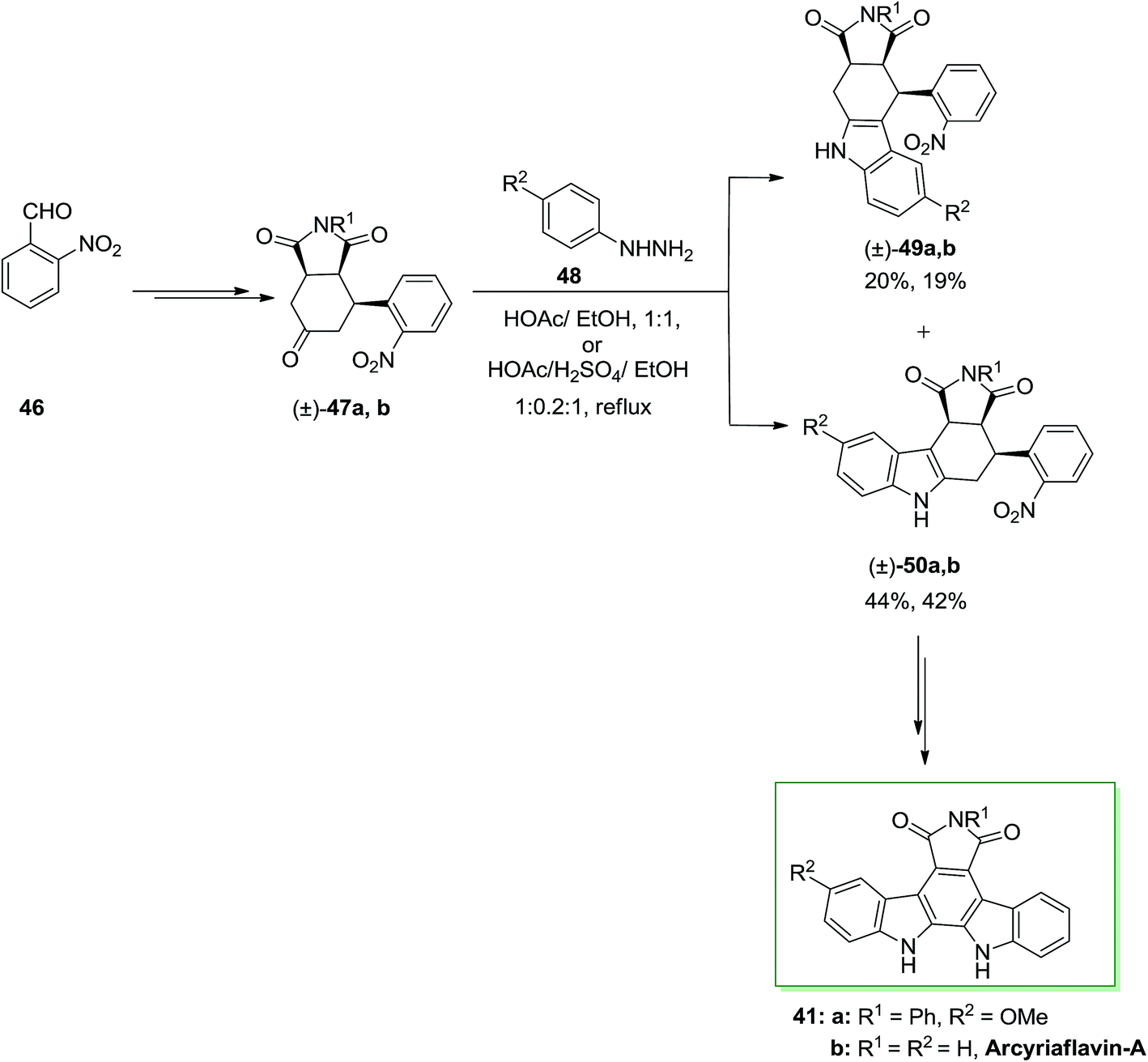 | ||
| Scheme 8 Total synthesis of arcyriaflavin-A 41. | ||
Physostigmine is an alkaloid which is extracted from the seeds of Physostigma venenosum (Calabal beans) and it is clinically effective as a anticholinergic drug. In addition, its enantiomer protects against organophosphate poisoning. Physostigmine's analogous have indicated a therapeutic power in Alzheimer's disease,2 and recently is in phase II efficacy trials. Additionally, physostigmine has an important role in neuroscience such as a powerful potent morphine-like narcotic agonist activity of (−)-eserine in vivo.74
Remarkably, there is significant attention in molecules bearing central stimulatory property including the anti-cholinergic Calabar bean alkaloids because of their therapeutic in cholinergic disorders and Alzheimer's disease. Enantioselective total synthesis of the Calabar bean alkaloid (−)-physostigmine 53 and (−)-physovenine 54 were accomplished in a short method in 1991 by using FIS under nonacidic conditions as the main stage.75 The total synthesis was initiated from the optically active tricyclic enone 56, which synthesized from racemic dicyclopentadiene 55 in four-steps. Then, alkylation reaction of the latter gave the monomethyl ketone 57 in 86% yields as a mixture of epimers. Polycyclic ketone 57 was as a stereochemical control parameter in the major FI step. Once monomethyl ketone 57 has been refluxed by using p-methoxyphenylhydrazine hydrochloride 39 in aqueous pyridine (1:10), a simple diastereoselective reaction happened to supply the carbinolamine 60 as a single product, in 82% chemical yield. Then, lactol 61 under reflux in MeOH with a trace of HCl produced concomitant deacetylation and cyclization to provide the tricyclic amino acetal 62. The reaction of 62 with boron tribromide continued by carbamylation of the obtained phenol 63 gave (−)-physovenine 54. This method created the first enantioselective synthesis of the natural products.
On the other hand, in another route, the lactol 61 upon several steps provided (−)-esermethole 51. Since 51 has formerly been converted into natural (−)-physostigmine 53 in two steps through (−)-eseroline 52, these reactions are considered as steps of a formal synthesis of the natural product (Scheme 9).75
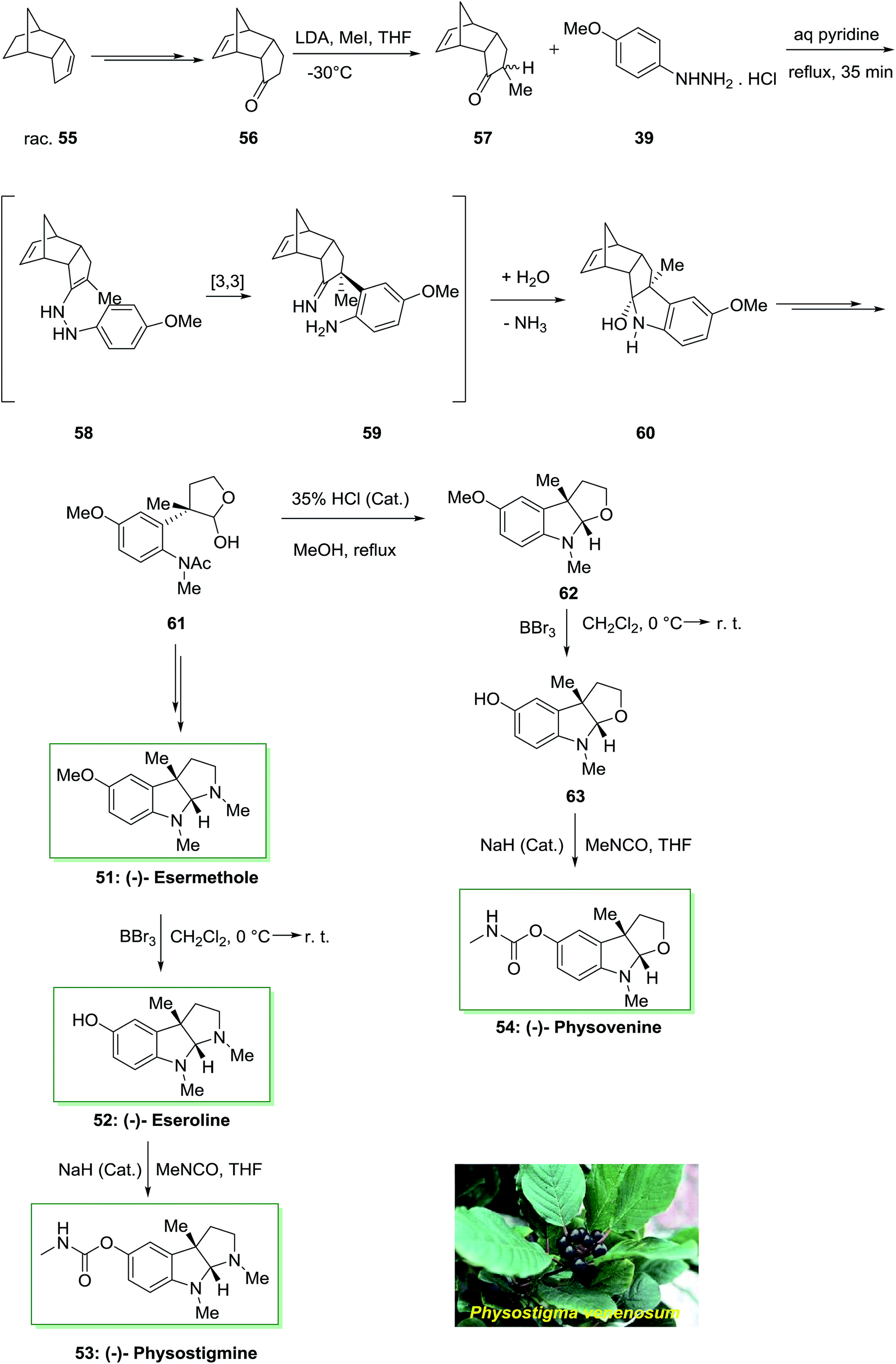 | ||
| Scheme 9 Total synthesis of (−)-esermethole 51, (−)-eseroline 52, (−)-physostigmine 53 and (−)-physovenine 54. | ||
Bioactivity-directed isolation of the obtain of the cyanophyte Tolypothrix tjipanasensis has resulted in the isolation of novel N-glycosides of indolo[2,3-a]carbazole derivatives planned tjipanazole derivatives Al, A2, B, Cl, C2, C3, C4, D, E, Fl, F2, Gl, G2, I and J. Tjipanazoles are known as novel antifungal agents extracted from the blue-green alga Tolypofhrix tjipanasensis. They showed moderate fungicidal property (strain DB-l-l) against Aspergillus flavus, Trichophyton mentagrophytes and Candida albicans.76
The total synthesis of tjipanazole D 64 and E 65 has been achieved by Bonjouklian and co-workers in 1991,75 which relied on FIS reaction. Initially, under air, two equivalents of p-chlorophenylhydrazine hydrochloride 66 and 1,2-cyclohexanedione 67 provided 6-chloro-1-(2-(4-chlorophenyl)hydrazono)-2,3,4,9-tetrahydro-1H-carbazole 68. The latter based on a FIS gave tjipanazole D 64 in 54% yield. Subsequently, tjipanazole E 65 has been extracted as a minor component through coupling reaction of 64 and 1-bromo-α-D-glucopyranosyl-2,3,4,6-tetraacetate 69 after elimination of the masking substituents (Scheme 10).76
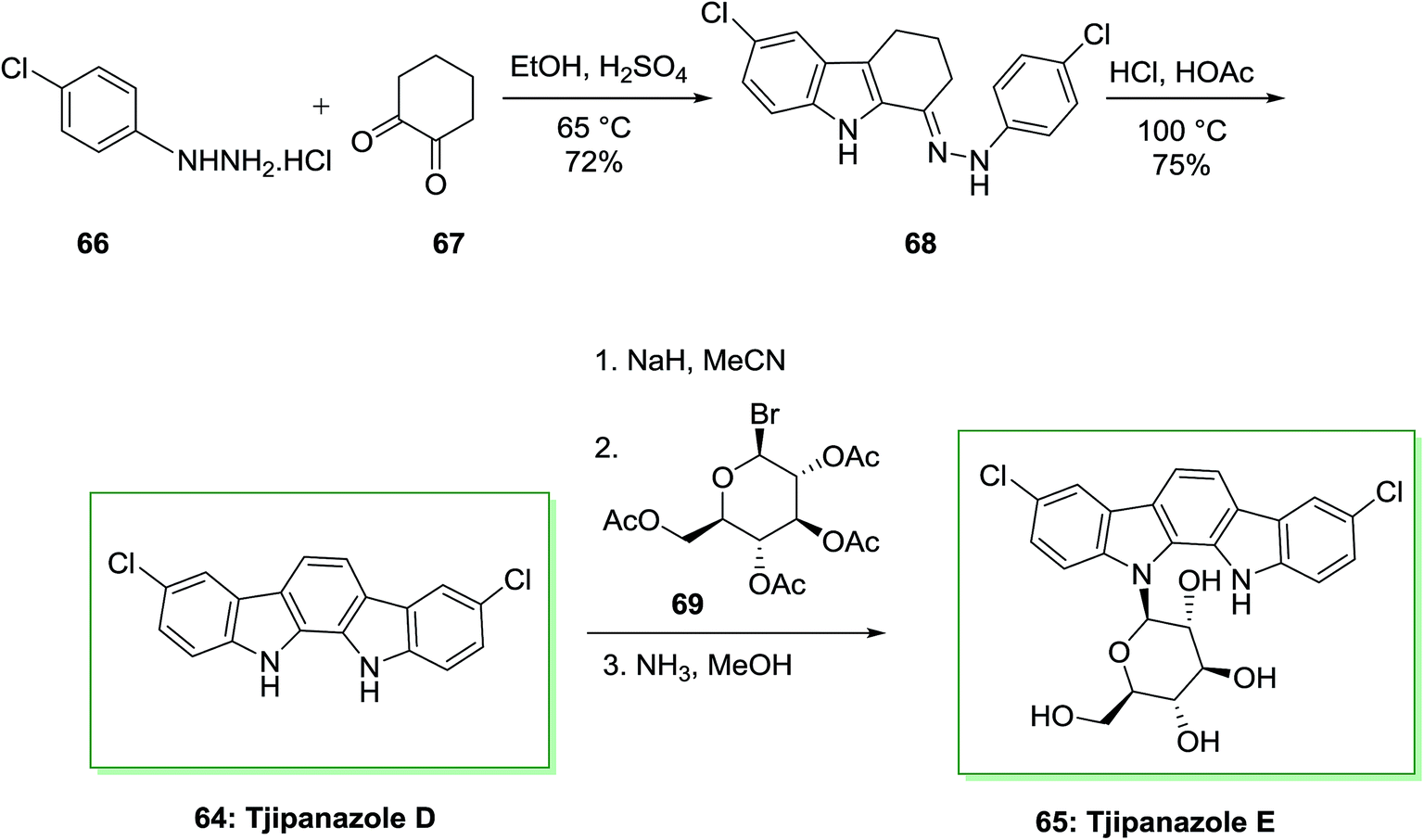 | ||
| Scheme 10 Total synthesis of tjipanazole D 64 and E 65. | ||
A novel and efficient synthesis of the pyrrolo[4,3,2-d,e] quinoline system was accomplished. It is a typical class of marine alkaloids contains the discorhabdins, prianosins and other antineoplastic. These include the discorhabdins, prianosins, damirones, iso-batzellines and batzellines which are extracted from wakayin and sponges, isolated from the Fijian ascidian Clauelina sp. Makaluvamine is a member of the pyrroloiminoquinone family and synthesis of makaluvamine D 70, a mammalian topoisomerase II inhibitor is discovered by the sponge Zyzzya cf. marsailis. They also showed power in vitro cytotoxicity in the direction of the human colon tumor cell line HCT 116 and lead into cancer chemotherapy.77
The present method to the total synthesis of makaluvamine D 70, a pyrroloiminoquinone containing a tyramine side chain at C7, was started from a FIS by applying (2,3-dimethoxyphenyl) hydrazine 72 and dihydrofuran 73. By this route, a pyrroloiminoquinone containing a tyramine side chain at C7 undergoes FIS that instantaneously developed two-carbon side chains. That prevents the requirement for lengthy synthetic approach at the indole 3-position, providing reasonably direct pathway to obtain an appropriate precursor for cyclization to 70. As a result, the starting material for this method was 2,3-dimethoxybenzoic acid 71 that has been transformed to crystalline (3,4-dimethoxyphenyl)hydrazine 72 upon several steps. Then, this hydrazine has been reacted with dihydrofuran 73 according to a procedure established by McKittrick78 and gave a 1:1 mixture of the tetrahydrofuran 74 and the hydrazone 75 (an E/Z mixture). The mixture has been exposed to FIS by using ZnCl2 to afford the desired tryptophol 76. Lastly, after several steps makaluvamine D 70 has been isolated as its trifluoroacetate 77 that was identical through comparison of its lH and 13C NMR spectra, mass spectrum and IR spectrum, and with an example of natural makaluvamine D trifluoroacetate (Scheme 11).77
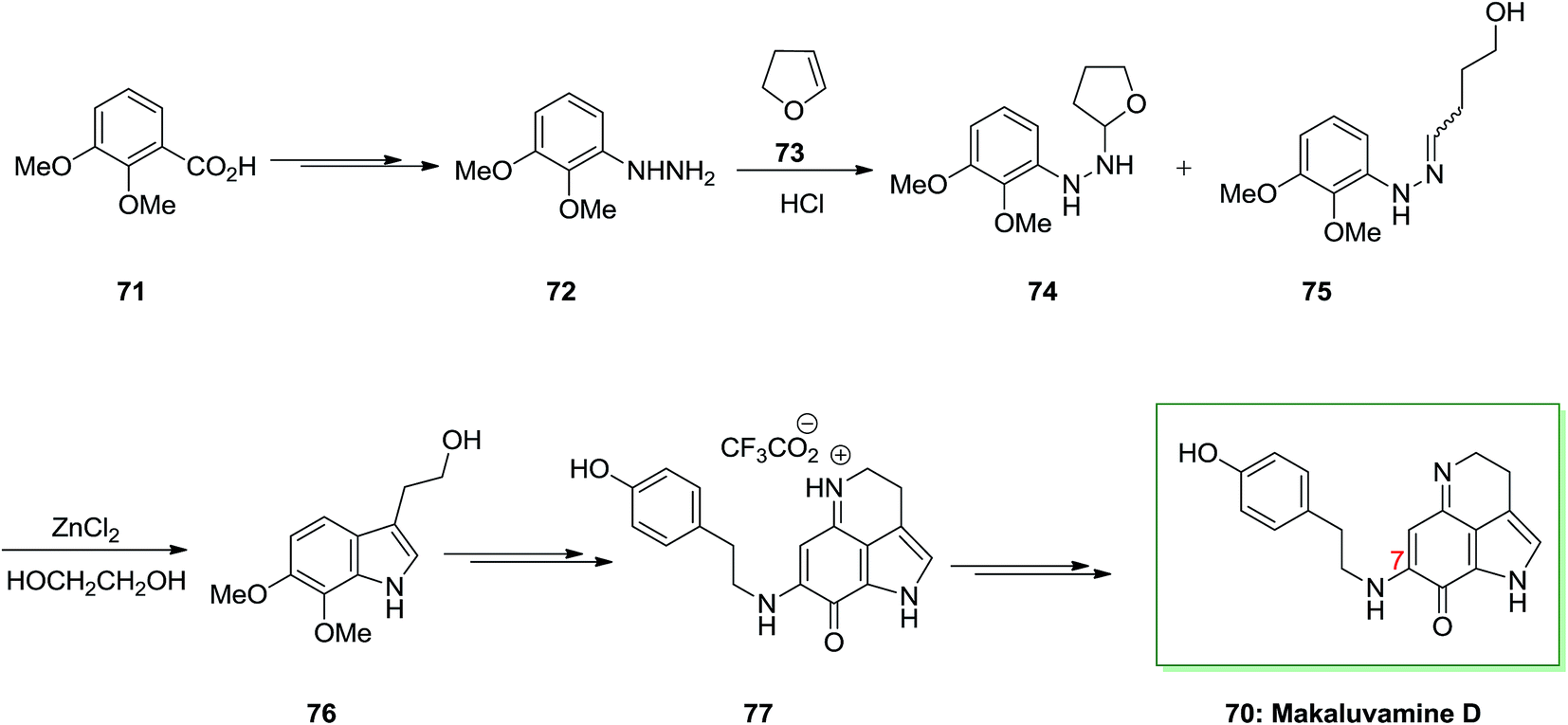 | ||
| Scheme 11 Total synthesis of makaluvamine D 70. | ||
The ibophyllidine alkaloids compose a small group of indole alkaloids of the ibogan type which is explored during the last twenty years and identified by the presence of the structurally unusual pyrrolizino [l,7-c,d] carbazole ring system. It actually containes a pyrrolidine D-nor ring instead of the piperidine ring that usually existed in the monoterpenoid indole alkaloids. Much research has been typifieded the synthesis of alkaloids with the former feature, by the Strychnos alkaloids and Aspidosperma although less attention has been performed to ibophyllidine alkaloids5 but deethylibophyllidine is chosen as the synthetic target.79
The total synthesis of (±)-deethylibophyllidine 78 was accomplished in eight steps starting from O-methyltyramine 79 in 5.5% overall yield in that regioselective FIS to a tetracyclic ring system as a main step. The synthesis was initiated by market purchasable O-methyltyramine (4-methoxyphenethylamine) 79 that has transformed into cis-octahydroindolone 80 by an enantioselective method in 54% overall yield in several steps. The FIS of the phenylhydrazone and ketone 80 occurred regioselectively in acetic acid as an acid catalyst and solvent to give the tetracyclic 81 in 60% yield. It is remarkable that the sulfoxide substituent did not endure Pummerer rearrangement in the presence of the acidic conditions needed for both the hydrolytic elimination of the enol ether and the FIS. β-Amino sulfoxide scaffold is more reactive than those sulfoxides bearing an electron-withdrawing group at the α-position. Thus, β-amino sulfoxide is useful from the synthetic point of view, thus employed in initial step of the synthesis with the requisite oxidation level at the methylene carbon linked to the nitrogen atom. In the following, after several steps the tetracyclic 81 was transformed into (±)-deethylibophyllidine 78 in 50% yield (Scheme 12).80,81
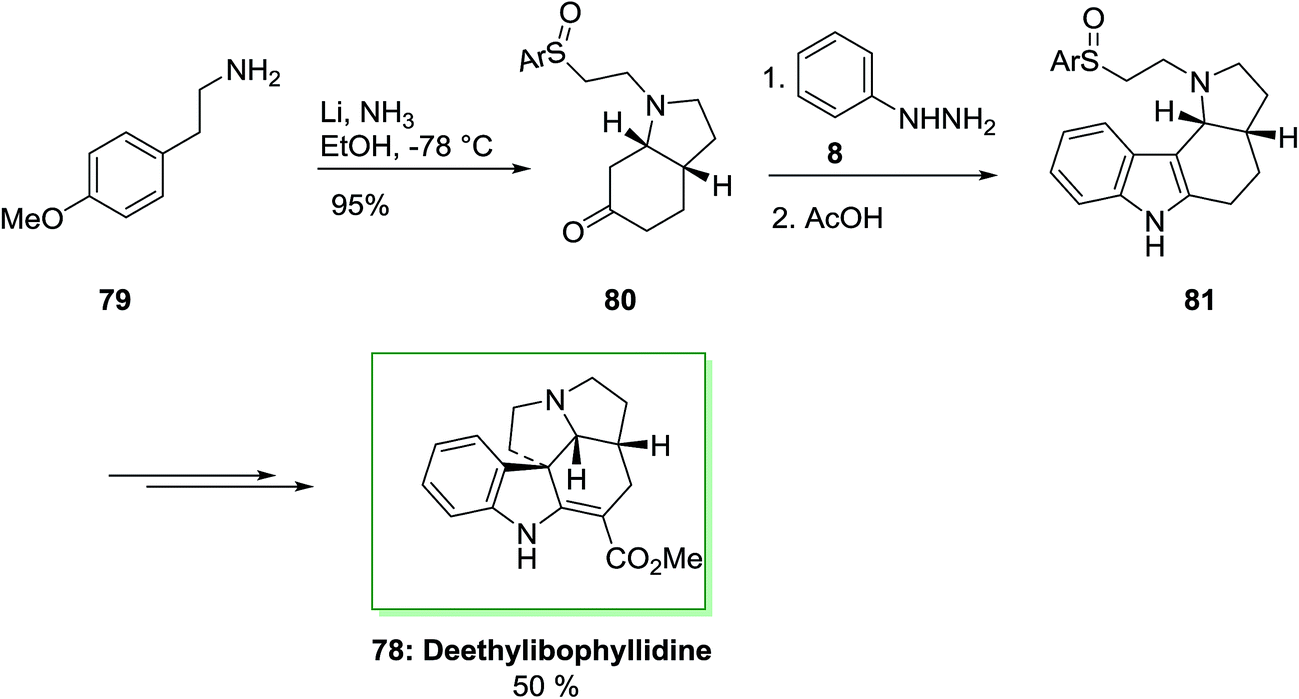 | ||
| Scheme 12 Total synthesis of deethylibophyllidine 78. | ||
Murrayafoline A 82, extracted from the root of various species of the genus Glycosmis, Murraya and Clausena (Rutaceae), displays potent fungicidal property against Cladosporium cucumerinum and growth inhibitory property on cell cycle M-phase inhibitory, human fibrosarcoma HT-1080 cells and apoptosis inducing properties on mouse tsFT210 cells.82
In 1998, Murakami and co-workers described the six-step total synthesis of murrayafoline A 82 with 40% yield.83 The current method initiated by the treatment of aminophenol 83 through the 2-hydrazino-5-methylphenol 84 to give the O-methanesulfonyl (mesyl) derivative 86. Then, the provided O-mesylphenylhydrazone 86 has been exposed to FIS to give the tetrahydrocarbazole derivatives 87 and 88 in 63% and 3% yield, respectively. Lastly, after multi reactions the mesyloxy compound 87 gave murrayafoline A 82 (Scheme 13).83
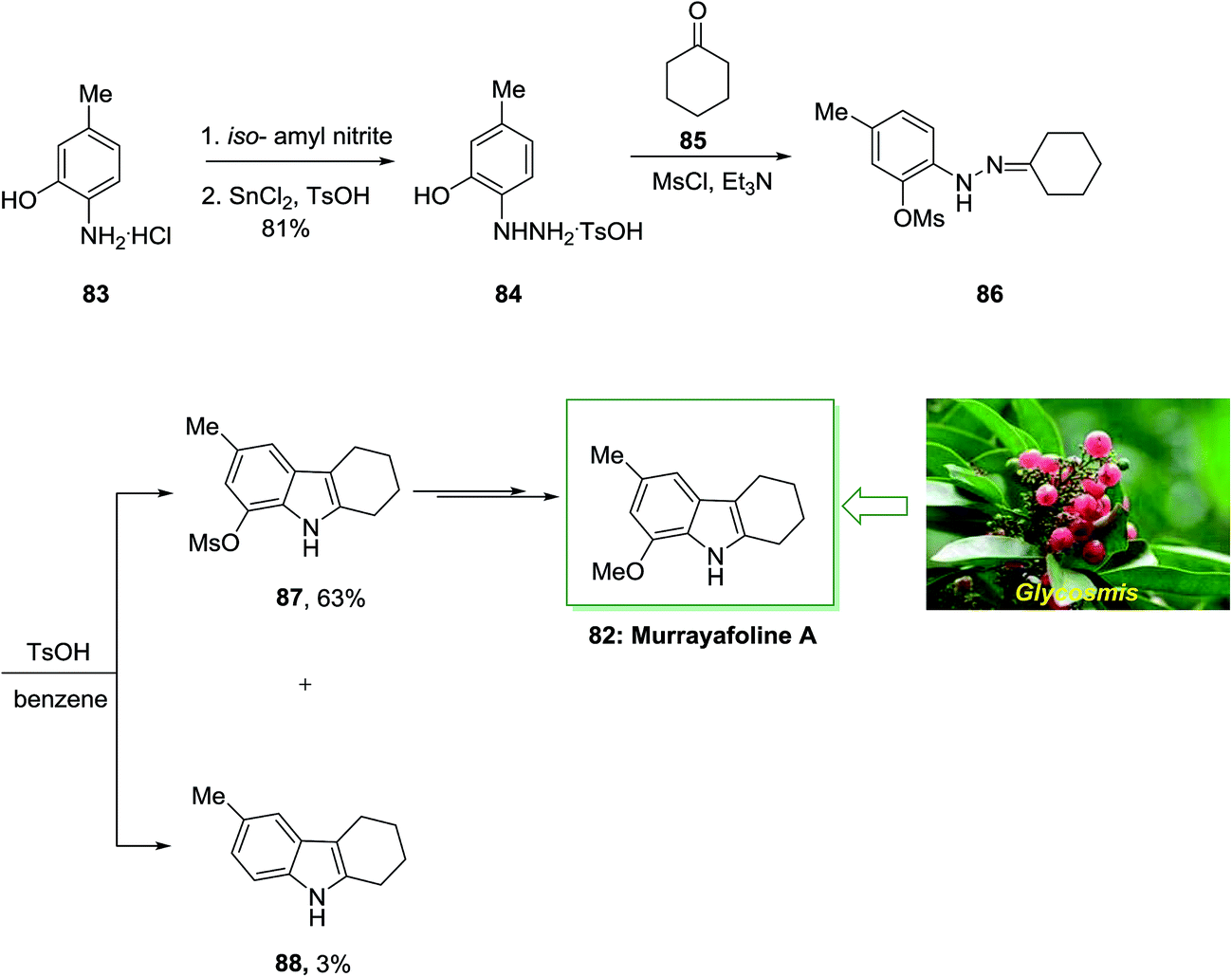 | ||
| Scheme 13 Total synthesis of murrayafoline A 132. | ||
A catalytic enantioselective synthesis of 20-deethyltubifolidine 89 by using the heterobimetallic enantioselective catalyst (ALB-KO-t-Bu-MS 4A) has been achieved in 1998.84 Initially, the catalytic enantioselective Michael addition of cyclohexenone 90 and dimethyl malonate 91, by utilizing AlLibis(binaphthoxide) complex (ALB) as catalyst, provided optically pure 92 in 99% ee and 94% yield even at ambient temperature. Then, optically pure compound 92 transformed into the indole derivative 93 in 92% yield, via an extremely regioselective FI approach continued through decarbalkoxylation (the ee of 93 has been shown to be 99%). Then, the latter has been transformed to 20-deethyltubifolidine 89, through a multi-step reaction with an overall yield of 27% (Scheme 14).84
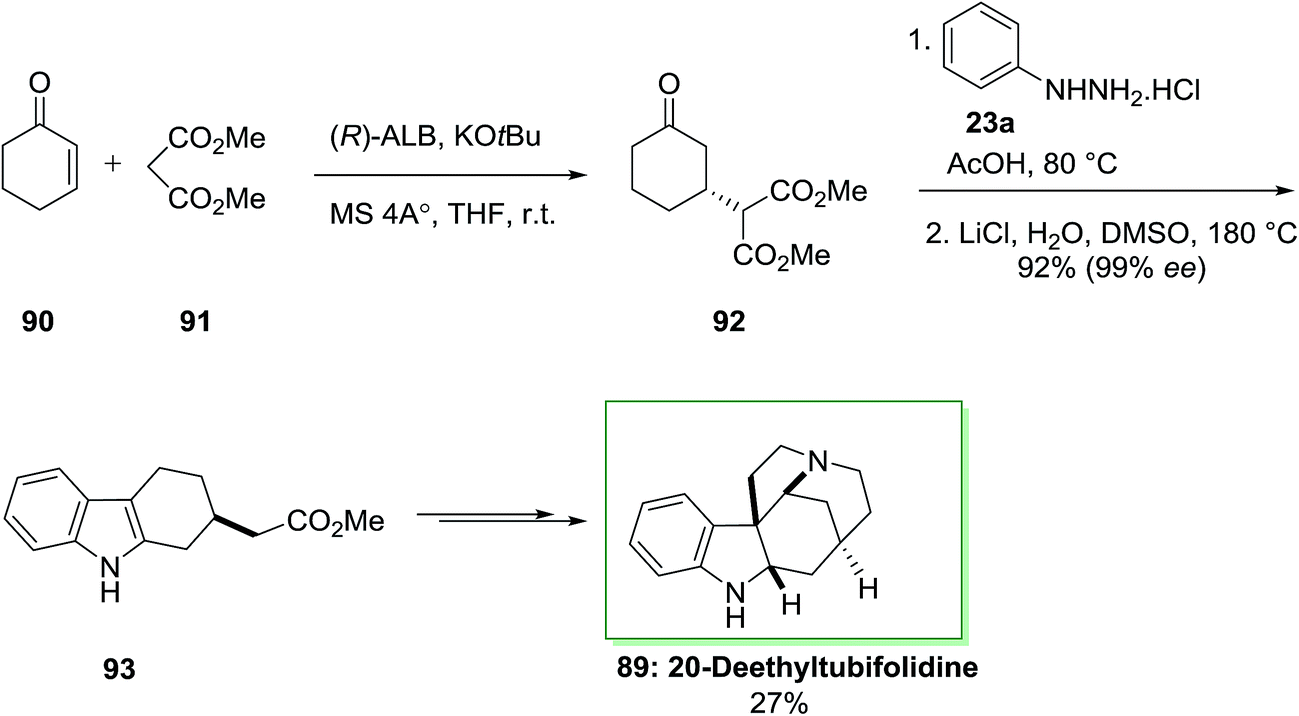 | ||
| Scheme 14 Total synthesis of 20-deethyltubifolidine 89. | ||
A catalytic enantioselective synthesis of the strychnos alkaloid tubifolidine 94, was extracted from the leaves of pleiocarpa tubicina, has been accomplished in an extremely stereocontrolled method.84 This total synthesis based on the chiral indoles 93 as a main intermediate to put the remaining enantioselective centers by using substrate control. This compound has been provided through an extremely regioselective FIS between ketone 92, which has been provided through enantioselective Michael reaction, and phenylhydrazine hydrochloride in AcOH at 80 °C. The latter has lastly converted to the tubifolidine 94 via a multi-step synthesis in 24% overal yield (Scheme 15).84
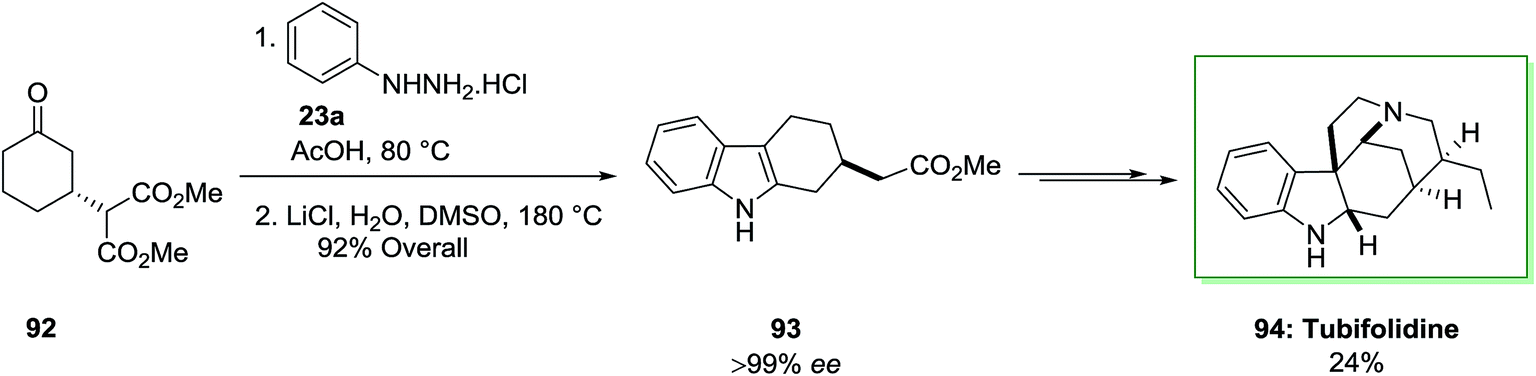 | ||
| Scheme 15 Total synthesis of tubifolidine 94. | ||
Several indole alkaloids contain the basic tetracyclic framework with eventually groups for example a methyl substituent on nitrogen 5 or 12 and/or hydroxy or methoxy substituents on carbon 1, 2, 3 or 4. A 9-azabicyclo [3.3.1] nonanone was applied during a tried synthesis of ajmaline via a FI reaction. (endo, endo)-9-benzyl-9-azabicyclo [3.3.1] nonane-2,6-diol 96 that already gave a simple admittance to enantioselective synthesis of indolizidine and quinolizidine alkaloids, can also be applied in principle for the formation of macroline/sarpagine type alkaloids. Ketone 97 has been produced in 37% yield from readily accessible (endo, endo)-9-benzyl-9-azabicyclo [3.3.1] nonane-2,6-diol 96 after several steps.
Next, ketone 97 was treated with functionalized phenylhydrazines 98 under reflux in hydrochloric acid-saturated MeOH. The desired derivatives 99 were readily provided via an abnormal FI. Then two intermediates 95a and 95b were directly produced through Swern oxidation of 99a and 99b, respectively. These two intermediates can be utilized for the formation of macroline/sarpagine kind alkaloids (Scheme 16).85
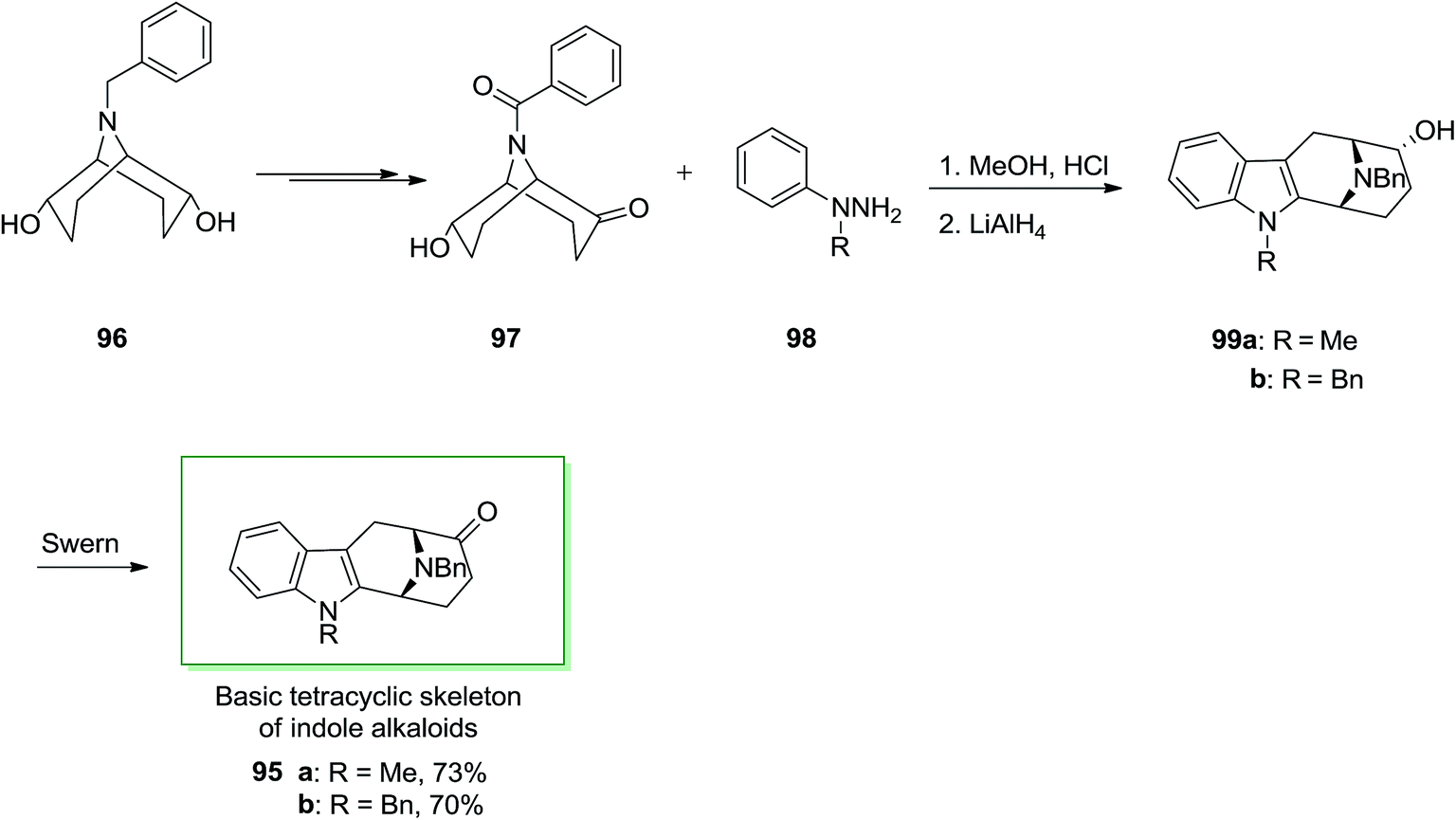 | ||
| Scheme 16 Total synthesis of basic teracyclic skeleton of macroline/sarpagine type alkaloids. | ||
Next, in another route, ketone 97 has been treated with meta-functionalized phenylhydrazine derivatives 101 noticeably should result in a mixture of 1- and 3-functionalized compounds: for example, with meta-tolylhydrazine a 1:1 mixture of 102a and 102b has been provided. To create this FIS regioselective, a transitory substitution by bromine at one ortho-position has been employed. After FI and hydrogenolysis by using Pd/C and potassium carbonate, the 1- or 3-functionalized 100a and 100b, respectively were provided (Scheme 17).85 These consequences exhibit that (endo, endo)-9-benzyl-9-azabicyclo[3.3.1]nonane-2,6-diol 96 provided a simple condition to enantioselective synthesis of quinolizidine and indolizidine alkaloids. This method can also basically apply for the synthesis of macroline/sarpagine alkaloids.
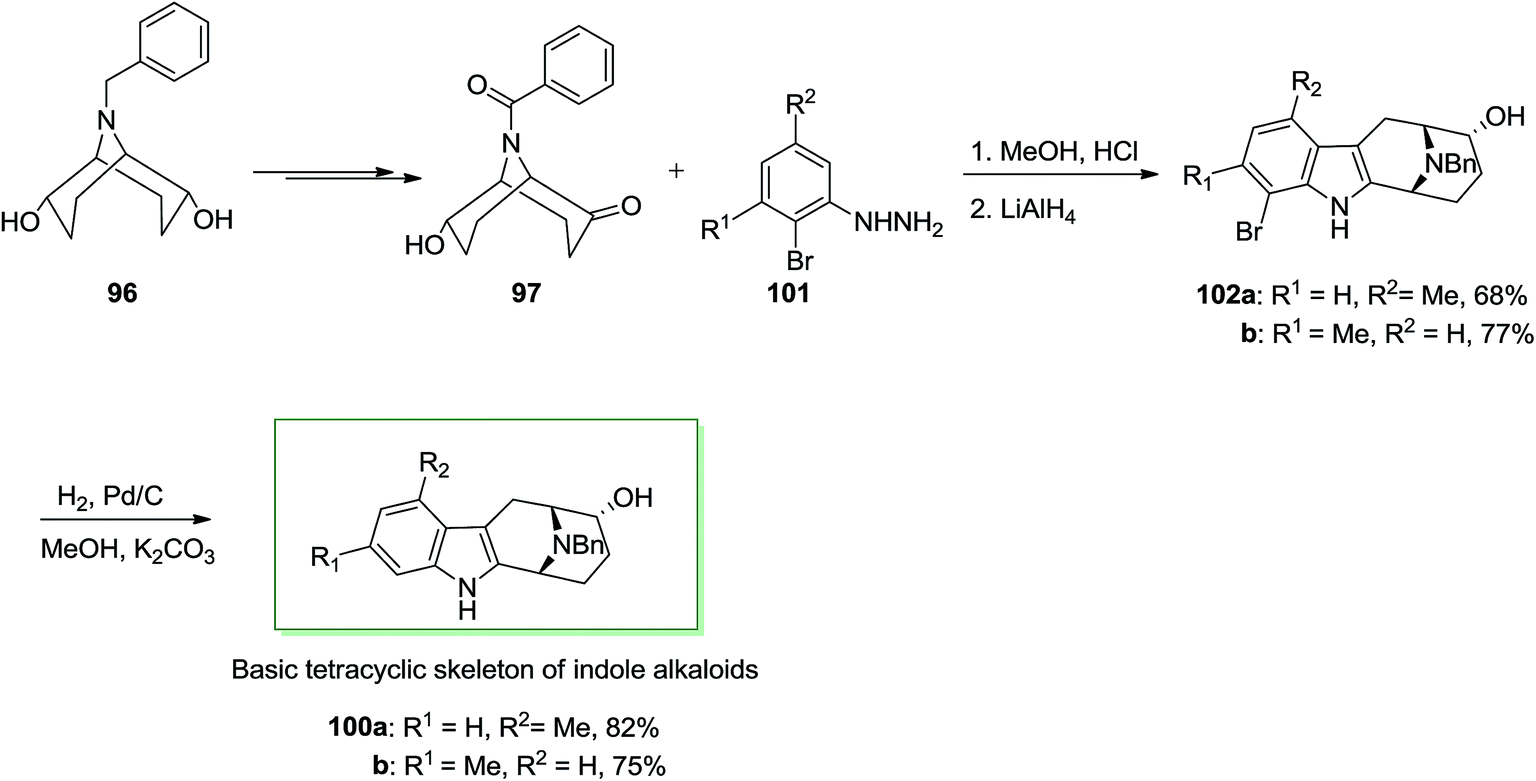 | ||
| Scheme 17 Total synthesis of basic tetracyclic of indole alkaloids 100. | ||
The Aspidosperma group demonstrates one of the widest class of indole alkaloids, having more than 250 products extracted from different biological sources. An important member of this group is tabersonine 103 that exhibits a main role in the synthetic chemistry of Aspidosperma alkaloids and biosynthesis. Initially, tabersonine has been extracted from Amsonia tabernaemontana in 1954 by Le Men and co-workers.86 Soon upon the first report, the alkaloid has been extracted from various other natural sources, demonstrating its relative biological abundance.
In 2001, Rawal and his group described the twelve-step enantioselective total synthesis of (±)-tabersonine 103 with overall yield of 70–80%.87 The enantioselective total synthesis of racemic tabersonine was initiated from market purchasable monoacetal 104. Upon several steps, carbamate 105 has been provided in 88% yield. Subsequent, reaction of the silyl enol ether 105 with dilute HCl provided a clean hydrolysis to bicyclic ketone 106, with the cis stereochemistry intact. This transformation, sacrificed the double bond position, which has been accomplished via the first cycloaddition. In providing for FIS, ketone 106 has been transformed into the phenylhydrazone via heating it with phenylhydrazine hydrochloride 23a by using Na2CO3. In the following, the crude hydrazone has been refluxed at 95 °C in glacial AcOH. A weakly acidic medium found to favor idolization toward the more functionalized carbon. Based on these reaction conditions, the indolization provided efficiently and in satisfactory yield (93% overall) but gave both possible indole isomers in roughly equal quantities. The two isomers have been easily isolated via column chromatography. Hence, by using FI reaction, this reaction didn't have stereocontrol. Next, the tetracyclic indole 107, upon several steps, afforded (±)-tabersonine (rac-103), in 70–80% yield (Scheme 18).87
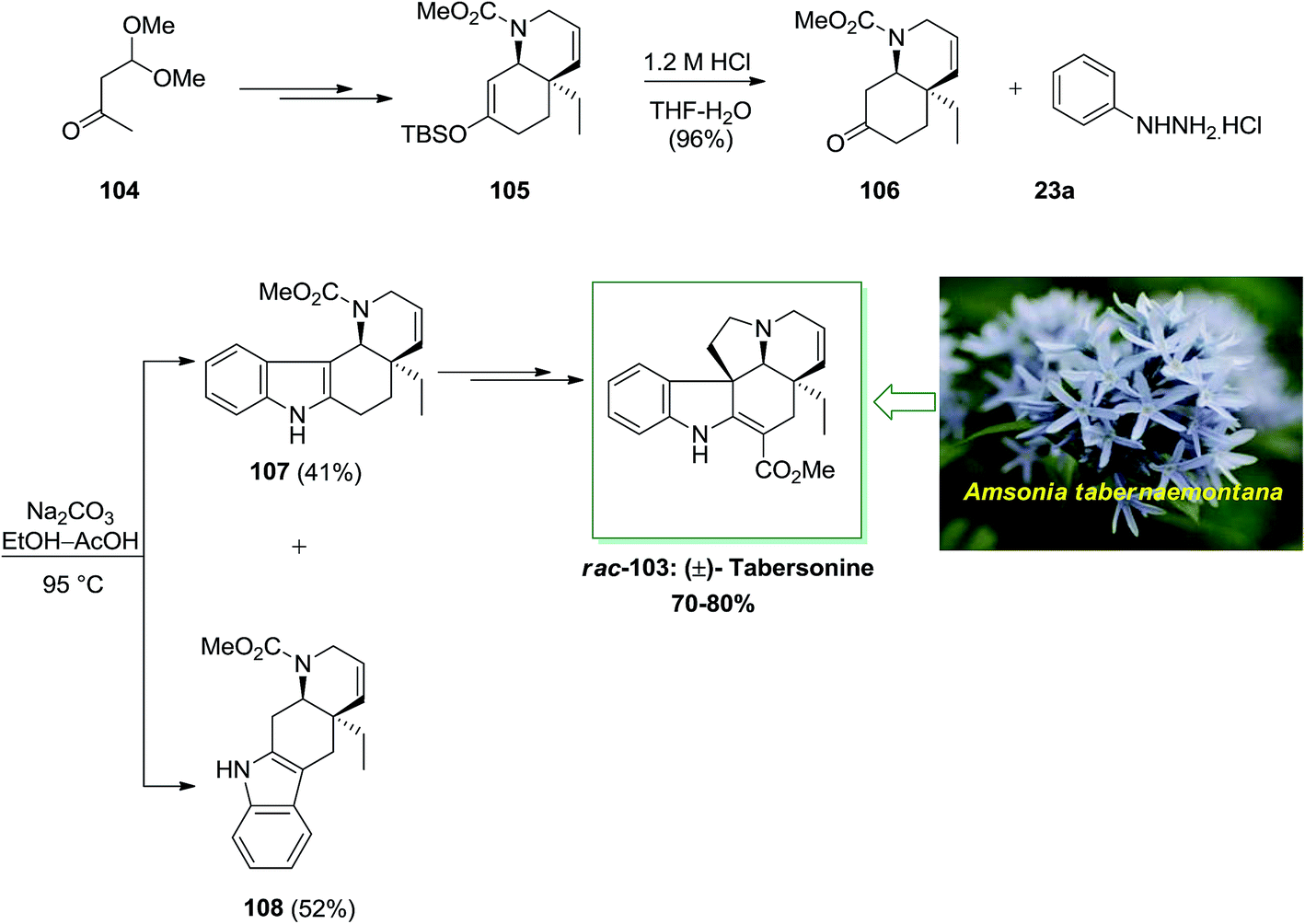 | ||
| Scheme 18 Total synthesis of (±)-tabersonine 103. | ||
Peduncularine 109 is a member of the indole alkaloids class with a monoterpene unit like the aliphatic portion, which firstly was extracted by Bick and co-workers in 1971, from the Tasmanian shrub Aristotelia peduncularis.88 Alkaloids have been derived from some of other elaeocarpaceous plants from A. serrata (New Zealand), A. chilensis (Chile) and also mostly from New Guinea. Its configuration associated alkaloids aristoteline, peduncularine, tasmanine, aristoserratine, sorelline and hobartine. That is also re-divided biogenetically pattern with a rearranged geranyl and tryptamine subunit. These natural productions such as peduncularine have indicated cytotoxic activity against cell lines of breast cancer and other biological activities.
In 2002, Roberson and co-workers achieved a relatively short total synthesis of (±)-peduncularine 109 in sixteen steps from market purchasable 1,4-cyclohexadiene 110.89 This compound could provide acetate 111, after several steps. The latter has been exposed to FIS to supply alcohol 112 with concomitant deprotection of the C-8 alcohol. The latter upon several steps produced (±)-peduncularine 109 with appropriate yield. The main step of in the current total synthesis is the [3 + 2] annulation reaction of an allylic silane by using chlorosulfonyl isocyanate, that transported the desired bicyclic nucleus of the naturally occurring compounds (Scheme 19).89
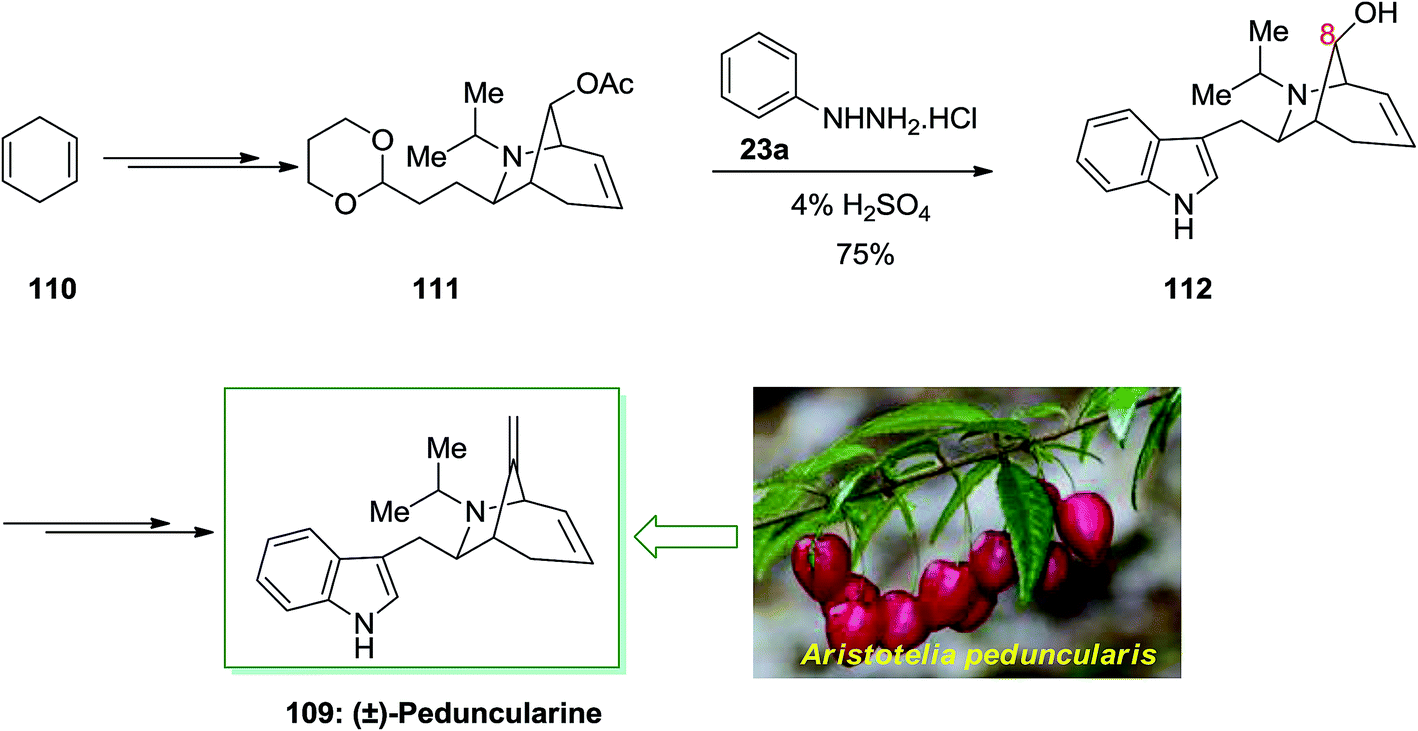 | ||
| Scheme 19 Total synthesis of (±)-peduncularine 109. | ||
A novel group of 5-heteroaryl-functionalized 1-(4-fluorophenyl)-3-(4-piperidinyl)-1H-indoles as enormously selective and potentially CNS-active α1-adrenoceptor antagonists was shown by Eilbracht and co-workers.90 The corresponding products were provided from the antipsychotic sertindole 113. The structure–affinity relationships of the 5-heteroaryl substituents and the groups on the piperidine nitrogen atom were optimized with respect to affinity for α1 adrenoceptors and selectivity in respect to dopamine (D1–4) and serotonin (5-HT1A–1B and 5-HT2A,2C) receptors.91
A usual aspect of unusual antipsychotics for example clozapine, olanzapine, sertindole 113 and seroquel is nanomolar attraction for α1 adrenoceptors additionally to their attractions for serotonin 5-HT2A and dopamine D2 and receptors. The real balanced attractions for these receptors might underlie the enhanced form of these drugs (enhanced rate between doses containing antipsychotic property and extrapyramidal side effects) as contrasted to classical antipsychotic drugs, for example haloperidol. The phenylindole framework of sertindole 113 is a promising pattern for the growth of main acting α1 antagonists. Replacing of the 5-chloro atom in sertindole 113 with polar substituents including functionalized aminomethyl and carbamoyl groups provided a novel group of particular α1 adrenoceptor antagonists. This research demonstrated that 5-substituents mixing hydrogen bond acceptor possessions with steric bulk in the plane of the indole core are necessary to provide high affinity for adrenergic α1 receptors mixed with satisfactory selectivity in respect to dopamine D2 and serotonin 5-HT2A and 5-HT2C receptors.
Tryptamine imitative is specifically included in many biological processes, such as melatonin in serotonin in neurological processes or in the circadian rhythm control. Therefore, tryptamine containing the nucleus indole and its imitative were employed for the reaction of various diseases such as migraine (for example Sumatriptan), schizophrenia (e.g. Sertindole) and depression (e.g. D-tryptophan). In this approach, firstly, tandem hydroformylation-FIS gave appropriate admittance to indole 116 initiating from the readily accessible olefin 114 through transformation with market purchasable 4-bromophenylhydrazine 115 and afforded indole 116 in 39% yield. The latter has been then transformed into the corresponding sertindole 113, upon several reactions (Scheme 20).90,91
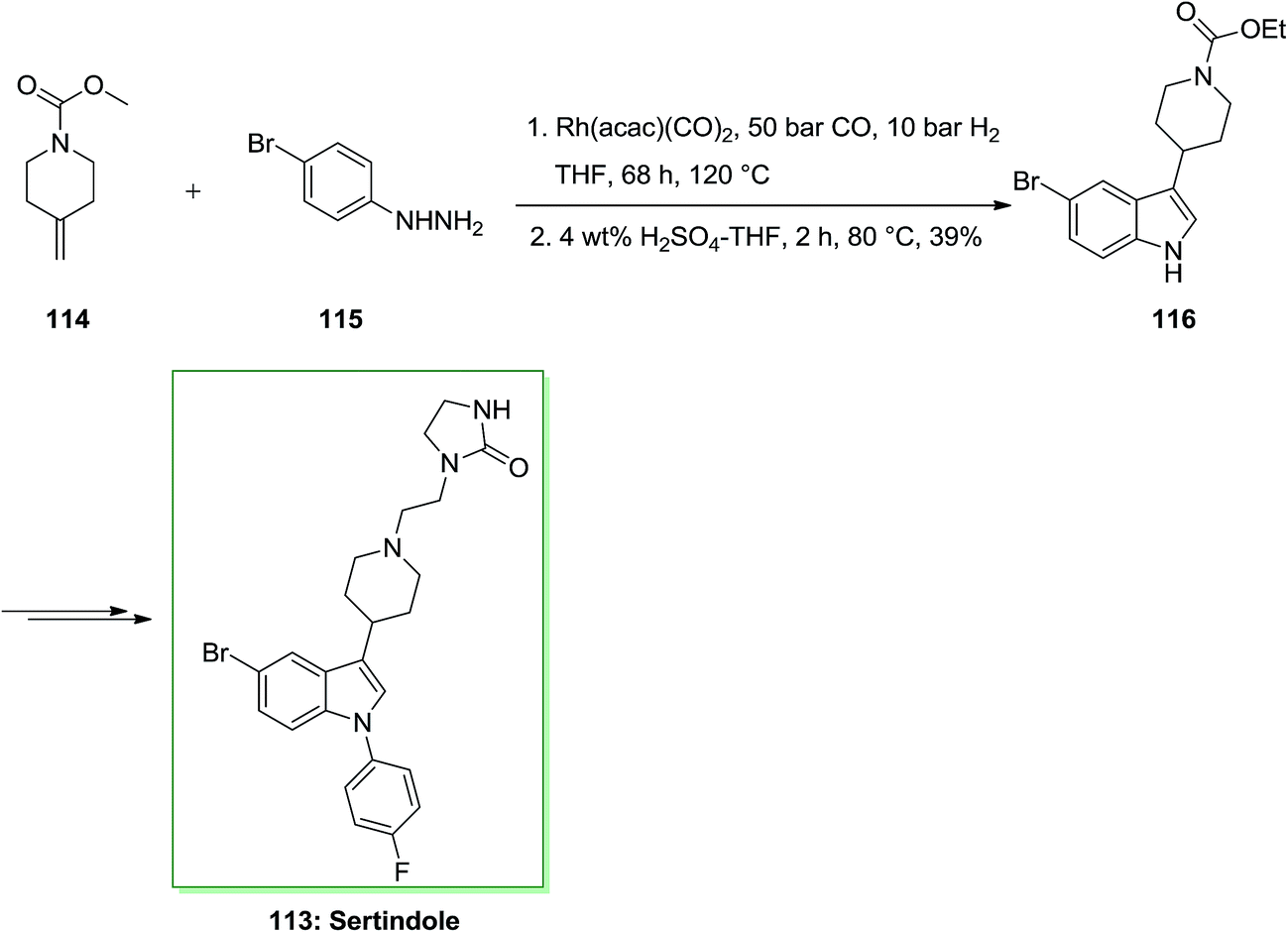 | ||
| Scheme 20 Total synthesis of sertindole 113. | ||
Meridianins are brominated 3-(2-aminopyrimidine)-indoles, which are purified from Aplidium meridianum, an Ascidian from the South Atlantic (South Georgia Islands). Meridianins prevent cell proliferation and induce apoptosis, a demonstration of their ability to enter cells and to interfere with the activity of kinases important for cell division and cell death. These results suggest that meridianins constitute a promising scaffold from which more potent and selective protein kinase inhibitors could be designed.92
The compounds, except meridianin G and the related iso-meridianins C, were found to inhibit CDKs, GSK-3, PKA and other protein kinases in the low micromolar range. Meridianins B and meridianin E were the most potent inhibitors while meridianin G, isomeridianin C and G were essentially inactive. Meridianins B and E were selected for further studies on selectivity and cellular effects.93
Franco and co-workers described synthesis of iso-meridianin derivatives 117 via microwave irradiation (MWI).93 The synthetic method contains six steps, in which FIS is considered as the main reaction. This group selected isocytosine 118 as a suitable starting compound, since the hydroxyl group at position 4 permits an appropriate functionalization and introduction of the requisite C-2 carbonyl group. In this procedure, isocytosine 118 has been transformed into the corresponding methyl ketone 119 upon different steps. The latter based on normal conditions afforded the desired phenylhydrazone derivatives 120a and 120b in 90% yield. The desired products 120a and 120b have been employed without more purification. The FIS was the main step of this method, so many efforts by utilizing various catalysts, solvents and heating conditions were attempted. As a result, the usage of zinc chloride and MWI provided a quick, clean and quantitative elimination of the Boc group. Although, addition of a small amount of dimethylformamide prior to MWI with zinc chloride into 120a and 120b provided the desired iso-meridianin G 117a and iso-meridianin C 117b in satisfactory yields, respectively (Scheme 21).93
 | ||
| Scheme 21 Total synthesis of iso-meridianins G 117a and C 117b. | ||
The total synthesis of 8-desbromohinckdentine A 121 has been effectively achieved and described by Liu and co-workers in 2003.94 In this route, 2-(2-bromophenyl)-indole 123, has been formed through the FIS from phenylhydrazine hydrochloride 23a and 2-bromoacetophenone 122. Upon several steps, indole 123 has been converted into the corresponding single dibrominated natural product 121 (Scheme 22).94
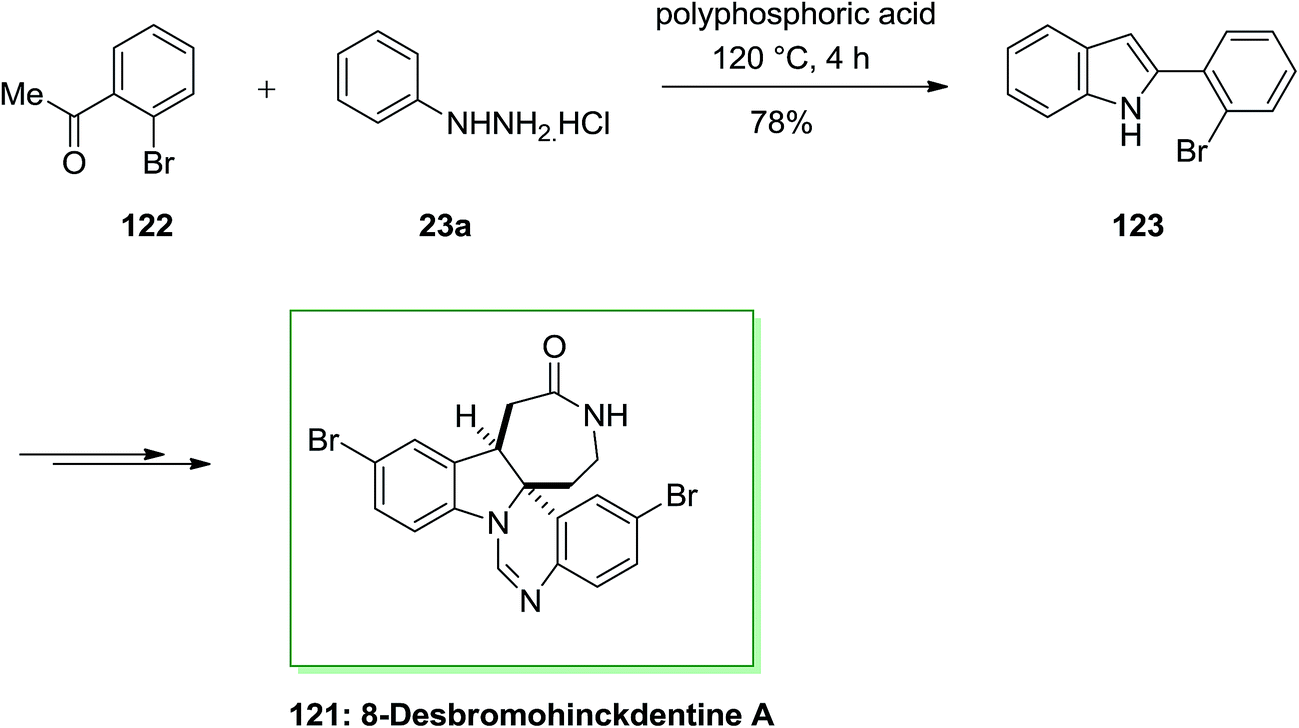 | ||
| Scheme 22 Total synthesis of 8-desbromohinckdentine A 121. | ||
(±)-Aspidospermidine 124 is one of the aspidosperma alkaloids family. Aspidosperma alkaloids structurally have pentacyclic [6.5.6.6.5] ABCDE ring system skeleton with a common structural feature which is cis-stereocenters at C-7, C-21, and C-20 (all carbon quaternary). Some parts of this kind of alkaloids like vinblastine and vincristine have been used as cancer chemotherapy medications. Additionally tabersonine (possess inhibitory effect against SK-BR-3 human cancer cell lines which is better than cisplatin), jerantinine-E (more potent in vitro cytotoxicity against human KB cells, IC50 <1 μg mL−1), and vincadifformine (cytotoxic), are pharmacologically important alkaloids.95
Total synthesis of (+)-aspidospermidine 124 has been described by Aube and co-workers in 2005.96 The main reactions used in the total synthesis of this pentacyclic Aspidosperma alkaloid contain a deracemizing imine alkylation/Robinson annulation sequence, a selective “redox ketalization”, and also an intramolecular Schmidt reaction. A FIS happened on a tricyclic ketone similar to the sequence described in their aspidospermine synthesis. The synthetic method utilized 2-ethylcyclopentanone 125 as the precursor. The latter provided tricyclic lactam 126 as a single diastereomer in 82% chemical yield and 84% enantioselectivity. Compound 126 did not show amenable to a direct FIS reaction and so has been reduced in a three-step procedure to the desired ketoamine 13 by using of selective protection of the ketone carbonyl continued through a reduction/deprotection reaction. The reaction of ketoamine 13 with phenylhydrazine 8, acid and lastly lithium aluminium hydride gave the target (+)-aspidospermidine (124, 51% yield from 13) along with 13% of a by-product 127. This by-product probably arose through the FI of the less-functionalized enamine isomer (Scheme 23).96
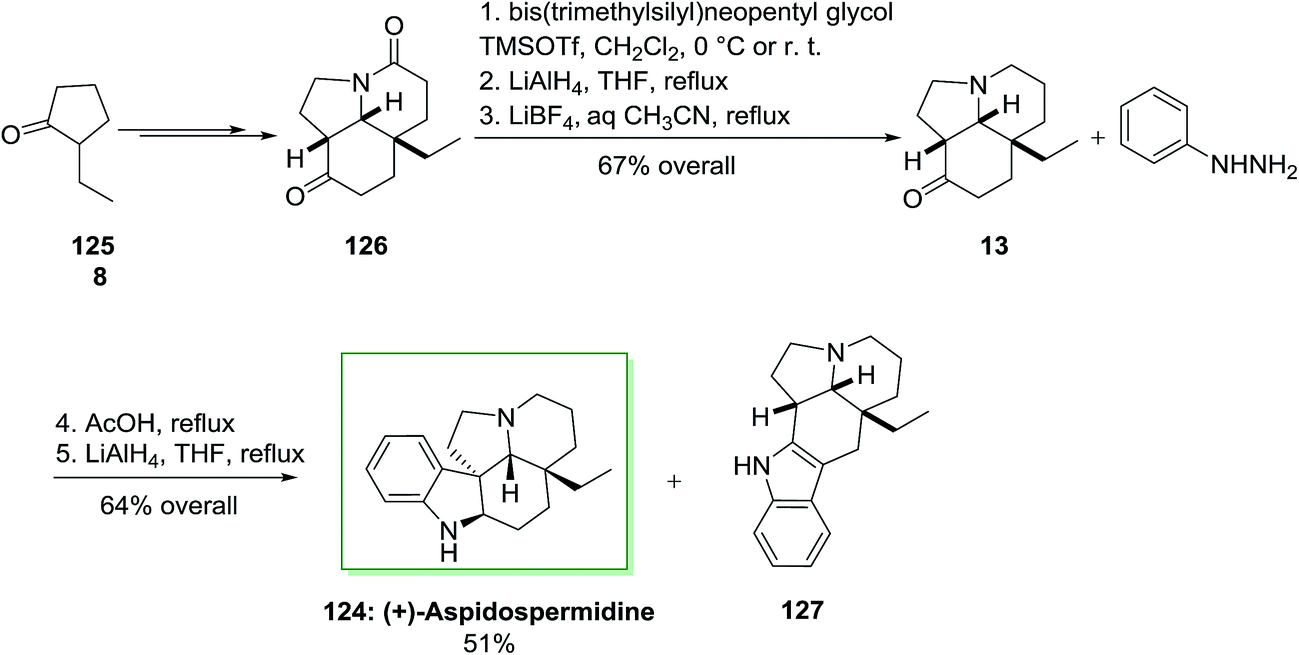 | ||
| Scheme 23 Total synthesis of (+)-aspidospermidine 124. | ||
Another approach has been described by Canesi and co-workers for the synthesis of (±)-aspidospermidine 124 in 12 steps.97 This method, relied on ‘‘aromatic ring umpolung’’ was initiated from a polyfunctionalized phenol 128. Upon diverse steps, this compound can be transformed into main intermediate 13. In the following, FIS of tricycle 13 and phenylhydrazine 8 under reflux condition resulted in the hydrazone 129 that is transformed to imine 130 in AcOH. The latter is reduced in the same pot by using lithium aluminium hydride to provide (±)-aspidospermidine 124, in 43% yield (Scheme 24).97
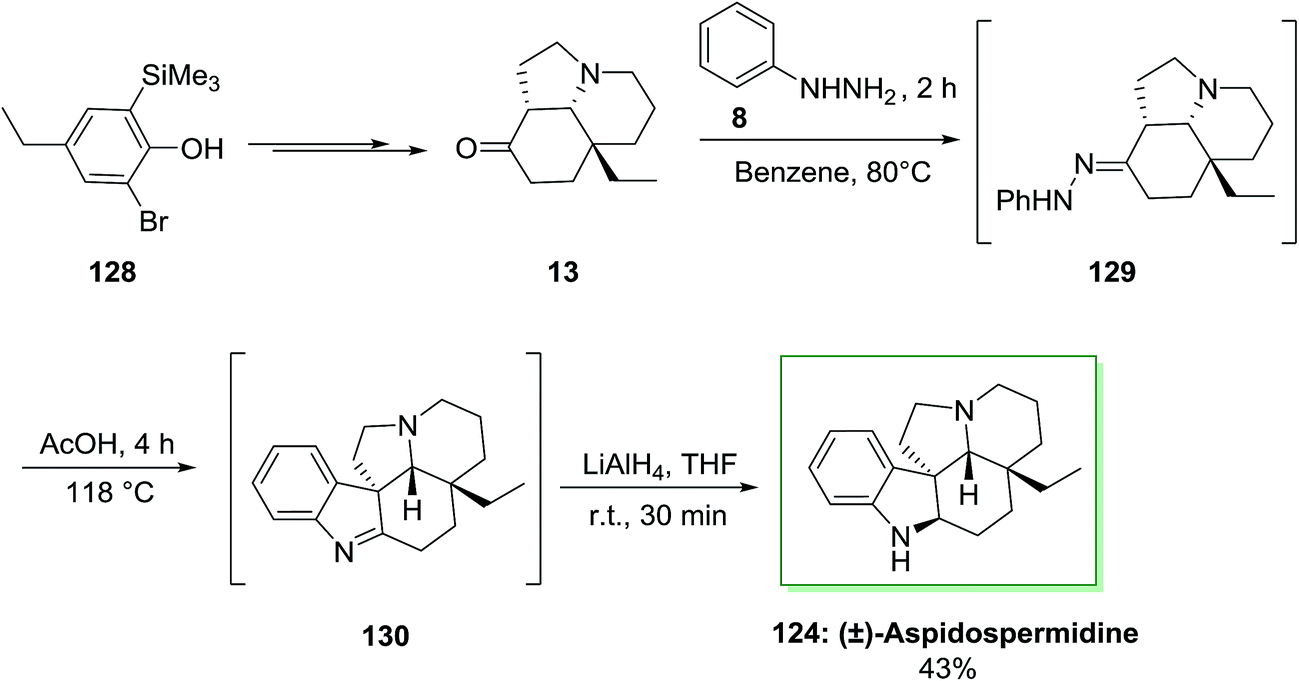 | ||
| Scheme 24 Total synthesis of (±)-aspidospermidine 124. | ||
Also, in another method, for the total synthesis of (+)-aspidospermidine 124, 1-(p-methoxy benzyl)piperidin-2-one 131 upon several steps provided tricyclic core 13. The latter is a privileged tricyclic nucleus having the crucial C-20 all-carbon quaternary stereocenter would be a significant issue in providing enantioselective synthesis of 124 and structurally related bisindole alkaloids. Also, FI cyclization reaction of 13 produced dehydroaspidospermidine that on reduction by using lithium aluminium hydride afforded corresponding naturally occurring compound (+)-aspidospermidine 124 in 50% yield (Scheme 25).95
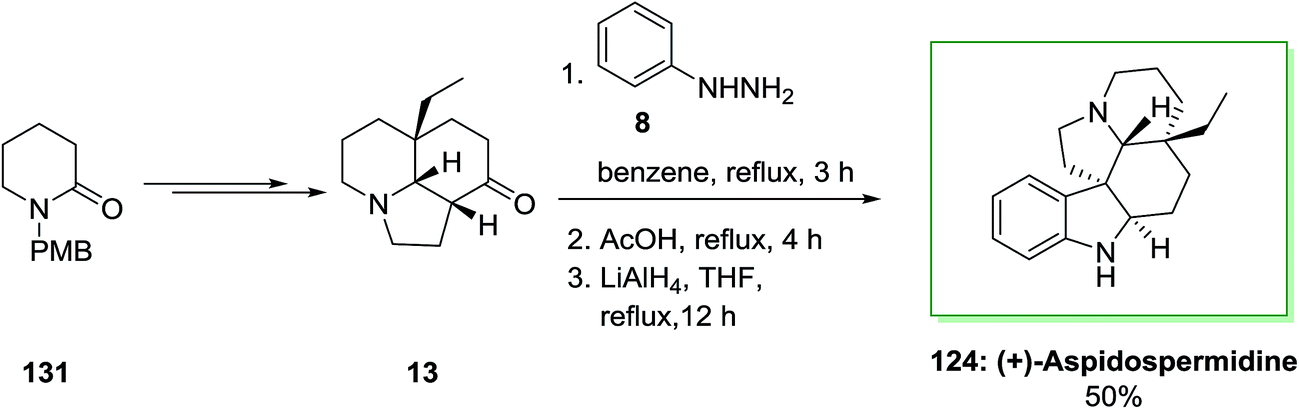 | ||
| Scheme 25 Total synthesis of (+)-aspidospermidine 124. | ||
Cryptosanguinolentine 132 or called isocryptolepine is the most important member of indoloquinoline alkaloids, which was separated from the roots of the West African plant Cryptolepis sanguinolenta. In traditional folk medicine was used for treatment of fevers such as fever of malaria. Various types of hetero-aromatic alkaloids have potential applications in the medicinal field. Many such alkaloids intercalate in the DNA double helix resulting in changes in DNA conformation so they can inhibit DNA transcription and replication. X-ray analysis and spectroscopy can investigate the mode and strength of binding of these alkaloids to DNA. In addition, some N-methyl derivatives of these ring systems exhibit important cytotoxic and antimicrobial activities.98
A novel and appropriate synthesis of cryptosanguinolentine 132, have been described by Dhanabal and co-workers in 2005.99 In this total synthesis, an improved FI cyclization reaction has been employed in the initial step. 4-Hydroxy-1-methyl-1H-quinolin-2-one 133 and phenylhydrazine hydrochloride 23a reacted by using a mixture of glacial AcOH and concentrated HCl in the ratio of 4:1 to afford the indoloquinoline scaffold 134 in a single step. Then, the corresponding compound 134 in several steps gave cryptosanguinolentine 132 in high yields (Scheme 26).99
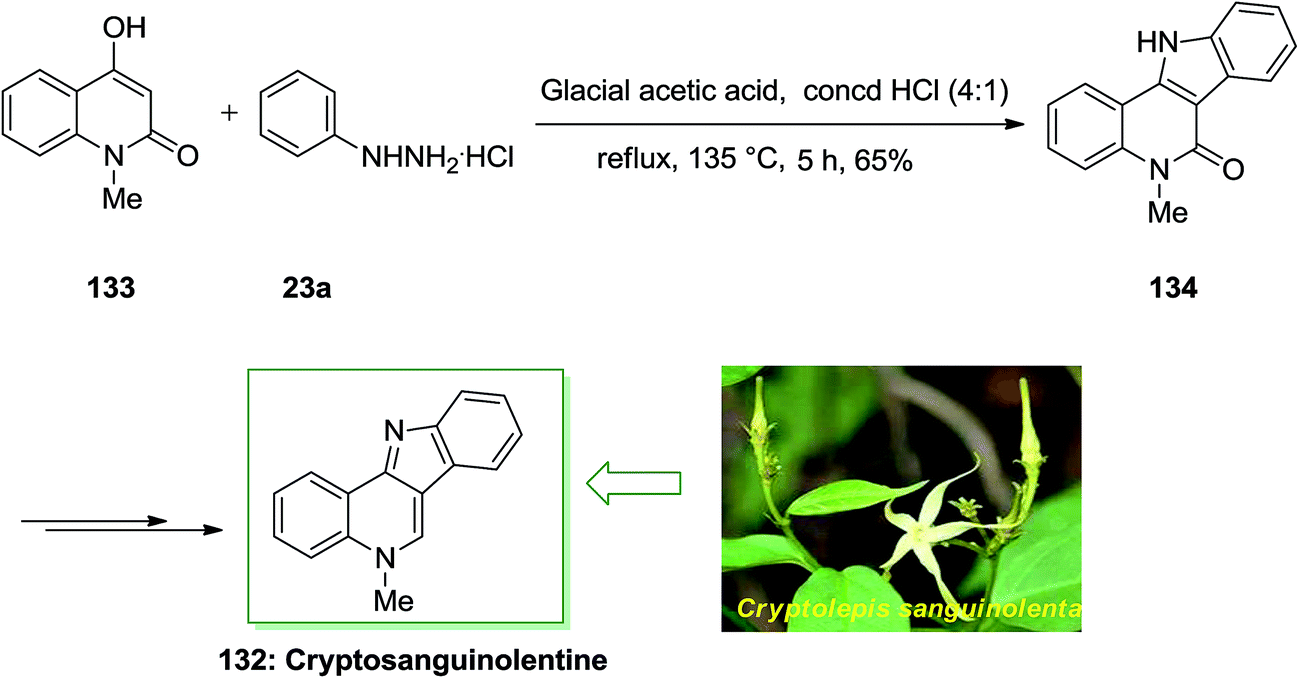 | ||
| Scheme 26 Total synthesis of cryptosanguinolentine 132. | ||
Haplophytine 135 (C27H31O5N3), the important indole alkaloid, and cimicidine (C23H28O5N2) were isolated from the Mexican plant Haplophyton cimicidum's dried leaves, first time in 1952 by Snyder and co-workers.100 Huplophyton cimicidum produces the aspidospermine and the biogenetically related eburnamine kinds of alkaloids. Haplophytine 135 is constituted of two parts that are joined by the forming of a quaternary carbon center. The right-half part is a hexacyclic aspidosperma class of alkaloid, known as aspidophytine, which is acquired by the acidic degradation of (+)-haplophytine. The left-half part has an unique structure, that includes a bicyclo[3.3.1] framework which possesses bridged aminal and ketone activities. Both alkaloids are toxic to insects, but most of the toxicity is owed by the haplophytine conLent (I) also they are toxic to German roaches on contact, injection and ingestion. The LD/50 dosage of cimicidine is about 60 γ/g (contact, 48 h) and for haplophytine is 18 γ/g. Haplophytine caused extended paralysis at dosage levels under the LD/50 value. The total crude alkaloid has been discovered to be toxic to an extensive range of insects containing Mexican bean beetle larvae, European corn borers, egg-plant lace bugs, Colorado potato beetle larvae and adults, codling moths and grasshoppers.101
The first total synthesis of (+)-haplophytine 135 has been reported by using several key steps including intramolecular Mannich reaction, oxidative rearrangement, the FIS and Friedel–Crafts alkylation reaction.102 Ueda and co-workers demonstrated the first total synthesis of (+)-haplophytine 135 in 2009 via an effective method.102 Haplophytine is contained of two segments that are linked by the construction of a quaternary carbon center. Total synthesis of (+)-haplophytine 135 was initiated from market purchasable 7-benzyloxyindole 136 and upon various steps afforded 137 as the main precursor of the FI reaction. The reaction of hydrazine 137 with tricyclic ketone 138, synthesized by d’Angelo and co-workers by using 50% H2SO4 provided the desired hydrazone.103 Upon widespread optimization, this group ultimately known that cautious control of the reaction temperature and the suitable selection of solvent and acid were necessary to favorably attain the corresponding indolenine 139 over the indole 140 in satisfactory yield. Therefore, the reaction with p-toluenesulfonic acid in tert-butanol at 80 °C provided indolenine 139 in 47% yield accompanied with indole 140 in 29% yield. Lastly, imine 139 after different steps provided (+)-haplophytine 218 (Scheme 27).102
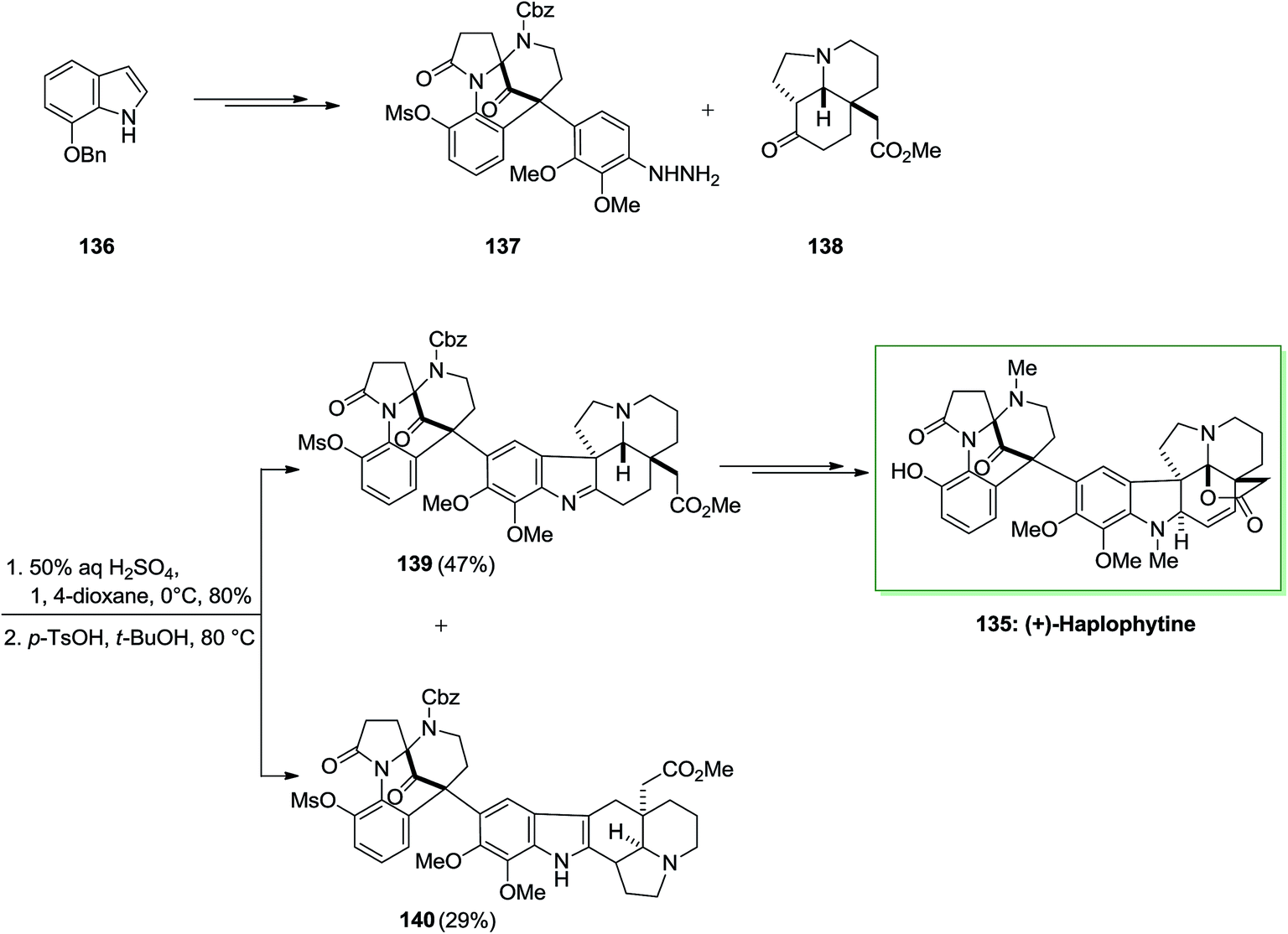 | ||
| Scheme 27 Total synthesis of (+)-haplophytine 135. | ||
Minfiensine is an indole alkaloid which is extracted from the African plant named Strychnos minfiensis by Massiot and co-workers in 1989.104 Minfiensine has significant biological functions containing anticancer activities. Additionally, various types of the Strychnos indole alkaloids have interesting anticancer functions too.104
In 2011, a short total synthesis of minfiensine 141 was accomplished in ten steps.105 The assembly of minfiensine 141 started with the FIS. Therefore, the reaction of cheap and market purchasable phenylhydrazine 8 and 1,4-cyclohexanedione monoethylene acetal 142 at ambient temperature continued by heating at 190 °C provided the corresponding indole product 143 in 89% yield. Next, the corresponding indole after several steps afforded (±)-minfiensine 141 in 95% yield (Scheme 28).105
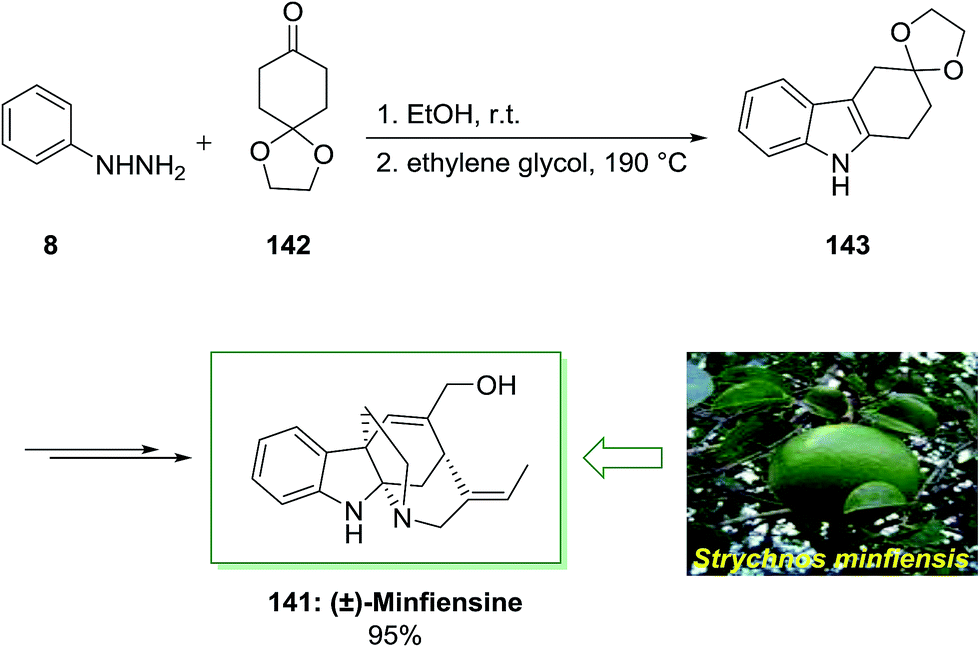 | ||
| Scheme 28 Total synthesis of minfiensine 141. | ||
(+)-Aspidoalbidine (or (+)-fendleridine) is an Aspidosperma alkaloid separated from the seeds of Aspidosperma fendleri. The name fendleridine was related to the name of the tree, by Burnell and co-workers in 1966.106 Brown found (+)-N-acetylaspidoalbidine in 1963,107 they gave the name “aspidoalbidine” to the postulated simplest member by analogy to aspidospermidine, discovered by Biemann two years before. Acetylaspidoalbidine is a member of Aspidosperma family, which is isolated from the Venezuelan tree species and Aspidosperma rhombeosignatum markgraf and Aspidosperma fendleri woodson. The Aspidosperma family is one of the indole alkaloid families also many of them have significant biological activities.108
The total synthesis of acetylaspidoalbidine 144, an alkaloid isolated from the Aspidosperma family, has been achieved by Guerard and co-workers in 2012.109 As depicted in Scheme 29, the total synthesis of 144 was initiated from different benzylic alcohol systems 145, that has been converted into alcohol 146 through a multi-step synthesis. Then, the pentacyclic scaffold has been generated by using a FIS method followed by treatment with lithium aluminium hydride. This approach resulted in a mixture of the indoline nucleus 147 and an unanticipated indole, in a 2:1 ratio favoring the corresponding 147. The corresponding compound 147 upon several steps transformed into acetylaspidoalbidine 144. A significant enantioselective form of this procedure is the ability to rapidly convert a cheap and simple phenol into an extremely substituted nucleus including a prochiral dienone in the form of a quaternary carbon center linked to different sp2 carbons. The provided structures are current in various naturally occurring compounds containing imperative biological properties. A fast method to diverse polycyclic molecules, involving the main tricyclic or tetracyclic systems of alkaloids belonging to the Aspidosperma group and a novel formal synthesis of acetylaspidoalbidine have been demonstrated. The consequences exhibit the potential of these oxidative transposition methods and the efficacy of the “aromatic ring Umpolung” concept. The reaction happens in valuable yields by using allyl groups (up to 67%) and is less significant with phenyl, vinyl, or alkyl substituents (Scheme 29).109
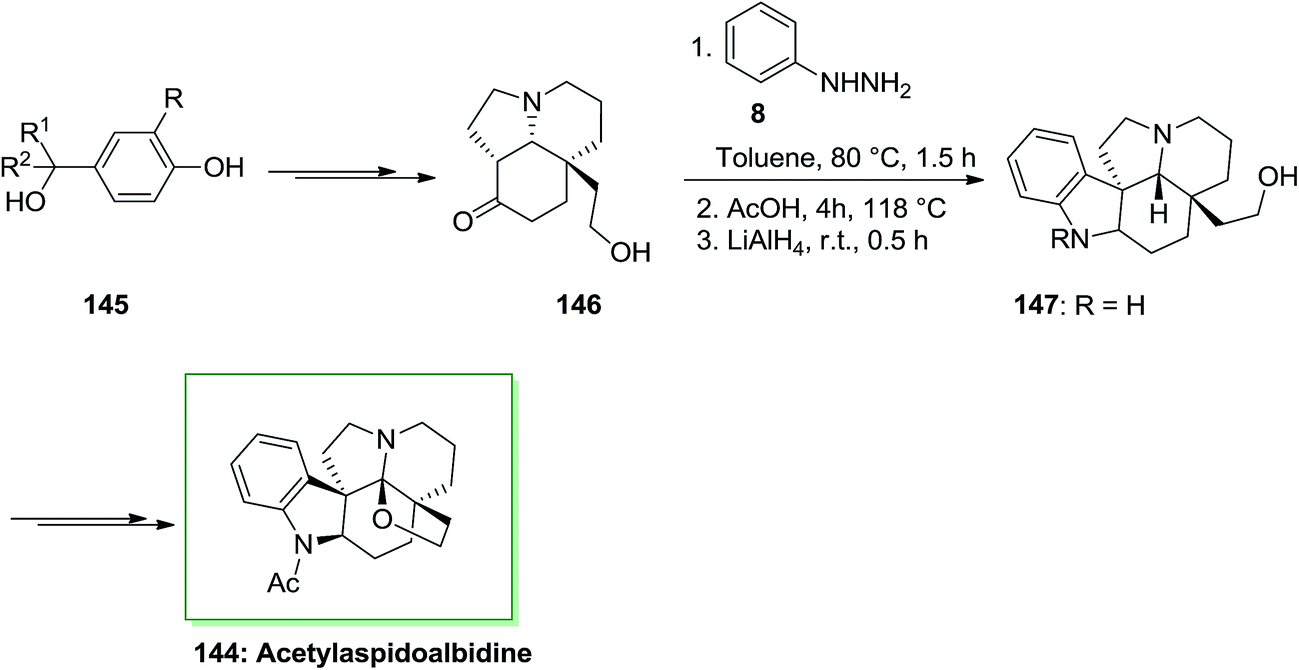 | ||
| Scheme 29 Total synthesis of acetylaspidoalbidine 144. | ||
In 2004, Kam and co-workers isolated Mersicarpine 148 from the stem-bark of the Kopsia arborea and Kopsia fruticosa.110 The tetracyclic dihydroindole mersicarpine was exhibited in another kind of Kopsia, viz., K. singapurensis too, which has a typical seven-membered cyclic imine joined with indoline and δ-lactam. The Kopsia has intriguing biological activities. Additionally other alkaloids were obtained from kopsia such as mersicarpine, pericidine, arboricinine, valparicine, arboflorine, arboricine and arboloscine.111
In 2013, Iwama and co-workers accomplished an extremely significant enantioselective total synthesis of (±)-mersicarpine 148 by using an 8-pot/11-step sequence in 21% chemical yield that initiated from market purchasable 2-ethylcyclohexanone.112 The features of this procedure were FIS, simple admittance to the azepinoindole framework, the short-step and significant synthesis. This method was started with the FIS by utilizing optically active ketoester 149, which synthesized in 99% enantioselectivity based on the procedure by d’Angelo and Desmaële.113 The latter has been reacted with phenylhydrazine 8 in AcOH at 120 °C to provide the desired tetracyclic tetrahydrocarbazole 150 in excellent yield without isolation of the transient tricyclic tetrahydrocarbazole 151. After several steps by using various routes, the corresponding compounds 150 and 151 produced benzyloxycarbonyl (Cbz) carbamate 152, that is as a main intermediate to form (±)-mersicarpine 148. This group tried to examine the reaction conditions of FIS reaction that afforded tricyclic tetrahydrocarbazole 150 without the production of the lactam ring. Relied on the previous observation, using phenylhydrazine hydrochloride in MeOH and in the presence of acetic acid afforded the corresponding tricyclic tetrahydrocarbazole 150, selectively. The effect of acid on the ratio of 150 to 151 was examined. As a result, methanesulfonic acid was found as a suitable acid for the improvement in selectivity of the FIS. More outstandingly, it was found that the product ratio was extremely sensitive to the quantity of acid employed. Therefore, the reaction in the presence of phenylhydrazine 8 and methanesulfonic acid afforded 151 in excellent yield and selectivity. Finally, the (−)-mersicarpine 148 was synthesized in 21% yield (Scheme 30).112
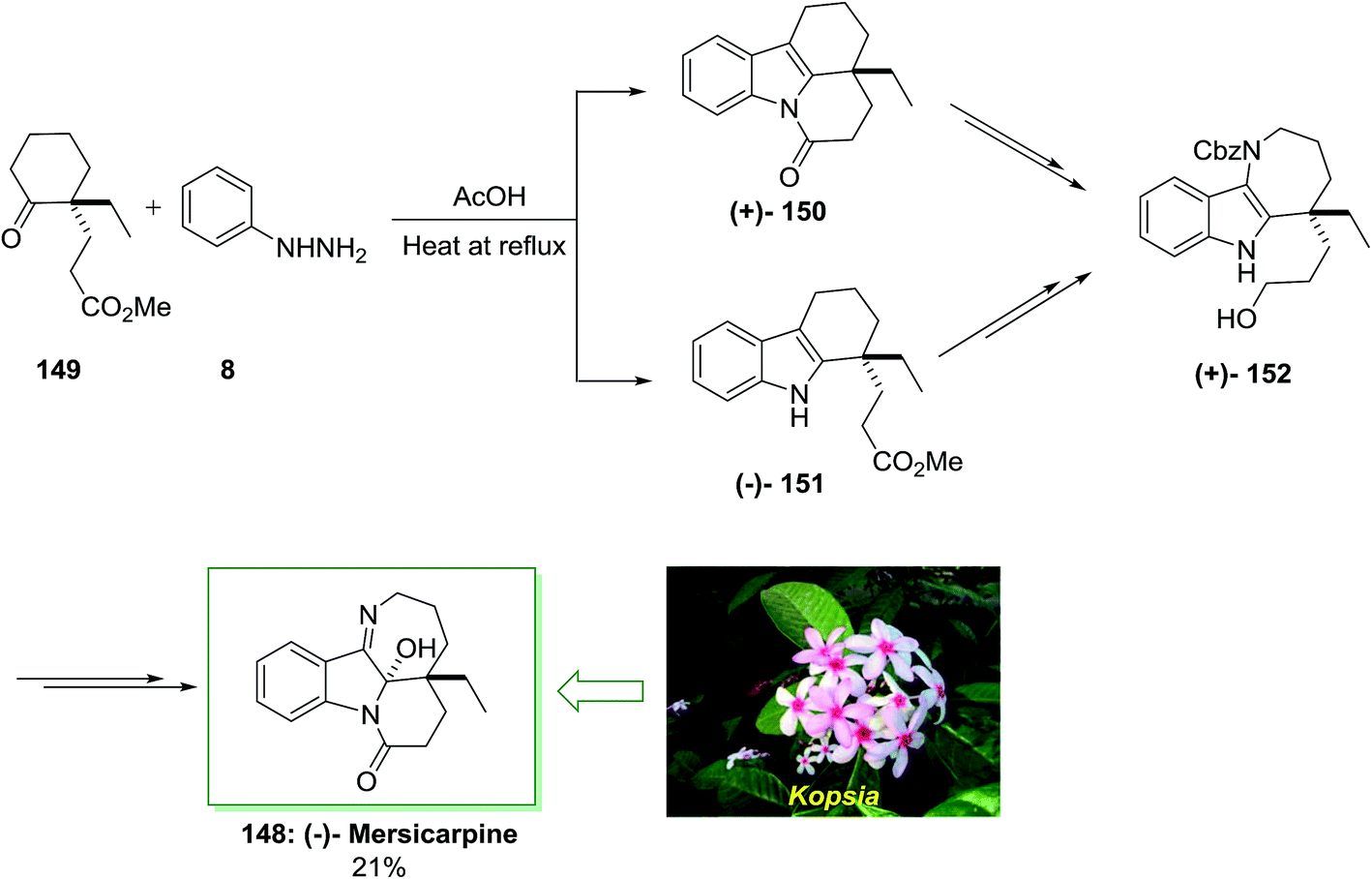 | ||
| Scheme 30 Total synthesis of (−)-mersicarpine 148. | ||
The Aspidosperma alkaloids own a main place in natural chemistry inventions due to their different biological activities and extensive range of compound structural variations. Acid cleavage of haplophytine (a dimeric indole alkaloid was found in the Haplophyton cimicidum's leaves) guided to aspidophytine, a lactonic aspidospermine member of alkaloid that has been recommended to be used in its synthesis and be a biosynthetic pioneer of haplophytine.114
In 2013 Satoh and co-workers described a total synthesis of (−)-aspidophytine 153 and also the initial total synthesis of its congeners, (+)-cimicidine 154 and (+)-cimicine 155, have been achieved in a divergent method. Preparation of the aspidosperma scaffold has been performed via FIS.115 The regiochemistry of the FIS was powerfully reliant on the type of acid, and a weak acid, including AcOH gave the corresponding indolenine isomer in satisfactory selectivity. In this method, total synthesis of (−)-aspidophytine 153 and (+)-cimicidine 154 was initiated from market purchasable ketoester 156, which has been provided from optically active tricyclic aminoketone 138 upon several steps.102 The latter compound 138 treated with functionalized phenylhydrazine 72 via FIS under reflux in benzene to afford hydrazone 157 which has been exposed to different acidic conditions. Finally, this group provided the corresponding indolenine 158 in 48% accompained with 5% of indole 159 once reaction has been completed in AcOH at 100 °C. Pentacyclic indolenine 158 provided (−)-aspidophytine 153 after several steps. Subsequent, they demonstrated the total synthesis of (+)-cimicidine 154 by using the usual intermediate 158 through diverse reactions (Scheme 31).115
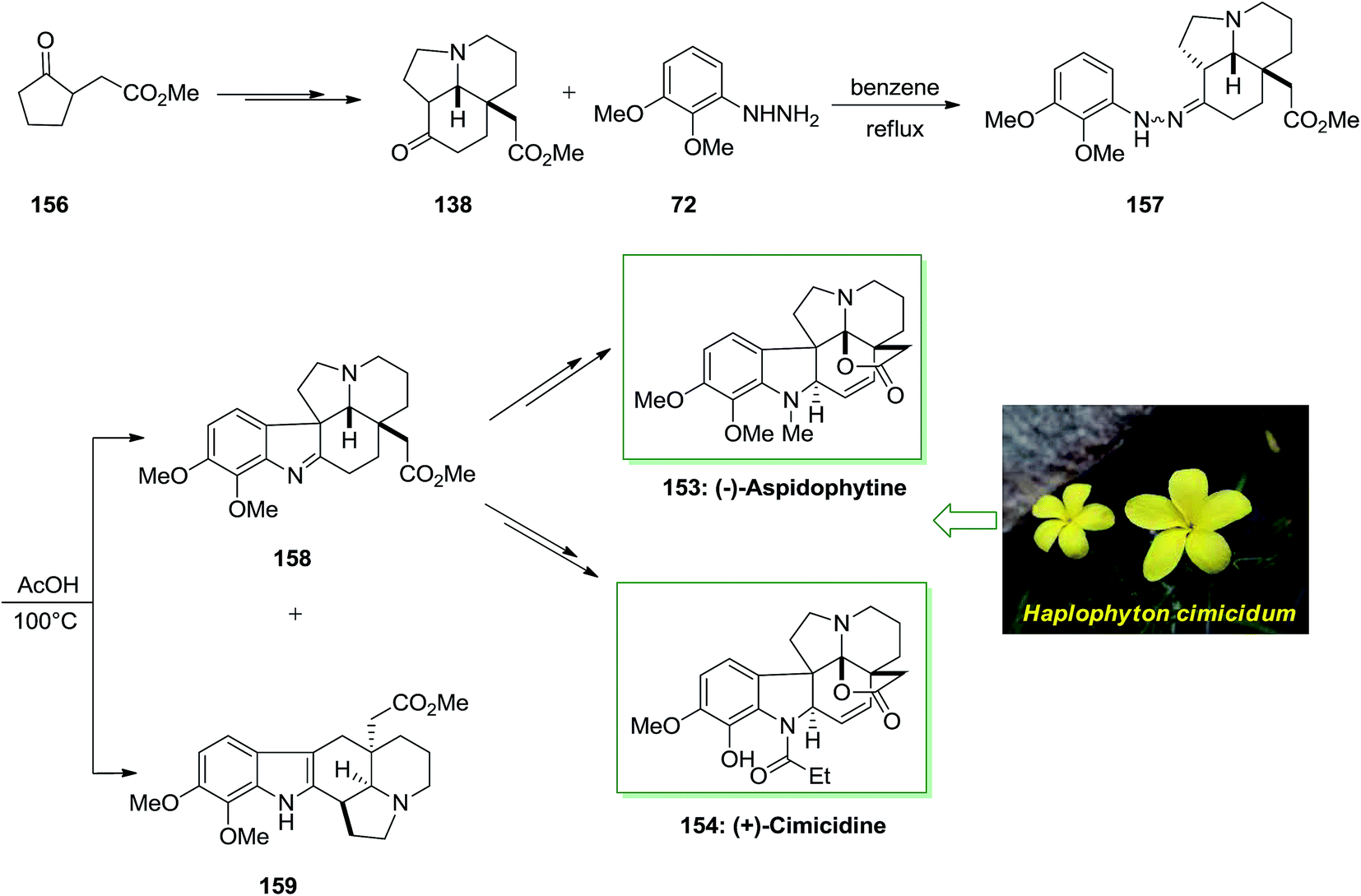 | ||
| Scheme 31 Total synthesis of (−)-aspidophytine 153 and (+)-cimicidine 154. | ||
The efficacy of the convergent synthetic approach has been shown by achievement of the first total synthesis of (+)-cimicine 155. Therefore, FIS through 2-methoxyphenylhydrazine 14 and aminoketone 138 provided hydrazine 160 which provided 41% of the corresponding indolenine compound 161 in satisfactory regioselectivity and 6% of compound 162 Next, upon different steps, imine 161 gave (+)-cimicine 155 (Scheme 32).115
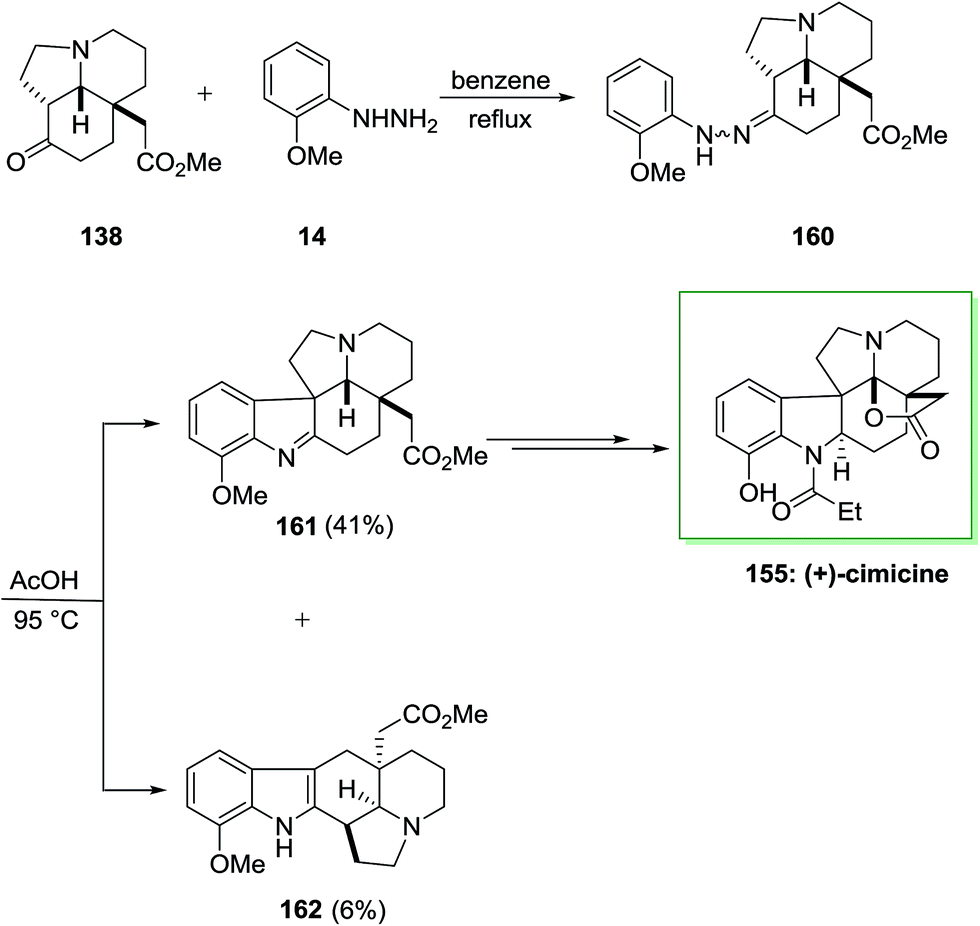 | ||
| Scheme 32 Total synthesis of (+)-cimicine 155. | ||
The efficiency of this synthetic approach for manufacturing extremely substituted aspidosperma alkaloids has been completely exhibited via the divergent synthesis of these three aspidosperma alkaloids from the usual tricyclic aminoketone intermediate. Furthermore, in this research, the regiochemistry of the FIS has been powerfully affected by acidity of acids, which produced the corresponding indolenine isomer in satisfactory selectivity (Scheme 32).115
Akuammilines have an indolenine or indoline core, which is combined to a polycyclic framework. The complicated structures of these molecules have biosynthetic source and biological efficacy that can show activities for combating plasmodial, viral, and cancerous diseases. The alkaloids fraction of alstonia scholarisleaf, vallesamine, picrinine and scholaricine, may make the analgesic and anti-inflammatory effects based on in vivo and in vitro screening. Picrinine is a component of the akuammiline family of alkaloids, which has six stereogenic centers, five of them are contiguous, and includes two N,O-acetal linkages within its polycyclic skeleton. Picrinine 163 reveals anti-inflammatory activity via inhibition of the 5-lipoxygenase enzyme.17 Picrinine was firstly extracted from the leaves of Alstonia scholaris in 1965.116 The plant Alstonia scholaris, also called the Dita Bark tree, has been a rich origin of alkaloids. They can be extracted from its flowers, bark, seeds, leaves, fruitpods and roots. These alkoldes have been used to treat chronic respiratory diseases, for treatment of malaria and dysentery in Southeast Asia, and have been used as traditional medicines to treat various ailments in livestock and humans for the centuries. Even in china it is used for their antitussive and antiasthmatic properties also to release tracheitis and cold symptom. In addition to picrinine and scholaricine, three new indole alkaloids, 5-epi-nareline ethyl ether, nareline ethyl ether and scholarine-V(4)-oxide were separated from the leaf extract of Alstonia scholaris.117
Smith and co-workers described the first total synthesis of the akuammiline alkaloid picrinine 163 in which a main step of their total synthesis was FIS.118 Synthesis of this natural product has been initiated from sulfonamide 164, which is available from market purchasable or can be easily synthesized. Then, sulfonamide 164 upon various steps produced carbonate 165 that was an appropriate initiating compound for FIS. In the critical FI, trifluoroacetic acid stimulated reaction of carbonate 165 and phenylhydrazine 8 gave the hexacyclic indolenine 166 with complete diastereoselectivity. This conversion is identified as one of the most complex cases of the FIS. It should be mentioned that indolenine 166 is in equilibrium with its hydrate, hence, purification and 2D NMR analysis were essential to assist structure clarification. However, upon numerous steps picrinine 163 has been provided. This synthetic method shows short assembly of a main FIS to forge the natural product's carbon scaffold, and a number of delicate late-stage conversions to furnish the total synthesis (Scheme 33).118
 | ||
| Scheme 33 Total synthesis of picrinine 163. | ||
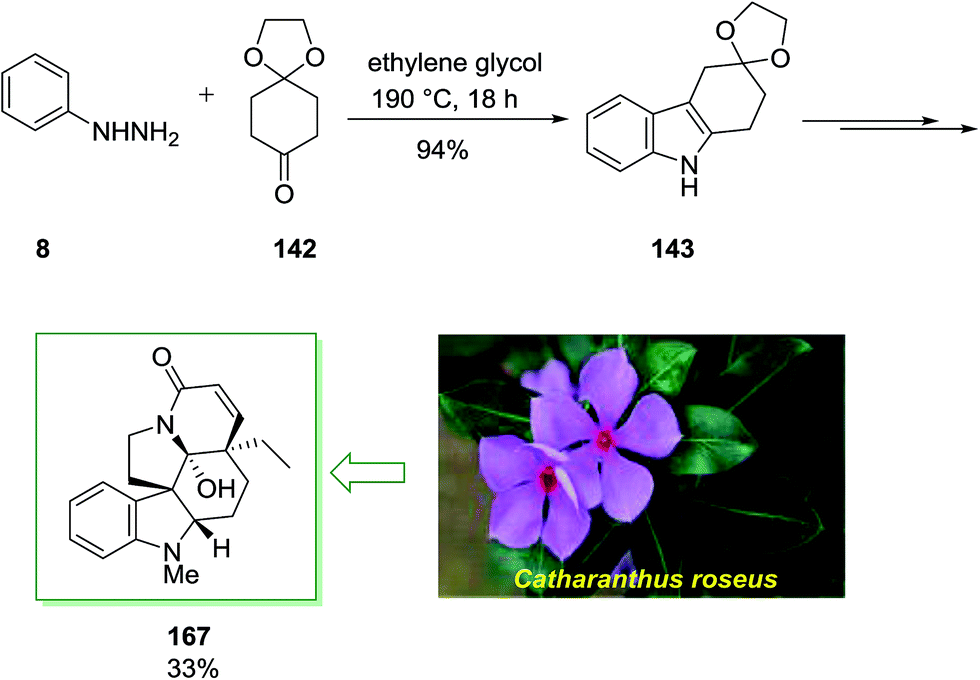
Family of the Vinca class of alkaloid was the subject of synthetic studies with attendant and important biological properties. For example the pentacyclic vindoline 167, which is the main alkaloid, isolated from the plant Catharanthus roseus. Due to its antimitotic properties compound 167 has been recently being used in the clinical treatment of different human cancers.119
Members of the Vinca group of alkaloid have been the topic of extensive synthetic studies. The total synthesis of 167 was initiated from phenylhydrazine 8 and cyclohexane-1,4-dione monoethylene ketal 142 as precursor for the first FIS to provide product 143 (94%). Next, the latter has been transformed into the pentacyclic compound 167 by using different steps in chiral, non-racemic form and in ca. 33% yield (Scheme 34).120
 | ||
| Scheme 34 Total synthesis of Vinca group of alkaloid 167. | ||
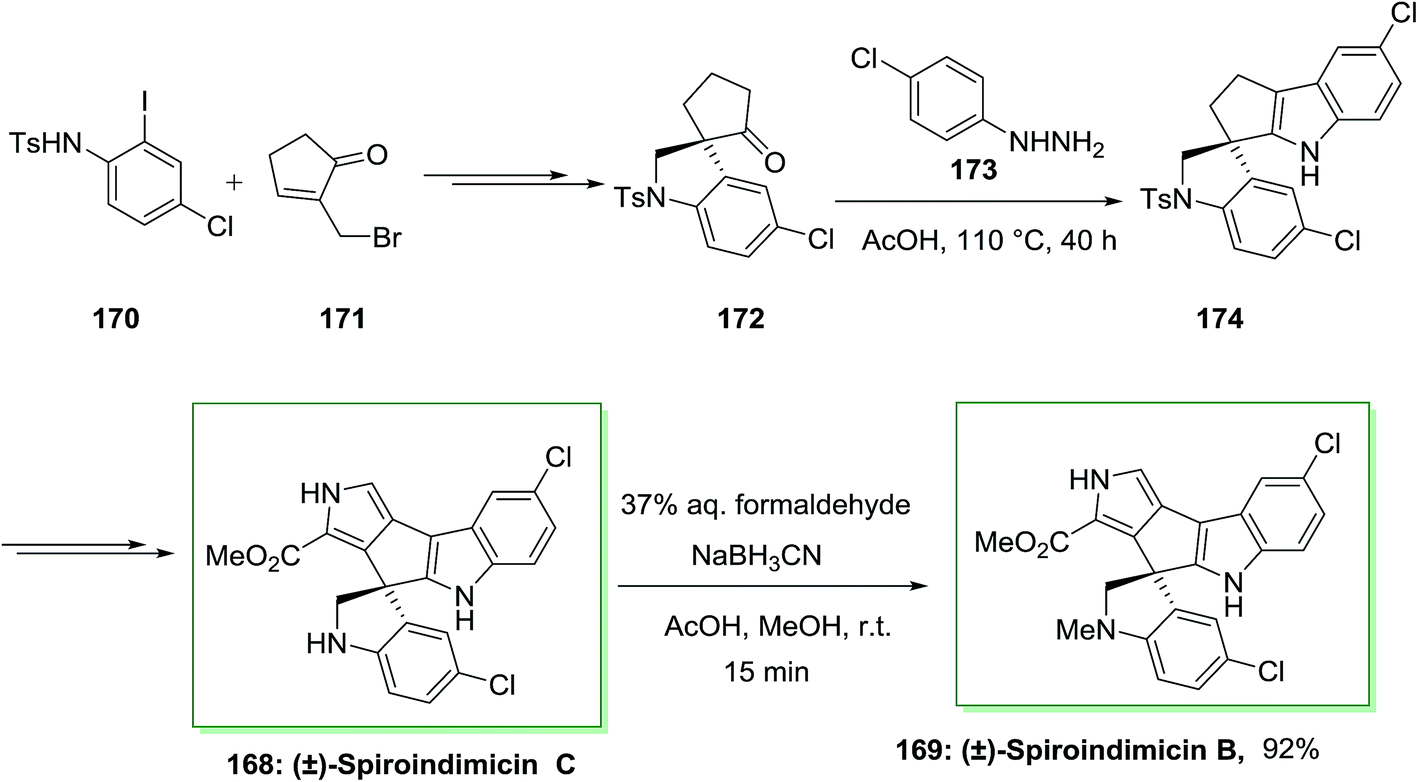
The spiroindimicins are a member of structurally alkaloids extracted from the deep-sea-derived marine actinomycete Streptomyces sp. SCSIO 03032. Marine actinomycete Streptomyces SCSIO 03032 can produce polyketide macrolactam heronamides, α-pyridone antibiotic piericidins, lynamicins and polyketide macrolactam heronamides. Deep-sea organisms have survived under their hard environment by adjusting an extensive range of their metabolic pathways and biochemical processes. The deep-sea-derived marine actinomycete Streptomyces sp. SCSIO 03032, that causes a range of structurally unprecedented natural products such as spiroindimicins A–D, dichlorinated bisindole alkaloids possessing unique heteroaromatic frameworks featuring or spiro-rings. Some analogues of bisindole alkaloids are DNA-topoisomerase I inhibitors and protein kinase inhibitors which have been used in cancer clinical trials.121
Sperry and co-workers in 2016 exhibited effective usage of the FIS to generate a pentacyclic spirobisindole.122 In this synthetic method, firstly, requisite spiroindolinyl pentanone 172 has been produced from the treatment of iodoaniline 170 and bromide 171 in high yield. Then, with the spiroindolinyl pentanone 172, the stage was set for the critical FI reaction. After heating a solution of 172 in hand, and 4-chlorophenylhydrazine in AcOH under reflux, the spirobisindole 174 has been provided in high yield, representing the timeless efficacy of this classic reaction in complex natural product synthesis. Subsequent, the latter transformed into the natural products (±)-spiroindimicin C 168, that after reductive amination produced (±)-spiroindimicin B 169 (Scheme 35).122
 | ||
| Scheme 35 Synthesis of spiroindimicins B 169 and C 168. | ||
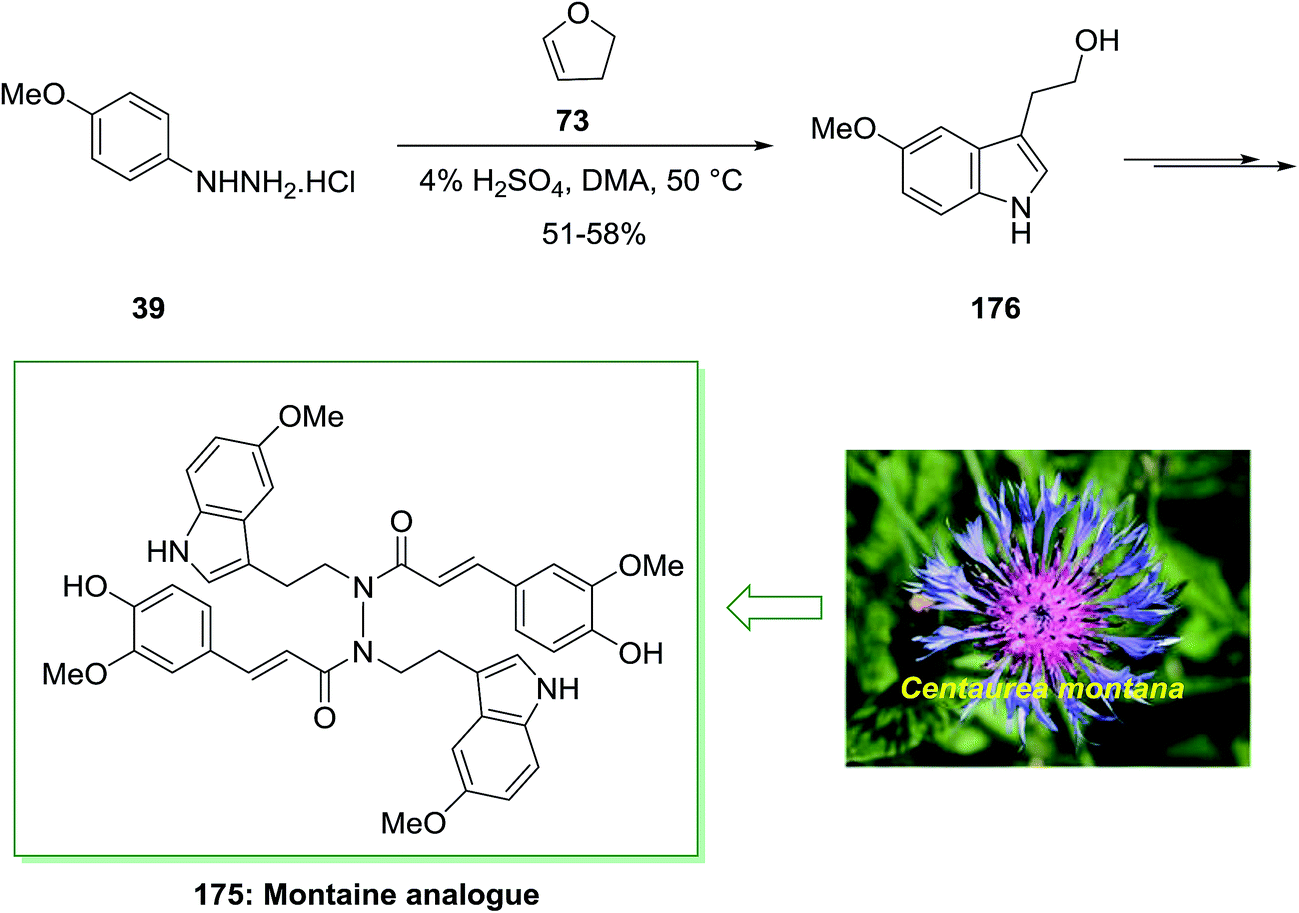
Montamine isolated from the seeds of Centaurea montana. The genus Centaurea have been applied in folk medicine for the treatment of different diseases. Centaurea montana, known as the mountain cornflower, is a native plant in Australia and Europe. Montamine has a unique dimeric N,N′-diacyl hydrazide structure and shows cytotoxicity against CaCo-2 colon cancer cells (IC50 = 43.9l M).123
In 2015, Xu and co-workers described the total synthesis of alkaloid natural product montamine analogue 175 in 55% yield.124 As depicted in Scheme 36, this group provided 3-(2-hydroxyethyl)-5-methoxyindole (usually found as 5-methoxytryptophol, 176) through the FIS with p-methoxyphenylhydrazine hydrochloride 39 and 2,3-dihydrofuran 73. Whereas the yield of this treatment was modest, it nevertheless gave a reasonable, one-step method to an indole suitably substituted having an oxygen group at the 5-position and ethyl alcohol scaffold at the 3-position. Alcohol 176 gave the advanced montaine analogue 175 in several steps (Scheme 36).124
 | ||
| Scheme 36 Total synthesis of montamine analogue 175. | ||
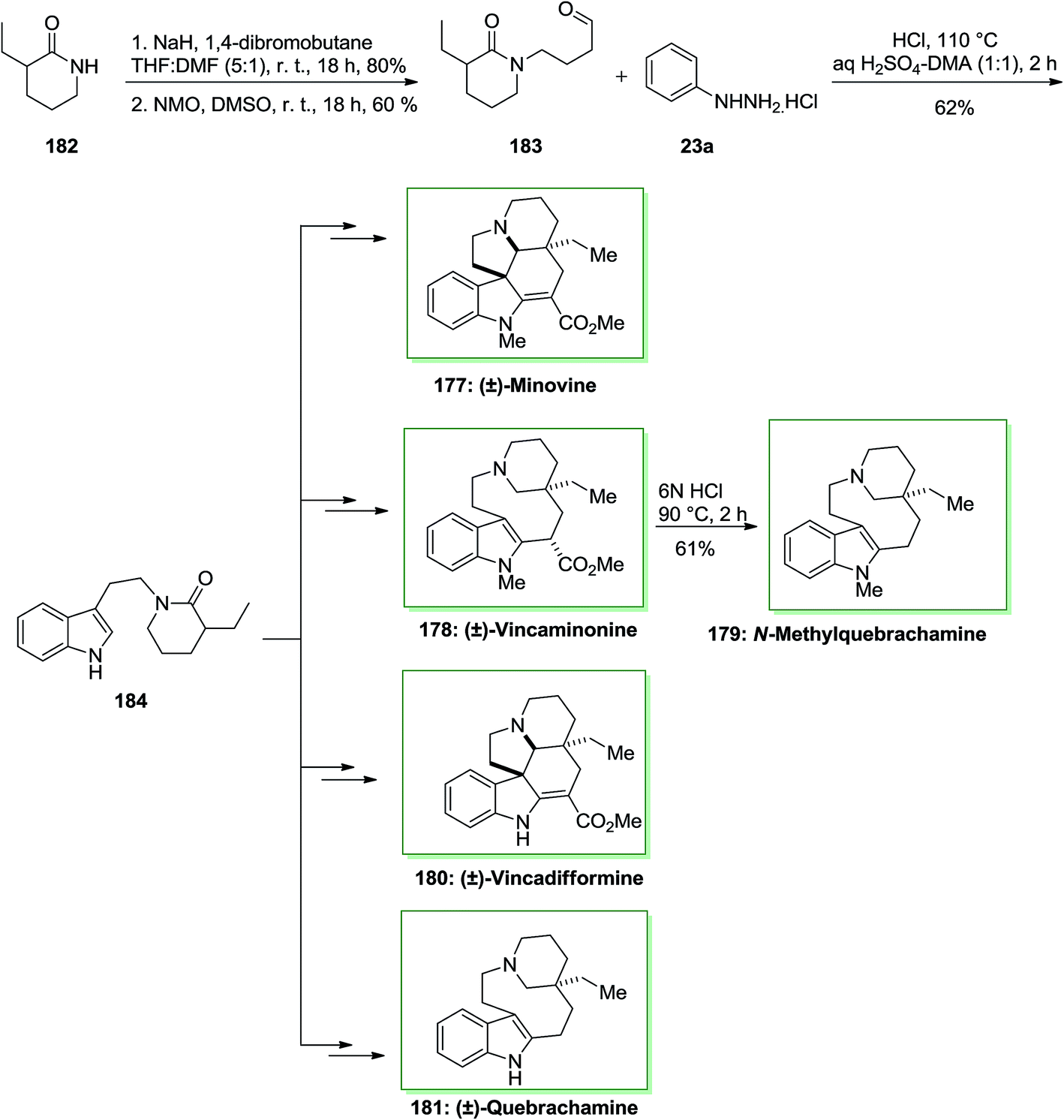
The monoterpene indole alkaloids are an extremely different group of naturally occurring compounds, which were provided in a widespread series of medicinal plants. They have natural structural complication and a number of significant biological properties, which qualifies a figure of them to be principle candidates for anti-arrhythmic, anti-malarial and anti-cancer agents.125 Dixon and co-workers, in 2016, achieved a novel method for the difference total synthesis of (±)-vincadifformine, (±)-vincaminorine, (±)-quebrachamine, (±)-N-methylquebrachamine and (±)-minovine and, each in slighter than 10 linear steps in perfect diastereoselectivities.126
Initially, for the synthesis of (±)-minovine 177, this route was started with the formation of aldehyde 183 from 3-ethyl-2-piperidone 182 upon two steps. The production of the indole functionality continued effectively by Stork's modification of the FIS of aldehyde 183 with phenylhydrazine hydrochloride 23a which produced indole 184 in 62% yield. The latter after different steps produced the desired alkaloid, (±)-minovine 177 in 52% yield. Another skeletally distinct alkaloid, (±)-vincaminorine 178 has been provided as a single diastereomer in the reaction vessel in 31% yield, which could be more transformed into N-methylquebrachamine 179 in 61% yield. Moreover, in another route, indole 184 can produce (±)-vincadifformine 180 and (±)-quebrachamine 181 with 84% and 71% yields, respectively.126 This approach that gives a concise and different synthetic method to various vincadifformine-type, quebrachamine-type and iboga-type alkaloids. Strategically, the novel method shows a key late-stage formation of reactive enamine functionality from stable indole-linked δ-lactam through an extremely chemoselective Ir(I) mediated reduction (Scheme 37).126
 | ||
| Scheme 37 Total synthesis of (±)-minovine 177, (±)-vincaminorine 178, (±)-N-methylquebrachamine 179, (±)-vincadifformine 180 and (±)-quebrachamine 181. | ||
Ergot alkaloids were in the group of the first fungal-derived naturally occurring compounds recognized, inspiring pharmaceutical requests in infective diseases, CNS disorders, cancer and migraine. Aurantioclavine 185 has been first extracted from the fungus Penicillium aurantiovirens in 1981. It has become an striking target for whole synthesis campaigns because of the interesting synthetic challenge presented by the fused azepinoindole nucleus and its jub as a biosynthetic pioneer to the communesin alkaloids, that show cytotoxicity against leukemia cell rules.127
In 2016, total synthesis of (−)-aurantioclavine has been accomplished by Cho and co-workers.128 However, the significant step of this total synthesis was the production of 3,4-fused tricyclic indole derivatives via intramolecular FIS of aryl hydrazides, that contain a carbonyl group including a side chain connected to the meta-position of the aromatic ring. The intramolecular FIS approach does not need cumbersome prefunctionalization, and therefore, it may act to simplify the formation of polycyclic indole alkaloids. The novel approach initiated with market purchasable (S)-β-amino-3-iodo-benzene ethanol 186. The latter after several steps gave aryl hydrazide 187 in 70% yield that exposed to the standard intramolecular FIS conditions and treated to generate the corresponding indole derivatives 188 in 72% yield. The latter underwent several chemical synthetic conversions to make the corresponding (−)-aurantioclavine 185 (Scheme 38).128
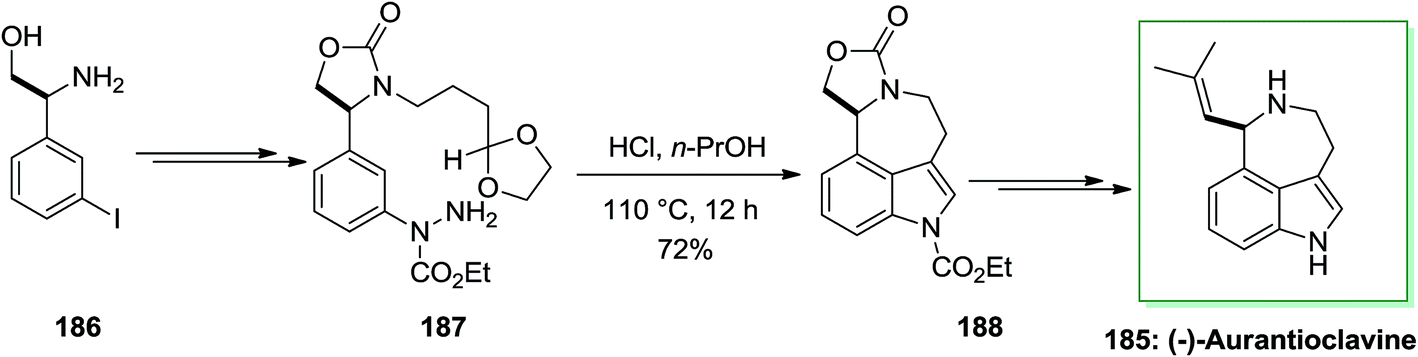 | ||
| Scheme 38 Total synthesis of (−)-aurantioclavine 185. | ||
2.2. Japp–Klingemann reaction
The Japp–Klingemann reaction presents a very useful alternative synthetic route to a number of arylhydrazones, employed in the Fischer indolization process. In 1969, Dutta and co-workers reported the total synthesis of (±)-mahanimbine 189 extracted from the stem bark of Murraya koenigii Spreng.129 This whole synthesis initiated by Japp–Klingemann of 2-hydroxymethylenecyclohexanone 191 and the diazoaryl complex 190, which afforded phenylhydrazone 192. Next, FIS of hydrazone 33 in the presence of acetic acid/hydrochloric acid under reflux gave 7-hydroxy-6-methoxy-2,3,4,9-tetrahydro-1H-carbazole-1-one 193. Then, the latter has been transformed into (±)-mahanimbine 189 in several steps (Scheme 39).129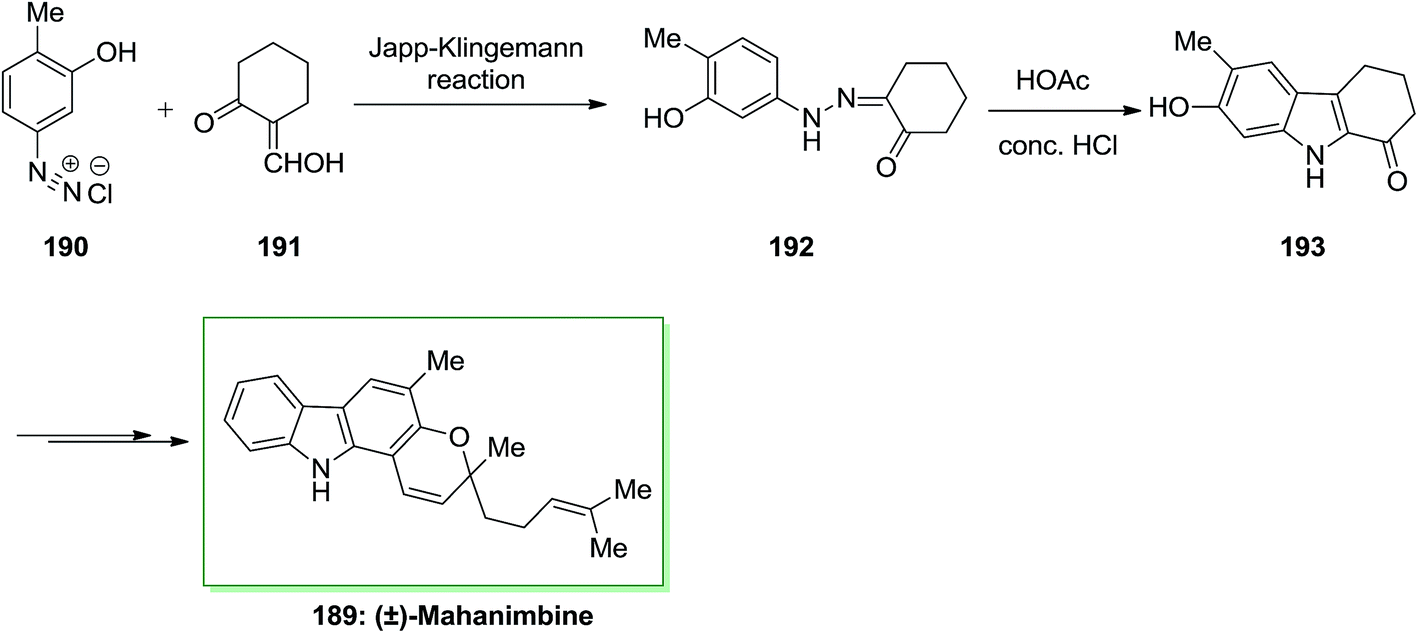 | ||
| Scheme 39 Total synthesis of (±)-mahanimbine 189. | ||
The first naturally occurring carbazole alkaloid extracted from Murraya koenigii Spreng (Rutaceae) was Murrayanine 194. However, later from other species of the genus Clausena and Murraya and displays important biological property. For example, it has exhibited to powerfully prevent the aggregation of platelet, serve as an antifungal and antibacterial agent, and contain cytotoxic property.130
Chakraborty and co-workers reported total synthesis of murrayanine 194 in 1968.131 In this approach, initial Japp–Klingemann reaction between phenyldiazonium chloride 195 and 2-hydroxymethylene-5-methylcyclohexanone 196 led to hydrazone 197. Next, FIS of hydrazone 197 in the presence of acetic acid/hydrochloric acid under reflux afforded 1-oxo-3-methyl-1,2,3,4-tetrahydrocarbazole 198. Finally, the latter was then transformed into the corresponding natural product murrayanine 194 upon different chemical reactions (Scheme 40).131
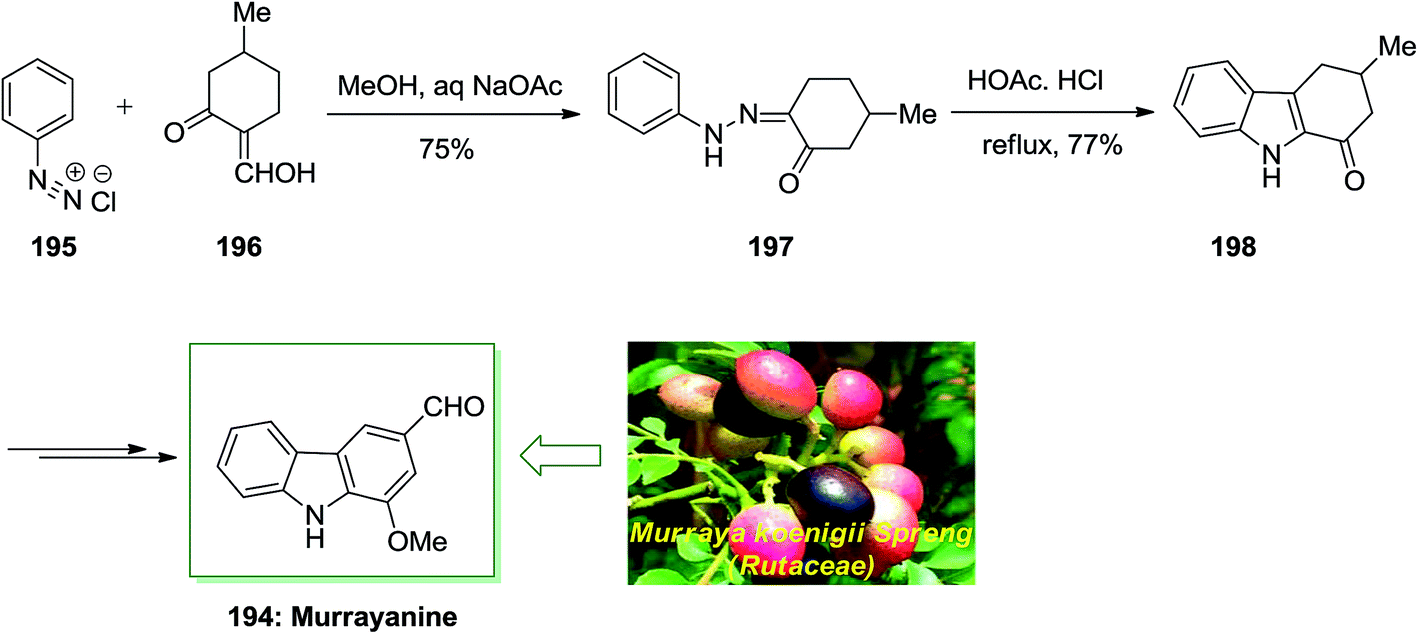 | ||
| Scheme 40 Total Synthesis of murrayanine 194. | ||
Murrayacine 199, that is an alkaloid extracted from Murraya koenigii stem bark and Clausena heptaphylla, is known in spices, herbs and (curryleaf tree).132 The total synthesis of murrayacine 199, has been demonstrated by Chakraborty and co-workers in 1973.133 7-Hydroxy-6-(hydroxymethyl)-2,3,4,9-tetrahydro-1H-carbazol-1-one 202. Lastly, the latter has been transformed to the corresponding murrayacine 199 (Scheme 41).133
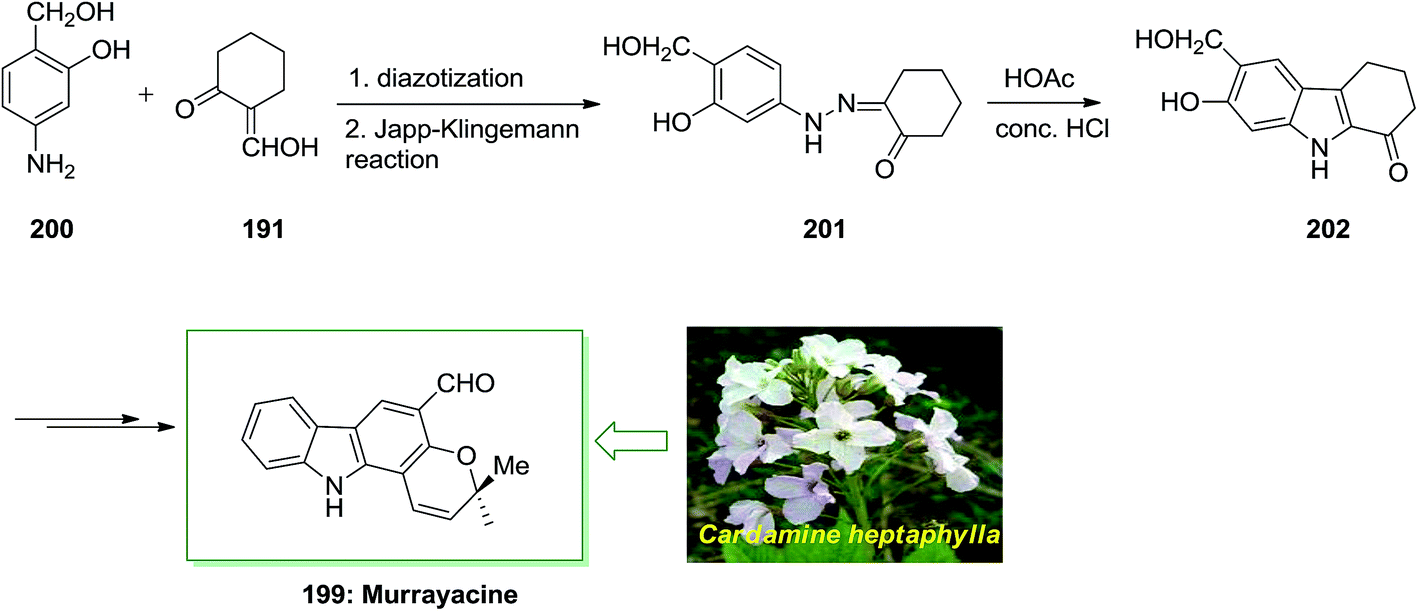 | ||
| Scheme 41 Total synthesis of murrayacine 199. | ||
Carbazole derivatives contain a significant group of heterocycles that are found for their powerful antibacterial, antitumor, anti-inflammatory, antihistamine and psychotropic, activities. Natural 1-oxygenated carbazole alkaloids are principally isolated from the genera Clausena and Murraya species, and in the case of 2- and 3-functionalized classes, their biogenesis were developed.134
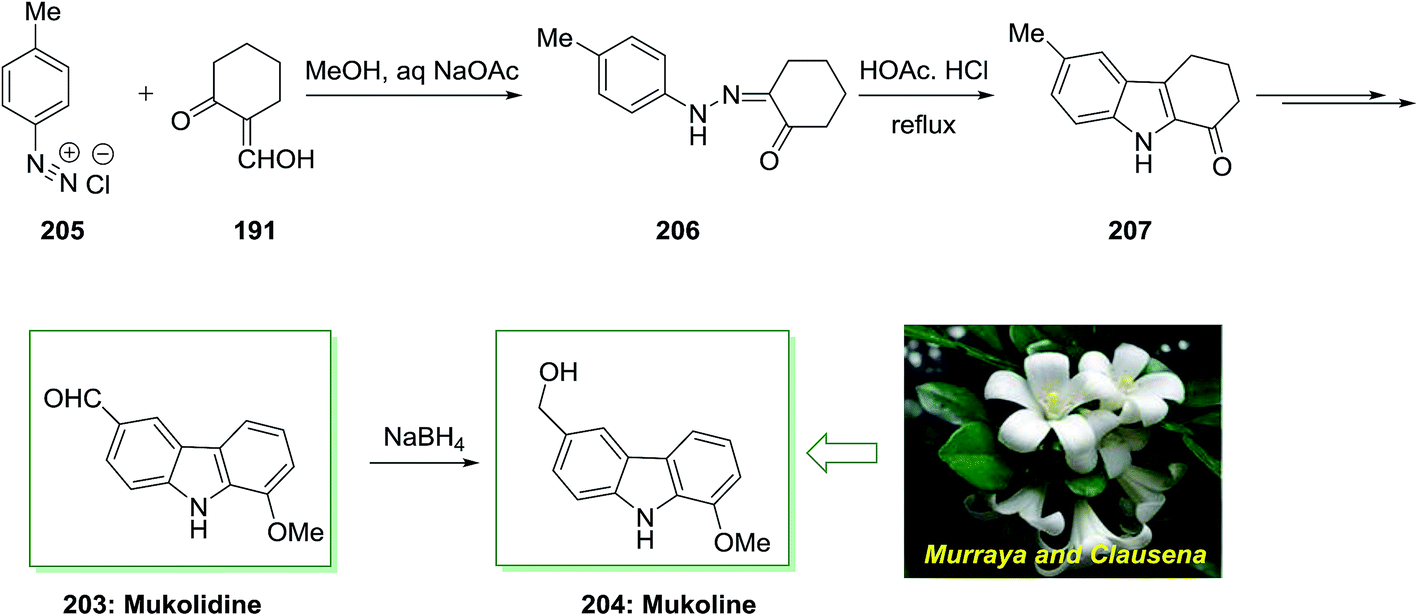
The total synthesis of natural products mukolidine 203 and mukoline 204 has been achieved by Chakraborty and co-workers in 1982.135 As depicted in Scheme 42, this total synthesis initiated through a Japp–Klingemann reaction of toluenediazonium chloride 205 and 2-hydroxymethylenecyclohexanone 191 to give hydrazone 206. Next, FIS of the latter afforded the 1-oxotetrahydrocarbazole 207. Subsequent, the desired 1-oxotetrahydrocarbazole 207 has been transformed into mukolidine 203 that reduced with NaBH4, to provide mukoline 204.135
 | ||
| Scheme 42 Total synthesis of mukolidine 203 and mukoline 204. | ||
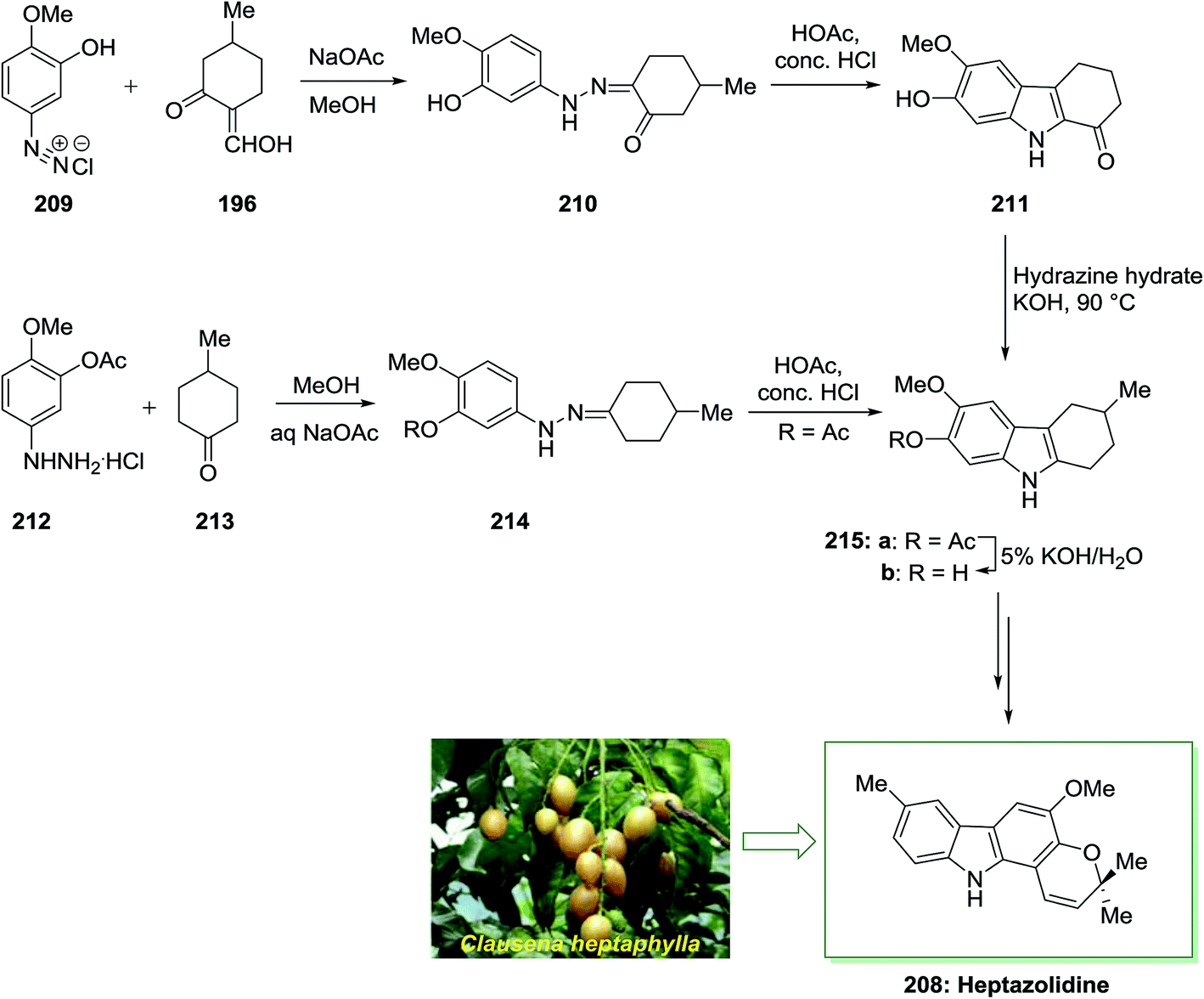
The total synthesis of heptazolidine 208, a carbazole alkaloid extracted from Clausena heptaphylla, has been achieved by Chakraborty and co-workers in 1985.136 In this approach, the main intermediate is 2-hydroxy-3-methoxy-6-methyltetrahydrocarbazole 215b. Initially, diazoaryl derivative 209 reacted with 2-hydroxymethylene-5-methylcyclohexanone 196 via Japp–Klingemann reaction to give the corresponding phenylhydrazone 210. The latter was then subjected into FIS to afford drocarbazole 211, which upon Wolff–Kishner reduction provided the tetrahydrocarbazole 215b. The tetrahydrocarbazole 215b has been produced via reaction between 4-methylcyclohexanone 213 and 3-acetoxy-4-methoxyphenylhydrazine hydrochloride 212 to the phenylhydrazone 214, FI reaction, and ester removal. The latter has been transformed into the corresponding natural product heptazolidine 208 through subjection to various chemical reactions (Scheme 43).136
 | ||
| Scheme 43 Total synthesis of heptazolidine 208. | ||
The carbazole alkaloids exhibit a big and constructional rich group of naturally occurring compounds, which are provided by a range of terrestrial plants. Especially, plants inside the Rutaceae group are remarkable producers of these products, with the genus Murraya affording the maximum figure of distinctive structures. Usually known in the external Himalayas and on the Indian peninsula, the leaves of M. koenigii (L.) Spreng are with other things, broadly used as a spice flavoring and giving it the name “curry-leaf tree” by the people of these areas. Extracts obtained of the plant are employed in local medicine due to their antimicrobial property.137
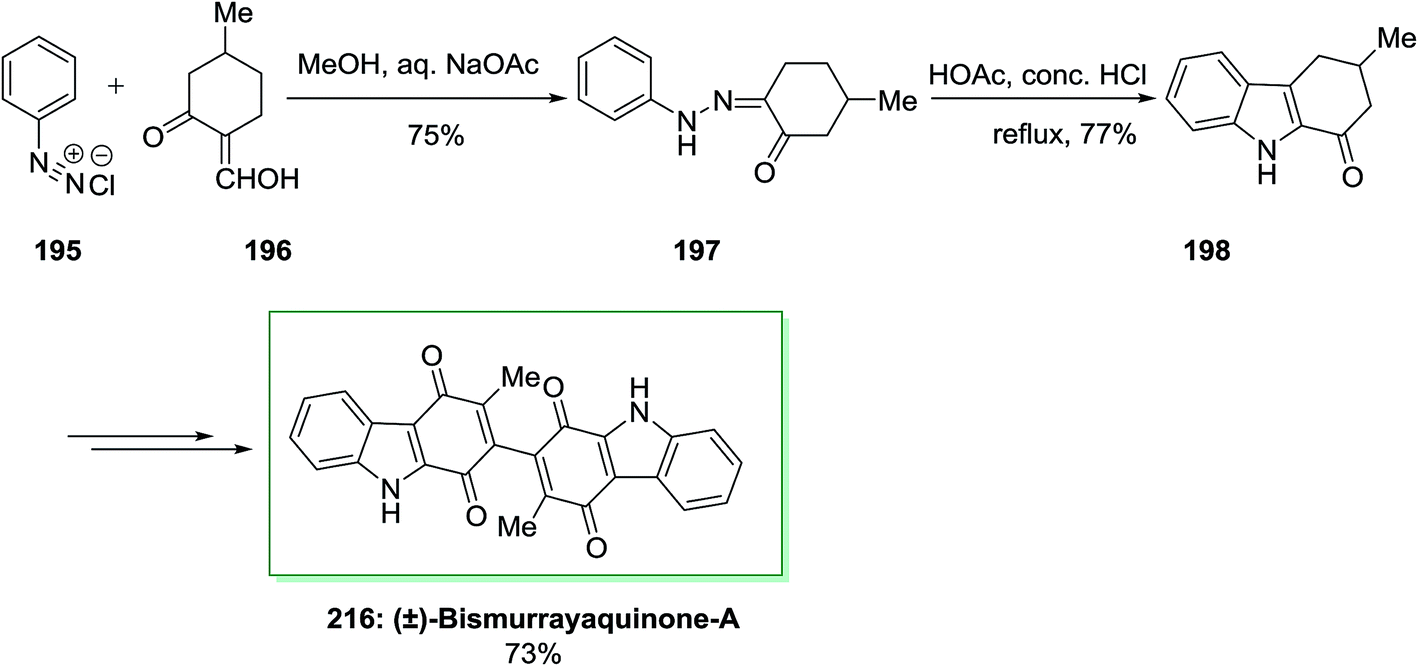
The first total synthesis of a natural dimeric carbazole alkaloid, (±)-bismurrayaquinone-A 216 was demonstrated by Bringmann and co-workers in 1995.138 In this synthetic method, firstly, the desired hydrazone 197 has been produced from phenyldiazonium chloride 195 and 2-hydroxymethylene-5-methylcyclohexanone 196 through Japp–Klingemann reaction. Then, the desired hydrazone 197 has been exposed to FIS by using acetic acid/hydrochloric acid to give 1-oxo-3-methyl-1,2,3,4-tetrahydrocarbazole 198. Subsequent, the latter has been transformed into the desired 216 in 73% yield, after several steps. Through chromatography on a chiral phase, the two enantiomers of (±)-bismurrayaquinone-A 216 have been separated (Scheme 44).138
 | ||
| Scheme 44 Total synthesis of (±)-bismurrayaquinone-A 216. | ||
Indole alkaloids are significant natural products due to their structural association to the important amino acid, tryptophan and the important metabolites of tryptophan, for instance the neurotransmitter serotonin. One class of indole alkaloids was extracted from Alstonia species.139 Tryprostatin A 217 was extracted as secondary metabolites of a marine fungal strain BM939 and depicted to entirely prevent respectively cell cycle progression of tsFT210 cells in the G2/M phase at a final concentration of 50 μg mL−1 of 217. Tryprostatins A 217 include a 2-isoprenyltryptophan scaffold and a proline residue that include the diketopiperazine unit.140
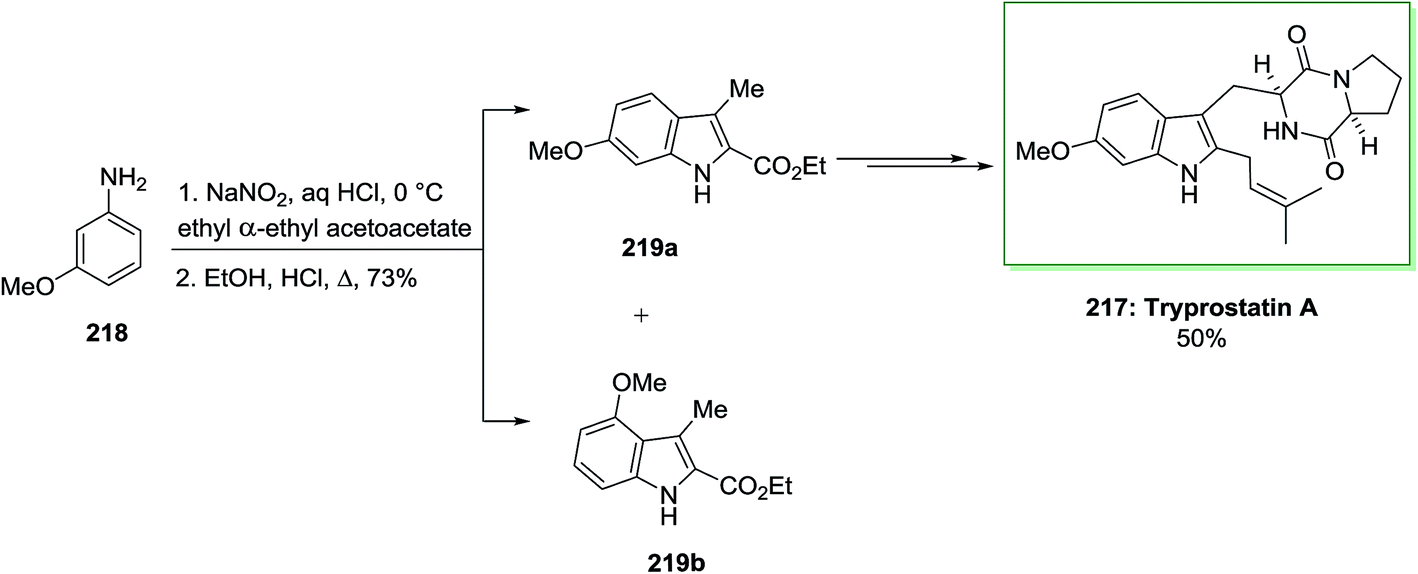
The first stereoselective total synthesis of tryprostatin A 217 has been described by Gan and co-workers in 1997.141 The synthesis was started by using the FI cyclization reaction through a Japp–Klingemann azo-ester intermediate. Once m-anisidine 218 has been reacted with NaNO2 and concentrated aqueous hydrochloric acid, continued by the addition of the anion of ethyl α-ethylacetoacetate, the Japp–Klingemann azo-ester intermediate has been synthesized. Once this intermediate has been heated in a solution of ethanolic hydrochloric acid, a mixture of ethyl 6-methoxy-3-methylindole-2-carboxylate 219a and its 4-methoxy isomer 219b has been provided in a ratio of 10:1. The corresponding 6-methoxyindole isomer 219a has been isolated from the mixture by simple crystallization. While this reaction was exothermic, this approach was far safer to execute on large scale than the Moody azide pyrolysis. Then, compound 219a was transformed into the corresponding indole alkaloid tryprostatin A 217 in 50% yield (Scheme 45).141
 | ||
| Scheme 45 Total synthesis of tryprostatin A 217. | ||
Chowdhury and co-workers extracted two novel carbazole alkaloids planned as koenigine-quinone A and koenigine-quinone B from the alcoholic extract of the stem bark of Murraya koenigii Spreng.142 In 1998, they have demonstrated the structures given for koeniginequinone A 220a and B 220b using a FI of the desired phenylhydrazones 222a,b as the main step. The phenylhydrazone derivatives 222 have been provided via a Japp–Klingemann reaction between 2-hydroxymethylene 5-methylcyclohexanone 196 and the aryldiazonium chloride derivatives 221a and 221b, respectively and transform into the 1-oxotetrahydrocarbazole 223a and 223b in the presence of acid. Next, koeniginequinone A 220a A and 220b B have been produced in 65.5% and 72% yield, respectively, through a multi-step reaction (Scheme 46).142
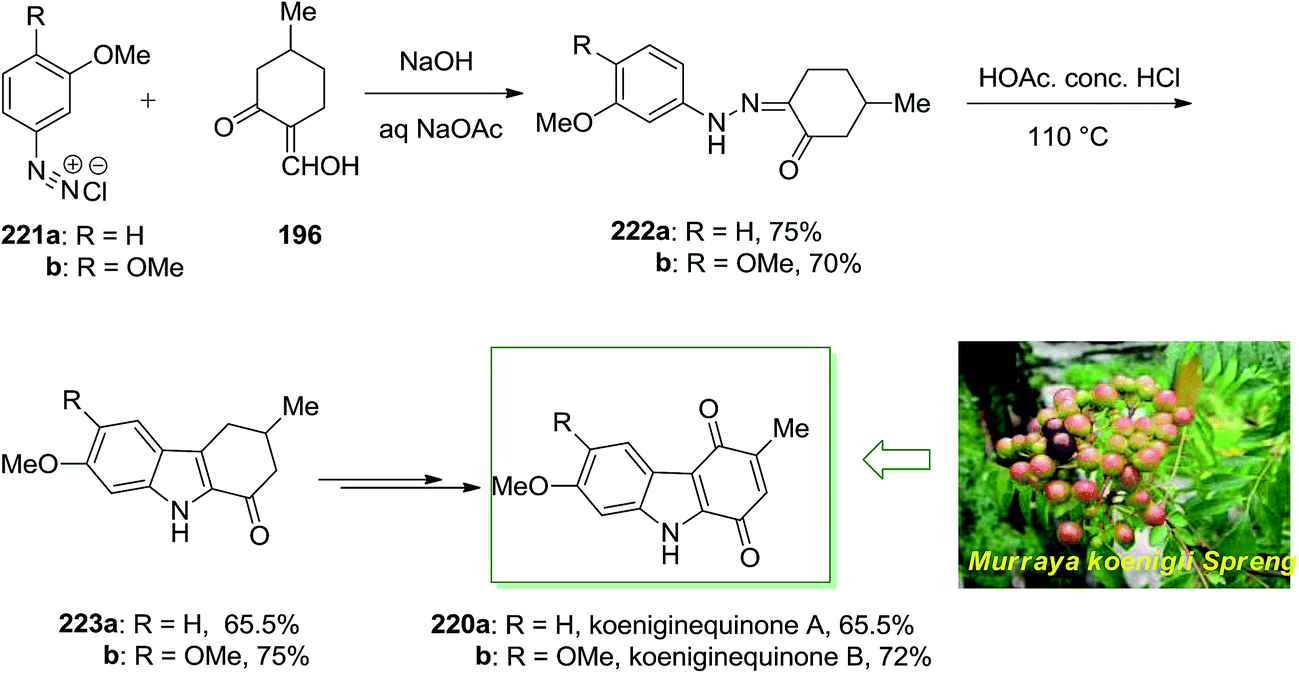 | ||
| Scheme 46 Total synthesis of koeniginequinone A 220a and B 220b. | ||
Melatonin (N-acetyl-5-methoxy tryptamine) 3 (darkness hormone) is an indolamine hormone which is made by the photoreceptor cells of the retina and the pineal gland in vertebrates, additionally it was first isolated in 1958 by Lerner and Case at Yale University.143 They found the light-related features within the skin cells of amphibians. The molecule produced the collection of the pigment melanin inside the melanocytes which is causing the skin to lighten. The hormone is increased at night and has been conducting as a time signal for an organism's annual (circannual) and daily (circadian) biological rhythms. Melatonin regulates sleep/wake patterns and also synchronizes the release of other hormone. Furthermore, melatonin has been displayed medicinally functions as inhibitor of the onset of Alzheimer's disease, treatment of sleep disorders and in protection against oxudative stress.143
In recent years, much was appealed about the therapeutic possessions of the hormone melatonin. This interest has resulted in the of publication of two general scientific books preserving that the hormone can cure the symptoms of some kinds of cancer, acting as hypertensive in case of high blood pressure, treating Alzheimer's disease, AIDS, and coronary heart disease as well as being used as sleep aid, sexual vivacity, and durability. Therefore, making it a phenomenon drug of the 1990s. Melatonin is mostly formed in the pineal gland, a peasized organ placed in the center of the brain, and to a minor extent in the retina. Melatonin is found valuable in some problems for example coronary heart disease and aging.144
The total synthesis of melatonin 3 needed phenylhydrazone 226, which is synthesized through coupling diazotized 4-methoxyaniline 224 and 2-oxopiperidine-3-carboxylic acid 225. This has been continued via FI cyclization reaction to give 6-methoxy-1-oxotetrahydro-β-carboline 227. The latter afforded the corresponding melatonin 3 in several reaction steps in 41% yield (Scheme 47).145
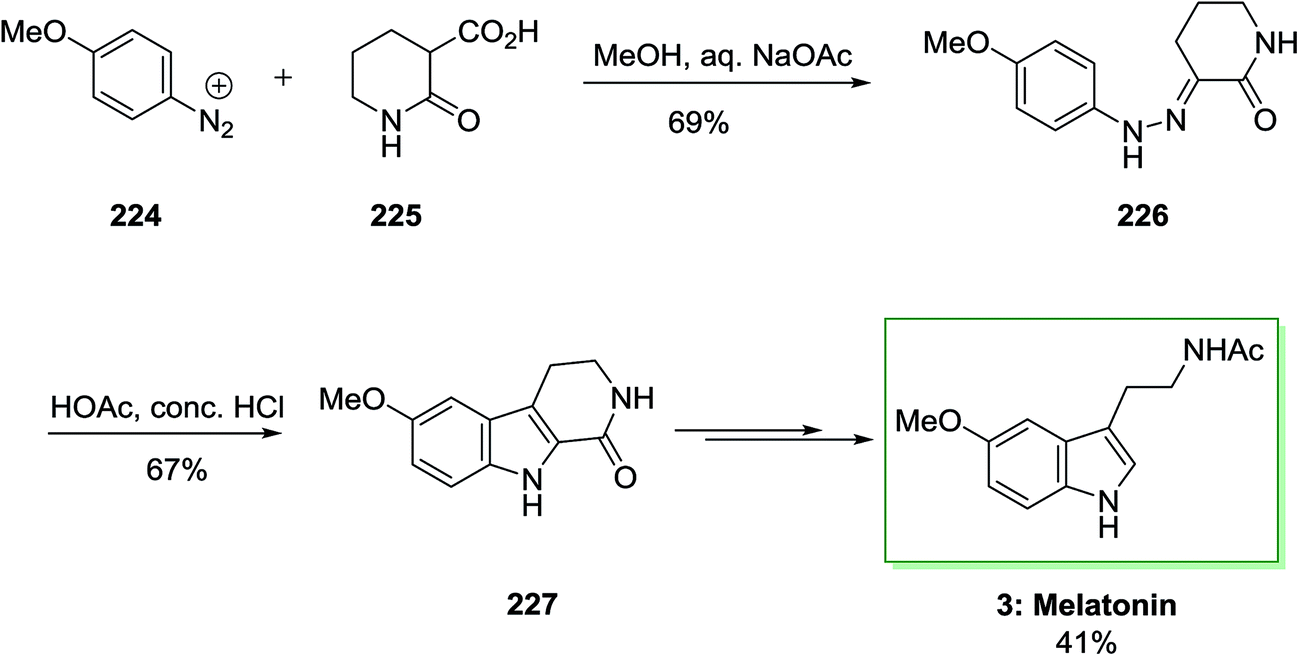 | ||
| Scheme 47 Total synthesis of melatonin 3. | ||
The first asymmetric total synthesis of (−)-gilbertine 228, a member of the uleine type indole alkaloids, has been demonstrated in 2004 by Blechert and Jiricek in a seventeen-steps reaction with a 5.5% yield.146 The uleine alkaloid (−)-gilbertine 228 has been extracted by Miranda and Blechert in 1982 from the Brazilian tree Aspidosperma gilbertii (A. P. Duarte).147 This method was initiated from 2-allylcyclohexenone 229, that could be readily synthesized from o-anisic acid through Birch conditions on a large scale, and dimethylmalonate as a market purchasable starting compound. Upon a multi-step reaction, 2-allylcyclohexenone 229 has been converted into compound 230, which was an appropriate precursor for FIS. That products have been isolated from the FIS depended powerfully on both the solvent and the pKa of the acid promoter. Pivalic acid afforded no transformation, and formic acid reaction led to deprotection of the alcohol and production of the formylester 231b, while trifluoracetic acid afforded the deprotected hydroxyindole 231a. Also, p-TsOH in tetrahydrofuran afforded only decomposition; although, in toluene the corresponding indole 231c could be provided in 72% yield. The Japp–Klingemann FI procedure has been utilized effectively as a convergent synthetic method for the formation of the corresponding tetrahydrocarbazole 231c. Then, the latter has been transformed into the corresponding natural product (−)-gilbertine 228 (Scheme 48).147
 | ||
| Scheme 48 Total synthesis of (−)-gilbertine 228. | ||
3. Miscellaneous
Another approach for the synthesis of ellipticine 232 has been advanced by Stillwell and Woodward with the key B-ring production through FIS.73,79 In this synthetic method, firstly, the requisite phenylhydrazone derivative 235 has been synthesized from the reaction of (Z)-pent-3-en-2-one 233 and 1-methyl-4-piperidone 234 and next exposed to FI reaction to give 2-methyl-1,2,3,4,5,5a,11,11a-octahydroellipticine 236 (82%). Lastly, dehydrogenation by using Pd on carbon afforded ellipticine 232 in a very poor yield of 0.3% (Scheme 49).62,66 | ||
| Scheme 49 Total synthesis of ellipticine 232. | ||
Alkaloids, for example manzamines and lamellarins, having diversely structures and significant biological properties provided by marine plants, microbes and invertebrates importantly encouraged interdisciplinary studies by biologists and chemists worldwide. Among the marine alkaloids, the structurally and discorhabdins associated alkaloids are a special group of nitrogenous pigments belonging to the pyrroloiminoquinone-kind alkaloids family. This class of naturally occurring compounds contains a typical nucleus pyrrolo[2,3,4-d,e]quinoline tetracyclic framework linked to a spiro-substituent at the C-6 location.148
The pyrroloquinoline alkaloids, found as the discorhabdins, are known in the sponges of the genus Latrunculia du Bocage along the New Zealand coast. These quinonimine alkaloids are responsible for the pigmentation possessed by the sponges, and several of the compounds in this group, together with the structurally related makaluvamines, prianosins, and epinardins, show antitumor activity.149
In 1997, Heathcock and co-workers revealed the approach relied on the FIS to make 7-hydroxy-indole derivatives in satisfactory yield.150 The unusual FIS is a common problem once an ortho group is current on the phenylhydrazone. Mixtures of indole derivatives are constantly provided, and yields of the corresponding indole are commonly low. They could evade this problem by using constrain the hydrazine that it could merely endure electrocyclization in the expected way. Such a limitation would result if the oxygen and nitrogen atoms could be connected by a short tether. Finally, this group examined the formation and usage of an appropriately-substituted benzoxazine. The requisite phenylhydrazine 242 has been formed in four steps from 2-amino-4-nitrophenol 241 in 60% yield. Next, the reaction with 4-benzyloxybutanal 243 gave hydrazone 244 in 93% yield. There are several formerly found instances of employing hydrazones of 4-aminobenzoxazines identical to hydrazone 244 in the FI reaction. In each of these cases, two carbon tether was an essential structural part in the desired compound. In this approach, they explored to employ the tether to suppress the unusual FI reaction, then eliminated it when the indole had been synthesized. The similar approach has been employed to form other indole derivatives containing different groups on the benzene ring. In each situation, no proof of contaminating indole products has been realized. Since the dimethylene chain is easily provided and then cleaved, this “tether” approach may show to be an important approach for overpowering the unusual FI reaction. Next, after several steps, indole 245 afforded the pyrrolo[2,3,4-d,e]quinoline core 246, that is a structural part known in a range of naturally occurring compounds involving the discorhabdin alkaloids, the isobatzellines, and wakayin, batzellines, the makaluvamines, damir ones A and B, and terrestrially-obtained haematopodin.150 The latter after several steps afforded discorhabdins C 237, E 238, D 239 and the dethia analogue 240 (Scheme 50).149
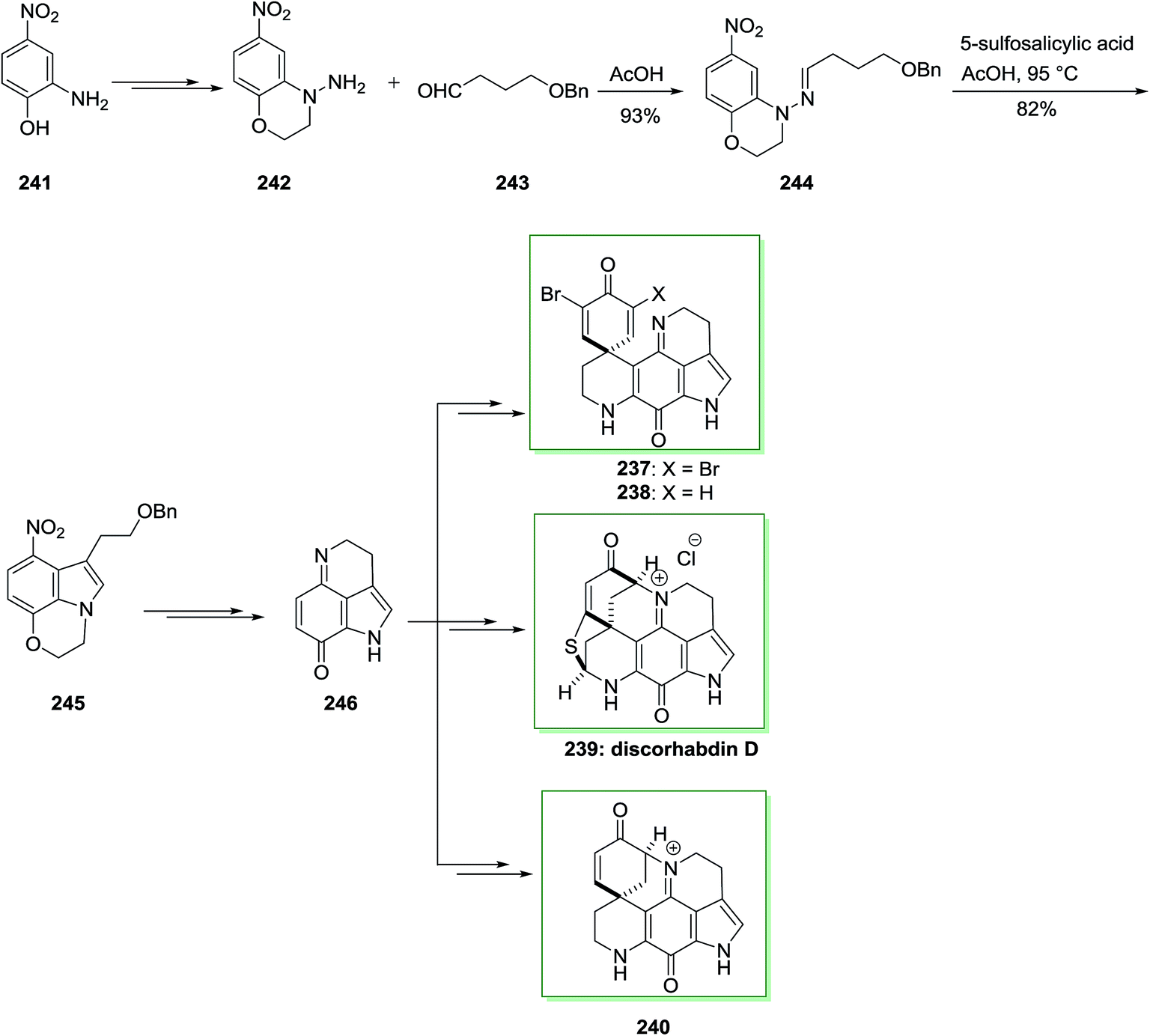 | ||
| Scheme 50 Total synthesis of discorhabdins C 237, E 238, D 239 and the dethia analogue 240. | ||
Eudistomidin-A is an alkaloid separated from Okinawan tunicate, Eudistoma glaucus, which has a calmodulin antagonistic effect. Recently, calmodulin antagonists have been effective as device for investigating physiological activities of calmodulin, a ubiquitous Ca2+-binding protein that acts as a main mediator regulating cellular function and a difference of cellular enzyme system.151
In 1998, the initial total synthesis of the marine alkaloid eudistomidin-A 247 in 72% yield presented a FIS as a key step has been described by Murakami and co-workers.152,153 This synthetic method was started from 2-amino-5-bromophenol 248 that has been transformed into ethyl pyruvate 2-(4-bromo-2-tosyloxyphenyl)hydrazone 249. FIS of the hydrazone 249 has effectively occurred with polyphosphoric acid (PPA) to give the desired ethyl 5-bromo-7-tosyloxyindole-2-carboxylate 250 as the only separable product (41% yield). The latter afforded eudistomidin-A 247 after several steps (Scheme 51).152,153
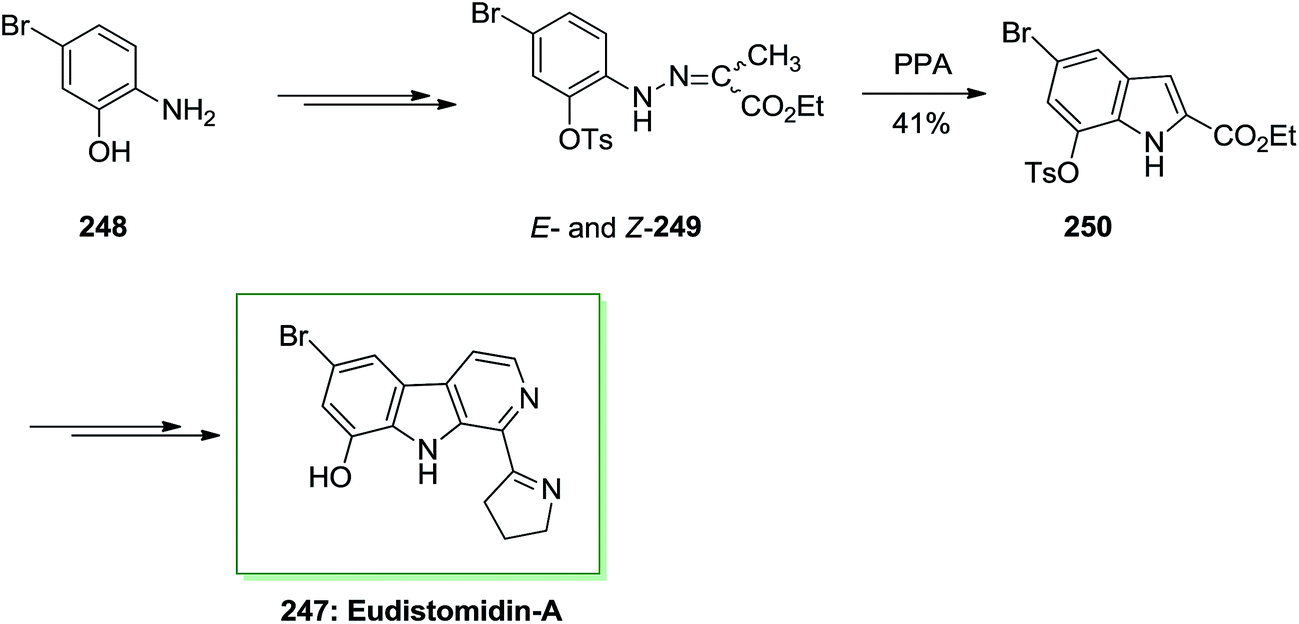 | ||
| Scheme 51 Total synthesis of eudistomidin-A 247. | ||
seco-Duocarmycins show a great guarantee as ultrapotent cytotoxins but due to poverty of therapeutic index, they are unsuccessful in advance clinical. seco-Duocarmycins include spirocyclization of a deep-embedded chloromethylindoline fragment to prompt production of an N3-adenine covalent. This spiracyclization can be stopped by blocking the seco-duocarmycins OH group.154 The natural antibiotic (+)-duocarmycin SA 251 is a powerful cytostatic agent (Scheme 52). (+)-Duocarmycin SA with an IC50 of 10 pM (cancer line L1210) shows high cytotoxicity power, which is candidate for cancer treatment. The cytotoxicity of 251, the biologically and structurally of related (+)-CC-1065 are created by an alkylation of N-3 of adenine in AT-rich parts of the minor groove of the DNA by reaction with the spiro-[cyclopropane-cyclohexadienone] moiety in 251 and (+)-CC-1065. Duocarmycin SA 251 looks better than CC-1065 because it hasn't deadly hepatotoxic side effect which is appeared with (+)-CC-1065. Besides (+)-duocarmycin SA 251 is a strongest one in cytotoxic potency and solvolytic stability and the most stable part is this class agents additionally glycosylated seco analogues of duocarmycin and CC-1065 are extremely encouraging for the critical cancer's treatment in an antibody-directed enzyme prodrug therapy.155
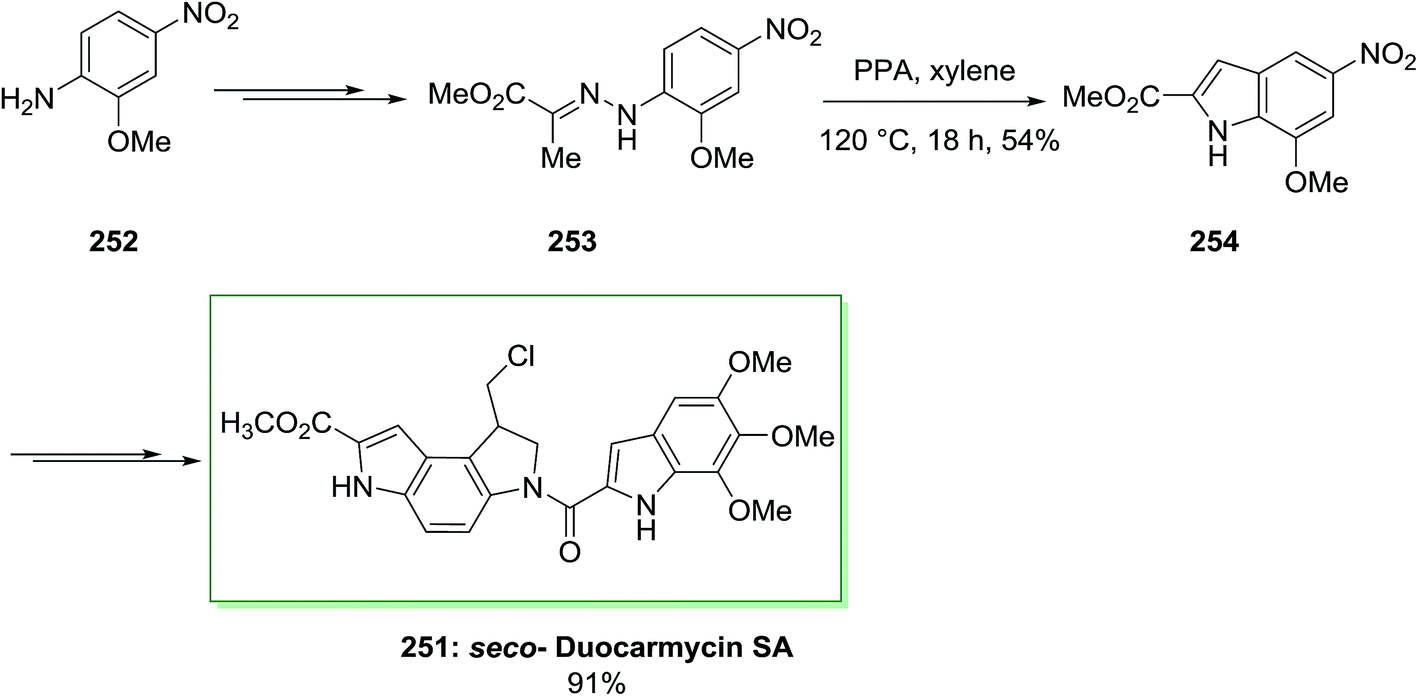 | ||
| Scheme 52 Total synthesis of seco-duocarmycin SA 251. | ||
A concise and significant synthesis of seco-duocarmycin SA 251, an extremely strong cytostatic agent and direct precursor of the natural product duocarmycin SA 251, was accomplished in 2003 by Tietze and co-workers.155 The synthetic procedure includes a FIS to show the heterocyclic moiety as a main reaction. The total synthesis of seco-duocarmycin SA 251 was initiated by diazotation of market purchasable 2-methoxy-4-nitroaniline 251, which upon two steps produced the hydrazone 253 extremely easily with an overall yield of 69%. Then, FI reaction happened by heating at 120 °C by using of polyphosphoric acid and xylene as co-solvent to provide methyl 7-methoxy-5-nitro-1H-indole-2-carboxylate 254 in 64% yield. In the following, upon several steps seco-duocarymcin SA 251 has been synthesized in 91% yield (Scheme 52).155
Such as Evodia officinalis and Evodia rutaecarpa, rutaecarpine 255 is the major of indoloquinazoline alkaloid, which is extracted from Rutaceous plants. Traditional medicinal has long used this plant for treatment of inflammation-related symptoms. Studies currently shows this anti-inflammatory function is related to its component rutaecarpine, exhibiting a selective and powerful COX-2 inhibited activity. Furthermore, rutaecarpine has other functions such as the analgesic, antianoxic, vasorelaxing, cytotoxic and antiplatelet.156
A wide range of quinazolinone alkaloids were extracted from numerous animals, plants, and microorganisms and formed because of their well-developed pharmacological properties. The dried fruits of Evodia rutaecarpa were employed in traditional Chinese medicine under the name Wu-Chu-ru and Shih-Hu as a treatment for cholera, dysentery, headache, postpartum and worm infections. The extracted drug includes quinazolinocarboline alkaloid rutaecarpine 255.156 In year 2004, the total synthesis of rutaecarpine 255 has been accomplished in several steps through FIS as a main step by Argade and co-workers.157 The treatment of anthranilamide 256 and glutaric anhydride 257 in benzene/1,4-dioxane (2:1) at ambient temperature gave the desired o-amidoglutaranilic acid 258 in quantitative yield. Upon various steps, 6-phenylhydrazono-6,7,8,9-tetrahydro-11H-pyrido-[2,1-b]quinazolin-11-one 259 has been produced in 98% yield from 258. The corresponding hydrazone 259 on zeolite (H-Mordenite) through FIS under reflux in glacial AcOH afforded the biologically active natural product rutaecarpine 255 in 82% yield (Scheme 53).157
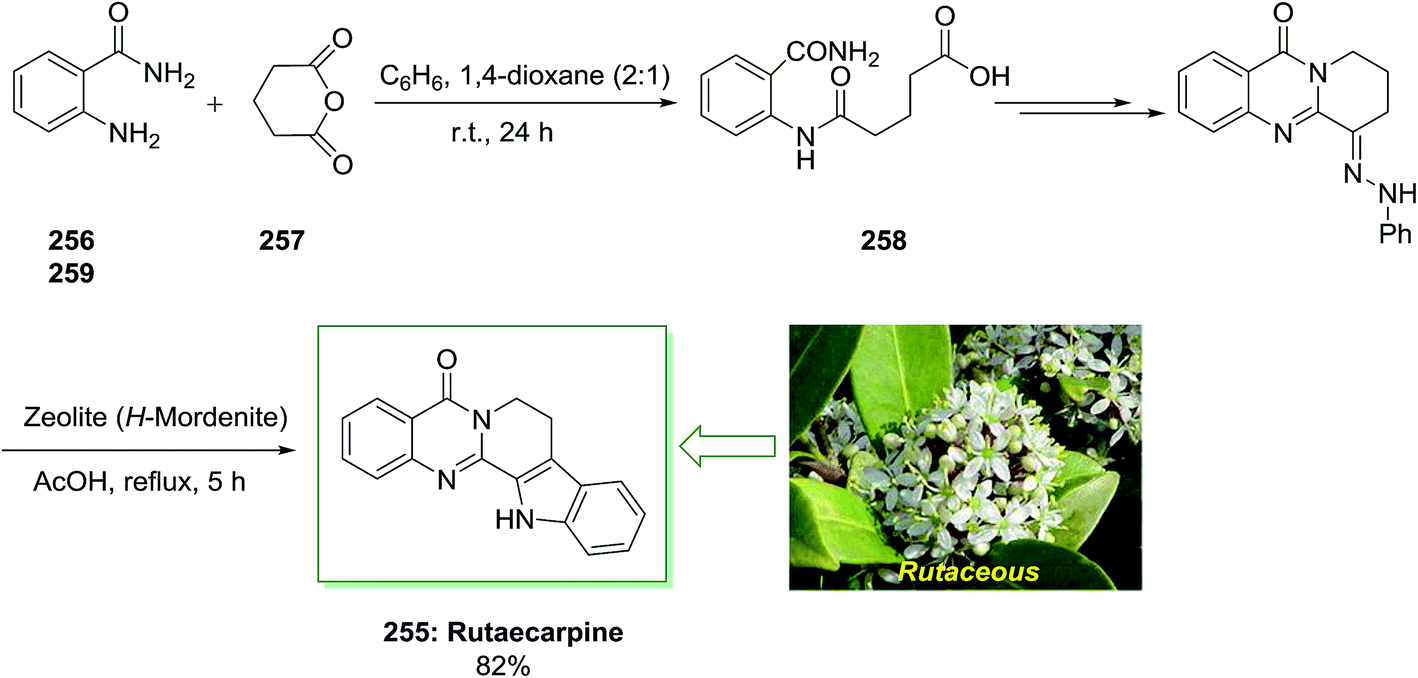 | ||
| Scheme 53 Total synthesis of rutaecarpine 255. | ||
Thiopeptide antibiotics are extremely strong and structurally complex secondary metabolites from soil bacteria (Actinomycetes), and they are provided by ribosomal peptide biosynthesis.158 The antibiotic nosiheptide 260 (RP9671) has been initially extracted by French and co-workers in the early 1960s from Streptomyces actuous 40037.159 Nosiheptide, that is equal to multhiomycin extracted from Streptomyces antibioticus 8446CC, is a part of the thiopeptide antibiotics. They have been topic of comprehensive biosynthetic studies. Some thiopeptides have known biological property against anaerobes and Gram-positive bacteria, involving pathogens resistant to antibiotics.
The total synthesis of nosiheptide 260 was achieved by Bentley and co-workers and reported in 2004. This approach has been accomplished through a FIS as a main step.160 The starting hydrazine was synthesized from the market purchasable 3-amino-4-chlorobenzoic acid 261 via diazotisation and reduction with SnCl2, and has been instantaneously reacted with methyl 2-oxobutanoate to provide the hydrazone 262. In the following, FI cyclisation reaction of hydrazone 262 by using PPA in AcOH produced the indole 263 in 87% yield. The corresponding indole 263 has been transformed into the nosiheptide 260 via several chemical transformations (Scheme 54).160
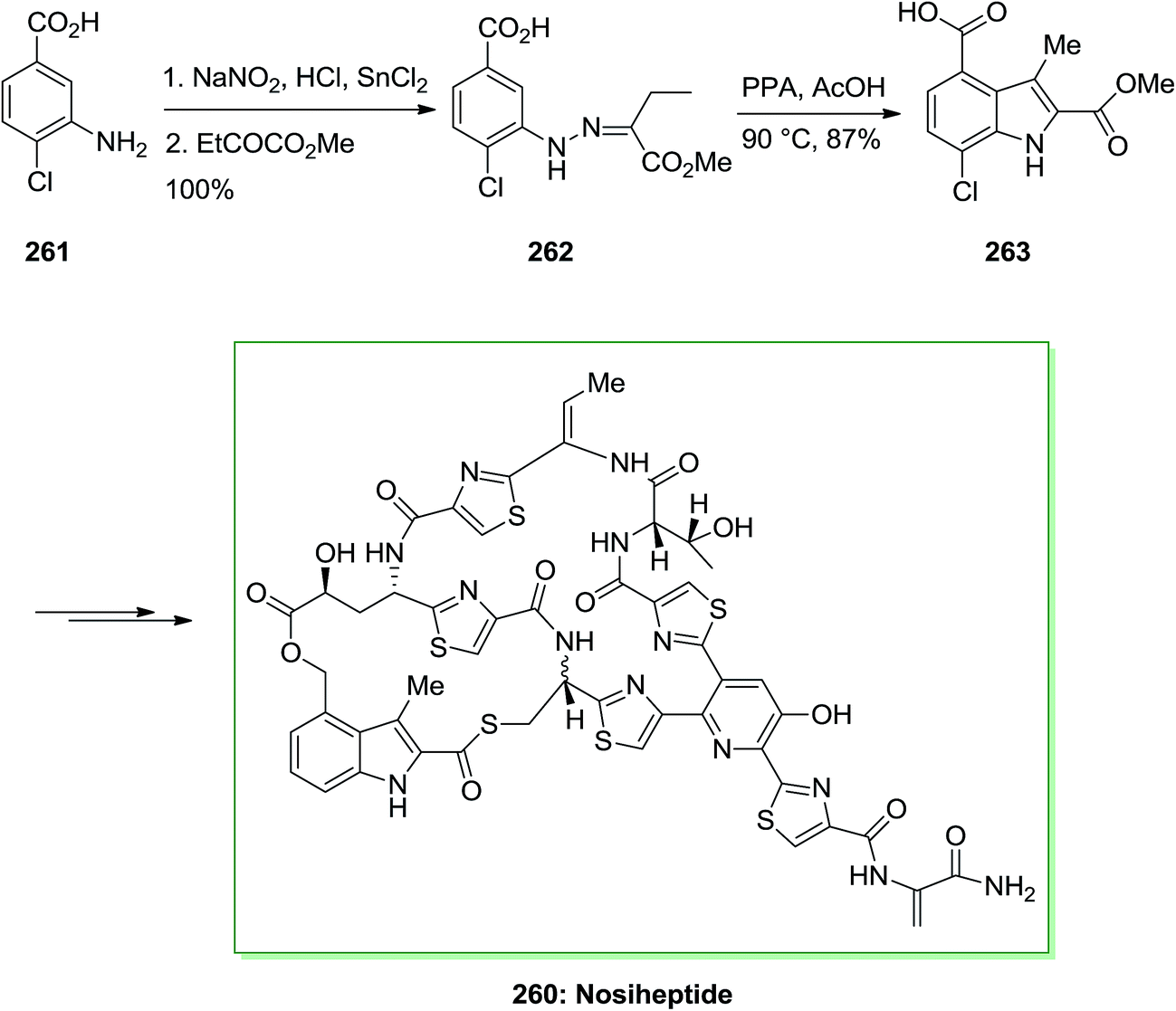 | ||
| Scheme 54 Total synthesis of nosiheptide 206. | ||
Sempervirine 264, an alkaloid isolated from the roots of Gelsemium sempervirens, in 1916 is known as an antiproliferative agent both in vitro and in vivo.161 Earlier in a high throughput screening (HTS) campaign of natural products, sempervirine was discovered as a MDM2 E3 ubiquitin ligase inhibitor. Sempervirine is known to stabilize p53 tumor suppressor protein levels by blocking its proteasomal degradation via an ubiquitin-dependent pathway. It inhibits both murine double minutes-2 (MDM2) dependent p53 ubiquitinylation and MDM2 auto-ubiquitinylation. Thus, cancer cells carrying wild-type p53 when treated with this compound induce stabilization of p53 leading to apoptosis. Sempervirine is also known to intercalate DNA, and inhibits DNA topoisomerase I; therefore, it is considered as a potential lead in anticancer therapeutics. Some of the known members of this family such as flavopereirine, serpentine and alstonine exhibit a variety of biological activities, for example anti-HIV, antipsychotic, sedative and immunostimulant activities together with notable cytostatic effects.162
In 2006, Lipinska and co-workers reported a unified synthetic approach for the total synthesis of zwitterionic indolo[2,3-a]quinolizine alkaloid in five steps via FIS as one of the main steps.1 This total synthesis was initiated from the accessible 5-acetyl-3-methylthio-1,2,4-triazine 268 that has been synthesized in 40% yield in two-step reaction from 3-methylthio-1,2,4-triazine 267 which can be provided on a large laboratory-scale by using the glioxal 265 and S-methylthiosemicarbazide hydroiodide 266. In the following, the corresponding compound 268, was transformed into 3-acetyl-1-methylthiocycloalka[c]pyridine 270 via various stages. The acetyl substituent stays in compound 270 that affords admittance to the synthesis of the indole scaffold through the FIS. The FIS of the phenylhydrazone 270 into 271 requisite MWI of the reaction mixture (substrate with zinc chloride (ZnCl2) solution in triethylene glycol (TEG)). The latter can then be converted into the sempervirine 264 (Scheme 55).1
 | ||
| Scheme 55 Total synthesis of sempervirine 211. | ||
4. Conclusion
Indole derivatives exhibit a very imperative family of compounds that show a key role in cell biology and are potential natural products. There has been a growing attention in the usage of indoles and their derivatives as bioactive molecules against microbes, cancer cells, and different types of disorder in the human body. The FIS is possibly the most widespread and extremely explored method to indole derivatives. It gives a significant and useful approach for manufacturing indole derivatives, a group of molecules with much significance in biological chemistry. The reaction's greatest application lies in the production of commercialized antimigraine drugs and in pharmaceutical chemistry. Parallelly, with the refinement of the synthetic methods the isolation of novel molecules from natural sources containing indole scaffolds is another continuing and increasing investigation field. The study of these novel compounds and the examination of their properties and potential usage in the reaction of different diseases is another synergistic aspect of the importance of the organic synthesis of indoles. This review highlights the significance and importance of applicability of FIS as a key step in total synthesis of natural products, particularly those showing biological activities.Conflicts of interest
The author has confirmed there are no conflicts to declare.References
- T. M. Lipińska, Tetrahedron, 2006, 62, 5736–5747 CrossRef.
- D. R. Stuart, M. Bertrand-Laperle, K. M. Burgess and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 16474–16475 CrossRef CAS PubMed.
- F. Zhan and G. Liang, Angew. Chem., Int. Ed., 2013, 52, 1266–1269 CrossRef CAS PubMed.
- D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893–930 CrossRef CAS PubMed.
- N. K. Kaushik, N. Kaushik, P. Attri, N. Kumar, C. H. Kim, A. K. Verma and E. H. Choi, Molecules, 2013, 18, 6620–6662 CrossRef CAS PubMed.
- D. F. Taber and P. K. Tirunahari, Tetrahedron, 2011, 67, 7195–7210 CrossRef CAS PubMed.
- E. Fischer and O. Hess, Eur. J. Inorg. Chem., 1884, 17, 559–568 Search PubMed.
- R. Larock, E. Yum and M. Refvik, J. Org. Chem., 1998, 63, 7652–7662 CrossRef CAS.
- C.-y. Chen, D. R. Lieberman, R. D. Larsen, T. R. Verhoeven and P. J. Reider, J. Org. Chem., 1997, 62, 2676–2677 CrossRef CAS.
- Y. Dong and C. A. Busacca, J. Org. Chem., 1997, 62, 6464–6465 CrossRef CAS.
- T. Sakamoto, Y. Kondo and H. Yamanaka, Heterocycles, 1988, 27, 2225–2249 CrossRef CAS.
- L. S. Hegedus, Angew. Chem., Int. Ed., 1988, 27, 1113–1126 CrossRef.
- S. Wagaw, B. H. Yang and S. L. Buchwald, J. Am. Chem. Soc., 1998, 120, 6621–6622 CrossRef CAS.
- S. Wagaw, B. H. Yang and S. L. Buchwald, J. Am. Chem. Soc., 1999, 121, 10251–10263 CrossRef CAS.
- E. Fischer and F. Jourdan, Eur. J. Inorg. Chem., 1883, 16, 2241–2245 Search PubMed.
- M. Shiri, M. A. Zolfigol, H. G. Kruger and Z. Tanbakouchian, Chem. Rev., 2009, 110, 2250–2293 CrossRef PubMed.
- P. A. Roussel, J. Chem. Educ., 1953, 30, 122 CrossRef CAS.
- B. Robinson, The Fischer indole synthesis, John Wiley & Sons Inc, New York, Chichester, 1982, pp. 48–59 Search PubMed.
- D. Q. Xu, W. L. Yang, S. P. Luo, B. T. Wang, J. Wu and Z. Y. Xu, Eur. J. Org. Chem., 2007, 2007, 1007–1012 CrossRef.
- D.-Q. Xu, J. Wu, S.-P. Luo, J.-X. Zhang, J.-Y. Wu, X.-H. Du and Z.-Y. Xu, Green Chem., 2009, 11, 1239–1246 RSC.
- G. L. Rebeiro and B. M. Khadilkar, Synthesis, 2001, 2001, 0370–0372 CrossRef.
- T. M. Lipińska and S. J. Czarnocki, Org. Lett., 2006, 8, 367–370 CrossRef PubMed.
- G. W. Gribble, J. Chem. Soc., Perkin Trans. 1, 2000, 7, 1045–1075 RSC.
- R. R. Phillips, Org. React., 1959, 10, 143–178 CAS.
- T. Sravanthi and S. Manju, Eur. J. Pharm. Sci., 2016, 91, 1–10 CrossRef CAS PubMed.
- Z. Bian, C. C. Marvin, M. Pettersson and S. F. Martin, J. Am. Chem. Soc., 2014, 136, 14184–14192 CrossRef CAS PubMed.
- E. V. Mercado-Marin, P. Garcia-Reynaga, S. Romminger, E. F. Pimenta, D. K. Romney, M. W. Lodewyk, D. E. Williams, R. J. Andersen, S. J. Miller and D. J. Tantillo, Nature, 2014, 509, 318–234 CrossRef CAS PubMed.
- V. Sharma, P. Kumar and D. Pathak, J. Heterocycl. Chem., 2010, 47, 491–502 CAS.
- M. Shiri, Chem. Rev., 2012, 112, 3508–3549 CrossRef CAS PubMed.
- G. R. Humphrey and J. T. Kuethe, Chem. Rev., 2006, 106, 2875–2911 CrossRef CAS PubMed.
- G. W. Gribble, Indole ring synthesis: From natural products to drug discovery, Wiley−VCH, 2016, p. 704 Search PubMed.
- B. Robinson, Chem. Rev., 1963, 63, 373–401 CrossRef.
- M. Inman and C. J. Moody, Chem. Sci., 2013, 4, 29–41 RSC.
- D. L. Hughes, Org. Prep. Proced. Int., 1993, 25, 607–632 CrossRef CAS.
- M. M. Heravi, S. Khaghaninejad and N. Nazari, Adv. Heterocycl. Chem., 2015, 112, 183–226 CrossRef.
- M. M. Heravi, S. Khaghaninejad and M. Mostofi, Adv. Heterocycl. Chem., 2014, 112, 1–50 CrossRef CAS.
- S. Sadjadi, M. M. Heravi and N. Nazari, RSC Adv., 2016, 6, 53203–53272 RSC.
- M. M. Heravi, E. Hashemi and N. Nazari, Mol. Diversity, 2014, 18, 441–472 CrossRef CAS PubMed.
- M. M. Heravi, E. Hashemi and F. Azimian, Tetrahedron, 2014, 70, 7–21 CrossRef CAS.
- M. M Heravi, E. Hashemi and N. Ghobadi, Curr. Org. Chem., 2013, 17, 2192–2224 CrossRef.
- M. M. Heravi and E. Hashemi, Tetrahedron, 2012, 68, 9145–9178 CrossRef CAS.
- M. M. Heravi, A. Bakhtiari and Z. Faghihi, Curr. Org. Chem., 2014, 11, 787–823 CAS.
- M. M. Heravi and V. Zadsirjan, Tetrahedron: Asymmetry, 2013, 24, 1149–1188 CrossRef CAS.
- M. M. Heravi and V. F. Vavsari, RSC Adv., 2015, 5, 50890–50912 RSC.
- M. M. Heravi and N. Nazari, Curr. Org. Chem., 2015, 19, 2358–2408 CrossRef CAS.
- M. M. Heravi, T. Ahmadi, A. Fazeli and N. M. Kalkhorani, Curr. Org. Synth., 2015, 12, 328–357 CrossRef CAS.
- M. M. Heravi and A. Fazeli, Heterocycles, 2010, 81, 1979–2026 CrossRef CAS.
- M. M. Heravi, B. Baghernejad and H. A. Oskooie, Curr. Org. Chem., 2009, 13, 1002–1014 CrossRef CAS.
- M. Heravi and S. Sadjadi, J. Iran. Chem. Soc., 2009, 6, 1–54 CrossRef CAS.
- M. M. Heravi and P. Hajiabbasi, Monatsh. Chem., 2012, 143, 1575–1592 CrossRef CAS.
- M. M Heravi, H. Hamidi and V. Zadsirjan, Curr. Org. Chem., 2014, 11, 647–675 Search PubMed.
- M. M. Heravi and E. Hashemi, Monatsh. Chem., 2012, 143, 861–880 CrossRef CAS.
- P. Pelletier and J. B. Caventou, Ann. Chim. Phys., 1818, 104, 323–324 Search PubMed.
- P. Pelletier and J. B. Caventou, Ann. Chim. Phys., 1819, 10, 142–177 Search PubMed.
- R. B. Woodward, M. P. Cava, W. D. Ollis, A. Hunger, H. U. Daeniker and K. Schenker, J. Am. Chem. Soc., 1954, 76, 4749–4751 CrossRef CAS.
- J. Bonjoch and D. Solé, Chem. Rev., 2000, 100, 3455–3482 CrossRef CAS PubMed.
- R. B. Woodward, M. P. Cava, W. D. Ollis, A. Hunger, H. U. Daeniker and K. Schenker, Tetrahedron, 1963, 19, 247–288 CrossRef CAS.
- S. M. Babu and S. Ranganathan, Resonance, 2014, 19, 641–644 CrossRef CAS.
- K. Biemann, M. Spiteller-Friedmann and G. Spiteller, J. Am. Chem. Soc., 1963, 85, 631–638 CrossRef CAS.
- J. D. Medina and J. A. Hurtado, Planta Med., 1977, 32, 130–132 CrossRef CAS PubMed.
- G. Stork and J. E. Dolfini, J. Am. Chem. Soc., 1963, 85, 2872–2873 CrossRef CAS.
- M. Sainsbury, Synthesis, 1977, 1977, 437–448 CrossRef.
- S. Goodwin, A. F. Smith and E. C. Horning, J. Am. Chem. Soc., 1959, 81, 1903–1908 CrossRef CAS.
- V. K. Kansal and P. Potier, Tetrahedron, 1986, 42, 2389–2408 CrossRef CAS.
- Y. Langlois, N. Langlois and P. Potier, Tetrahedron Lett., 1975, 16, 955–958 CrossRef.
- E. Von Angerer and J. Prekajac, J. Med. Chem., 1986, 29, 380–386 CrossRef CAS PubMed.
- M. Amat, A. Linares and J. Bosch, J. Org. Chem., 1990, 55, 6299–6312 CrossRef CAS.
- J. Bonjoch, N. Casamitjana, J. Quirante, M. Rodriguez and J. Bosch, J. Org. Chem., 1987, 52, 267–275 CrossRef CAS.
- J. Gracia, N. Casamitjana, J. Bonjoch and J. Bosch, J. Org. Chem., 1994, 59, 3939–3951 CrossRef CAS.
- S. Archer, B. S. Ross, L. Pica-Mattoccia and D. Cioli, J. Med. Chem., 1987, 30, 1204–1210 CrossRef CAS PubMed.
- G. W. Gribble and S. J. Berthel, Studies in natural products chemistry, Atta-ur-Rahman, Ed., Elsevier, Amsterdam, 1993, vol. 12, pp. 365–409 Search PubMed.
- M. Adeva, F. Buono, E. Caballero, M. Medarde and F. Tomé, Synlett, 2000, 2000, 0832–0834 CrossRef.
- J. Bergman and B. Pelcman, J. Org. Chem., 1989, 54, 824–828 CrossRef CAS.
- R. Tsuji, M. Nakagawa and A. Nishida, Heterocycles, 2002, 58, 587–593 CrossRef CAS.
- S. Takano, M. Moriya and K. Ogasawara, J. Org. Chem., 1991, 56, 5982–5984 CrossRef CAS.
- R. Bonjouklian, T. A. Smitka, L. E. Doolin, R. M. Molloy, M. Debono, S. A. Shaffer, R. E. Moore, J. B. Stewart and G. M. Patterson, Tetrahedron, 1991, 47, 7739–7750 CrossRef CAS.
- J. D. White, K. M. Yager and T. Yakura, J. Am. Chem. Soc., 1994, 116, 1831–1838 CrossRef CAS.
- B. Mckittrick, A. Failli, R. J. Steffan, R. M. Soll, P. Hughes, J. Schmid, A. A. Asselin, C. Shaw, R. Noureldin and G. Gavin, J. Heterocycl. Chem., 1990, 27, 2151–2163 CrossRef CAS.
- J.-C. Fernàndez, N. Valls, J. Bosch and J. Bonjoch, J. Chem. Soc., Chem. Commun., 1995, 22, 2317–2318 RSC.
- J. Bonjoch, J. Catena and N. Valls, J. Org. Chem., 1996, 61, 7106–7115 CrossRef CAS PubMed.
- J. Catena, N. Valls, J. Bosch and J. Bonjoch, Tetrahedron Lett., 1994, 35, 4433–4436 CrossRef CAS.
- T. T. T. Thuy, N. M. Cuong, T. Q. Toan, N. N. Thang, B. H. Tai, N. X. Nhiem, H.-J. Hong, S. Kim, S. Legoupy and Y. S. Koh, Arch. Pharmacal Res., 2013, 36, 832–839 CrossRef CAS PubMed.
- Y. Murakami, H. Yokoo and T. Watanabe, Heterocycles, 1998, 49, 127–132 CrossRef CAS.
- S. Shimizu, K. Ohori, T. Arai, H. Sasai and M. Shibasaki, J. Org. Chem., 1998, 63, 7547–7551 CrossRef CAS PubMed.
- D. Gennet, P. Michel and A. Rassat, Synthesis, 2000, 2000, 447–451 CrossRef.
- M. M. Janot, H. Pourrat and M. J. Le, Bull. Soc. Chim. Fr., 1954, 707–708 CAS.
- S. A. Kozmin, T. Iwama, Y. Huang and V. H. Rawal, J. Am. Chem. Soc., 2002, 124, 4628–4641 CrossRef CAS PubMed.
- I. Bick, J. Bremner, N. Preston and I. Calder, J. Chem. Soc., Chem. Commun., 1971, 19, 1155–1156 RSC.
- C. W. Roberson and K. Woerpel, J. Am. Chem. Soc., 2002, 124, 11342–11348 CrossRef CAS PubMed.
- A. M. Schmidt and P. Eilbracht, Org. Biomol. Chem., 2005, 3, 2333–2343 CAS.
- T. Balle, J. Perregaard, M. T. Ramirez, A. K. Larsen, K. K. Søby, T. Liljefors and K. Andersen, J. Med. Chem., 2003, 46, 265–283 CrossRef CAS PubMed.
- M. Gompel, M. Leost, E. B. D. K. Joffe, L. Puricelli, L. H. Franco, J. Palermo and L. Meijer, Bioorg. Med. Chem. Lett., 2004, 14, 1703–1707 CrossRef CAS PubMed.
- L. H. Franco and J. A. Palermo, Chem. Pharm. Bull., 2003, 51, 975–977 CrossRef CAS PubMed.
- Y. Liu and W. W. McWhorter, J. Am. Chem. Soc., 2003, 125, 4240–4252 CrossRef CAS PubMed.
- G. Pandey, S. K. Burugu and P. Singh, Org. Lett., 2016, 18, 1558–1561 CrossRef CAS PubMed.
- R. Iyengar, K. Schildknegt, M. Morton and J. Aubé, J. Am. Chem. Soc., 2005, 70, 10645–10652 CAS.
- C. Sabot, K. C. Guérard and S. Canesi, Chem. Commun., 2009, 20, 2941–2943 RSC.
- K. Cimanga, T. De Bruyne, L. Pieters, M. Claeys and A. Vlietinck, Tetrahedron Lett., 1996, 37, 1703–1706 CrossRef CAS.
- T. Dhanabal, R. Sangeetha and P. Mohan, Tetrahedron Lett., 2005, 46, 4509–4510 CrossRef CAS.
- E. F. Rogers, H. Snyder and R. F. Fischer, J. Am. Chem. Soc., 1952, 74, 1987–1989 CrossRef CAS.
- J. E. Saxton, Alkaloids: Chemistry and Physiology, 1965, vol. 8, pp. 673–678 Search PubMed.
- H. Ueda, H. Satoh, K. Matsumoto, K. Sugimoto, T. Fukuyama and H. Tokuyama, Angew. Chem., 2009, 121, 7736–7739 CrossRef.
- J. d'Angelo, D. Desmaële, F. Dumas and A. Guingant, Tetrahedron: Asymmetry, 1992, 3, 459–505 CrossRef.
- G. Massiot, P. Thépenier, M.-J. Jacquier, L. Le Men-Olivier and C. Delaude, Heterocycles, 1989, 29, 1435–1438 CrossRef CAS.
- P. Liu, J. Wang, J. Zhang and F. G. Qiu, Org. Lett., 2011, 13, 6426–6428 CrossRef CAS PubMed.
- R. Burnell, J. Medina and W. Ayer, Can. J. Chem., 1966, 44, 28–31 CrossRef CAS.
- K. Brown Jr, H. Budzikiewicz and C. Djerassi, Tetrahedron Lett., 1963, 4, 1731–1736 CrossRef.
- E. L. Campbell, A. M. Zuhl, C. M. Liu and D. L. Boger, J. Am. Chem. Soc., 2010, 132, 3009–3012 CrossRef CAS PubMed.
- K. C. Guérard, A. Guérinot, C. Bouchard-Aubin, M.-A. Ménard, M. Lepage, M. A. Beaulieu and S. Canesi, J. Org. Chem., 2012, 77, 2121–2133 CrossRef PubMed.
- T.-S. Kam, G. Subramaniam, K.-H. Lim and Y.-M. Choo, Tetrahedron Lett., 2004, 45, 5995–5998 CrossRef CAS.
- K.-H. Lim, O. Hiraku, K. Komiyama, T. Koyano, M. Hayashi and T.-S. Kam, J. Nat. Prod., 2007, 70, 1302–1307 CrossRef CAS PubMed.
- Y. Iwama, K. Okano, K. Sugimoto and H. Tokuyama, Chem.–Eur. J., 2013, 19, 9325–9334 CrossRef CAS PubMed.
- D. Desmaeele and J. d'Angelo, J. Org. Chem., 1994, 59, 2292–2303 CrossRef CAS.
- J. M. Lopchuk, Recent advances in the synthesis of aspidosperma-type alkaloids, Prog. Heterocycl. Chem., Eds, Elsevier Science, New York, 2011, vol. 23, pp. 1–25 Search PubMed.
- H. Satoh, H. Ueda and H. Tokuyama, Tetrahedron, 2013, 69, 89–95 CrossRef CAS.
- A. Chatterjee, B. Mukherjee, A. Ray and B. Das, Tetrahedron Lett., 1965, 6, 3633–3637 CrossRef.
- J. M. Smith, J. Moreno, B. W. Boal and N. K. Garg, J. Org. Chem., 2015, 80, 8954–8967 CrossRef CAS PubMed.
- J. M. Smith, J. Moreno, B. W. Boal and N. K. Garg, J. Am. Chem. Soc., 2014, 136, 4504–4507 CrossRef CAS PubMed.
- J. D. White and Y. Li, Heterocycles, 2014, 88, 899–910 CrossRef CAS.
- L. V. White, M. G. Banwell and A. C. Willis, Heterocycles, 2015, 90, 298–315 CrossRef CAS.
- W. Zhang, Z. Liu, S. Li, T. Yang, Q. Zhang, L. Ma, X. Tian, H. Zhang, C. Huang and S. Zhang, Org. Lett., 2012, 14, 3364–3367 CrossRef CAS PubMed.
- L. M. Blair and J. Sperry, Chem. Commun., 2016, 52, 800–802 RSC.
- M. Shoeb, S. M. MacManus, M. Jaspars, J. Trevidu, L. Nahar, P. Kong-Thoo-Lin and S. D. Sarker, Tetrahedron, 2006, 62, 11172–11177 CrossRef CAS.
- M. B. Freitas, K. A. Simollardes, C. M. Rufo, C. N. McLellan, G. J. Dugas, L. E. Lupien and E. A. C. Davie, Tetrahedron Lett., 2013, 54, 5489–5491 CrossRef CAS.
- B. Gigant, C. Wang, R. B. Ravelli and F. Roussi, Nature, 2005, 435, 519–522 CrossRef CAS PubMed.
- P. W. Tan, J. Seayad and D. J. Dixon, Angew. Chem., Int. Ed., 2016, 55, 13436–13440 CrossRef CAS PubMed.
- S. R. McCabe and P. Wipf, Org. Biomol. Chem., 2016, 14, 5894–5913 CAS.
- J. Park, D.-H. Kim, T. Das and C.-G. Cho, Org. Lett., 2016, 18, 5098–5101 CrossRef CAS PubMed.
- N. L. Dutta, C. Quasim and M. S. Wadia, Indian J. Chem., 1969, 7, 1061–1062 CAS.
- P. Bernal and J. Tamariz, Helv. Chim. Acta, 2007, 90, 1449–1454 CrossRef CAS.
- D. P. Chakraborty and B. Chowdhury, J. Org. Chem., 1968, 33, 1265–1268 CrossRef CAS.
- S. Ray and D. P. Chakraborty, Phytochemistry, 1976, 15, 356–360 CrossRef CAS.
- D. P. Chakraborty, A. Islam and P. Bhattacharyya, J. Org. Chem., 1973, 38, 2728–2729 CrossRef CAS.
- K. Prabakaran and K. J. Rajendra Prasad, Synth. Commun., 2012, 42, 2966–2980 CrossRef CAS.
- S. Roy, L. Bhattacharyya and D. P. Chakraborty, J. Indian Chem. Soc., 1982, 59, 1369–1371 CAS.
- S. Roy, L. Bhattacharyya and D. P. Chakraborty, J. Indian Chem. Soc., 1985, 62, 673–680 CAS.
- L. C. Konkol, F. Guo, A. A. Sarjeant and R. J. Thomson, Angew. Chem., Int. Ed., 2011, 50, 9931–9934 CrossRef CAS PubMed.
- G. Bringmann, A. Ledermann, M. Stahl and K. P. Gulden, Tetrahedron, 1995, 51, 9353–9360 CrossRef CAS.
- Y. Bi, L. K. Hamaker and J. M. Cook, Studies in Natural Products Chemistry, Bioactive Natural Products, Part A, ed. F. Z. Basha and A. Rahman, Elsevier Science, Amsterdam, 1993, vol. 13, p. 383 Search PubMed.
- C. B. Cui, H. Kakeya and H. Osada, J. Antibiot., 1996, 49, 534–540 CrossRef CAS PubMed.
- T. Gan, R. Liu, P. Yu, S. Zhao and J. M. Cook, J. Org. Chem., 1997, 62, 9298–9304 CrossRef CAS.
- C. Saha and B. Chowdhury, Phytochemistry, 1998, 48, 363–366 CrossRef CAS.
- C. Prabhakar, N. V. Kumar, M. R. Reddy, M. Sarma and G. O. Reddy, Org. Process Res. Dev., 1999, 3, 155–160 CrossRef CAS.
- J. Arendt, Melatonin and the Mammalian Pineal Gland, Chapman & Hall, London, 1st edn, 1995, vol. 60, pp. 17–109 Search PubMed.
- R. A. Abramovitch and D. Shapiro, J. Chem. Soc., 1956, 4589–4592 RSC.
- J. Jiricek and S. Blechert, J. Am. Chem. Soc., 2004, 126, 3534–3538 CrossRef CAS PubMed.
- F. Tang, M. G. Banwell and A. C. Willis, J. Org. Chem., 2016, 81, 10551–10557 CrossRef CAS PubMed.
- J.-F. Hu, H. Fan, J. Xiong and S.-B. Wu, Chem. Rev., 2011, 111, 5465–5491 CrossRef CAS PubMed.
- K. M. Aubart and C. H. Heathcock, J. Org. Chem., 1999, 64, 16–22 CrossRef CAS PubMed.
- B. G. Szczepankiewicz and C. H. Heathcock, Tetrahedron, 1997, 53, 8853–8870 CrossRef CAS.
- J. i. Kobayashi, H. Nakamura, Y. Ohizumi and Y. Hirata, Tetrahedron Lett., 1986, 27, 1191–1194 CrossRef CAS.
- Y. Murakami, H. Takahashi, Y. Nakazawa, M. Koshimizu, T. Watanabe and Y. Yokoyama, Tetrahedron Lett., 1989, 30, 2099–2100 CrossRef CAS.
- Y. Murakami, T. Watanabe, H. Takahashi, H. Yokoo, Y. Nakazawa, M. Koshimizu, N. Adachi, M. Kurita, T. Yoshino and T. Inagaki, Tetrahedron, 1998, 54, 45–64 CrossRef CAS.
- H. M. Sheldrake, S. Travica, I. Johansson, P. M. Loadman, M. Sutherland, L. Elsalem, N. Illingworth, A. J. Cresswell, T. Reuillon and S. D. Shnyder, J. Med. Chem., 2013, 56, 6273–6277 CrossRef CAS PubMed.
- L. F. Tietze, F. Haunert, T. Feuerstein and T. Herzig, Eur. J. Org. Chem., 2003, 2003, 562–566 CrossRef.
- J.-F. Liao, W.-F. Chiou, Y.-C. Shen, G.-J. Wang and C.-F. Chen, Chin. Med., 2011, 6, 6–13 CrossRef PubMed.
- S. B. Mhaske and N. P. Argade, Tetrahedron, 2004, 60, 3417–3420 CrossRef CAS.
- K. P. Wojtas, M. Riedrich, J. Y. Lu, P. Winter, T. Winkler, S. Walter and H. D. Arndt, Angew. Chem., Int. Ed., 2016, 55, 9772–9776 CrossRef CAS PubMed.
- F. Benazet, M. Cartier, J. Florent, C. Godard, G. Jung, J. Lunel, D. Mancy, C. Pascal, J. Renaut and P. Tarridec, Experientia, 1980, 36, 414–416 CrossRef CAS PubMed.
- D. J. Bentley, J. Fairhurst, P. T. Gallagher, A. K. Manteuffel, C. J. Moody and J. L. Pinder, Org. Biomol. Chem., 2004, 2, 701–708 CAS.
- T. Lipińska, Tetrahedron Lett., 2002, 43, 9565–9567 CrossRef.
- T. C. Rao, S. Saha, G. B. Raolji, B. Patro, P. Risbood, M. J. Difilippantonio, J. E. Tomaszewski and S. V. Malhotra, Tetrahedron Lett., 2013, 54, 487–490 CrossRef.
| This journal is © The Royal Society of Chemistry 2017 |