
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Improving the cyclability performance of lithium-ion batteries by introducing lithium difluorophosphate (LiPO2F2) additive†
Guanghua Yang‡
ab,
Junli Shi‡a,
Cai Shen a,
Shuwei Wangab,
Lan Xiaa,
Huasheng Hua,
Hao Luoa,
Yonggao Xia*a and
Zhaoping Liu*a
a,
Shuwei Wangab,
Lan Xiaa,
Huasheng Hua,
Hao Luoa,
Yonggao Xia*a and
Zhaoping Liu*a
aNingbo Institute of Materials Technology & Engineering (NIMTE), Chinese Academy of Sciences, Ningbo, Zhejiang 315201, China. E-mail: xiayg@nimte.ac.cn; liuzp@nimte.ac.cn
bDepartment of Polymer Materials, Shanghai University, Shangda Street 99, Mailbox 152, Shanghai 200444, China
First published on 17th May 2017
Abstract
The cyclability of lithium-ion batteries (LIBs) is often affected by the components of the solid electrolyte interphase (SEI) layer which is generated from electrochemical decomposition of electrolyte. Here, lithium difluorophosphate (LiPO2F2) is studied in this work. When 1.6 wt% LiPO2F2 additive is incorporated into the reference electrolyte, the capacity retention of graphite/Li half-cell is increased from 82.53% to 98.04% and the capacity retention of LiCoO2/Li half-cell is increased from 89.60% to 97.53% after 160 cycles. Electrochemical impedance spectroscopy (EIS) indicates that the SEI layer containing LiPO2F2 can decrease the surface impedance of cells in the last stage cycle. In situ atomic force microscopy (AFM), DFT calculations and X-ray photoelectron spectroscopy (XPS) results show that LiPO2F2 is deposited on the surface of both LiCoO2 and graphite electrodes, which effectively protects the graphite anode and suppresses the degradation of the cathode during the long-term cycling of LIBs.
Introduction
From the first lithium-ion batteries (LIBs) produced in 1991, LIBs have been become an important part of our daily life.1,2 However, the cyclability of LIBs remains one of the main obstructions to be resolved.3 It is known that the cyclability of LIBs is primarily limited by parasitic reactions in the solid electrolyte interphase (SEI) layer and deterioration of the SEI layer.4–6 During cycling, the degradation of lithium salts and electrolyte solvents can occur and lead to the formation or growth of SEI on both electrodes. When LIBs are used for long-term cycling, the formed SEI layers either grow in thickness or become compromised at the last stage. This process can lead to a decrease in capacity and even cell failure.7,8Up to now, several methods have been proposed to inhibit the detrimental reactions in SEI layer and improve the interphasial stability between electrode and liquid electrolyte. Among them, there are mainly two approaches. Firstly, some electrolyte additives such as vinylene carbonate (VC),9 fluoroethylene carbonate (FEC)10–12 have been used to suppress unwanted decomposition of electrolyte. The use of electrolyte additives is one of the most effective and economical ways to improve the cyclability of LIBs. Secondly, new solvents such as nitriles,13,14 sulfoxides15 and ionic liquids16 solvents have been developed to replace conventional carbonate solvents to modify the properties of the SEI layer. These efforts motivate researches on correlating the structure and property of each SEI component to the performance of LIBs. It has generally been accepted that there are two types of components in SEI layer: inorganic components such as LiF, Li2O, LixPOyFz, LixPFy, Li2CO3, and organic components such as ROCO2Li, (CH2OCO2Li)2, ROLi.17,18 Their mechanical and transport properties have been investigated by simulations and their contributions to the cycling performance have been revealed by experiments.19 Among these components, LixPOyFz has been repeatedly mentioned. Andersson et al.20 reported that LixPOyFz-type component was hydrolysis product of LiPF6. Zhang et al.21 found that the increase of the concentration of LixPFyOz in SEI layer helped to promote the discharge capacity retention and coulombic efficiency when 1,3-propanesultone and VC were used as a bifunctional additive. Li et al.22 found that the SEI layer was covered by less inorganic components and more LixPFyOz when they used prop-1-ene-1,3-sultone as an electrolyte additive, which effectively enhanced the cycle stability of LiCoO2/graphite battery under 4.5 V. Based on previous literatures, it can be inferred that a dynamically stable protective SEI layer usually contains LixPOyFz, which inhibits side reactions between electrode and electrolyte and improves the cyclability of LIBs.
In this paper, we used the lithium difluorophosphate (LiPO2F2) as an electrolyte additive to improve content of LixPOyFz in SEI components and enhance the cyclability of graphite and LiCoO2 electrodes. According to the work by Kim et al.23 LiPO2F2 and VC could modify the SEI film formation on graphite, which led to the high rate performance of graphite electrodes. Yang et al.24 found a conductive and stable SEI film formed on the graphite by the sacrifice of LiPO2F2, which led to a low impedance and richer content of LiF and Li2CO3 in SEI components. We introduced LiPO2F2 into a reference electrolyte of 1 mol (M) LiPF6 in ethylene carbonate (EC)/dimethyl carbonate (DMC) (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7, v/v). For convenience, the reference electrolyte was called A0, 0.8 wt% (mass faction) LiPO2F2 in reference electrolyte was called A1 and 1.6 wt% LiPO2F2 in reference electrolyte was called A2. The maximum content of LiPO2F2 additive in reference electrolyte was 1.6 wt%. The properties of the electrolyte containing LiPO2F2 and their influences on cyclability of LIBs were detailedly explored. The experimental results indicate that cells containing LiPO2F2 display superior capacity retention.
:7, v/v). For convenience, the reference electrolyte was called A0, 0.8 wt% (mass faction) LiPO2F2 in reference electrolyte was called A1 and 1.6 wt% LiPO2F2 in reference electrolyte was called A2. The maximum content of LiPO2F2 additive in reference electrolyte was 1.6 wt%. The properties of the electrolyte containing LiPO2F2 and their influences on cyclability of LIBs were detailedly explored. The experimental results indicate that cells containing LiPO2F2 display superior capacity retention.
Experimental
Preparation of electrode and electrolyte
LiPF6, EC, DMC, and commercial electrolyte (the basic recipe was 1 M LiPF6 in EC/DMC (3/7, v/v)) were purchased from Guotaihuarong New Chemical Materials Co. Ltd (Zhangjiagang, China). 1 M LiPF6 in EC/DMC (3/7, v/v) was as a reference electrolyte. LiPO2F2 was purchased from Sigma. All the reagents do not need further purification. For convenience, the reference electrolyte was called A0, 0.8 wt% (mass faction) LiPO2F2 in reference electrolyte was called A1 and 1.6 wt% LiPO2F2 in reference electrolyte was called A2. The maximum content of LiPO2F2 additive in reference electrolyte was 1.6 wt%. All processes were prepared in a dry argon-filled glovebox (MBraun LABmaster glovebox workstation), and the glovebox with the mass factions of oxygen and water below 0.1 ppm.LiCoO2 was purchased from Ningbo Veken Battery Co., Ltd (Ningbo, China). LiCoO2 electrode was prepared by coating the N-methyl pyrrolidone (NMP)-based slurry containing LiCoO2 (80 wt%), PVDF (10 wt%) and Super P carbon (10 wt%) in NMP solvent on the aluminum foil. Thickness of slurry was about 15 μm. For the cell performance test, CR-2032 coin cells of Li/LiCoO2 and graphite/Li were investigated. The counter electrodes and reference electrodes were lithium sheets. The Celgard-2400 polypropylene membrane (25 μm thick) was used as the separator. All the cells were assembled in the argon-filled glovebox.
DFT calculations and electrochemical measurements
The Kohn–Sham density functional theory (DFT) equation was calculated with the B3PW91 functional25 and 6-311++G(d,p) basis sets of diffuse-augmented triple-z quality. The ground-state structures of the molecules were fully optimized within C1 symmetry by means of DFT methods. All the DFT calculations were performed using the Gaussian 09 program package.26Cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) experiments were performed on an Autolab Electrochemical Analytical Instrument (ECO CHEMIE, B. V. Utrecht, The Netherlands).
CV was also measured in three-electrode electrochemical cell with a Pt disk (0.1 mm diameter) was used as working electrode, Li foil was as both reference electrode and counter electrode. The scan rate was 0.1 mV s−1. EIS of cells were conducted with oscillation amplitude of 5 mV in the frequency range from 10−2 Hz to 106 Hz.
The charge–discharge experiment was carried out using a LAND battery program-control test system (CT2001ALand Battery Testing System Wuhan, China) at 25 °C. The graphite/Li cell was cycled over the range of 0–2 V with a constant current rate of 0.2C. The LiCoO2/Li cell was over the range of 3.0–4.3 V at 1.0C, respectively.
The 18650 full batteries (APR18650M, 1.56Ah) were provided by A123 Systems with a graphite anode and a LiCoO2 cathode. All the full batteries in the experiment were from the same batch which ensures that all materials and manufacturing condition are the same.
Material characterization
After charge–discharge test, the cells were disassembled in a glovebox to collect graphite and LiCoO2 electrodes, which were rinsed with DMC many times to remove the residual electrolyte salts on the surface. All the tests must avoid ingress of air.The surface morphology and element content were characterized using SEM and energy dispersive X-ray analysis (EDX) analyses on a JEOL-5900 SEM field emission scanning electron microscopy (FESEM, S-4800, Hitachi, Japan).
In situ atomic force microscopy (AFM) (AFM Bruker Icon) analysis was conducted in the argon-filled glovebox at 25 °C. The Li/HOPG (highly oriented pyrolytic graphite) cell was composed of HOPG substrate (working electrode) and Li wire (counter and reference electrodes). HOPG (Bruker Corporation, ZYB grade, 12 × 12 × 2 mm) was cleaved with adhesive tape to obtain a flat basal plane. In order to study the SEI layer formation on HOPG, the cell was studied by CV at a scanning rate of 2 mV s−1 between 3.0 and 0 V. AFM topography was collected simultaneously in ScanAsyst mode using nitride coated silicon probes (tip model: Scanasyst fluid with k = 0.7 N m−1, Bruker Corporation). Contact mode was applied to scratch the surface using the same probe.
XPS measurement was carried out with an X-ray photoelectron spectrometer (Kratos AXIS Ultra DLD) using focused monochromatized Al Kα radiation (1486.6 eV), the X-ray anode was run at 250 W and the high voltage was kept at 14.0 kV with a detection angle at 54°. The pass energy was fixed at 93.90 eV to ensure sufficient resolution and sensitivity. Curve fitting of slow-scanned XPS spectra was carried out using a peak-fit program with a Gaussian–Lorentzian sum function. The binding energy scale was calibrated from the hydrocarbon contamination using the C 1s peak at 284.8 eV.
Results and discussion
DFT-calculated HOMO/LUMO energies and experimental potentials
The density functional theory (DFT) calculations are performed to quantitatively evaluate the effection of LiPO2F2 in liquid electrolyte. The highest occupied molecular orbital (HOMO) energies, lowest unoccupied molecular orbital (LUMO) energies of LiPF6 and LiPO2F2 and experimental potentials of the reference electrolyte and electrolyte containing LiPO2F2 are listed in Table 1. It can be seen that the LUMO energy level of LiPF6 is lower than that of LiPO2F2. Based on the molecular orbital theory, molecule with lower LUMO energy level should be a better electron acceptor and more reactive on anode surface. Therefore, LiPF6 would be reduced prior to LiPO2F2 during the first charge process. The HOMO energy level of LiPF6 is upper than that of LiPO2F2. LiPF6 would be oxidized prior to LiPO2F2 during the first discharge process. These results suggest that LiPO2F2 is expected to be beneficial in terms of cathodic stability and anodic stability. The DFT results exactly match with CV (Fig. S1 ESI†). In Fig. S1,† there are not obvious difference among the first three CV cycles of cells.| Molecule | DFT | Experiment | ||
|---|---|---|---|---|
| HOMO/eV | LUMO/eV | Ere/Li+/Li/V | Eox/Li+/Li/V | |
| LiPF6 | −6.13 | −1.52 | 0.15 | 4.25 |
| LiPO2F2 | −8.57 | −1.10 | 0.15 | 4.25 |
The electrochemical performances of the electrolyte containing LiPO2F2
Fig. 1a shows the cycling performance of graphite/Li cells with electrolytes A0, A1, and A2 at 0.2C. The charge capacity of cell with electrolyte A0 declines from 332 mA h g−1 to 273.9 mA h g−1, with a capacity loss of 17.47% at the 160th cycle. The charge capacity of cells with electrolytes A1 and A2 are 370.1 mA h g−1 and 377.4 mA h g−1, respectively, and show only 5.70% and 1.96% capacity loss after 160 cycles. The cycling performances of LiCoO2/Li cells are investigated in Fig. 1b, it can be seen that cells with electrolytes A1 and A2 exhibit excellent capacity retention at 1.0C, and retain 95.21% and 97.53% of the initial capacity after 160 cycles, respectively. By contrast, the capacity retention of cell without LiPO2F2 is 89.60% after 160 cycles. To further evaluate the suitability of electrolyte containing LiPO2F2. In Fig. 1c, the cycle performances of LiCoO2/graphite 18650 full batteries with electrolyte A1, A2 and a commercial electrolyte (3015A, the basic recipe was 1 M LiPF6 in EC/DMC 3/7, v/v) are investigated, the discharge capacity retention of batteries with electrolyte A1, A2 are 81.5% and 88.6%, respectively, while it is 75.7% for battery with the commercial electrolyte after 2400 cycles.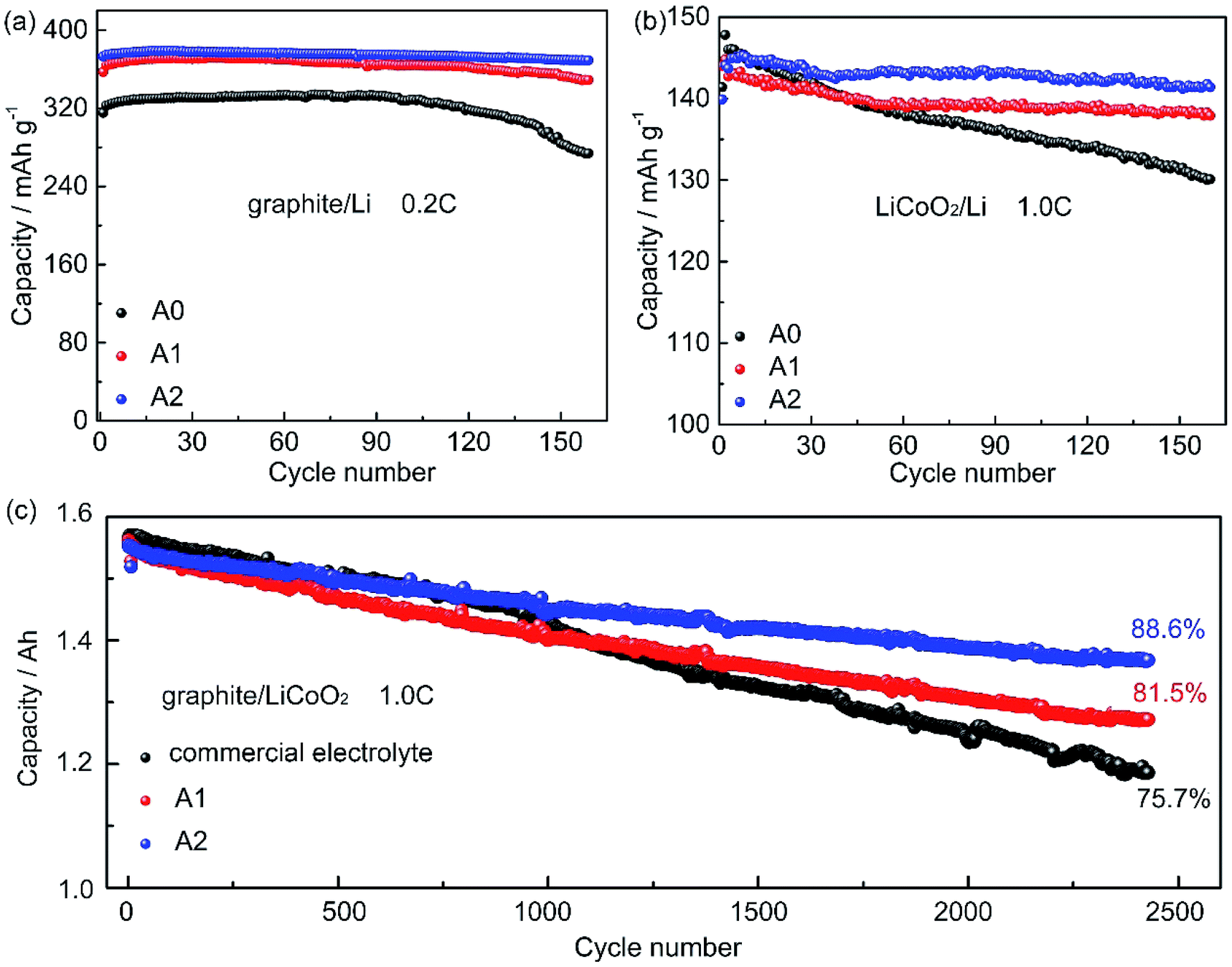 | ||
| Fig. 1 (a) The cycling performance of the graphite/Li cells using electrolytes A0, A1, and A2 that charge–discharge rate was 0.2C in the potential range of 0–2 V at 25 °C. (b) The cycling performances of LiCoO2/Li cells with electrolytes A0, A1, and A2 at current density of 1.0C in the potential range of 3.0–4.3 V at 25 °C. (c) The cycling performance of LiCoO2/graphite 18650 full batteries with electrolyte A1, A2 and the commercial electrolyte at 1.0C in the potential range of 3.0–4.3 V at 25 °C. | ||
These results demonstrate that LiPO2F2 can effectively improve the cyclability of cells. Moreover, the cyclability of the electrolyte containing LiPO2F2 is much better than that of electrolyte with some additives.27–29 For example, using the typical additives FEC (3 wt%), VC (3 wt%), and ethylene sulphite (ES 3 wt%) in 1 M LiClO4/PC electrolyte in the Li/graphite half-cells,29 the best electrochemical cyclability in graphite/Li half-cells by Jeong et al. was the electrolyte containing VC with about 96% capacity retention after 50 cycles. While the graphite/Li half-cell with electrolyte containing LiPO2F2 can retain 99.53% of the initial capacity after 50 cycles. Jeong et al.30 reported 2-(triphenylphosphoranylidene) succinic anhydride (TPSA) to improve the cycle performance of LiCoO2/Li cells. The cell containing 3 wt% of TPSA retain 85% of the initial discharge capacity after 100 cycles, which was not so stable as the electrolyte containing LiPO2F2 (98.9%). Wang et al.3 used additives VC, LiBOB, VEC in 1 M LiPF6/(EC + EMC, 3:7, v/v) in the LiCoO2/graphite pouch cells. The best electrochemical cyclability was the electrolyte containing 2 wt% VC and 1 wt% LiBOB with about 97.74% capacity retention after 70 cycles. While electrolyte containing LiPO2F2 can retain 98.5% of the initial capacity after 70 cycles (Table S1, ESI†).
EIS studies of half-cells were done in order to clarify the capacity fade of anode and cathode electrodes. EIS measurements of graphite/Li cells are performed under full charged, while LiCoO2/Li cells are done under full discharge. The diameter of the semicircle appearing in the high-frequency region represents the sum of the charge-transfer resistances at both two electrodes. What is called the charge transfer resistance (Rct) in this paper for convenience includes the active particle-current collector contact resistance of both electrodes (small), the resistance to the transfer of Li+ from the electrolyte to the electrode through the solid electrolyte interface (SEI) of both electrodes, and the electron transfer to the active material of both electrodes.12 Rct of graphite/Li and LiCoO2/Li half-cells with different electrolytes are shown in Fig. 2. We list Rct values of graphite/Li and LiCoO2/Li half-cells in the Table S2 and S3 (ESI†). In Fig. 2a, Rct values of cells after storage for 24 h with electrolytes A0, A1 and A2 are 11, 5 and 6 Ω cm2, respectively, which indicate that the property of SEI layer is altered by LiPO2F2. Rct value of cell with electrolyte A0 decreases from 42 Ω cm2 (after 1 cycle) to 60 Ω cm2 after 100 cycles, then rapidly increases to 1700 Ω cm2 after 160 cycles. It is possible that the reference electrolyte is not enough to form effective SEI layers at the electrode surfaces. Meanwhile, Rct values of cells with electrolytes A1 and A2 are 17 Ω cm2 and 15 Ω cm2 after the first cycle and lower than that of cell with electrolyte A0 at all stages. In Fig. 2b, the Rct value of LiCoO2/Li cell with electrolyte A0 increases more than twenty times from 80 to 2200 Ω cm2 after 160 cycles. However, Rct values of cells with electrolytes A1 and A2 remain stable at all stages (40 Ω cm2 and 70 Ω cm2 respectively). These results nicely underscore that LiPO2F2 is enough to form effective SEI layers at electrode surfaces.
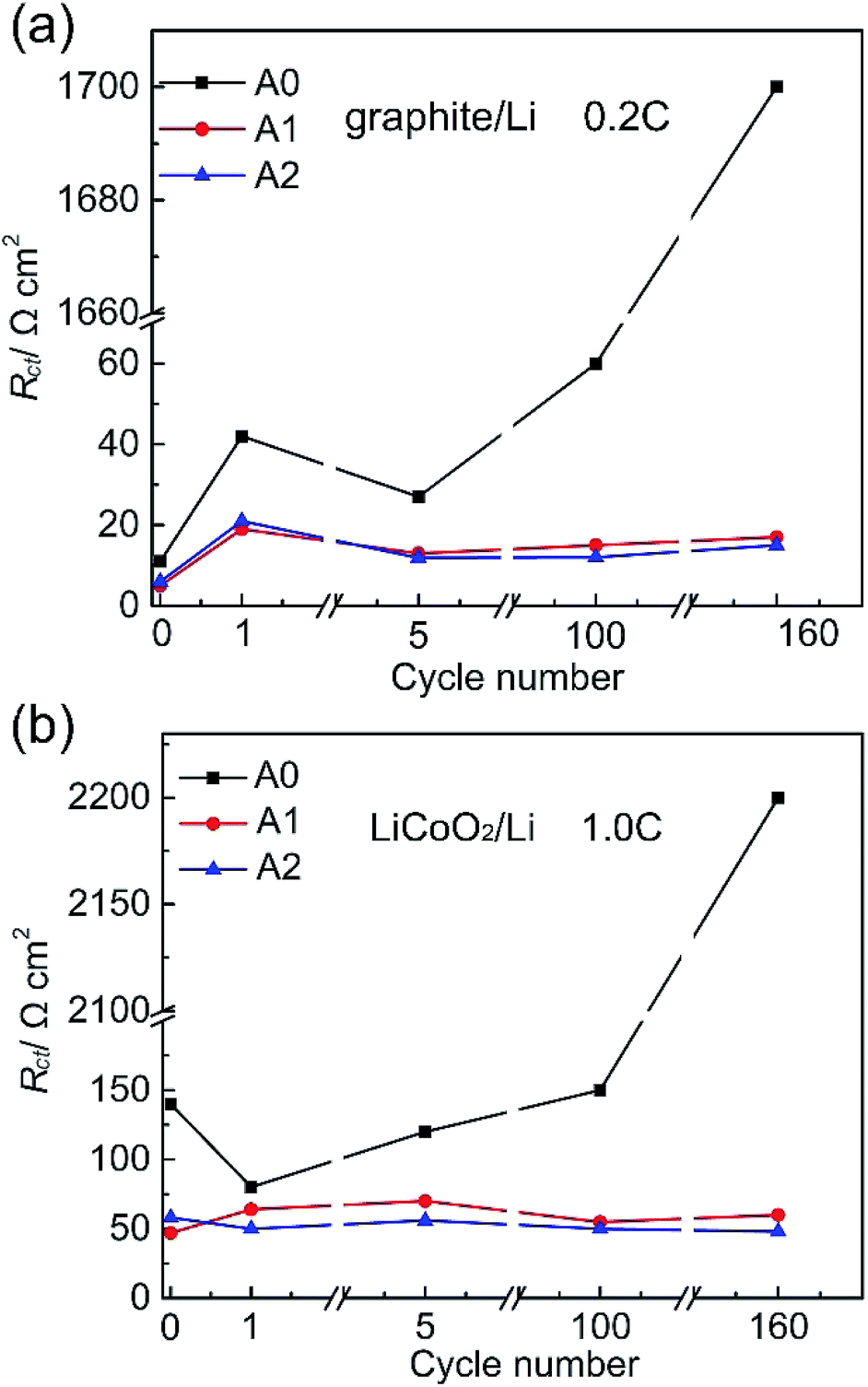 | ||
| Fig. 2 EIS spectra of the graphite/Li and LiCoO2/Li half-cells with electrolytes A0, A1 and A2, after storage for 24 h, 1st, 5th, 100th and 160th cycles at 25 °C. Frequency range of 10−2 Hz to 105 Hz with a potential amplitude of 5 mV. (a) The change of the charge-transfer resistances (Rct) in the graphite/Li cells. (b) The change of Rct in the LiCoO2/Li cells. | ||
The properties of the SEI layers formed on electrode surfaces
In situ AFM is performed to visually explore the morphology of SEI layer. Fig. 3 shows in situ AFM images of HOPG electrodes cycled between 3.0 and 0.0 V with electrolytes A0, A1, and A2. In the CV cycles, the sharp cathodic peak starts around 0.8 V and disappears in the following cycles (Fig. S1a–c, ESI†), which is attributed to the formation of SEI by the irreversible decomposition of electrolyte. Fig. 3a–c show the surface structural evolution of HOPG electrode during the first lithiation–delithiation cycle. In Fig. 3a-I, there are some small particles with the height of few nanometres formed on the HOPG surface at the stage of open circuit voltage (OCV) due to the decomposition of electrolyte on the surface of HOPG electrode. A large amount of SEI begins to grow after voltage sweep down to ∼0.8 V (Fig. 3a-II), the SEI particles grow bigger, and the sizes of these particles are in the range of ∼100–200 nm. Further the decline of the potential raises a larger scale of deposition which covered the whole surface of HOPG. The surface keeps unchanged during the anodic sweeping (Fig. 3a-III and IV). In Fig. 3d, line profile clearly reveals that the height of SEI layer is in the range of 10–20 nm, and SEI layer is thin and scattered. For electrolyte A1, there is not particle (Fig. 3b-I) on the surface of HOPG at the OCV stage. This result is consistent with our speculation that the reaction between electrolyte and lithium is restrained by LiPO2F2 which decreases the Rct before cycle. SEI layer starts to appear after voltage sweep down to ∼0.9 V (Fig. 3b-II). The particles are in a much smaller sizes (in the range of 10 to 20 nm) in comparison to those of electrolyte A0. A dense and compact SEI layer is found to be cover the whole surface after the potential is reduced to zero. The surface remains unchanged during the anodic sweeping (Fig. 3b-III and IV), and the height of SEI layer is in the range of 20 to 40 nm (Fig. 3d). For electrolyte A2 (Fig. 3c and d), similar to the observation with electrolyte A1, it is found that a thicker and more compact SEI is obtained. The height of SEI layer is in the range of 50–250 nm. These results indicate that the LiPO2F2 would increase the thickness of the SEI layer.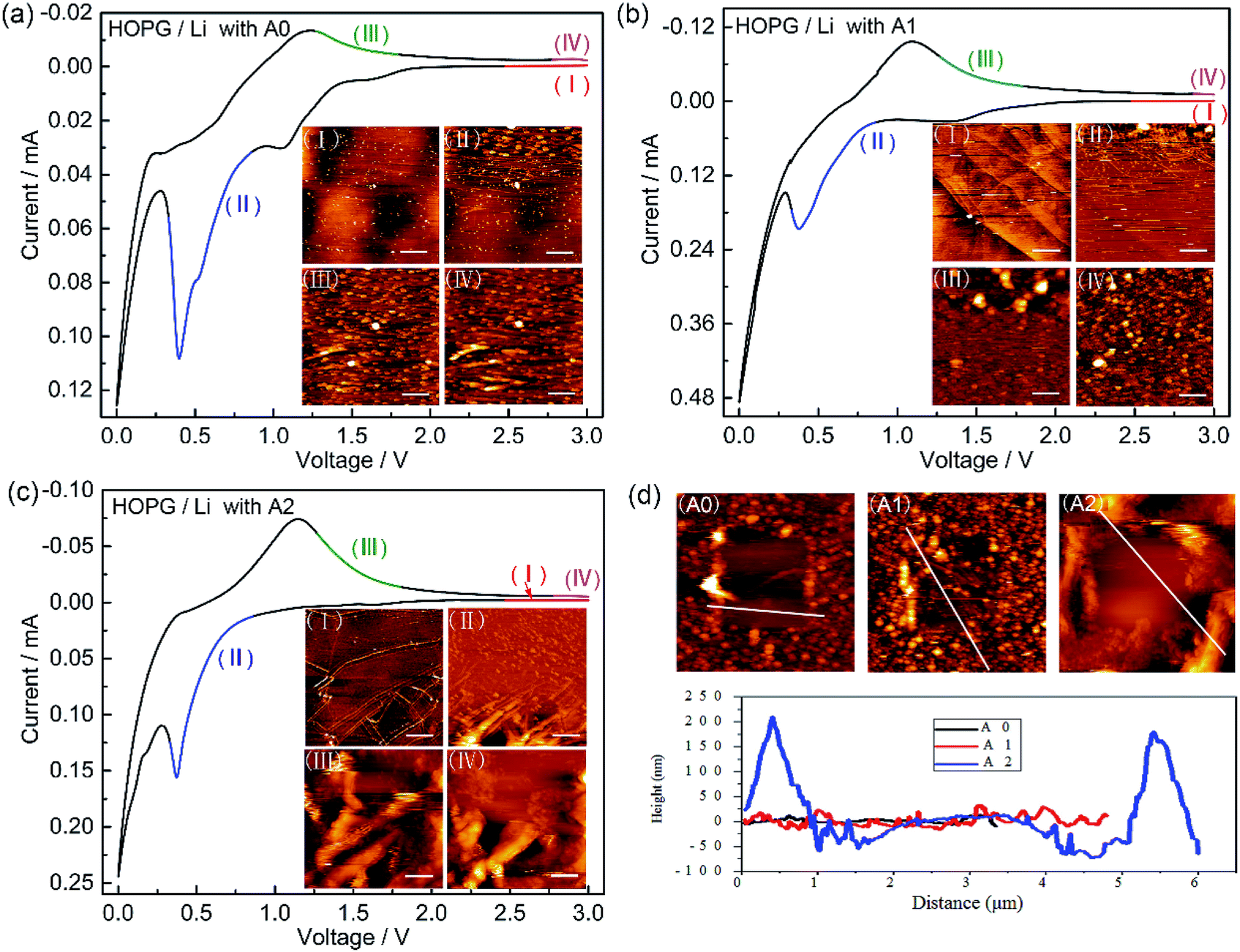 | ||
| Fig. 3 In situ AFM images of HOPG electrode cycled at a scanning rate of 2 mV s−1 between 3.0 and 0.0 V with electrolytes A0 (a), A1 (b) and A2 (c). (I) 3.0–2.47 V; (II) 0.86–0.33 V; (III) 1.27–1.80 V; (IV) 2.87–3.0 V. The scanning directions are from bottom to top. The scales bars are 1 μm. (d) AFM images after scratching a 2.5 × 2.5 μm2 area on the EEI covered surface from electrolytes A0, A1, and A2. The bottom shows line profiles indicated in (A0), (A1), and (A2). | ||
Fig. 4 shows SEM images of the pristine LiCoO2 electrode and LiCoO2 electrodes surface taken out from the cell after 160 cycles at 1.0C. In Fig. 4a, the LiCoO2 particles with layered structure and clean surface can be identified on the fresh cathode. The LiCoO2 particle is broken (Fig. 4b) when cycled in the reference electrolyte. Fig. 4c and d show the image of the LiCoO2 electrode cycled in electrolyte A1 and A2, it can be found that both the surface of cycled electrodes show significant change. There are some substances covered onto the surface of the cathode, which produced from electrolyte decomposition, forming thick residue layer. These results suggest that LiPO2F2 improves the stability of electrodeposition and builds up a more stable interface layer.
 | ||
| Fig. 4 (a) SEM image of pristine LiCoO2 electrode surface. (b) With electrolyte A0 after 160 cycles; (c) with electrolyte A1 after 160 cycles; (d) with electrolyte A2 after 160 cycles. | ||
The XPS spectra of LiPO2F2 and graphite anodes after 160 cycles are depicted in Fig. 5. There are three characteristic peaks in the C 1s spectrum of graphite anode. The peak at 284.8 eV is assigned to C–C bond in carbon black. The peak at 287.1 eV is attributed to C–O bond,31 and the peak at 290.0 eV is classified to OCO2− group, respectively. These groups indicate that ROCO2Li, ROLi and Li2CO3 species are formed on the anode surface, which are originated from the decomposition of LiPF6 and carbonate solvents.31 There is one characteristic peak at 685.5 eV in F 1s spectra of pristine LiPO2F2, but two peaks are appeared on graphite anode surfaces. The peak at 682.1 eV is assigned to LiF,32 and the peak at 685.5 eV is attributed to LiPO2F2, which is in accordance with the F 1s spectra of pristine LiPO2F2. Intensity of the peak at 685.5 eV is enhanced, but the peak intensity of LiF is weakened with increasing concentration of LiPO2F2 in reference electrolyte. In the P 2p spectra, there is a characteristic peak at 133.6 eV for LiPO2F2. Its intensity also increases with increasing LiPO2F2 concentration. These results are consistent with the EDX observations (Fig. S3, ESI†) on graphite surfaces. The surface compositions of LiCoO2 electrode after 160 cycles are analysed by XPS (Fig. S4 ESI†), from the F 1s spectra and P 2p spectra, we get the same information with that from the spectra of graphite surfaces. Combined with the DFT results, we believe that LiPO2F2 could be deposited on the surface of both the cathode and anode surfaces and help to form more stable SEI layers, which inhibit the increase of the impedance of cell during cycles and improve the cyclability performance of LIBs.
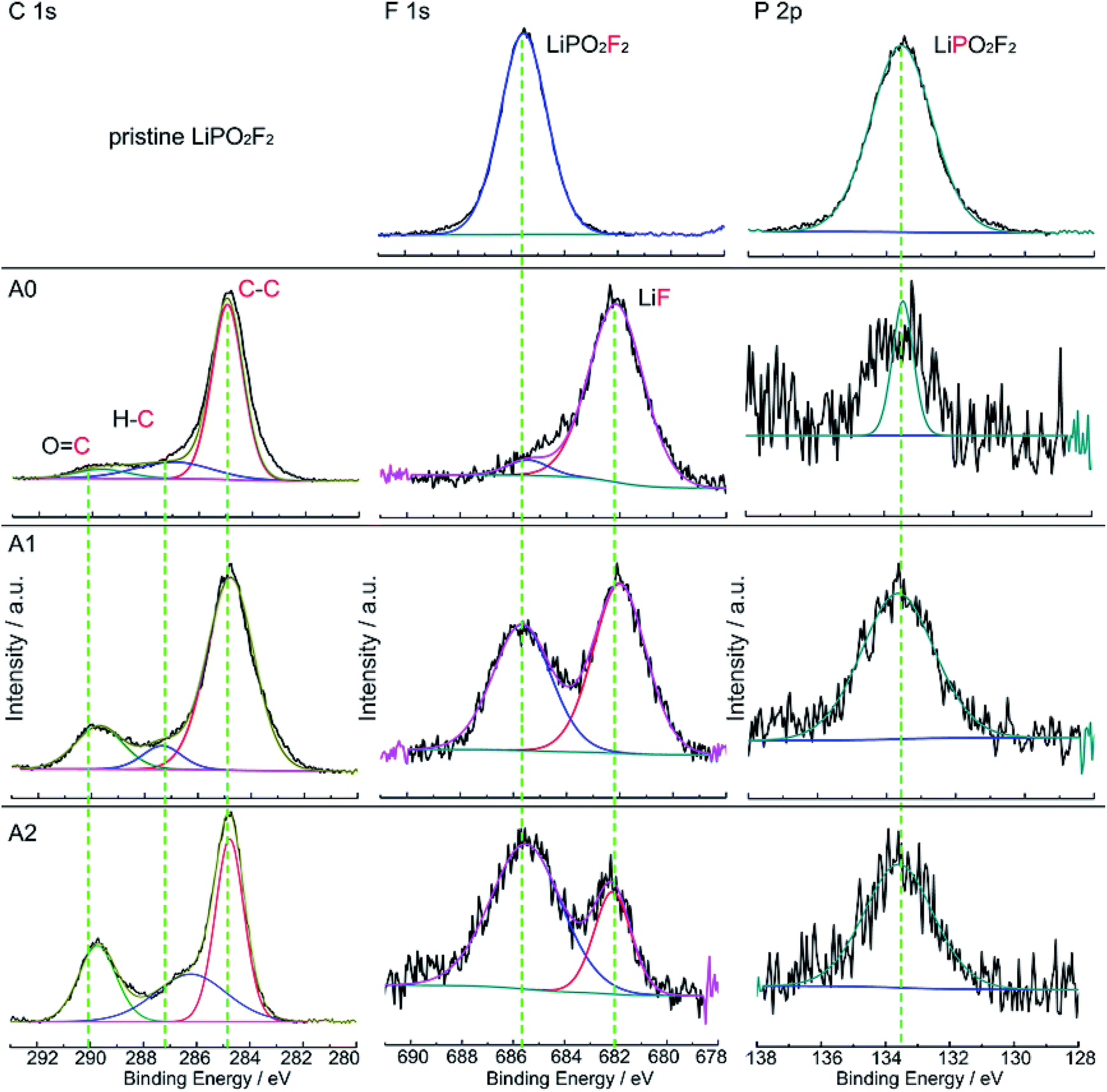 | ||
| Fig. 5 C 1s, F 1s and P 2p XPS spectra of graphite electrodes from the pristine LiPO2F2 (top row), the cell with electrolyte A0, the cell with electrolyte A1, and the cell with electrolyte A2 (bottom row) after 160 cycles at 25 °C. | ||
Discussion for the possible function mechanism
Based on the above evidence, in Fig. 6, we propose a cathode and anode SEI form mechanism with electrolyte containing LiPO2F2 in LiCoO2/graphite 18650 full batteries during charge process. LixPO2F2 (x = 2 or 3) reaches the graphite surface by the electric field, and LiPO2F2 additive would be deposited on graphite surface since LiPO2F2 would not be reduced. Li intercalates into graphite host electrode. During the discharge process, PO2F2− carries many Li ions (LixPO2F2, x = 2 or 3) to the Li1−xCoO2 surface, a majority of Li ions intercalate into Li1−xCoO2 host electrode which can reduce interfacial resistance and form LiCoO2, and LiPO2F2 is left on the surface. The SEI layer containing LiPO2F2 protects LiCoO2 from destruction and suppresses the subsequent decomposition of electrolyte.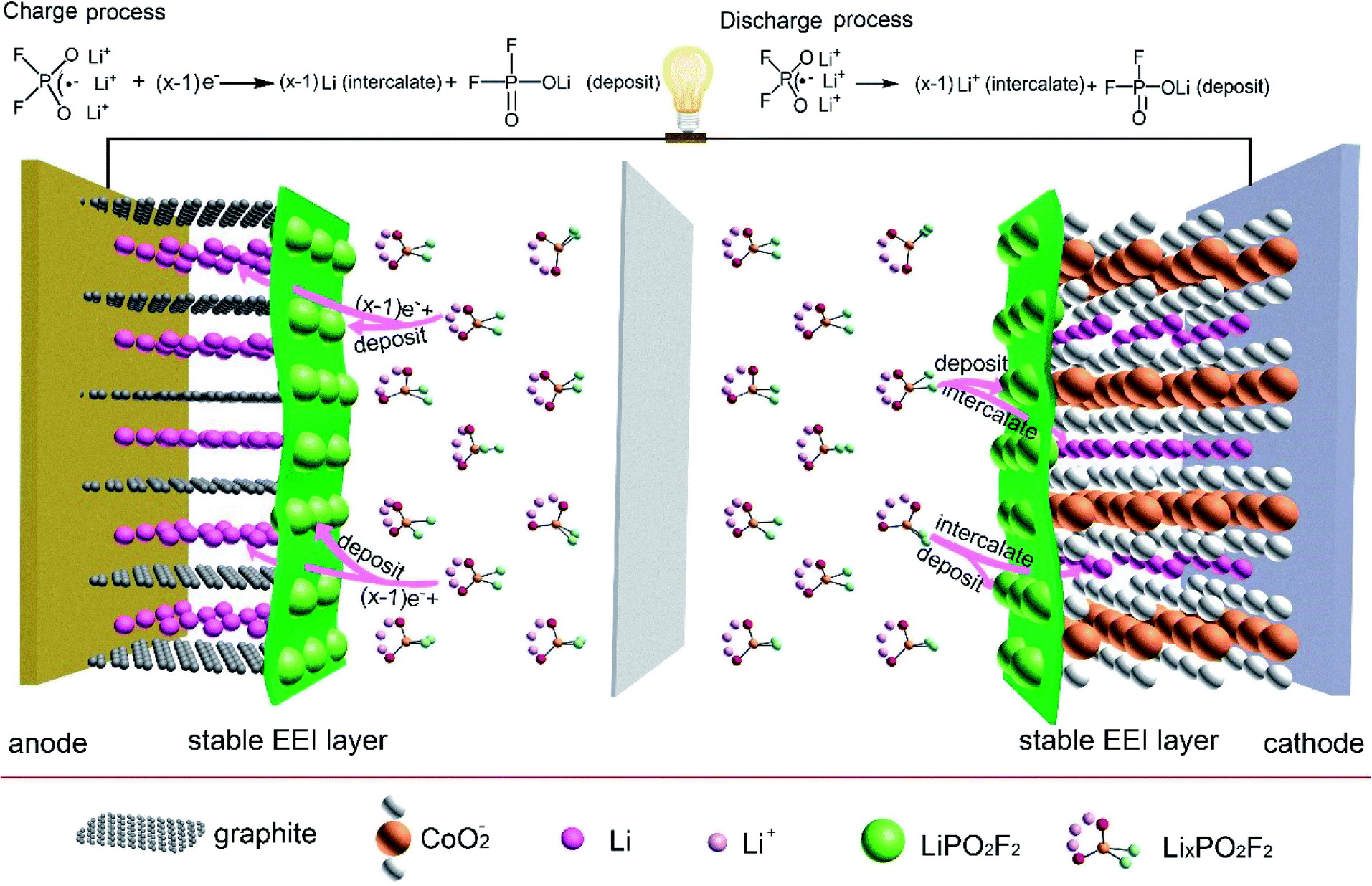 | ||
| Fig. 6 The schematic of the forming process of SEI on the anode and cathode surfaces in the LiCoO2/graphite 18650 full battery with electrolyte containing LiPO2F2, and the reaction on the anode and cathode surfaces during cycle process. | ||
Conclusion
In this work, we used LiPO2F2 as the electrolyte additive. The electrolyte containing LiPO2F2 can effectively improve the cyclability of LIBs. EIS indicated that LiPO2F2 can reduce the surface impedance of cells. DFT calculations and CV confirmed that LiPO2F2 is beneficial in terms of cathodic stability and anodic stability. XPS and EDX showed that LiPO2F2 can be formed an SEI layer with higher content of LiPO2F2 and lower content of LiF. We propose that LiPO2F2 can be deposited in the SEI layer on the anode and cathode surfaces, optimize compositions in the SEI layer and help to form a less impedance and more stable interfacial layer. The SEI layer containing LiPO2F2 can prohibit the successive decomposition of electrolyte and suppress degradation of cathode electrode. Moreover, the maximum amount of LiPO2F2 used in reference electrolyte is only 1.6 wt%, suggesting that it is a very efficient additive. As said in the introduction, we can use the other components in SEI layer, such as LiF, Li2O, LixPOyFz, LixPFy, Li2CO3, ROCO2Li, (CH2OCO2Li)2, ROLi to design electrolyte additives, electrolyte salts, solid polymer electrolytes even artificial SEI layer to improve cyclability, cycle life, or safety of LIBs. We believe that this work provides a new idea for the optimum design of electrolyte and other energy storage equipment.Acknowledgements
The authors acknowledgement C. Shen for in situ AFM determination. This work was financially supported by the National Natural Science Foundation of China (Grant No. 51403227), Strategic Priority Research Program of Chinese Academy of Sciences (CAS, Grant No. XDA09010101), the National Key Research and Development Program of China (Grant No. 2016YFB0100100), the National High-tech R&D program (863 Program) (2015AA033905), and China Postdoctoral Science Foundation (Grant No. 2015T80641).Notes and references
- A. Yoshino, Angew. Chem., Int. Ed. Engl., 2012, 51, 5798–5800 CrossRef CAS PubMed
.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed
- D. Y. Wang, N. N. Sinha, J. C. Burns, R. Petibon and J. R. Dahn, J. Power Sources, 2014, 270, 68–78 CrossRef CAS
- J. Shim, R. Kostecki, T. Richardson, X. Song and K. A. Striebel, J. Power Sources, 2002, 112, 222–230 CrossRef CAS
- S. Menkin, D. Golodnitsky and E. Peled, Electrochem. Commun., 2009, 11, 1789–1791 CrossRef CAS
- A. J. Smith, J. C. Burns, S. Trussler and J. R. Dahn, J. Electrochem. Soc., 2010, 157, A196–A202 CrossRef CAS
- T. Waldmann, M. Wilka, M. Kasper, M. Fleischhammer and M. Wohlfahrt-Mehrens, J. Power Sources, 2014, 262, 129–135 CrossRef CAS
- J. Vetter, P. Novak, M. R. Wagner, C. Veit, K. C. Moller, J. O. Besenhard, M. Winter, M. Wohlfahrt-Mehrens, C. Vogler and A. Hammouche, J. Power Sources, 2005, 147, 269–281 CrossRef CAS
- D. Aurbach, K. Gamolsky, B. Markovsky, Y. Gofer, M. Schmidt and U. Heider, Electrochim. Acta, 2002, 47, 1423–1439 CrossRef CAS
- Y. Lu, Z. Tu and L. A. Archer, Nat. Mater., 2014, 13, 961–969 CrossRef CAS PubMed
- N. N. Sinha, J. C. Burns and J. R. Dahn, J. Electrochem. Soc., 2013, 160, A709–A714 CrossRef CAS
- J. Xia, L. Ma and J. R. Dahn, J. Power Sources, 2015, 287, 377–385 CrossRef CAS
- M. Nagahama, N. Hasegawa and S. Okada, J. Electrochem. Soc., 2010, 157, A748–A752 CrossRef CAS
- H. Duncan, N. Salem and Y. Abu-Lebdeh, J. Electrochem. Soc., 2013, 160, A838–A848 CrossRef CAS
- Y. Yamada, Y. Takazawa, K. Miyazaki and T. Abe, J. Phys. Chem. C, 2010, 114, 11680–11685 CAS
- R. Hayes, G. G. Warr and R. Atkin, Chem. Rev., 2015, 115, 6357–6426 CrossRef CAS PubMed
- M. Gauthier, T. J. Carney, A. Grimaud, L. Giordano, N. Pour, H. H. Chang, D. P. Fenning, S. F. Lux, O. Paschos, C. Bauer, F. Maglia, S. Lupart, P. Lamp and Y. Shao-Horn, J. Phys. Chem. Lett., 2015, 6, 4653–4672 CrossRef CAS PubMed
- K. Xu, Chem. Rev., 2004, 104, 4303–4417 CrossRef CAS PubMed
- M. Y. Nie, D. Chalasani, D. P. Abraham, Y. J. Chen, A. Bose and B. L. Lucht, J. Phys. Chem. C, 2013, 117, 1257–1267 CAS
- A. M. Andersson, D. P. Abraham, R. Haasch, S. MacLaren, J. Liu and K. Amine, J. Electrochem. Soc., 2002, 149, A1358–A1369 CrossRef CAS
- B. Zhang, M. Metzger, S. Solchenbach, M. Payne, S. Meini, H. A. Gasteiger, A. Garsuch and B. L. Lucht, J. Phys. Chem. C, 2015, 119, 11337–11348 CAS
- B. Li, Y. Wang, W. Tu, Z. Wang, M. Xu, L. Xing and W. Li, Electrochim. Acta, 2014, 147, 636–642 CrossRef CAS
- K. E. Kim, J. Y. Jang, I. Park, M. H. Woo, M. H. Jeong, W. C. Shin, M. Ue and N. S. Choi, Electrochem. Commun., 2015, 61, 121–124 CrossRef CAS
- B. W. Yang, H. Zhang, L. Yu, W. Z. Fan and D. H. Huang, Electrochim. Acta, 2016, 221, 107–114 CrossRef CAS
- K. Raghavachari, Theor. Chem. Acc., 2000, 103, 361–363 CrossRef CAS
- L. A. Curtiss, K. Raghavachari, G. W. Trucks and J. A. Pople, J. Chem. Phys., 1991, 94, 7221–7230 CrossRef CAS
- R. McMillan, H. Slegr, Z. X. Shu and W. D. Wang, J. Power Sources, 1999, 81, 20–26 CrossRef
- G. H. Wrodnigg, J. O. Besenhard and M. Winter, J. Electrochem. Soc., 1999, 146, 470–472 CrossRef CAS
- S. K. Jeong, M. Inaba, R. Mogi, Y. Iriyama, T. Abe and Z. Ogumi, Langmuir, 2001, 17, 8281–8286 CrossRef CAS
- J. Jeong, J.-N. Lee, J.-K. Park, M.-H. Ryou and Y. M. Lee, Electrochim. Acta, 2012, 170, 353–359 CrossRef
- M. Xu, L. Hao, Y. Liu, W. Li, L. Xing and B. Li, J. Phys. Chem. C, 2011, 115, 6085–6094 CAS
- M. Q. Xu, Y. L. Liu, B. Li, W. S. Li, X. P. Li and S. J. Hu, Electrochem. Commun., 2012, 18, 123–126 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Additional characterisation data. See DOI: 10.1039/c7ra03926c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |