
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Discovery of new BTK inhibitors with B cell suppression activity bearing a 4,6-substituted thieno[3,2-d]pyrimidine scaffold†
Qiumeng Zhang‡
a,
Luyao Zhang‡bc,
Jie Yua,
Heng Libc,
Shijun Hebc,
Wei Tangbc,
Jianping Zuo*bc and
Wei Lu *a
*a
aSchool of Chemistry and Molecular Engineering, East China Normal University, 3663 North Zhongshan Road, Shanghai 200062, P. R. China. E-mail: wlu@chem.ecnu.edu.cn; Tel: +86-21-62238771
bLaboratory of Immunopharmacology, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, P. R. China. E-mail: jpzuo@simm.ac.cn; Tel: +86-21-50801659
cUniversity of Chinese Academy of Sciences, No. 19A Yuquan Road, Beijing 100049, P. R. China
First published on 16th May 2017
Abstract
A series of 4,6-substituted thieno[3,2-d]pyrimidine derivatives as Bruton's tyrosine kinase (BTK) inhibitors are designed, synthesized and evaluated for their enzymatic inhibition and immunosuppressive activities. These derivatives exhibit varying inhibitory activities against BTK in vitro. Compound 8 is a novel potent BTK inhibitor which has an IC50 value of 29.9 nM, exerts excellent immunosuppressive activity by inhibiting the proliferation of B cells (IC50 = 284 nM), and shows low cytotoxicity (CC50 = 53![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 632 nM) on murine splenocytes. In addition, compound 8 displays considerable selectivity between T cells (IC50 > 10 μM) and B cells. Furthermore, enzymatic assays on more than twenty kinases confirm that compound 8 is more selective than the reference compound Olmutinib. In summary, the results suggest that compound 8 is a potential BTK inhibitor for further evaluation and modification of the C-4 and C-6 position of the thieno[3,2-d]pyrimidine scaffold, which could be considered a new strategy in the development of BTK inhibitors.
632 nM) on murine splenocytes. In addition, compound 8 displays considerable selectivity between T cells (IC50 > 10 μM) and B cells. Furthermore, enzymatic assays on more than twenty kinases confirm that compound 8 is more selective than the reference compound Olmutinib. In summary, the results suggest that compound 8 is a potential BTK inhibitor for further evaluation and modification of the C-4 and C-6 position of the thieno[3,2-d]pyrimidine scaffold, which could be considered a new strategy in the development of BTK inhibitors.
Introduction
Bruton's tyrosine kinase (BTK) is a non-receptor tyrosine kinase that belongs to the Tec kinase family,1,2 which is expressed primarily in immune cells including B cells, monocytes, neutrophils, mast cells and macrophages.3,4 Recently, significant progress has been made in understanding the function and regulation of BTK in multiple signaling pathways,5–10 such as the B cells receptor (BCR) signaling11 and Fcγ receptor (FCR) signaling pathways.12 BTK plays a critical role in B cell development, including proliferation, differentiation, maturation, activation and survival. In the past decades, BTK inhibitors have been considered as a therapeutic option for the treatment of B cell malignancies.13–21 Ibrutinib, which is a selective irreversible BTK inhibitor, has been approved by the US Food and Drug Administration (FDA) for the treatment of patients with chronic lymphocytic leukemia (CLL), mantle cell lymphoma (MCL) and Waldenstrom's macroglobulinemia.9,12–24 Besides, B cells have been recognized to play a pivotal role in regulating immune responses and BTK inhibitors have been proven to be effective in treating autoimmune diseases such as rheumatoid arthritis (RA) and systemic lupus erythematous (SLE).25,26 Several BTK inhibitors have been reported in the clinical development for the treatment of RA or SLE,22,27–33 including Ibrutinib, Olmutinib, Spebrutinib, and Acalabrutinib (Fig. 1).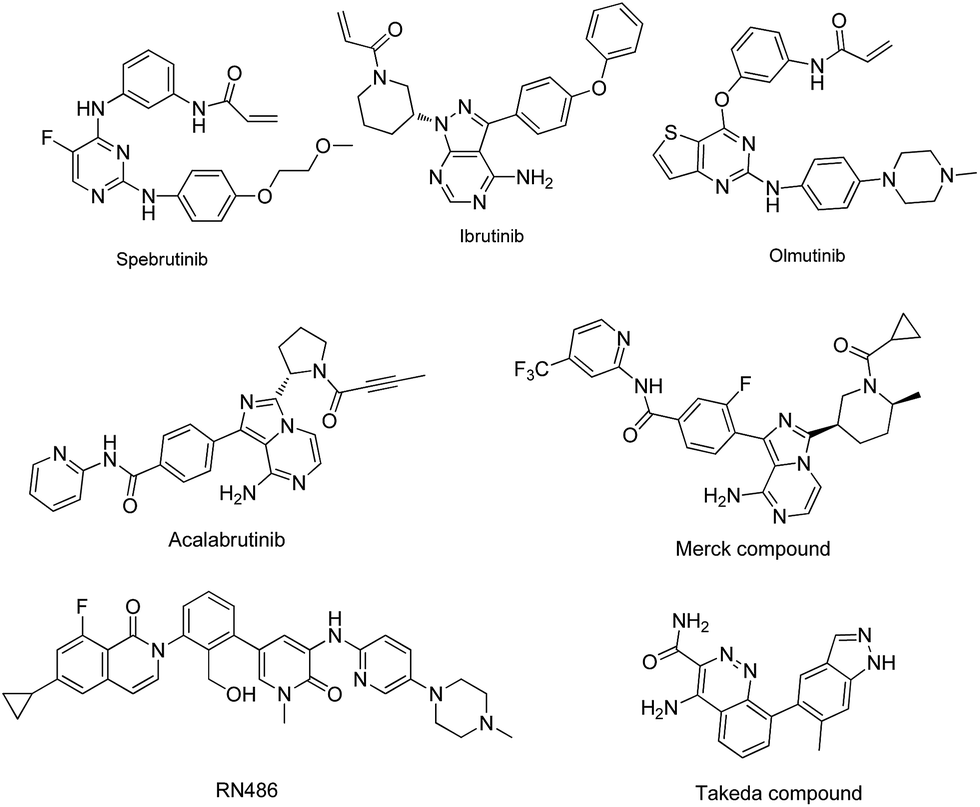 | ||
| Fig. 1 Chemical structures of some clinical BTK inhibitors used in the treatment RA or SLE. | ||
To date, thieno[3,2-d]pyrimidine derivatives have attracted a great deal of attention due to their broad spectrum of biological activities, including antitumor, anti-autoimmune, and antifungal activities. Over the past decades, numerous studies have resulted in vast literature on the modification of the thieno[3,2-d]pyrimidine core. These thieno[3,2-d]pyrimidines exhibit remarkable biological functions by targeting different kinases, such as EGFR, VEGFR, mTOR, PI3K, BTK, HDAC and SIRT (Fig. 2).34–46 Among the reported thieno[3,2-d]pyrimidine derivatives, Olmutinib, which is a 2,4-substituted thieno[3,2-d]pyrimidine, is being developed as an EGFR inhibitor47 for the treatment of non-small cell lung cancer and as a BTK inhibitor27 for autoimmune disorders.
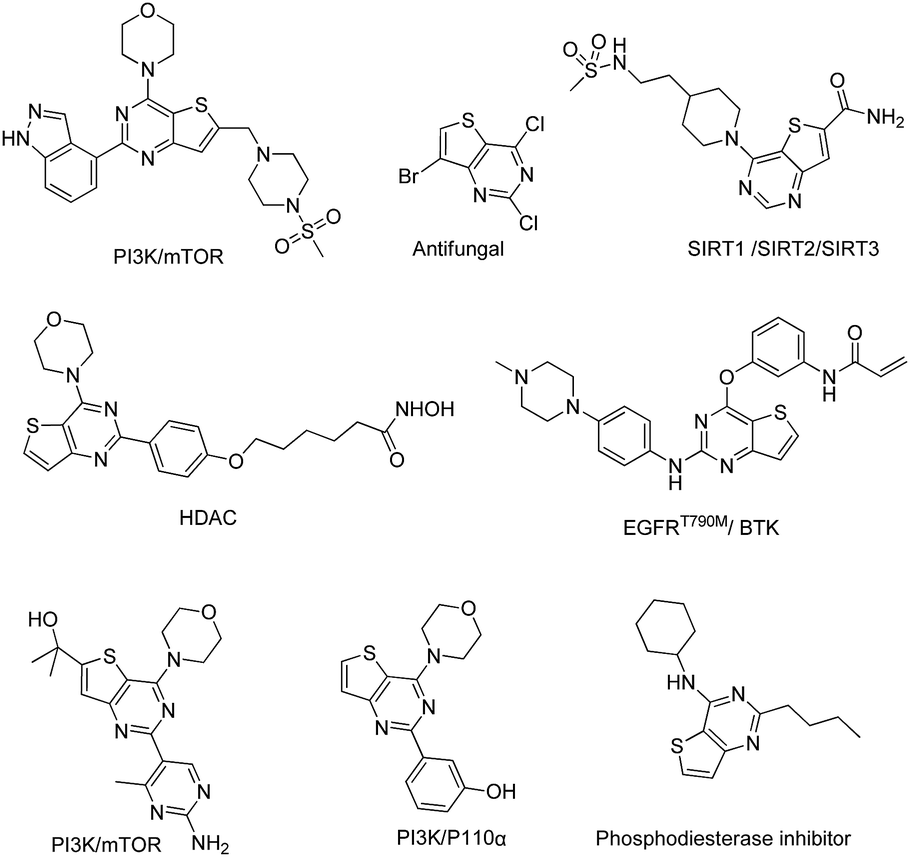 | ||
| Fig. 2 Chemical structures of some thieno[3,2-d]pyrimidines. | ||
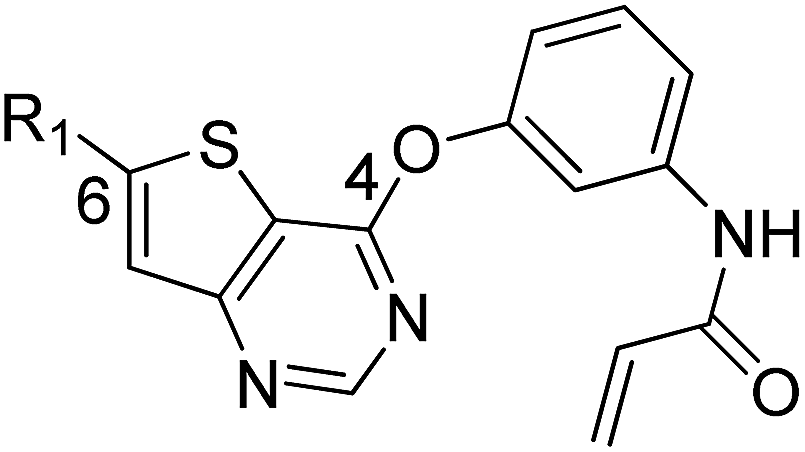
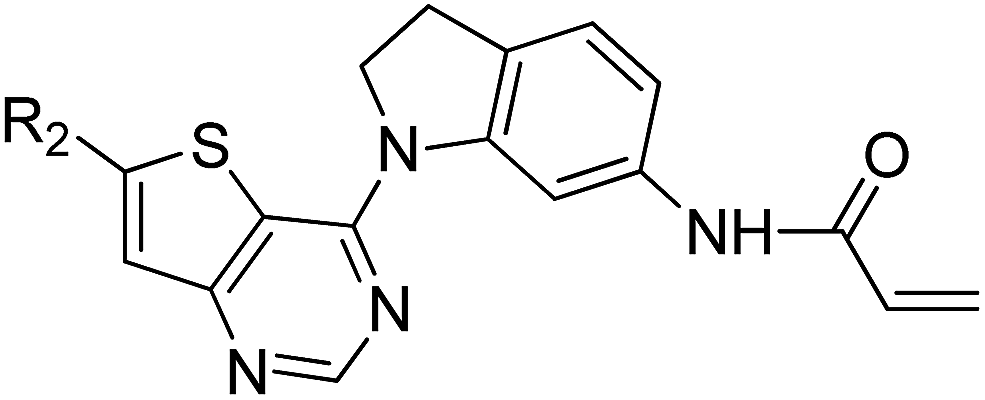
We are interested in developing BTK inhibitors for the treatment of autoimmune diseases and thus attracted to the broad spectrum activities of thieno[3,2-d]pyrimidine derivatives. Therefore, a series of novel thieno[3,2-d]pyrimidine based BTK inhibitors are designed by extending the C-2 substitution of Olmutinib to the C-6 position of our thieno[3,2-d]pyrimidine derivatives using an angle-changing strategy. Considering the space length, the substituents are attached to the thiophene ring through the C–C bond. An electrophilic warhead is essential for irreversible BTK inhibitors to form a covalent bond with Cys481 of BTK, thus the C-4 substitution of Olmutinib is kept on the C-4 position of our thieno[3,2-d]pyrimidine derivatives. The results show that most of the synthesized compounds exhibit weak potency against BTK, whereas compounds 7 and 8 are equipotent to Olmutinib. In addition, it is exciting that compounds 7 and 8 are ineffective in inhibiting EGFR. Thus, high selectivity between BTK and EGFR could be achieved by compounds with the 4,6-substituted thieno[3,2-d]pyrimidine scaffold. Herein, we describe the design, synthesis and preliminary activities of the compounds related to the general structure A (Fig. 3).
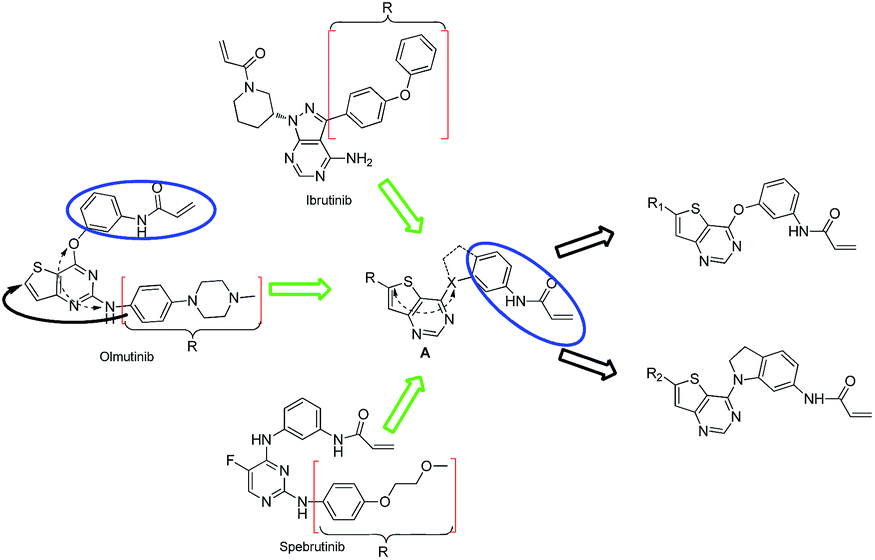 | ||
| Fig. 3 Design of 4,6-substituted thieno[3,2-d]pyrimidines. | ||
Results and discussion
Chemistry
The detailed synthetic strategy to target compounds 4–20 is illustrated in Scheme 1. Compound 1 was prepared according to published procedures.48,49 Intermediates 2 and 3 were obtained from compound 1 through nucleophilic substitution with 3-nitrophenol and 6-nitroindoline, respectively. Compounds 4a–20a were assembled via the following two steps from 2 or 3: (i) Suzuki coupling between intermediates 2/3 and the corresponding boric acid/boric acid ester and (ii) reduction of nitro with RANEY®-Ni under H2. Finally, 4a–20a were reacted with acryloyl chloride to obtain the targets 4–20. Some of the intermediates and final targets have poor solubility, especially 3, 14, 16, 17, 18 and 20.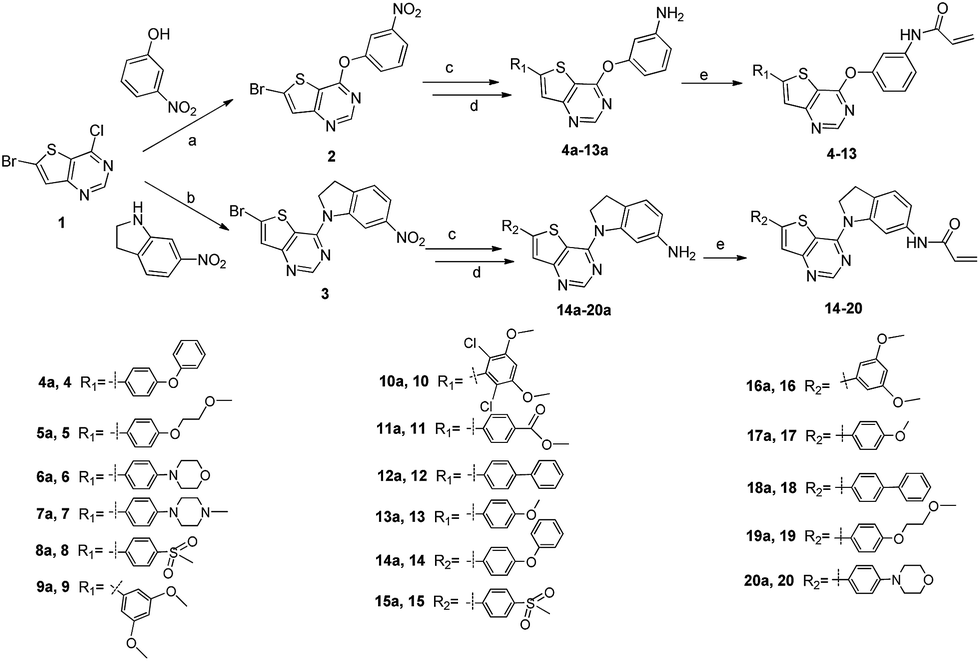 | ||
| Scheme 1 Synthesis of target compounds 4–20. Reagents and conditions: (a) 3-nitrophenol, Cs2CO3, anhydrous DMF, rt, 96%; (b) 6-nitroindoline, trifluoroacetic acid, isopropyl alcohol, 80 °C, 93%; (c) Pd(dppf)Cl2-DCM, Na2CO3, corresponding boric acid or boric acid ester, THF, water, 80 °C, N2, overnight; (d) RANEY®-Ni, H2, MeOH, rt, overnight; and (e) acryloyl chloride, NaHCO3, THF, water, 0 °C. | ||
Biological evaluation
First, the 4,6-substituted thieno[3,2-d]pyrimidine derivatives were evaluated for their activities against BTK using the LanthaScreen® Tb Kinase Activity Assay. The results were expressed as IC50 values and are summarized in Tables 1 and 2. Compound 4, which was obtained by evolving the C-2 substitution of Olmutinib to the C-6 position, was approximately 215-fold less potent than Olmutinib. Among the tested compounds, 7 and 8 were the most potent BTK inhibitors which had IC50 values of 11.9 nM and 29.9 nM, respectively. Apparently, they are equipotent to Olmutinib (IC50 = 13.9 nM) in suppressing BTK activity. Moreover, when the C-4 position of 8 was replaced by N-(indolin-6-yl)acrylamide, the BTK inhibitory activities of compound 15 disappeared (IC50 > 10000 nM). Besides, most of the compounds shown in Table 2 exhibit negligible activities against BTK. These results suggest that substitution of the indoline ring at the C-4 position is unfavorable for the interaction with BTK. In addition, the wide varieties of activities shown in Table 1, which range from 11.9 nM to more than 10000 nM, indicating that BTK is sensitive to the substitution at the C-6 position.
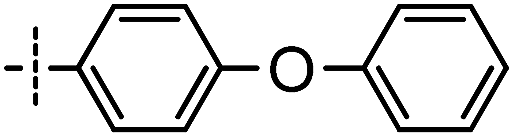
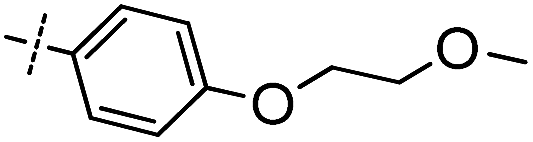
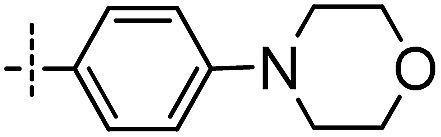
Based on the overall profile of this series, compounds 7 and 8 were selected for further profiling because of their excellent enzymatic potency. Since the structures of 7 and 8 were similar with the dual BTK and EGFR inhibitor Olmutinib, the EGFR kinase activity assay was performed. As shown in Table 3, compounds 7 and 8 were ineffective in inhibiting EGFR (IC50 > 10000 nM). The high selectivity between BTK and EGFR of 7 and 8 is beneficial in the treatment of autoimmune diseases.
In order to evaluate the immunosuppressive activity of compounds 7 and 8, the nonspecific toxicity of murine splenocytes was determined using a Cell Counting Kit-8 (CCK-8) and lymphocytes proliferation responses were determined using the 3H-TdR incorporation method. Also, concanavalin A (Con A) and lipopolysaccharide (LPS), which induce the proliferation of T cells and B cells, respectively, were used. The results are summarized in Table 4. Interestingly, compound 7 exhibits high cytotoxicity with a CC50 value of 557 nM, however, its toxicity was greatly reduced when the C-6 position substituents of p-methylpiperazinephenyl was converted into p-methanesulfonylphenyl (8, CC50 = 53632 nM). Besides, 7 and 8 show potent inhibition against B cells (184 nM and 284 nM, respectively) but are ineffective against T cells (4507 nM and >10000 nM, respectively). Considering that BTK mediates signaling pathways downstream of BCR in B cells,50 not T cells, the immunosuppressive results are reasonable. In summary, 7 is confirmed to possess no immunosuppressive activity against B cells due to its high cytotoxicity, and 8 exhibits excellent immunosuppressive activity by inhibiting B cells compared with Olmutinib and Ibrutinib.
Furthermore, we screened the inhibitory activities of compound 8 against twenty-five selected kinases related to the SRC kinase family, JAK family, MAPK family and others. Compound 8 and Olmutinib were measured at single concentration of 200 nM in duplicate. As presented in Fig. 4, Olmutinib exhibits potent inhibition of JAK3, EGFR, ITK, BLK and BTK at 200 nM. Compound 8 has a negligible impact on most of the tested kinases except BTK and BLK, which indicates that compound 8 has relatively better kinase selectivity than Olmutinib.
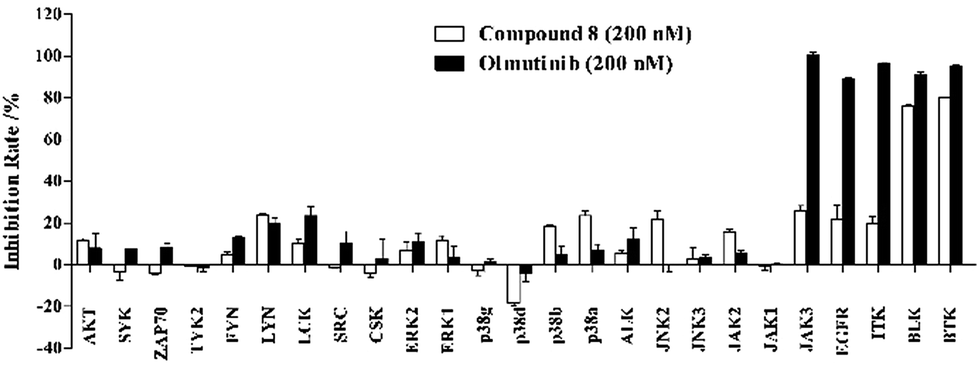 | ||
| Fig. 4 Enzyme inhibition screen of 8 and Olmutinib against twenty-five selected kinases. | ||
Conclusion
In summary, a series of 4,6-substituted thieno[3,2-d]pyrimidines were designed and synthesized. Among the derivatives, compounds 7 and 8 with IC50 values of 11.9 nM and 29.9 nM, respectively, exhibit potent anti-BTK activity that is equipotent to Olmutinib. Due to the fact that BTK plays a critical role in B cell derived diseases, the nonspecific toxicity and proliferation responses of murine splenocytes were analyzed. Compound 8 exhibits excellent immunosuppressive activity by inhibiting B cell proliferation (IC50 = 284 nM) and low cytotoxicity (CC50 = 53632 nM). Furthermore, the enzymatic inhibition screen assays on more than twenty kinases confirm that compound 8, which has negligible activities for EGFR, JAK3, and ITK, is more selective than the reference compound Olmutinib. These preliminary results suggest that compound 8 is a novel promising BTK inhibitor for further evaluation and modification of the 4,6-substituted thieno[3,2-d]pyrimidine scaffold could be applied in the development of novel BTK inhibitors.
Experimental
Material and methods
All reagents and solvents were purchased from suppliers and purified/dried if an anhydrous solvent was necessary. Mass spectra (MS) were recorded on a Waters SDQ mass spectrometer and high resolution mass spectra (HRMS) were recorded on a Waters SYNAPT G2 ESI-TOF-MS analyzer. 1H NMR spectra and 13C NMR spectra were recorded in CDCl3, MeOD, CF3COOD, D2O and DMSO-d6 on a Bruker DRX-400 (400 MHz) using TMS as the internal standard. Chemical shifts are reported as δ (ppm) and spin–spin coupling constants as J (Hz) values. Melting points were taken on a SGW X-4 melting point apparatus, uncorrected and reported in degrees centigrade. Column chromatography was performed with silica gel (200–300 mesh). The purity of all tested compounds was established by HPLC to be >95%.Synthetic procedures
Step 1. A solution of compound 2 or 3 (0.5 mmol), corresponding boric acid or boric acid ester (0.75 mmol) and Pd(dppf)Cl2-DCM (41 mg, 0.05 mmol) in THF (10 mL) was degassed with N2 for 5 min followed by the addition Na2CO3 (106 mg, 0.5 M in water) under a continuous flow of nitrogen. The reaction mixture was stirred at 70 °C until the reaction was complete (monitored by TLC). The reaction mixture was allowed to cool to room temperature and then filtered. The filter cake was washed with water and DCM several times to afford the crude product (poor solubility). The crude product was suspended in methanol (30 mL) and then the mixture was purged with N2. RANEY®-Ni was added and the mixture was purged with N2 again, then H2. The resulting solution was stirred at room temperature overnight. MeOH (20 mL) and DCM (50 mL) were added and the solution was filtered and concentrated in vacuo to afford 4a–20a, which were used directly in the next step.
Step 2. Corresponding 4a–20a (0.1 mmol) was dissolved or suspended in THF (10 mL) and then NaHCO3 (0.2 mmol, 2 M in water) was added. Subsequently, acryloyl chloride (0.11 mmol, 1 M in anhydrous THF) was added to the solution dropwise at 0 °C. The resulting mixture was stirred for 1 h (monitored by TLC). Then EA (20 mL) and water (30 mL) were added and the layers were partitioned and separated. The organic layers were washed with NH4Cl solution, then washed with water and brine, and dried over anhydrous sodium sulfate. The filtrate was concentrated and crystallized from MeOH/CH2Cl2/hexane to give the corresponding product 4–20.
TR-FRET LanthaScreen assay (enzyme inhibition assay)
Firstly, we optimized the LanthaScreen™ kinase assay for BTK (ThermoFisher Scientific, Waltham, MA, USA) according to the manufacture's specifications. TR-FRET assays were performed by incubating a diluted series of compound concentrations with ATP (ThermoFisher Scientific), fluorescein–poly GT substrate (ThermoFisher Scientific) and BTK Kinase (ThermoFisher Scientific) in kinase reaction buffer (ThermoFisher Scientific). The kinase reaction buffer consisted of 50 mM HEPES pH 7.5, 0.01% BRIJ-35, 10 mM MgCl2, and 1 mM EGTA. The kinase reaction mixtures were incubated at room temperature (23 ± 2 °C) for 1 h before stopping the kinase reaction by the addition of 10 mM EDTA (ThermoFisher Scientific). The phosphorylation of the substrate by BTK was detected using Tb-PY20 antibody (ThermoFisher Scientific) in TR-FRET dilution buffer at pH 7.5 (ThermoFisher Scientific) and then measured by determining the emission ratio of 520/495 nm on an microplate reader (SpectraMax M5, Molecular Devices). IC50 was estimated using the log(inhibitor) vs. response non-linear fit (GraphPad Prism 6.0). Additional assays were similarly carried out to determine selectivity over EGFR (ThermoFisher Scientific).Animals
Inbred 6–8 week-old female BALB/c mice were purchased from Shanghai Lingchang Biotechnology Co., Ltd. (certificate no. 2013-0018, Shanghai, China). Normal BALB/c mice (n = 6) were used for preparation of splenocytes and all mice were raised under specific pathogen-free conditions and kept in a 12 h light/dark cycle with controlled humidity (60–80%) and temperature (22 ± 1 °C). Experiments were carried out according to the National Institutes of Health Guides for the Care and Use of Laboratory Animals and were approved by the Bioethics Committee of the Shanghai Institute of Materia Medica Sciences.Cell viability assay
Preparation of splenocytes from BALB/c mice was performed as previously described.51 Splenocytes (1 × 106 cells) were cultured in 96-well plates in triplicate with 200 μL RPMI 1640 media containing 10% FBS, penicillin (100 U mL−1), and streptomycin (100 μg mL−1) in a humidified, 37 °C, 5% CO2-containing incubator for 48 h in the presence or absence of indicated concentrations of compounds. A total of 20 μL CCK-8 was added to each well. After 6–8 h incubation, the absorbance value at 450 nm (570 nm calibration) was measured using a microplate reader (Molecular Devices, Sunnyvale, CA, USA) and the cell viability was calculated.Proliferation responses of murine lymphocytes
Splenocytes (5 × 105 cells) were cultured in triplicate for 48 h with 5 μg mL−1 of ConA (Sigma-Aldrich, St Louis, MO) or 10 μg mL−1 of LPS (Sigma-Aldrich) in the presence or absence of indicated concentrations of compounds. Cells were pulsed with 0.5 μCi per well of [3H]-thymidine (Perkin Elmer, MA, USA) for 8 h and harvested. The incorporated radioactivity was then counted using a Beta Scintillation Counter (MicroBeta Trilux, Perkin Elmer Life Sciences, Boston, MA). The IC50 was estimated using the log(inhibitor) vs. response non-linear fit (GraphPad Prism 6.0).In vitro enzymatic screen assays
The selectivity of attractive compounds for BTK and non-BTK kinases was determined by testing the compounds at a single 200 nM concentration against a panel of 25 kinases using the Caliper assay at Shanghai ChemPartner Co. Ltd (Shanghai, China). Firstly, the compounds were incubated at the final concentration of 200 nM with ATP, enzyme and FAM-labeled peptide in kinase base buffer. The kinase base buffer contained 50 mM HEPES (pH 7.5), 0.0015% Brij-35, 10 mM MgCl2 and 2 mM DTT. The kinase reaction mixtures were then incubated at 28 °C for a specified period of time. Finally, 25 μL of stop buffer was added to stop the reaction. The stop buffer consisted of 100 mM HEPES (pH 7.5), 0.015% Brij-35, 0.2% coating reagent #3 and 50 mM EDTA. Data was collected on a Caliper and the conversion data copied from the Caliper program. The inhibition rate was calculated using following formula: percent inhibition = (max − conversion)/(max − min) × 100, where, max stands for the DMSO control and min stands for the low control.Acknowledgements
This work was supported by the “Personalized Medicines—Molecular Signature-based Drug Discovery and Development”, Strategic Priority Research Program of the Chinese Academy of Sciences, Grant No. XDA12020337.Notes and references
- C. I. Edvard Smith, T. C. Islam, P. T. Mattsson, A. J. Mohamed and B. F. Nore, BioEssays, 2001, 23, 436–446 CrossRef PubMed.
- J. G. Rodriguez, J. A. Readinger, I. C. Viorritto, K. L. Mueller, R. A. Houghtling and P. L. Schwartzberg, Immunol. Rev., 2007, 218, 45–64 CrossRef PubMed.
- S. Tsukada, D. C. Saffran, D. J. Rawlings, O. Parolini, R. C. Allen, I. Klisak, R. S. Sparkes, H. Kubagawa, T. Mohandas, S. Quan, J. W. Belmont, M. D. Cooper, M. E. Conley and O. N. Witte, Cell, 1993, 72, 279–290 CrossRef CAS PubMed.
- D. Vetrie, I. Vorechovsky, P. Sideras, J. Holland, A. Davies, F. Flinter, L. Hammarstrom, C. Kinnon, R. Levinsky, M. Bobrow, C. I. Edvard Smith and D. R. Bentley, Nature, 1993, 361, 226–233 CrossRef CAS PubMed.
- M. de Weers, M. C. Verschuren, M. E. Kraakman, R. G. Mensink, R. K. Schuurman, J. J. van Dongen and R. W. Hendriks, Eur. J. Immunol., 1993, 23, 3109–3114 CrossRef CAS PubMed.
- C. Brunner, B. Muller and T. Wirth, Histol. Histopathol., 2005, 20, 945–955 CAS.
- A. J. Mohamed, L. Yu, C. Bäckesjö, L. V. R. Faryal, A. Aints, B. Christensson, A. Berglöf, M. Vihinen, B. F. Nore and C. I. Edvard Smith, Immunol. Rev., 2009, 228, 58–73 CrossRef CAS PubMed.
- J. A. Woyach, E. Bojnik, A. S. Ruppert, M. R. Stefanovski, V. M. Goett, K. A. Smucker, L. L. Smith and J. A. Dubovsky, et al., Blood, 2014, 123, 1207–1213 CrossRef CAS PubMed.
- S. Ponader, S. S. Chen, J. J. Buggy, K. Balakrishnan, V. Gandhi, W. G. Wierda and M. J. Keatin, et al., Blood, 2012, 119, 1182–1189 CrossRef CAS PubMed.
- H. Mueller, A. Stadtmann, H. V. Aken, E. Hirsch, D. Wang, K. Ley and A. Zarbock, Blood, 2010, 115, 3118–3127 CrossRef CAS PubMed.
- C. U. Niemann and A. Wiestner, Semin. Cancer Biol., 2013, 23, 410–421 CrossRef CAS PubMed.
- L. Ren, A. Campbell, H. Q. Fang, S. Gautam, S. Elavazhagan and K. Fatehchand, et al., J. Biol. Chem., 2016, 291, 3043–3052 CrossRef CAS PubMed.
- J. A. Burger, Curr. Hematol. Malig. Rep., 2014, 9, 44–49 CrossRef PubMed.
- Y. Lou, Z. K. Sweeney, A. Kuglstatter, D. Davis, D. M. Goldstein, X. C. Han, J. Hong, B. Kocer and R. K. Kondru, et al., Bioorg. Med. Chem. Lett., 2015, 25, 367–371 CrossRef CAS PubMed.
- E. K. Evans, R. Tester, S. Aslanian, R. Karp, M. Sheets and M. T. Labenski, et al., J. Pharmacol. Exp. Ther., 2013, 346, 219–228 CrossRef CAS PubMed.
- Q. J. Liu, D. G. Batt, J. S. Lippy, N. Surti, A. J. Tebben and J. K. Muckelbauer, et al., Bioorg. Med. Chem. Lett., 2015, 25, 4265–4269 CrossRef CAS PubMed.
- H. L. Gardner, B. K. Harrington, R. Izumi, A. Hamdy, A. Kaptein, B. V. Lith, C. A. London, J. C. Byrd, A. J. Johnson and W. C. Kisseberth, AACR Annual Meeting, San Diego, CA, April 5–9, 2014 Search PubMed.
- X. L. Gao, J. Wang, J. Liu, D. Guiadeen, A. Krikorian and S. B. Boga, et al., Bioorg. Med. Chem. Lett., 2017, 27, 1471–1477 CrossRef CAS PubMed.
- D. Zhao, S. S. Huang, M. H. Qu, C. Y. Wang, Z. H. Liu, Z. Li, J. Y. Peng, K. X. Liu, Y. X. Li, X. D. Ma and X. H. Shu, Eur. J. Med. Chem., 2017, 126, 444–455 CrossRef CAS PubMed.
- E. D. Deeks, Drugs, 2017, 77, 225–236 CrossRef CAS PubMed.
- Y. Zou, J. Xiao, Z. Tu, Y. Zhang, K. Yao, M. Luo, K. Ding, Y. Zhang and Y. Lai, Bioorg. Med. Chem. Lett., 2016, 26, 3052–3059 CrossRef CAS PubMed.
- L. A. Honigberg, A. M. Smith, M. Sirisawad, E. Verner, D. Loury, B. Chang, S. Li, Z. Pan, D. H. Thamm, R. A. Miller and J. J. Buggy, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13075–13080 CrossRef CAS PubMed.
- A. Akinleye, Y. M. Chen, N. Mukhi, Y. P. Song and D. L. Liu, J. Hematol. Oncol., 2013, 6, 59 CrossRef CAS PubMed.
- Y. Ge, Y. Jin, C. Y. Wang, J. B. Zhang, Z. Y. Tang, J. Y. Peng, K. X. Liu, Y. X. Li, Y. W. Zhou and X. D. Ma, ACS Med. Chem. Lett., 2016, 7, 1050–1055 CrossRef CAS PubMed.
- G. López-Herrera, A. Vargas-Hernández, M. E. González-Serrano, L. Berrón-Ruiz, J. C. Rodríguez-Alba, F. Espinosa-Rosales and L. Santos-Argumedo, J. Leukocyte Biol., 2014, 95, 243–250 CrossRef PubMed.
- A. D. Chu and B. Y. Chang, OA Arthritis, 2013, 1, 17–23 CrossRef.
- P. Norman, Expert Opin. Invest. Drugs, 2016, 25, 891–899 CrossRef CAS PubMed.
- J. Liu, D. Guiadeen, A. Krikorian, X. L. Gao, J. Wang and S. B. Boga, et al., ACS Med. Chem. Lett., 2016, 7, 198–203 CrossRef CAS PubMed.
- D. G. Xu, Y. Kim, J. Postelnek, M. D. Vu, D. Q. Hu, C. Liao and M. Bradshaw, et al., J. Pharmacol. Exp. Ther., 2012, 341, 90–103 CrossRef CAS PubMed.
- Y. Lou, X. C. Han, A. Kuglstatter, R. K. Kondru, Z. K. Sweeney, M. Soth and J. McIntosh, et al., J. Med. Chem., 2015, 58, 512–516 CrossRef CAS PubMed.
- C. R. Smith, D. R. Dougan, M. Komandla, T. Kanouni, B. M. Knight, J. D. Lawson, M. Sabat, E. R. Taylor, P. Vu and C. Wyrick, J. Med. Chem., 2015, 58, 5437–5444 CrossRef CAS PubMed.
- K. H. Kim, A. Maderna, M. E. Schnute, M. Hegen, S. Mohan, J. Miyashiro, L. Lin, E. Li, S. Keegan, J. Lussier and C. Wrocklage, et al., Bioorg. Med. Chem. Lett., 2011, 21, 6258–6263 CrossRef CAS PubMed.
- Q. Shi, A. Tebben, A. J. Dyckman, H. Li, C. J. Liu, J. Lin, S. Spergel, J. R. Burke, K. W. McIntyre and G. C. Olini, et al., Bioorg. Med. Chem. Lett., 2014, 24, 2206–2211 CrossRef CAS PubMed.
- J. S. Lee, K. Park, J. Y. Han, K. H. Lee, J. H. Kim, E. K. Cho, J. Y. Cho, Y. J. Min, J. S. Kim, H. G. Kim, B. S. Kim, J. Jung and D. W. Kim, Ann. Oncol., 2015, 26(9), ix128–ix129 CrossRef PubMed.
- B. S. Safina, S. Baker, M. Baumgardner, P. M. Blaney, B. K. Chan, Y. H. Chen, M. W. Cartwright, G. Castanedo and C. Chabot, et al., J. Med. Chem., 2012, 55, 5887–5900 CrossRef CAS PubMed.
- D. P. Sutherlin, D. Sampath, M. Berry, G. Castanedo, Z. G. Chang, I. Chuckowree and J. Dotson, et al., J. Med. Chem., 2010, 53, 1086–1097 CrossRef CAS PubMed.
- K. W. Temburnikar, S. C. Zimmermann, N. T. Kim, C. R. Ross, C. Gelbmann, C. E. Salomon, G. M. Wilson, J. Balzarini and K. L. Seley-Radtke, Bioorg. Med. Chem., 2014, 22, 2113–2122 CrossRef CAS PubMed.
- Q. Tan, Z. T. Zhang, J. Hui, Y. Zhao and L. Zhu, Bioorg. Med. Chem., 2014, 22, 358–365 CrossRef CAS PubMed.
- E. Perspicace, V. Jouan-Hureaux, R. Ragno, F. Ballante, S. Sartini and C. L. Motta, et al., Eur. J. Med. Chem., 2013, 63, 765–781 CrossRef CAS PubMed.
- J. S. Disch, G. Evindar, C. H. Chiu, C. A. Blum, H. Dai and L. Jin, et al., J. Med. Chem., 2013, 56, 3666–3679 CrossRef CAS PubMed.
- W. F. Zhu, Y. J. Liu, X. Zhai, X. Wang, Y. Zhu, D. Wu, H. Y. Zhou, P. Gong and Y. F. Zhao, Eur. J. Med. Chem., 2012, 57, 162–175 CrossRef CAS PubMed.
- Z. J. Liu, S. S. Wu, Y. Wang, R. J. Li, J. Wang, L. H. Wang, Y. F. Zhao and P. Gong, Eur. J. Med. Chem., 2014, 87, 782–793 CrossRef CAS PubMed.
- A. J. Folkes, K. Ahmadi, W. K. Alderton, S. Alix, S. J. Baker, G. Box and I. S. Chuckowree, et al., J. Med. Chem., 2008, 51, 5522–5532 CrossRef CAS PubMed.
- M. Hayakawa, H. Kaizawa, H. Moritomo, T. Koizumi, T. Ohishi, M. Okada, M. Ohta, S. Tsukamoto, P. Parker, P. Workmanc and M. Waterfieldd, Bioorg. Med. Chem., 2006, 14, 6847–6858 CrossRef CAS PubMed.
- J. Wang, M. B. Su, T. T. Li, A. H. Gao, W. Yang, L. Sheng, Y. Zang, J. Li and H. Liu, Eur. J. Med. Chem., 2017, 128, 293–299 CrossRef CAS PubMed.
- S. Bugge, A. F. Buene, N. Jurisch-Yaksi, I. U. Moen, E. M. Skjønsfjell, E. Sundby and B. H. Hoff, Eur. J. Med. Chem., 2016, 107, 255–274 CrossRef CAS PubMed.
- E. S. Kim, Drugs, 2016, 76, 1153–1157 CrossRef CAS PubMed.
- J. L. Woodring, G. Patel, J. Erath, R. Behera, P. J. Lee, S. E. Leed, A. Rodriguez, R. J. Sciotti, K. Mensa-Wilmot and M. P. Pollastri, MedChemComm, 2015, 6, 339–346 RSC.
- W. Devine, J. L. Woodring, U. Swaminathan, E. Amata, G. Patel, J. Erath, N. E. Roncal, P. J. Lee, S. E. Leed, A. Rodriguez, K. Mensa-Wilmot, R. J. Sciotti and M. P. Pollastri, J. Med. Chem., 2015, 58, 5522–5537 CrossRef CAS PubMed.
- C. I. Smith, B. Baskin, P. Humire-Greiff, J. N. Zhou, P. G. Olsson, H. S. Maniar, P. Kjellén, J. D. Lambris, B. Christensson and L. Hammarström, J. Immunol., 1994, 152, 557–565 CAS.
- R. Zhou, F. Zhang and P. L. He, et al., Int. Immunopharmacol., 2005, 5, 1895–1903 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: General HPLC methods, spectrum of the corresponding compounds and the IC50 curves of compounds 7, 8 and Olmutinib against BTK and EGFR. See DOI: 10.1039/c7ra04261b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |