
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMOF-derived RuO2/Co3O4 heterojunctions as highly efficient bifunctional electrocatalysts for HER and OER in alkaline solutions†
Haizhen Liu a,
Guoliang Xiaa,
Ruirui Zhanga,
Peng Jianga,
Jitang Chena and
Qianwang Chen*ab
a,
Guoliang Xiaa,
Ruirui Zhanga,
Peng Jianga,
Jitang Chena and
Qianwang Chen*ab
aHefei National Laboratory for Physical Science at Microscale, Department of Materials Science & Engineering, Collaborative Innovation Center of Suzhou Nano Science and Technology, University of Science and Technology of China, Hefei 230026, China. E-mail: cqw@ustc.edu.cn
bHigh Magnetic Field Laboratory, Hefei Institutes of Physical Science, Chinese Academy of Sciences, Hefei 230031, China
First published on 13th January 2017
Abstract
The oxygen evolution reaction (OER) and hydrogen evolution reaction (HER) are key half reactions involved in electrochemical water splitting. The design of active and robust Co3O4-based electrocatalysts for overall water splitting in basic media is highly desirable but still remains a great challenge. Herein, a catalyst of a combined metal oxide heterojunction (RuO2/Co3O4) was synthesized by directly annealing a MOF-derived Co–Ru complex under an air atmosphere. The catalyst shows a low OER and HER overpotential of only 305 mV and 89 mV at 10 mA cm−2 in 1 M KOH solution, respectively. It contains only a small amount of precious metal oxides, however, and demonstrates a better performance than most of the other Co3O4-based electrocatalysts reported at the present stage.
Introduction
Nowadays, more and more attention has been paid to the increase in global energy consumption and environmental problems due to burning fossil fuels associated with transportation and industry.1,2 It is well known that splitting water into hydrogen and oxygen to store light or electric energy in the form of chemical bonds plays an important role in developing clean-energy technologies.3–6 The water splitting reaction can be divided into two half-reactions: the hydrogen evolution reaction (HER) and the oxygen evolution reaction (OER), both of which are essential for the overall efficiency of water splitting. It has been demonstrated that electrochemical water splitting can be completed both in acidic and alkaline media. Unfortunately, the acid electrolyzers are hindered by the lack of efficient and low-cost counter electrode catalysts in acidic electrolytes. Therefore, tremendous efforts have been made to develop bi-functional catalysts in basic media due to the available alkaline oxygen evolution electrocatalysts.7–10Precious-metal electrocatalysts, such as IrO2 and RuO2, are regarded as pioneering OER and HER catalysts, respectively, and can be implicated in a large window of solution pHs with high efficiencies.11,12 But their low abundance and high cost prevent them from large range of applications.13,14 Alternatively, the first-row transition metal oxides are currently intensively investigated to improve the intrinsic activity as potential earth-abundant material candidates for water splitting electrocatalysts.15–19 It has been reported that compounds of Co3O4-based compounds exhibit high activities toward OER,20,21 which can serve as a low-cost replacement for the state-of-the-art RuO2, due to its practical availability, environmentally benign nature and low cost.22 However, compared to the precious metals-based catalysts, Co3O4 still exhibits a relatively lower catalytic activity, which has unfortunately limited its large-scale applications.14 The easy accumulation and low conductivity of pure Co3O4 decrease the active sites and hinder the transport of electrons or protons.23–25 Fortunately, recent studies found that catalysts based absolutely on combined oxides can bring about an increase in the real electrocatalytic activity, which may be attributed to the synergetic or heterojunction effect between the oxide components.26 Some bi-functional materials have already been reported yet, such as NiFe LDH,27 Co/MoOx (ref. 28) and Co, CoOx@CN,25 with the overall water splitting activities of 1.7 V, 1.72 V and 1.60 V, respectively, to reach the current density of 10 mA cm−2. Therefore, the synergetic effect between combined oxide components needs to be further explored aiming to develop composite oxide catalysts with higher activity and stability.
Metal–organic frameworks (MOFs), with its high surface area, hybrid features and tunable porosity, multi-functionality and good catalytic selectivity, have drawn much more attention and are very attractive precursors for constructing nanostructured metal (oxide)/carbon and their nanocomposites by taking advantage of the unique thermal behavior and chemical reactivity.29–31 As a result, MOFs should be a class of ideal precursors to prepare metals, alloys, and metallic oxides, thus emerging as a new platform in various fields.30–32
Herein, we report a simple and facile strategy to synthesize a bi-functional MOF-derived RuO2/Co3O4 heterojunction electrocatalyst that is active for both HER and OER in alkaline solutions. A Co-based metal–organic framework (Co-BTC) was chosen as precursor for synthesizing designed RuO2/Co3O4 by a two-step transformation from MOFs. The high catalytic activity of RuO2/Co3O4 is originated from the fast charge-transfer kinetics resulting from the interface between Co3O4 and RuO2.
Experimental
Material preparation
All reagents used were of analytical grade and used as received. Cobalt(II) acetate tetrahydrate (C4H6CoO4·4H2O), 1,3,5-benzenetricarboxylic acid (H3BTC), polyvinylpyrrolidone ((C6H9NO)n, PVP), ethanol (CH3CH2OH) was purchased from Sinopharm Chemical Reagent Co. Ltd (Shanghai, China), Ruthenium(III) chloride trihydrate (RuCl3·H2O), Nafion@perfluorinated resin solution. Deionized water was used throughout this work.Syntheses of Co-BTC and Co3O4
Co-BTC used as MOF precursors was prepared according to the method described in the related literatures with some modifications.33,34 For synthesis of Co-BTC, 0.2 g C4H6CoO4·4H2O and 1.2 g PVP was dissolved into the mixed solution of ethanol (20 mL) and deionized water (20 mL), formed solution A, which was put on the magnetic stirrer with a low speed. 0.36 g H3BTC was dissolved into the mixed solution of ethanol (20 mL) and deionized water (20 mL), formed solution B. Then solution B was poured into solution A with a constant speed by using an injector (10 mL). The mixed solution was kept stirring until the precipitation formed. Finally, the product was centrifuged after 24 hours' standing and washed four times by ethanol before drying at 60 °C in vacuum. The powder of Co-BTC was placed in a tube furnace and heated to the desired temperature (500, 550, 600, and 650 °C) for 1 h under air atmosphere with a heating rate of 10 °C min−1 to obtain Co3O4-x, where “x” represents the carbonization temperature.Syntheses of RuO2/Co3O4
For synthesis of RuO2/Co3O4, the powder of Co-BTC (50 mg) was dispersed in the solution of ethanol (20 mL) and deionized water (20 mL), stirring for 30 minutes. Then the well-prepared RuCl3 (10 mg mL−1, dissolved in deionized water) was added to the Co-BTC dispersion with different mole ratios (where Co![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ru equal to 10:1, 8:1, 6:1, and 4:1, respectively). The whole system was kept stirring for 24 h, then centrifugal separation and washed four times by ethanol before drying at 60 °C in vacuum. Finally, the product was placed in a tube furnace and heated to desired temperature (500, 550, 600, and 650 °C) for 1 h under air atmosphere with a heating rate of 10 °C min−1.
:Ru equal to 10:1, 8:1, 6:1, and 4:1, respectively). The whole system was kept stirring for 24 h, then centrifugal separation and washed four times by ethanol before drying at 60 °C in vacuum. Finally, the product was placed in a tube furnace and heated to desired temperature (500, 550, 600, and 650 °C) for 1 h under air atmosphere with a heating rate of 10 °C min−1.
Materials characterization
The powder XRD patterns of the samples were recorded with an X-ray diffractometer (Japan Rigaku D/MAX-γA) using Cu-Kα radiation (λ = 1.54178 Å) with a 2θ range of 20–80°. SEM images were taken by a JEOL JSM-6700M scanning electron microscope. TEM images were obtained with HitachiH-800 transmission electron microscope using an accelerating voltage of 200 kV, and HRTEM (JEOL-2011) was operated at 200 kV. TGA of Co-BTC and RuO2/Co3O4 was carried out using a Shimadzu-50 thermoanalyser under flowing nitrogen at a heating rate of 10 °C min−1. XPS was measured on an ESCALAB 250 X-ray photoelectron spectrometer using Al Kα radiation. The binding energies were calibrated based on the graphite C 1s peak at 284.8 eV. The XPS peakfit program with a Gaussian–Lorentzian mixed function and Shirley background subtraction was used to analyses the spectra. Inductively coupled plasma (ICP) data was obtained by using an Optima 7300 DV instrument. The specific surface and pore diameters were obtained from the results of N2 physisorption at 77 K (Micromeritics ASAP 2020) by using BET (Brunauer–Emmet–Teller) and BJH (Barrett–Joyner–Halenda) methods, respectively. Raman spectra were obtained using a LabRAM HR Raman spectrometer.Electrochemical measurement
All of the electrochemical measurements were performed in a three-electrode system on an electrochemical workstation (CHI 660D) in 1 M KOH electrolyte. Typically, 4 mg of catalyst and 30 μL Nafion solution (Sigma Aldrich, 5 wt%) were dispersed in 1 mL ethanol solution by sonicating for 1 h to form a homogeneous ink. Then 5 μL of the dispersion (containing 20 μg of catalyst) was loaded onto a glassy carbon electrode with 3 mm diameter (loading 0.285 mg cm−2). While a Ag/AgCl (in 3 M KCl solution) electrode and a platinum foil were served as the reference electrode and counter electrodes, respectively. All of the potentials were calibrated to a reversible hydrogen electrode (RHE). The working electrode was polished with Al2O3 powders with size ranging from 1 μm down to 0.05 μm. Linear sweep voltammetry with a scan rate of 2 mV s−1 was conducted between −0.4 V and 0.1 V (vs. RHE) into 1 M KOH electrolyte with a stable flow of N2 gas maintained over the electrolyte during the HER experiment. Cyclic voltammetry (CV) was conducted in 1 M KOH solution in the potential region from −0.4 V and 0.1 V (vs. RHE) at a sweep rate of 100 mV s−1 for 10000 times to investigate the cycling stability. The same method is also applicable for OER process except that the LSV and CV curve was conducted between 1.20 V and 1.72 V (vs. RHE) with a scan rate of 5 mV s−1.
Results and discussion
Characterization of MOF-derived Co3O4
The synthesis process for the RuO2/Co3O4 heterojunction was illustrated in Scheme 1. The SEM and TEM images of the MOF-derived Co-BTC particles, illustrated respectively in Fig. 1a and b, suggest that it is a well-shaped rectangular bar in the range 15–20 μm in length and about 500 nm in diameter with a relatively smooth surface.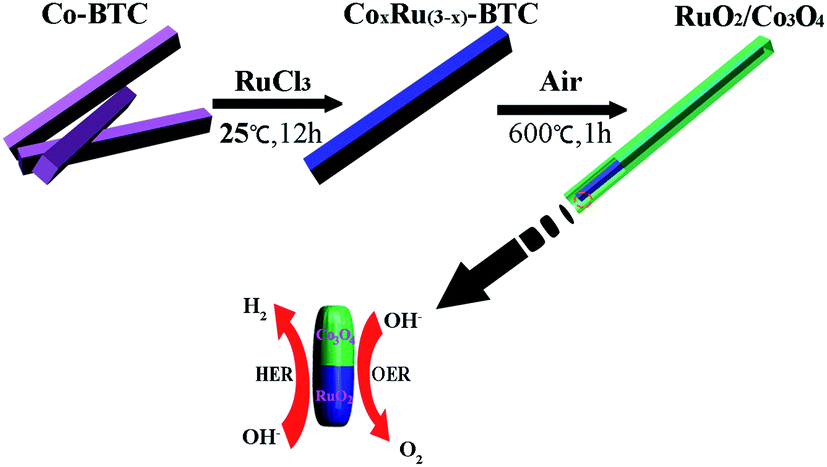 | ||
| Scheme 1 Synthetic route for RuO2/Co3O4. | ||
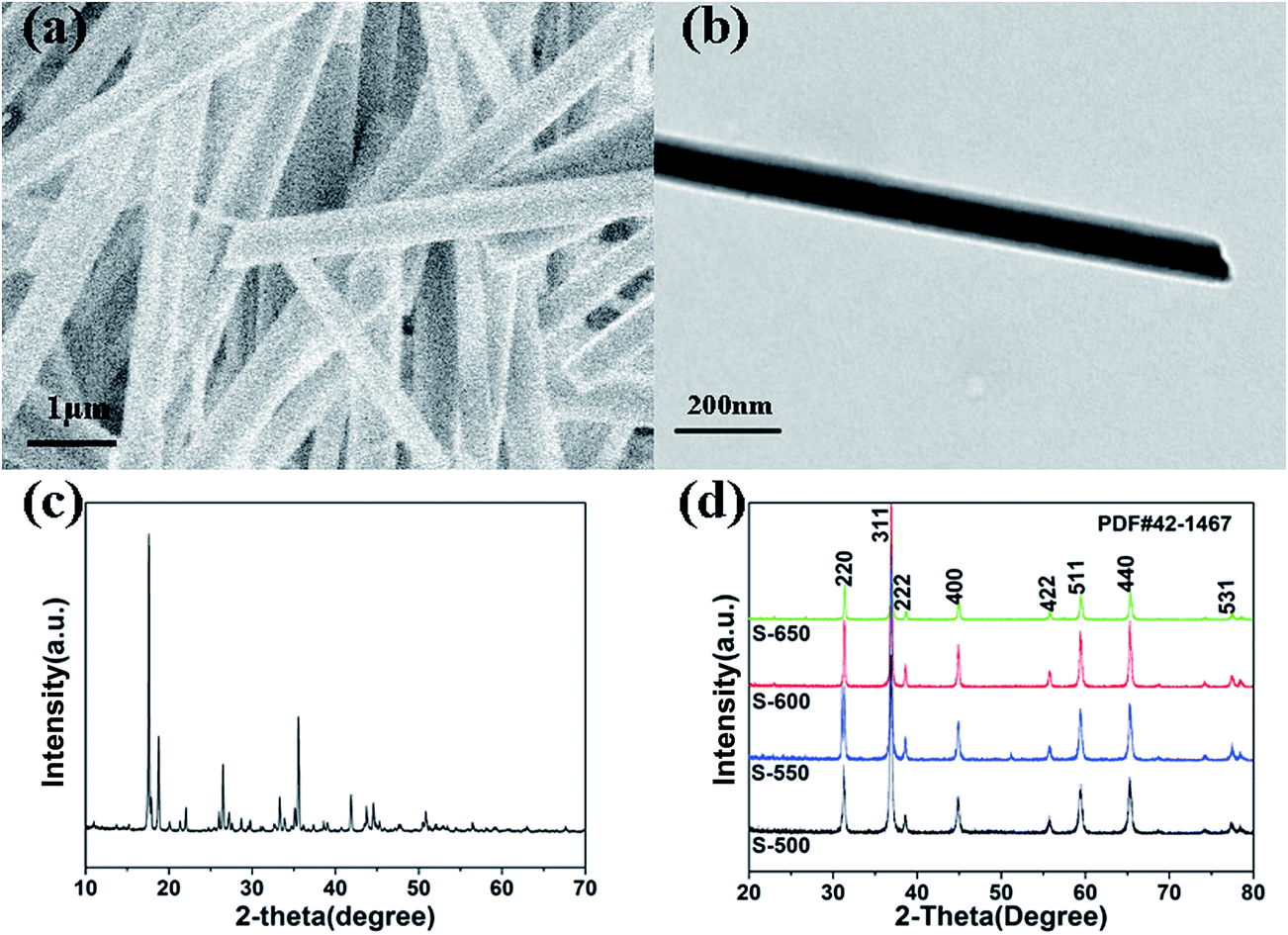 | ||
| Fig. 1 (a) SEM image of Co-BTC. (b) TEM image of Co-BTC. (c) XRD patterns of Co-BTC. (d) XRD patterns of the Co3O4 samples (S-650, S-600, S-550, S-500). | ||
The products of Co3O4 were prepared by annealing the precursors Co-BTC under air atmosphere. The sample annealed at 650 °C is named as S-650. Similar products obtained with the same annealing strategy under different temperatures refer to compounds S-600, S-550, and S-500.
The typical XRD patterns of precursors are shown in Fig. 1c. The diffraction peaks of the precursors are consistent with the values of Co-BTC reported by previous research.33,35 The XRD patterns of annealed products are shown in Fig. 1d. The strong and sharp reflection peaks and the smooth baseline of S-650 indicate that the sample is well crystallized. It can be seen that no other impurity is presented in the sample except for the Co3O4. Similar XRD patterns are also observed for compounds S-600, S-550, and S-500. The obvious diffraction peaks can be identified for the (220), (311), (222), (400), (422), (511), (440), and (531) planes of cubic Co3O4 with a space group of Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (227), which is consistent with the standard pattern of the Co3O4 (JCPDS: 42-1467). Raman spectroscopy has also been performed to determine the structural features that the sample is not carbonized (Fig. 2c, S-600).
m (227), which is consistent with the standard pattern of the Co3O4 (JCPDS: 42-1467). Raman spectroscopy has also been performed to determine the structural features that the sample is not carbonized (Fig. 2c, S-600).
 | ||
| Fig. 2 XRD patterns of the Co–Ru complex precursors (a) and the annealed samples (b) (S-4, S-6, S-8, S-10). Raman spectra of Co3O4 (S-600) and Co3O4/RuO2 (S-6) (c); XPS spectra of S-6 (d–f) (Co-in-S-6 (d); Ru-in-S-6 (e); O-in-S-6 (f)). | ||
Characterization of RuO2/Co3O4
We get the final products RuO2/Co3O4 by simply mixing Co-BTC with different amounts of RuCl3 and then annealing at 600 °C under air atmosphere. The sample prepared from nominal composition Ru:Co molar ratio of 1:4 is named as S-4. Similar structures have also been obtained with the same annealing strategy for reference compounds S-6, S-8, and S-10.
The corresponding XRD patterns of the Co–Ru complex precursors were shown in Fig. 2a. The characteristic peaks show little difference with pure Co-BTC (yellow), indicating that the structure of Co–Ru complex stay the same with MOF Co-BTC. The XRD patterns of annealed products are shown in Fig. 2b. We can see that all samples are well crystallized. XRD patterns of the four samples are similar with each other and the peaks of Co3O4 and RuO2 are both detected in four samples. The diffraction peaks appearing at 2θ = 31.27°, 36.85°, 38.54°, 44.81°, 55.65°, 59.35°, 65.23°, and 68.63° are respectively correspond to the same crystalline planes with standard pattern of Co3O4 (JCPDS: 42-1467). While the diffraction peaks occurring at 2θ = 28.02°, 35.07°, 40.04°, and 54.27° are indexed well to the (110), (101), (200), and (211) crystalline planes of RuO2 (JCPDS: 43-1027). Raman spectroscopy of S-6 is shown in Fig. 2c. As one can see, the peaks of S-6 have an obvious shift to the left compared with the pure Co3O4 (black line), suggesting that there may be some interaction between Co3O4 and RuO2.
The valence state of the components on the catalyst surface is identified by X-ray photoelectron spectroscopy (XPS) to confirm the formation of Co3O4 and RuO2 (Fig. 2d–f). As shown in Fig. 2d, the presence of the Co 2p3/2 peak at 779.89 eV and the Co 2p1/2 peak at 795 eV indicates the formation of the Co3O4 phase.22,36–39 The XPS spectrum of RuO2 in the Ru 3p region (Fig. 2e) shows the peaks for Ru 3p3/2 at 462.8 eV and Ru 3p1/2 at 486.1 eV, which are attributed to the photoemission from RuO2 (Ru4+).40 The O 1s peak shown in Fig. 2f corresponds to metal–oxygen bonds as we expected.39 The peaks centered at 530.09 eV and 531.22 eV correspond to oxygen species in the Co3O4 phase, while 531.91 eV correspond to oxygen species in the RuO2 phase.41,42 The shift of binding energy for Co and Ru reveals that there is an interaction between the metal atoms and O atoms, which is consistent with our previous results.
The SEM and TEM images of the Co–Ru complex precursor and annealed products S-6 were shown in Fig. 3a–c. It is clear to see that both the precursor and the air-annealed samples show the rectangular bar structure of Co–MOF, with some deposits on the surface. From Fig. 3c, we can get detailed structure information of S-6, which indicates that the annealed sample owns a long bar structure with metal oxides distributed along it. The dark part refers to RuO2 and the light part represents Co3O4. It can also be proved by further HRTEM analysis shown in Fig. 4a, which confirms that the nanocomposites are composed of noble metal oxide RuO2 and transition metal oxide Co3O4. The lattice fringes can be clearly observed in HRTEM image, in which the 2.43 Å is in agreement with the (311) plane of Co3O4, while the 3.18 Å belongs to the (110) plane of RuO2, respectively. It is worth noting that Fig. 4a also reveals the apparent crystal boundary between the Co3O4 nano-crystal and the RuO2 nanocrystal. The boundaries demonstrate the generation of RuO2/Co3O4 heterojunctions. In the heterojunctions, RuO2 and Co3O4 particles closely connect to each other on a nano-level. The images of both EDS line profiles along the yellow line recorded on a random single small particle (Fig. 4b) and the corresponding elemental mapping (Fig. 4c–e) reveal that the Co, Ru and O are closely connected and well-distributed to each other at the nano-level. Element contents of S-6 were determined by ICP. The result indicates that atom ratio of Co and Ru is exactly to be 29.3:0.79. This result is somewhat different from the EDS line scan result, which shows that Co:Ru = 39.09:2.96. RuO2 tends to be extremely stable in both acidic and alkaline solution although tremendous efforts have been made to dissolve it, which results in the large difference between them.
 | ||
| Fig. 3 SEM images of Co–Ru complex precursor (a) and S-6 (b). TEM image of S-6 (c). | ||
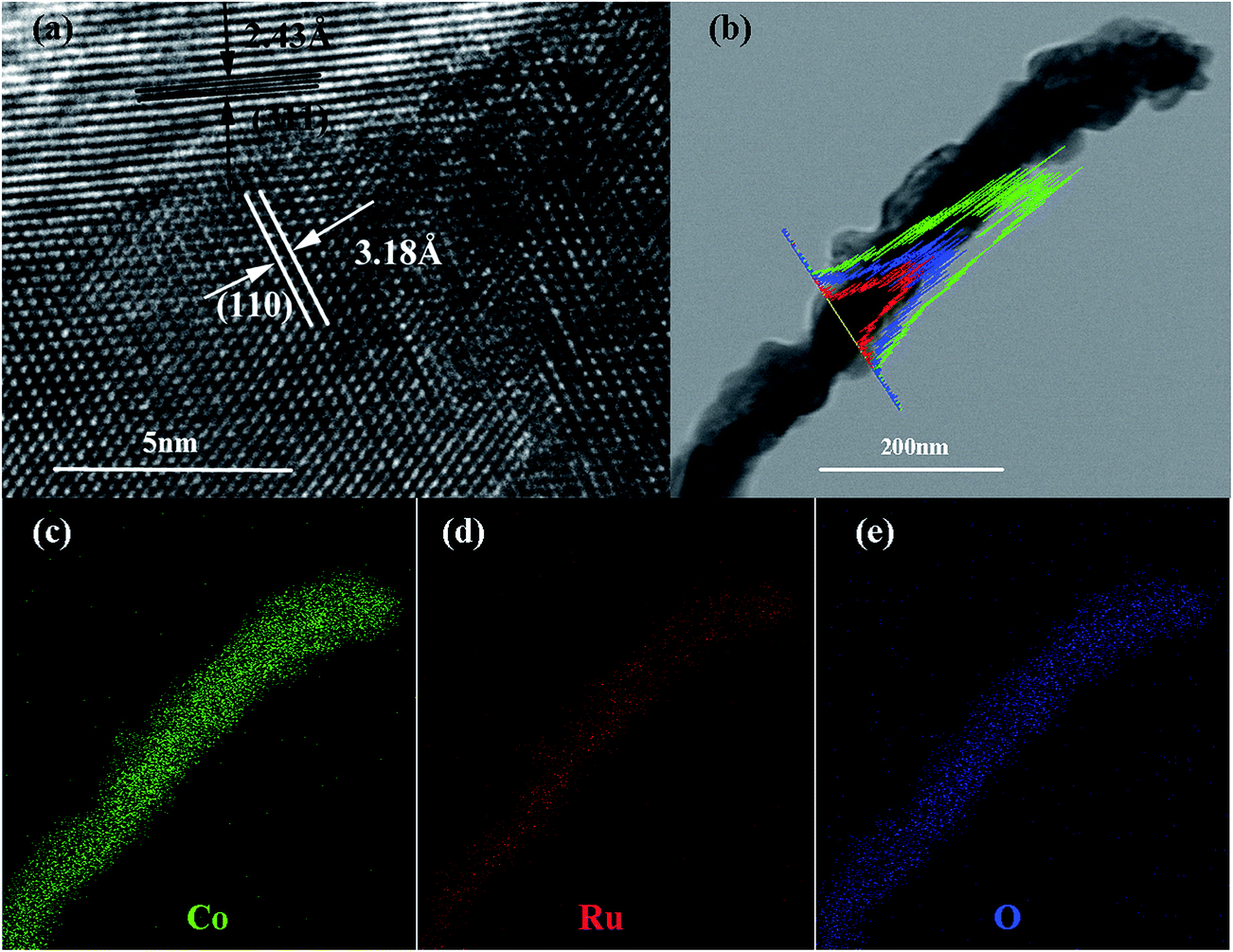 | ||
| Fig. 4 HRTEM of S-6 (a); TEM image of the RuO2/Co3O4 nanocrystalline-scan EDS analysis across the nanocrystal (b); elemental mappings of Co (c); Ru (d); O (e). | ||
Electrochemical properties of Co3O4 and Co3O4/RuO2
The electrochemical catalytic activity of the samples for OER is recorded using a typical three-electrode electrochemical cell in N2-saturated 1 M KOH electrolyte. The OER performance is evaluated by observing the overpotential at 10 mA cm−2 versus a reversible hydrogen electrode (RHE). The polarization curves of Co3O4 annealed at different temperatures are shown in Fig. 5a. The trend in the overpotential can be concluded as S-500 (482 mV) < S-550 (435 mV) < S-600 (423 mV) > S-650 (457 mV), suggesting that the S-600 catalyst has the highest OER activity among four catalysts with an overpotential of 423 mV without iR-correction. Fig. 5b displays the LSV curves of the final samples synthesized in different molar ratios and annealed at 600 °C, which throw light on the main overpotential changes against each other. That is S-10 (377 mV) < S-8 (353 mV) < S-6 (305 mV) > S-4 (338 mV). With the amount of RuO2 increase, the overpotential of the samples decreases until the molar ratio reaches an extreme point (nominal starting molar Co:Ru = 6:1). Further addition of RuO2 only received a counterproductive effect. We can also come to a conclusion as follows. Despite the small content of RuO2 (Co:Ru refers to 39.09:2.96), the overpotential of combined mental oxides still presents obvious ascension than the best performance of Co3O4 samples from the above data, which may be attributed to the synergetic effect between the oxide components. It is also much better than commercial RuO2 catalyst (377 mV) according to the previous research reported by our group.15 Tafel slope (Fig. 5d) of the S-6 sample is 69 mV dec−1, which is slightly better than the value of S-4 (78 mV dec−1), S-8 (87 mV dec−1) and S-10 (133 mV dec−1), indicating that S-6 drives the OER process at a lower overpotential than the other four samples.
 | ||
| Fig. 5 OER polarization curves of Co3O4 samples annealed at different temperature (a); and RuO2/Co3O4 complex with different molar ratios (b) in 1 M KOH; polarization curves of S-6 after the 1st and 10000th cycles (c); Tafel plots of S-4, S-6, S-8, and S-10 (d). | ||
In addition, durability is another important parameter in the exploration of electrocatalyst. A cycling test was applied with polarization curves at an accelerated scanning rate. The polarization curve of S-6 after 10000 cycles is displayed in Fig. 5c. The overpotential of S-6 increased by 79 mV to reach a result of 384 mV, which is obviously superior to the durability of commercial RuO2.15 Therefore, the S-6 sample has a higher activity and durability than commercial RuO2 catalysts.
HER activities are also examined by observing the overpotential at 10 mA cm−2 in N2-saturated 1 M KOH electrolyte under the same condition. The LSV curves of Co3O4 annealed at different temperatures are displayed in Fig. 6a. Apparently, the overpotential goes down with the anneal temperature increases. The overpotential of S-500, S-550, S-600 and S-650 are 387 mV, 375 mV, 351 mV and 379 mV, respectively, indicating that the best performance of the Co3O4 samples occurs at a anneal temperature of 600 °C. Further studies are done based on this result. Polarization curves of RuO2/Co3O4 metal oxide products were shown in Fig. 6b. To achieve a current density of 10 mA cm−2, the S-10, S-8, S-6 and S-4 require overpotential of 189 mV, 155 mV, 89 mV and 140 mV, respectively, which is much lower than the best performance of Co3O4 (351 mV) displayed above, revealing that our synthesized nanocomposites are effective catalysts for HER in alkaline solution.
 | ||
| Fig. 6 HER polarization curves of Co3O4 samples annealed at different temperature (a); and RuO2/Co3O4 with different molar ratios (b) in 1 M KOH; polarization curves of S-6 after the 1st and 10000th cycles (c); chronoamperometry measurements (I vs. t) recorded on S-6 at a constant applied potential of 89 mV vs. RHE (d) Tafel plots of S-4, S-6, S-8, and S-10 (e). | ||
The stability of S-6 is also tested by a cycling test of 10000 (Fig. 6c). There is a slight decrease emerging after 10000 cycles in alkaline environment, displaying an excellent durability. Besides, I–t curve is also taken for further validation. The stable current density over 10 h of continuous operation at an applied overpotential of 89 mV in alkaline media is presented in Fig. 6d. It remains almost steady for 40000 s.
To investigate the kinetics of the HER process promoted by as-prepared catalysts, Tafel plots analyses are carried out (Fig. 6e). The present study shows that S-6 gives a Tafel slope of 91 mV dec−1, lower than that of S-4 (103 mV dec−1), S-8 (121 mV dec−1) and S-10 (122 mV dec−1), implying its favorable HER catalytic kinetics in alkaline solution. As the rate-determining step (RDS) is reflected by Tafel slope of a catalyst, the variation of the Tafel slope can be explained by the change of RDS in the HER process. Based on the data of the four Tafel slope, Volmer–Heyrovsky mechanism can be determined.
With the aforementioned excellent HER and OER catalytic performance of RuO2/Co3O4 in hand, we are confident that it can act as an electrocatalyst for both HER and OER in alkaline solution. As is concluded above, this material shows a low OER and HER overpotential of only 305 mV and 89 mV at 10 mA cm−2 of current density in alkaline solution, respectively. Such a superior electrocatalytic performance may be attributed to its unique properties. Transition metal oxide with metal–organic frameworks are known to be very active HER electrocatalysts, while RuO2 owns a perfect activity towards OER process. Once they combined together, they will contribute to the overall water splitting. In order to confirm the synergetic effect of the nanointerfaces in RuO2/Co3O4, a group of controlled trials are taken by simply mix the two compounds (Co3O4 and RuO2) together and test the LSV values of the mixture. The HER and OER polarization curves are displayed in Fig. 7a and b. The mixture reaches a current density of 10 mA cm−2 at overpotentials of 413 mV for OER and 203 mV for HER, respectively. We can clearly find that the performance of RuO2/Co3O4 proves to be much better than the mixture. RuO2 may be responsible for the electrochemistry performance, however, the interface of Co3O4 and RuO2 enhance its electrochemical activity. Further electrochemically active surface area (ECSA) experiments have shown that the high performance of the product could be caused by the increased active sites at the interface of RuO2/Co3O4 heterojunction (Fig. S1†).
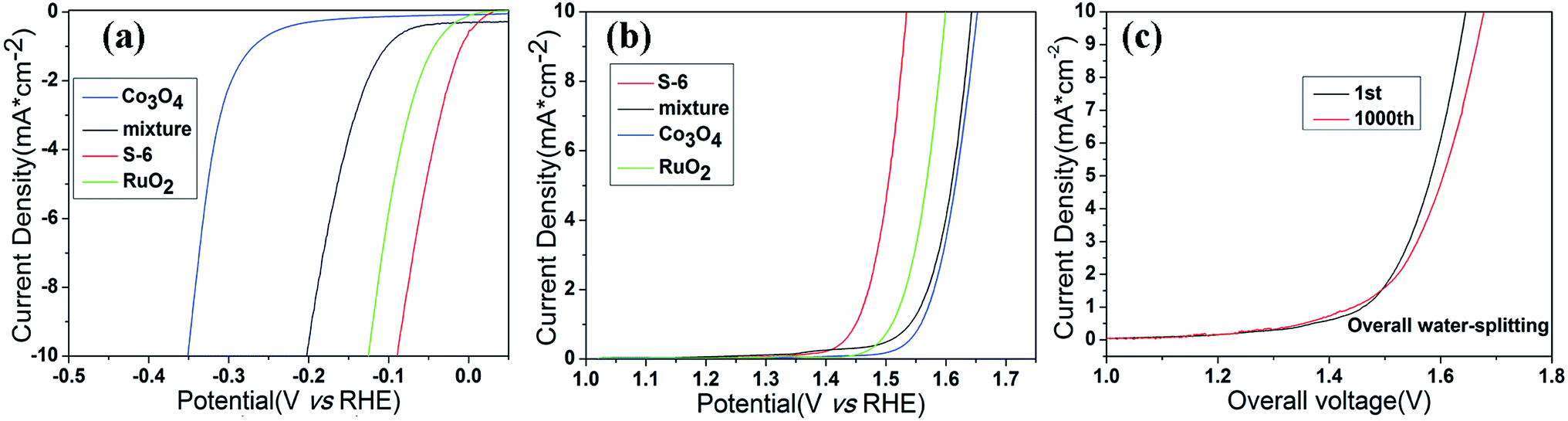 | ||
| Fig. 7 HER (a) and OER (b) polarization curves of RuO2/Co3O4, Co3O4 and the mixture of Co3O4 and RuO2. (c) Overall water-splitting characteristics of RuO2/Co3O4∥RuO2/Co3O4 in two-electrode configurations in 1 M KOH. All the data are without iR-compensation. | ||
The bifunctional RuO2/Co3O4 were further investigated as the same electrocatalyst on both electrodes with a two-electrode configuration to test the activity for overall water splitting in 1 M KOH (Fig. 7). The liner sweep voltammetry of the RuO2/Co3O4∥RuO2/Co3O4 system shows that the current density of 10 mA cm−2 is achieved at a voltage of 1.645 V. This value changes to 1.678 V after 1000 cycles, showing a decrease in activity. Also, we have compared the electrocatalytic performances of RuO2/Co3O4 with a series of related catalysts listed in Table 1. It is shown that no Co3O4-based material serves as bi-functional catalysts in water splitting progress at present stage. Moreover, Co3O4-based catalysts towards HER are not reported as much as that of OER. Even though the reported Co/Co3O4 owns a similar overpotential of 90 mV towards HER in 1 M KOH compared with our RuO2/Co3O4, its OER activity in alkaline solution is absent. Therefore, it is concluded that RuO2/Co3O4 has a competitive performance towards both HER and OER in alkaline solution. The combination of Co3O4 and RuO2 provides an interface that owns highly active catalytic sites in electrochemistry, which may be attributed to the structure of heterojunction.31 Previous density-functional theory (DFT) calculations revealed that interface show significantly increased DOS at the band edge with respect to combined oxides, resulting in improvement of the catalytic performance.15,31,56 This is the main reason why our material performs much better than many other Co3O4-based catalysts.
| Catalyst | Catalyst loading (mg cm−2) | Electrolyte | HER overpotential (mV) | OER overpotential (mV) | OWS potential (V) | Reference |
|---|---|---|---|---|---|---|
| CuxCo3−xO4 | — | 1 M KOH | — | 410 | 43 | |
| NixCo3−xO4 | 0.7 | 1 M KOH | — | 337 | — | 44 |
| NiCo/NiCoOx | 0.7 | 1 M KOH | 155 | — | — | 44 |
| ZnxCo3−xO4 | 1.0 | 1 M KOH | — | 320 | — | 45 |
| Co3O4 shell/Au core NPs | 0.2 | 0.1 M KOH | — | 390 | — | 46 |
| Plasma-engraved Co3O4 | — | 0.1 M KOH | — | 300 | — | 47 |
| Co/Co3O4 | 0.85 | 1 M KOH | 90 | — | — | 48 |
| Co3O4/N-graphene + Ni foam | 0.17 | 1 M KOH | — | 310 | — | 49 |
| Co3O4 NPs/Ni foam | 0.2 | 1 M KOH | — | 360 | — | 50 |
| meso-Co3O4 | 0.1 | 0.1 M KOH | — | 411 | — | 51 |
| CoxOy/NC | 0.21 | 0.1 M KOH | — | 430 | — | 52 |
| Co3O4–carbon | — | 0.1 M KOH | — | 290 | — | 53 |
| Co, CoOx@CN | 0.42 | 1 M KOH | 232 | 260 | 1.60 | 25 |
| Co/MoOx | 0.7 | 1 M KOH | 40(onset) | 230(onset) | 1.72 | 28 |
| Ni–RuO2 | 4.2 | 1 M NaOH | 115(100) | — | — | 54 |
| MnxOy/RuO2 | — | 1 M KOH | — | 312 | 55 | |
| RuO2/Co3O4 | 0.285 | 1 M KOH | 89 | 305 | 1.645 | This work |
Conclusions
In summary, we adopt a MOF-annealing strategy to synthesize a combined metal oxide RuO2/Co3O4 as an efficient bi-functional electrocatalyst towards OER and HER in alkaline solution. The catalyst exhibited excellent activity with low overpotential, small Tafel slope, as well as an outstanding electrochemical durability in HER evaluation even after the 10000th cycling. The stability in OER also performs even better than commercial RuO2. This work shows that a heterojunction composed by transition metal oxides with small amounts of noble metal oxides can provide more active sites for OER and HER electrocatalysts with high performance.
Acknowledgements
This work was supported by the National Natural Science Foundation (NSFC, 21271163, U1232211), CAS/SAFEA International Partnership Program for Creative Research Teams, Hefei Science Center CAS (2016HSC-IU011).References
- L. Yu, B. Y. Xia, X. Wang and X. W. Lou, Adv. Mater., 2016, 28, 92–97 CrossRef CAS PubMed.
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS PubMed.
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed.
- F. Song and X. Hu, Nat. Commun., 2014, 5, 4477 CAS.
- B. You, N. Jiang, M. Sheng, S. Gul, J. Yano and Y. Sun, Chem. Mater., 2015, 27, 7636–7642 CrossRef CAS.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- A. J. Esswein, M. J. McMurdo, P. N. Ross, A. T. Bell and T. D. Tilley, J. Phys. Chem. C, 2009, 113, 15068–15072 CAS.
- B. Seo, Y. J. Sa, J. Woo, K. Kwon, J. Park, T. J. Shin, H. Y. Jeong and S. H. Joo, ACS Catal., 2016, 6, 4347–4355 CrossRef CAS.
- A. Indra, P. W. Menezes, N. R. Sahraie, A. Bergmann, C. Das, M. Tallarida, D. Schmeisser, P. Strasser and M. Driess, J. Am. Chem. Soc., 2014, 136, 17530–17536 CrossRef CAS PubMed.
- T. Maiyalagan, K. A. Jarvis, S. Therese, P. J. Ferreira and A. Manthiram, Nat. Commun., 2014, 5, 3949 CAS.
- J. Chang, Y. Xiao, M. Xiao, J. Ge, C. Liu and W. Xing, ACS Catal., 2015, 5, 6874–6878 CrossRef CAS.
- J. Li, Y. Wang, T. Zhou, H. Zhang, X. Sun, J. Tang, L. Zhang, A. M. Al-Enizi, Z. Yang and G. Zheng, J. Am. Chem. Soc., 2015, 137, 14305–14312 CrossRef CAS PubMed.
- J. B. Gerken, J. G. McAlpin, J. Y. C. Chen, M. L. Rigsby, W. H. Casey, R. D. Britt and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 14431–14442 CrossRef CAS PubMed.
- Y. Sun, S. Gao, F. Lei, J. Liu, L. Liang and Y. Xie, Chem. Sci., 2014, 5, 3976 RSC.
- J. Su, G. Xia, R. Li, Y. Yang, J. Chen, R. Shi, P. Jiang and Q. Chen, J. Mater. Chem. A, 2016, 4, 9204–9212 CAS.
- J. Deng, P. Ren, D. Deng and X. Bao, Angew. Chem., 2015, 54, 2100–2104 CrossRef CAS PubMed.
- J. Deng, P. Ren, D. Deng, L. Yu, F. Yang and X. Bao, Energy Environ. Sci., 2014, 7, 1919 CAS.
- Y. Yang, Z. Lun, G. Xia, F. Zheng, M. He and Q. Chen, Energy Environ. Sci., 2015, 8, 3563–3571 CAS.
- M. Tavakkoli, T. Kallio, O. Reynaud, A. G. Nasibulin, C. Johans, J. Sainio, H. Jiang, E. I. Kauppinen and K. Laasonen, Angew. Chem., 2015, 54, 4535–4538 CrossRef CAS PubMed.
- Z. Lu, H. Wang, D. Kong, K. Yan, P.-C. Hsu, G. Zheng, H. Yao, Z. Liang, X. Sun and Y. Cui, Nat. Commun., 2014, 5, 4345 CAS.
- J. Rosen, G. S. Hutchings and F. Jiao, J. Am. Chem. Soc., 2013, 135, 4516–4521 CrossRef CAS PubMed.
- C. Yuan, L. Yang, L. Hou, L. Shen, F. Zhang, D. Li. and X. Zhang, J. Mater. Chem., 2011, 45, 18183–18185 RSC.
- Y. Fan, H. Shao, J. Wang, L. Liu, J. Zhang and C. Cao, Chem. Commun., 2011, 47, 3469–3471 RSC.
- Y. Xu, R. Yi, B. Yuan, X. Wu, M. Dunwell, Q. Lin, L. Fei, S. Deng, P. Andersen, D. Wang and H. Luo, J. Phys. Chem. Lett., 2012, 3, 309–314 CrossRef CAS PubMed.
- H. Jin, J. Wang, D. Su, Z. Wei, Z. Pang and Y. Wang, J. Am. Chem. Soc., 2015, 137, 2688–2694 CrossRef CAS PubMed.
- L. M. Da Silva, J. F. C. Boodts and L. A. De Faria, Electrochim. Acta, 2001, 46, 1369–1375 CrossRef CAS.
- J. Luo, J.-H. Im, M. T. Mayer, M. Schreier, M. K. Nazeeruddin, N.-G. Park, S. D. Tilley, H. J. Fan and M. Grätzel, Science, 2014, 345, 1593–1596 CrossRef CAS PubMed.
- X. Yan, L. Tian, S. Atkins, Y. Liu, J. Murowchick and X. Chen, ACS Sustainable Chem. Eng., 2016, 4, 3743–3749 CrossRef CAS.
- H. Hu, L. Han, M. Yu, Z. Wang and X. W. Lou, Energy Environ. Sci., 2016, 9, 107–111 CAS.
- C. Wang, D. Liu and W. Lin, J. Am. Chem. Soc., 2013, 135, 13222–13234 CrossRef CAS PubMed.
- R. Zhang, L. Hu, S. Bao, R. Li, L. Gao, R. Li and Q. Chen, J. Mater. Chem. A, 2016, 4, 8412–8420 CAS.
- Y. Z. Chen, C. Wang, Z. Y. Wu, Y. Xiong, Q. Xu, S. H. Yu and H. L. Jiang, Adv. Mater., 2015, 27, 5010–5016 CrossRef CAS PubMed.
- N. Yang, H. Song, X. Wan, X. Fan, Y. Su and Y. Lv, Analyst, 2015, 140, 2656–2663 RSC.
- F. Zheng, G. Xia, Y. Yang and Q. Chen, Nanoscale, 2015, 7, 9637–9645 RSC.
- O. M. Yaghi, H. L. Li and T. L. Groy, J. Am. Chem. Soc., 1996, 118, 9096–9101 CrossRef CAS.
- J. Wu, Y. Xue, X. Yan, W. S. Yan, Q. M. Cheng and Y. Xie, Nano Res., 2012, 5, 521–530 CrossRef CAS.
- H. Tuysuz, Y. Liu, C. Weidenthaler and F. Schuth, J. Am. Chem. Soc., 2008, 130, 14108–14110 CrossRef PubMed.
- D. Oh, J. Qi, B. Han, G. Zhang, T. J. Carney, J. Ohmura, Y. Zhang, Y. Shao-Horn and A. M. Belcher, Nano Lett., 2014, 14, 4837–4845 CrossRef CAS PubMed.
- L. Huang, Y. Mao, G. Wang, X. Xia, J. Xie, S. Zhang, G. Du, G. Cao and X. Zhao, New J. Chem., 2016, 40, 6812–6818 RSC.
- M. Gopiraman, R. Karvembu and I. S. Kim, ACS Catal., 2014, 4, 2118–2129 CrossRef CAS.
- S. Xiong, C. Yuan, X. Zhang, B. Xi and Y. Qian, Chem.–Eur. J., 2009, 15, 5320–5326 CrossRef CAS PubMed.
- D. Susanti, D. S. Tsai, Y. S. Huang, A. Korotcov and W. H. Chung, J. Phys. Chem. C, 2007, 111, 9530–9537 CAS.
- P. Bothra and S. K. Pati, ACS Energy Lett., 2016, 858–862 CrossRef CAS.
- X. Yan, K. Li, L. Lyu, F. Song, J. He, D. Niu, L. Liu, X. Hu and X. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 3208–3214 CAS.
- X. Liu, Z. Chang, L. Luo, T. Xu, X. Lei, J. Liu and X. Sun, Chem. Mater., 2014, 26, 1889–1895 CrossRef CAS.
- Z. Zhuang, W. Sheng and Y. Yan, Adv. Mater., 2014, 26, 3950–3955 CrossRef CAS PubMed.
- L. Xu, Q. Jiang, Z. Xiao, X. Li, J. Huo, S. Wang and L. Dai, Angew. Chem., 2016, 55, 5277–5281 CrossRef CAS PubMed.
- X. Yan, L. Tian, M. He and X. Chen, Nano Lett., 2015, 15, 6015–6021 CrossRef CAS PubMed.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS PubMed.
- A. Mendoza-Garcia, D. Su and S. Sun, Nanoscale, 2016, 8, 3244–3247 RSC.
- Y. J. Sa, K. Kwon, J. Y. Cheon, F. Kleitz and S. H. Joo, J. Mater. Chem. A, 2013, 1, 9992–10001 CAS.
- J. Masa, W. Xia, I. Sinev, A. Zhao, Z. Sun, S. Grutzke, P. Weide, M. Muhler and W. Schuhmann, Angew. Chem., 2014, 53, 8508–8512 CrossRef CAS PubMed.
- T. Y. Ma, S. Dai, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2014, 136, 13925–13931 CrossRef CAS PubMed.
- N. Spataru, J. G. LeHelloco and R. Durand, J. Appl. Electrochem., 1996, 26, 397–402 CrossRef CAS.
- A. J. Jeevagan, Y. Suzuki, T. Gunji, G. Saravanan, Y. Irii, T. Tsuda, T. Onobuchi, S. Kaneko, G. Kobayashi and F. Matsumoto, ECS Trans., 2014, 58, 9–18 CrossRef.
- S. Bao, N. Yan, X. Shi, R. Li and Q. Chen, Appl. Catal., A, 2014, 487, 189–194 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra25810g |
| This journal is © The Royal Society of Chemistry 2017 |