
ortho-Quinone methide (o-QM): a highly reactive, ephemeral and versatile intermediate in organic synthesis
Maya Shankar Singh
*,
Anugula Nagaraju
,
Namrata Anand
and
Sushobhan Chowdhury
Department of Chemistry, Faculty of Science, Banaras Hindu University, Varanasi 221 005, India. E-mail: mssinghbhu@yahoo.co.in; Fax: +91-542-2368127; Tel: +91-542-6702502
First published on 7th October 2014
Abstract
Since its first observation in 1907, ortho-quinone methide (o-QM) has occupied a strategic place within the framework of reactive intermediates in organic synthesis. In recent years, o-QM has enhanced its importance as a versatile reactive intermediate, and its applications in organic synthesis, material chemistry, fine chemicals, and pharmaceuticals are increasing rapidly. This critical review summarizes the key concepts behind o-QMs and provides an overview of current applications in organic synthesis to provide an appropriate background for synthetic, medicinal and combinatorial developments. This review covers the literature from its origin to the mid of 2014 (112 references).
 Maya Shankar Singh | Maya Shankar Singh born in 1960, received his MSc degree in Organic Chemistry from Banaras Hindu University, Varanasi, India in 1981, where he also earned his PhD degree in 1986 working under the tutelage of Professor K. N. Mehrotra. After his postdoctoral work, he joined Vikram University Ujjain in 1990 as Assistant Professor in Organic Chemistry, and moved to Gorakhpur University as Associate Professor in 1998, then to Banaras Hindu University in 2004, where he is currently Professor in Organic Chemistry since 2006. During his sabbatical, he visited the Chemistry Department of the University of Arizona, Tucson, USA; Nagoya Institute of Technology, Nagoya, Japan; Loughborough University, UK and University of Leicester, UK. His research interests are centered on synthetic Organic chemistry with special emphasis in the development of novel building block precursors, new eco-compatible synthetic methods, multicomponent domino/tandem reactions, and structural studies. Fifteen students have completed their PhD degrees under his supervision. He has published 125 research articles and four reviews in journals of high status. Additionally, Prof. Singh has also authored three textbooks in organic chemistry published from Pearson-Education and Wiley-VCH, Weinheim, Germany. He has been elected as a fellow of the Indian Academy of Sciences, India in 2013. |
 Anugula Nagaraju | Anugula Nagaraju was born in Chandurthi, Karimnagar, Andhra Pradesh, India, in 1986. He received his BSc and MSc degrees in Chemistry from Kakatiya University, Warangal in 2007 and 2010, respectively. Currently, he is pursuing his Ph.D. under the supervision of Professor Maya Shankar Singh at Banaras Hindu University, Varanasi, India. His major research interest is to develop new synthetic methodologies and novel reagents for the synthesis of bioactive molecules. |
 Namrata Anand | Namrata Anand obtained her BSc (2005) and MSc (2007) degrees in Organic Chemistry from Deen Dayal Upadhyay Gorakhpur University, Gorakhpur (India). In 2013, she completed her Ph.D. at the Central Drug Research Institute, Lucknow (India) with Dr R. P. Tripathi. Since 2013, she is a Postdoctoral Researcher at Banaras Hindu University, Varanasi (India) with Prof. M.S. Singh. Her current research interests focus on the carbohydrate chemistry and the development of transition-metal catalysed C–H bond activation. |
 Sushobhan Chowdhury | Sushobhan Chowdhury was born in South 24 Parganas, West Bengal, India, in 1985. He received his B. Sc. degree from the University of Calcutta in 2007 and moved to the Banaras Hindu University, Varanasi for post-graduate studies. There he obtained his M. Sc. degree in 2010 and completed Ph.D. under the supervision of Prof. Maya Shankar Singh in 2014. His doctoral study was mainly themed on the development of synthetic methods based on the dithioester chemistry. Presently, he is working as a postdoctoral fellow at UNIST, Ulsan, South Korea. |
1. Introduction
ortho-Quinone methides (o-QMs) are short-lived, highly reactive and versatile intermediates in organic synthesis, material chemistry, fine chemicals, and pharmaceuticals.1 The ortho-quinone methide (o-QM) was first suggested by Fries in 1907 and is generally formed by the reaction of phenol with aldehyde in the presence of an acid or a base. The first direct evidence was given by Gardner in 1963 by trapping it at −100 °C. After 1963, its use as a versatile intermediate in organic synthesis increased enormously, particularly in many tandem [4 + 2] cycloaddition reactions with a variety of dienophiles. However, there are abundant indirect evidences for the in situ generation of ortho-quinone methides. Most indirect evidence comes from the structural identification of the products that result from dimerization, trimerization, intramolecular and intermolecular [2 + 2] cycloadditions as well as the nucleophilic trapping of o-QMs.2 The chemical behaviour of o-QMs resembles that of α, β-unsaturated ketones due to the presence of a 1,3-cyclohexadiene core substituted with a carbonyl and an exo-methylene group. They react very rapidly with nucleophiles and undergo efficient Diels–Alder reactions with electron-rich olefins. Expansive reactivity of the o-QM can be used in the linchpin reaction for the construction of various natural products.3Compared to carbocations, carbanions, radicals, carbenes, nitrenes and so forth, ortho-quinone methides are relative newcomers to the family of inherently reactive species. Quinone methides exist in three isomeric forms o-, m-, and p-quinone methides (also known as o-, m-, and p-QMs) (Fig. 1). meta-Quinone methide is actually a resonance hybrid of two canonical forms, one of which is a zwitterionic form stabilized by aromatic conjugation, increasing its polarity and thus enhancing its reactivity. The importance of o- and p-quinone methides in organic synthesis and their role in biochemistry have been studied in detail. Unlike benzoquinones, o- and p-quinone methide derivatives are highly polarized compounds, usually observed with difficulty or postulated as reactive intermediates because of the facile reactions driven by the formation of aromatized phenol derivatives.
 | ||
| Fig. 1 Isomeric forms of quinone methides. | ||
ortho-Quinone methides are highly versatile intermediates that have been extensively harnessed by nature (Fig. 2). A variety of plants, animals, and insects capitalize upon these types of compounds as a mean of defence. However, despite general knowledge of o-QMs for over a century, these intermediates still lie outside the synthetic mainstream. Pettus and Water1a reviewed o-QMs, particularly its preparation, the benefits and limitations associated with each method as well as the applications in total synthesis.
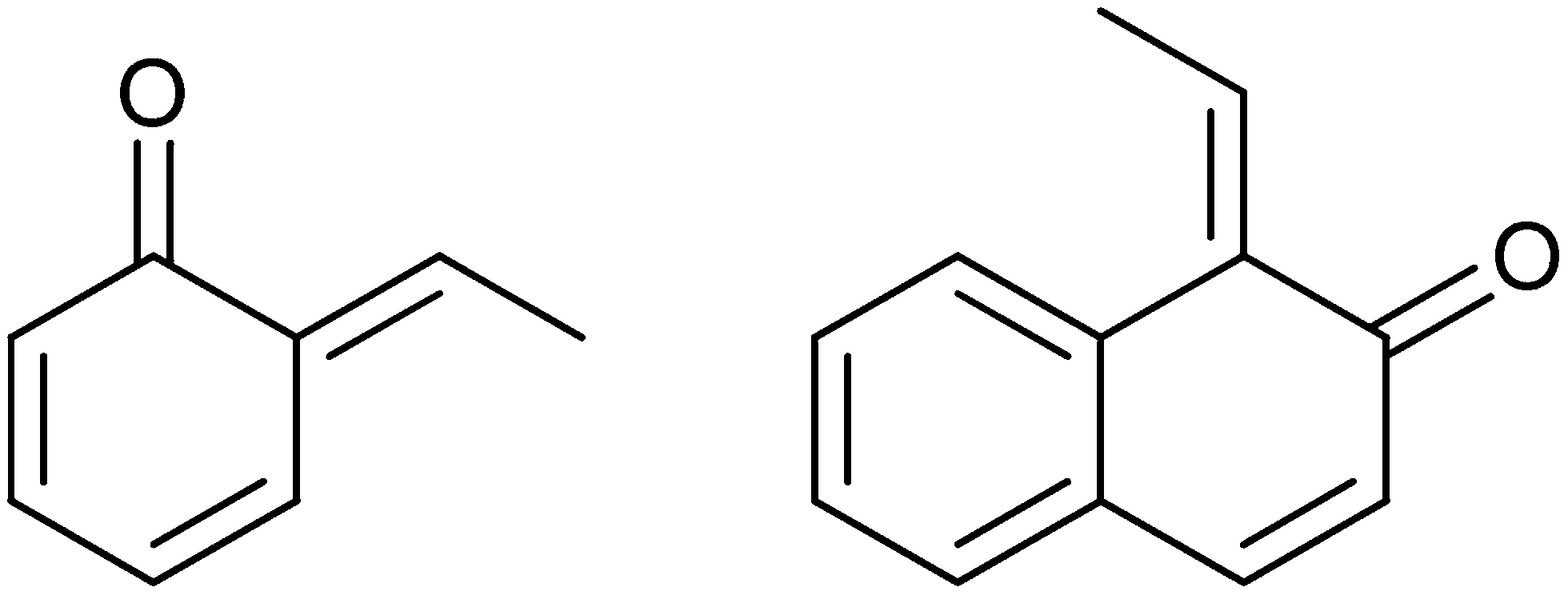 | ||
| Fig. 2 ortho-Quinone methides. | ||
ortho-Quinone methide exists in E/Z geometric isomers that are fluxional in nature, and behaves as a combination of a charged zwitterionic and biradical structures (Fig. 3). The distribution among these geometric isomers is believed to result from the differences between non-bonded interactions. If from a steric point of view, R1 substituent is smaller than oxygen, then the E-configuration is preferred. However, increasing the size of R1 substituent can cause the Z-configuration to predominate. The E/Z ratio proves important in governing the diastereoselective outcome for Diels–Alder cycloadditions.
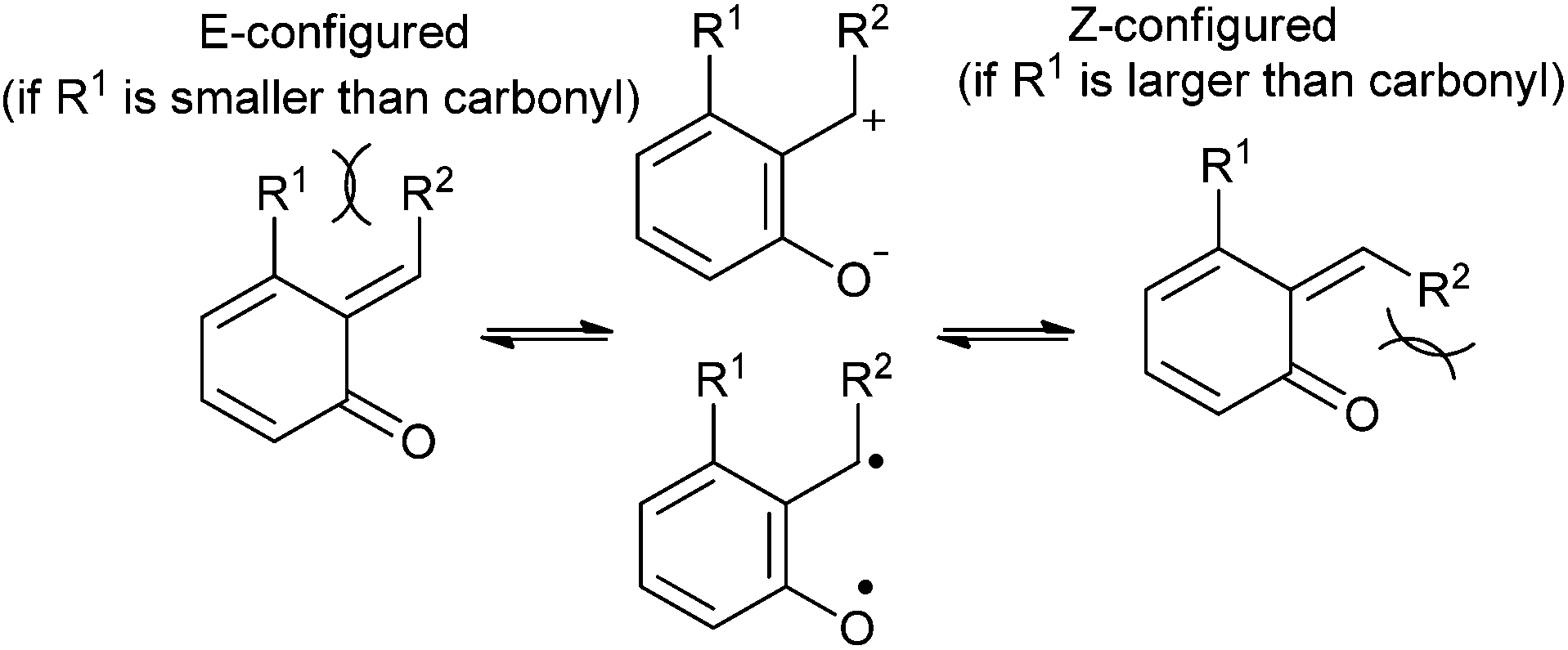 | ||
| Fig. 3 Geometric, zwitterionic and biradical structures of o-QM. | ||
Quinone methides (QMs) are common reactive intermediates in chemistry, as well as in the photochemistry of phenols, attracting considerable attention recently owing to their biological activity.4 Although the partial zwitterionic character of QMs makes them both electrophilic and nucleophilic, their reactivity with nucleophiles is particularly important in biological systems. It has been demonstrated that QMs react with amino acids5 and proteins,6 and inhibit the action of some enzymes. Moreover, they have been implicated as the ultimate cytotoxins responsible for the function of agents such as antitumor drugs, antibiotics, and DNA alkylators in biological chemistry.7,8 Later on, it was shown that QMs are not the responsible reagents for cross-linking DNA, but they are probably involved in the metabolic formation of formaldehyde.9 To understand the chemistry of anthracycline antitumor antibiotic, a simple stable o-QM has been constructed and entirely characterized. Reaction of quinone methide with nucleophiles, including 2′,3′-isopropylideneadenosine has been examined.10
Several important clinical drugs like cisplatin, psoralens, and mitomycin C are known to induce DNA ISC (interstrand cross-links) formation, which can disrupt cell maintenance and replication.11 Both reductive and oxidative metabolisms form quinone methides and have become a very important topic in drug design and drug safety. Even though ortho-quinone methides were investigated in detail 50 years ago, the last few years have brought about a renaissance in their chemistry. This review summarizes the recent developments in this field and highlights key historical contributions.
2. Generation of ortho-quinone methides
ortho-Quinone methides are highly reactive species that have attracted considerable attention because of their intriguing structure and properties. A number of methods for the generation of o-QMs have been developed till date, enabling the straightforward construction of diverse aromatic molecules with successful applications to a number of complete syntheses of complex natural products. In the past decade, the most popular methods were photochemical initiation of o-(α-phenyl) substituted phenols or thermal initiation of various substituents on the benzene ring of ortho-methyleneacetoxy phenols. Lewis acid, base, and chemical oxidants were also used to generate o-QMs. Thus, the method of generation of the o-QM becomes intimately linked with the manner in which it is to be utilized.2.1. Thermal generation
Among the used procedures to generate o-QMs, the most common ones are thermal and base initiations, in which good to excellent stereoselectivity with excellent yields of the corresponding products have been obtained. Only a limited number of accounts have been reported where acid is used to generate the o-QMs. This is mainly due to the low compatibility of acid with the dienophiles, and because of the greater ionic character of the reaction conditions that result in a product with low stereoselectivity. Baldwin and co-workers12 introduced an efficient method for the generation of o-QM from ortho-methyleneacetoxy phenols that was used for the biomimetic synthesis of (±)-alboatrin (Scheme 1). Alboatrin 1 is a phytotoxic natural product, and its biosynthesis has been realized through hetero Diels–Alder reaction of an orcinol derived o-quinone methide 2 and (R)-4,5-dihydro-2,4-dimethylfuran 3. | ||
| Scheme 1 Retrosynthesis of Alboatrin. | ||
Baldwin assumed that dehydration of the corresponding hydroxymethyl orcinol derivative 4 would give o-QM. Compound 4 was obtained by the reduction of precursor 6b, which was found to be unstable and rapidly decomposed under the reaction conditions. Then a diacetate 5 was prepared, which was reduced to its alcohol and consequently underwent transesterification by itself to give an ortho-methyleneacetoxy phenol 6a under thermal conditions (Scheme 2). Subsequently, simple heating of 6a in the presence of dimethylfuran 3 afforded acetylalboatrin 7 as the major product (63%), acetyl epi-alboatrin (5%), and an inseparable mixture of diastereoisomers 8 (25%). Deacylation of 7 yielded the desired (±)-alboatrin, for which spectral data was found to be identical to the natural (±)-alboatrin. Intramolecular hydrogen bonding might facilitate the elimination of acetic acid upon heating through a six-member transition state, furnishing the o-quinone methide.
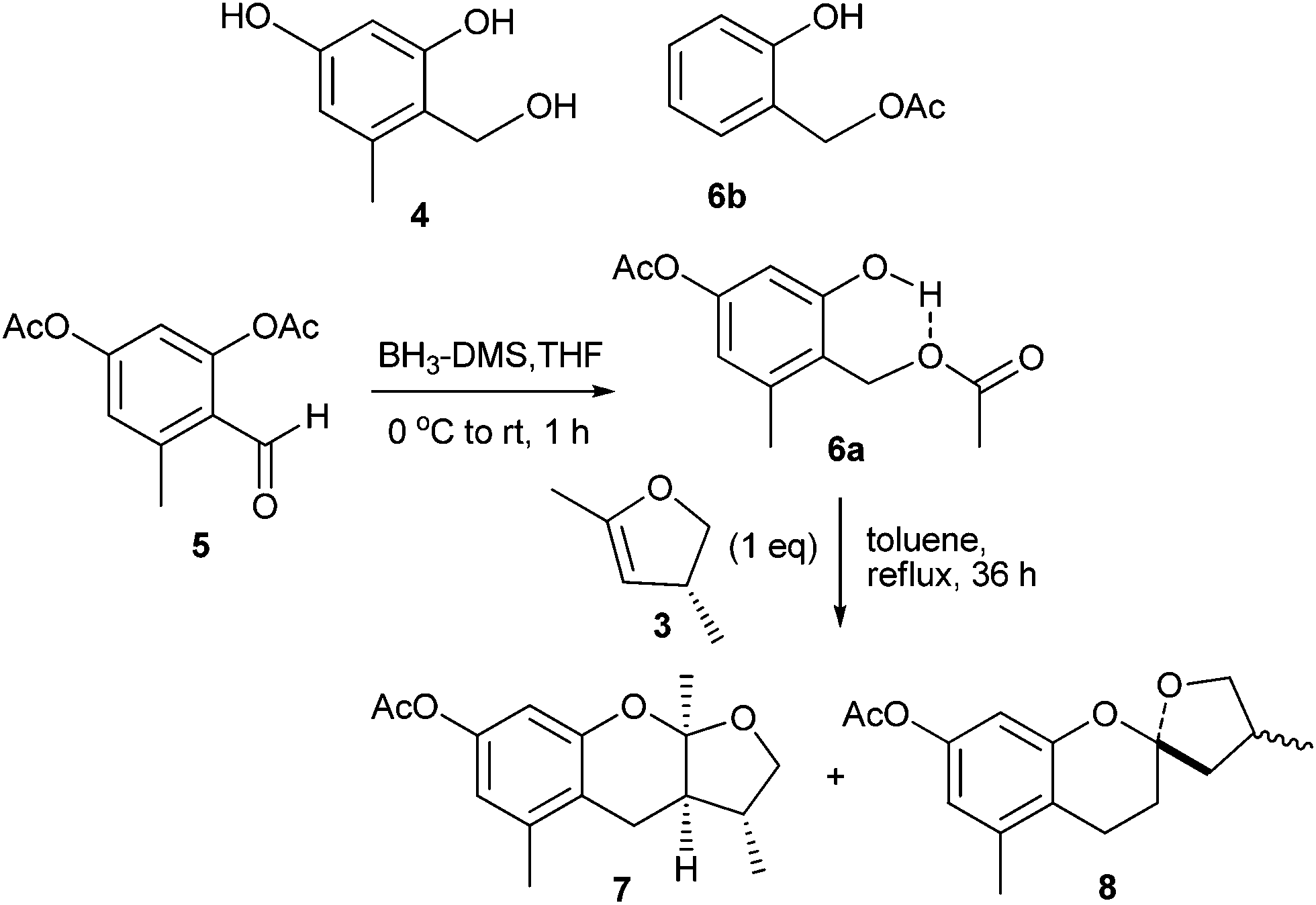 | ||
| Scheme 2 Synthesis of acetylated (±)-alboatrin (7). | ||
Christopher D. Bray13 reported the generation of o-QM from deprotonation of ortho-hydroxybenzyl acetate 9 with iPrMgCl under mild anionic reaction conditions, which reacts with exo-enol ethers 10 as 2π partners in hetero Diels–Alder (HDA) reactions, furnishing mono-benzannelated spiroketals 11 (Scheme 3). This rapid and simple strategy is clearly applicable to the synthesis of a wide range of natural products, including berkelic acid, chaetoquadrin A and cephalostatin.
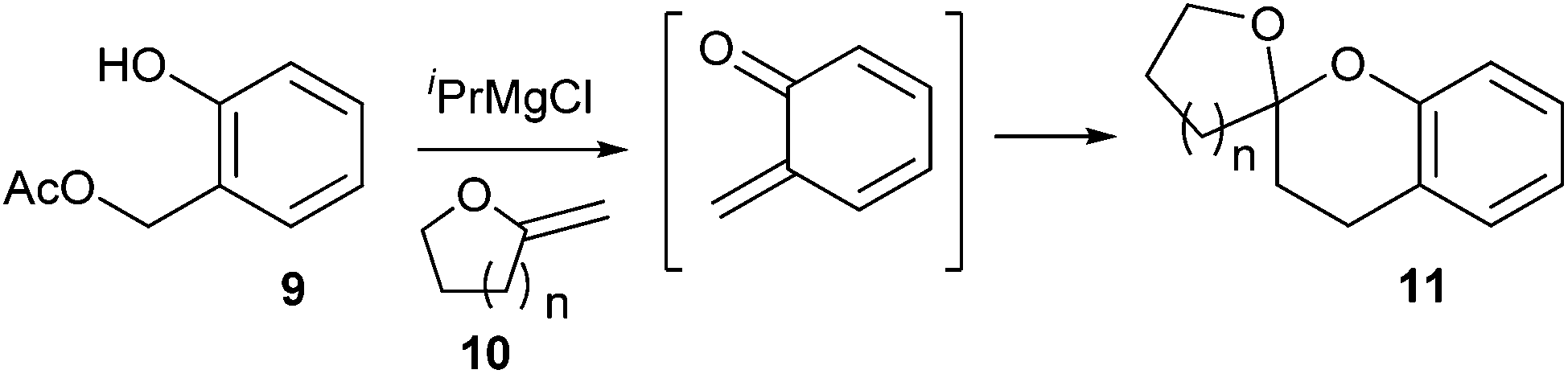 | ||
| Scheme 3 Generation of o-QM using o-hydroxybenzyl acetate and iPrMgCl. | ||
Rosellon and co-workers14 developed a method for the generation of ortho-quinone methides 13 via fluoride-induced desilylation of silyl derivatives of o-hydroxybenzyl iodides 12 under mild experimental conditions (Scheme 4).
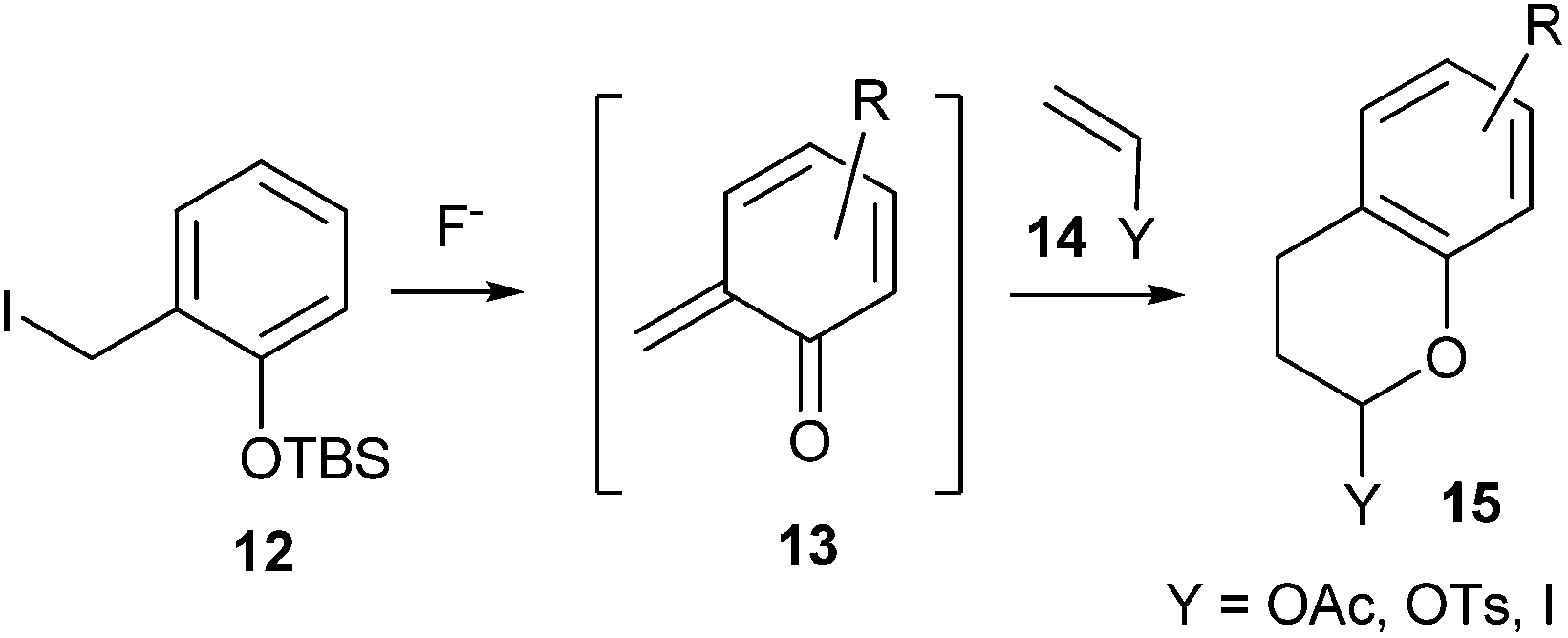 | ||
| Scheme 4 Formation of o-QMs via fluoride-induced desilylation. | ||
Dolatowska and Wojciechowski15 reported the thermal extrusion of sulfur dioxide from 1,3-dihydrobenzo[c]thiophene-2,2-dioxides 16a and 1,3-dihydro-2,1-benzisothiazole-2,2-dioxides 16b to generate the corresponding quinodimethanes 17a and quinonemethyleneimines 17b, which undergo Diels–Alder reaction with alkene 18 to give the compound 19 (Scheme 5).
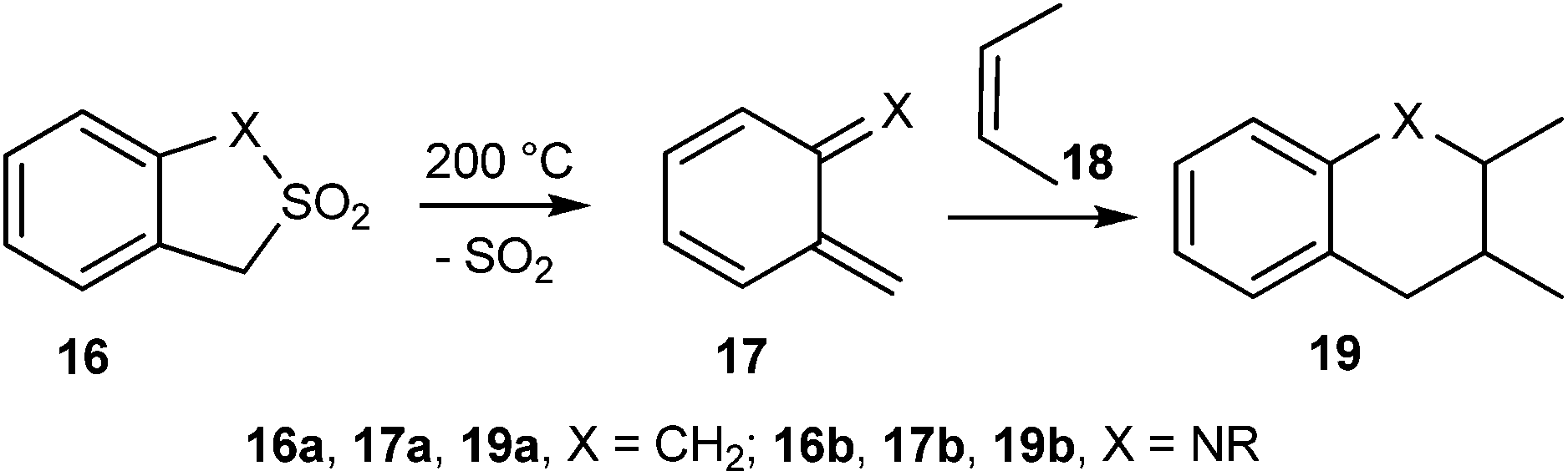 | ||
| Scheme 5 Generation of o-QMs 17 by thermal extrusion of sulfur dioxide. | ||
The sultone of 2-hydroxytoluene-α-sulfonic acid 20 after extrusion of SO2 upon photochemical irradiation gave o-quinone methide 21, which upon reaction with N-phenyl maleimides 22 afforded the respective adducts 23 (Scheme 6).
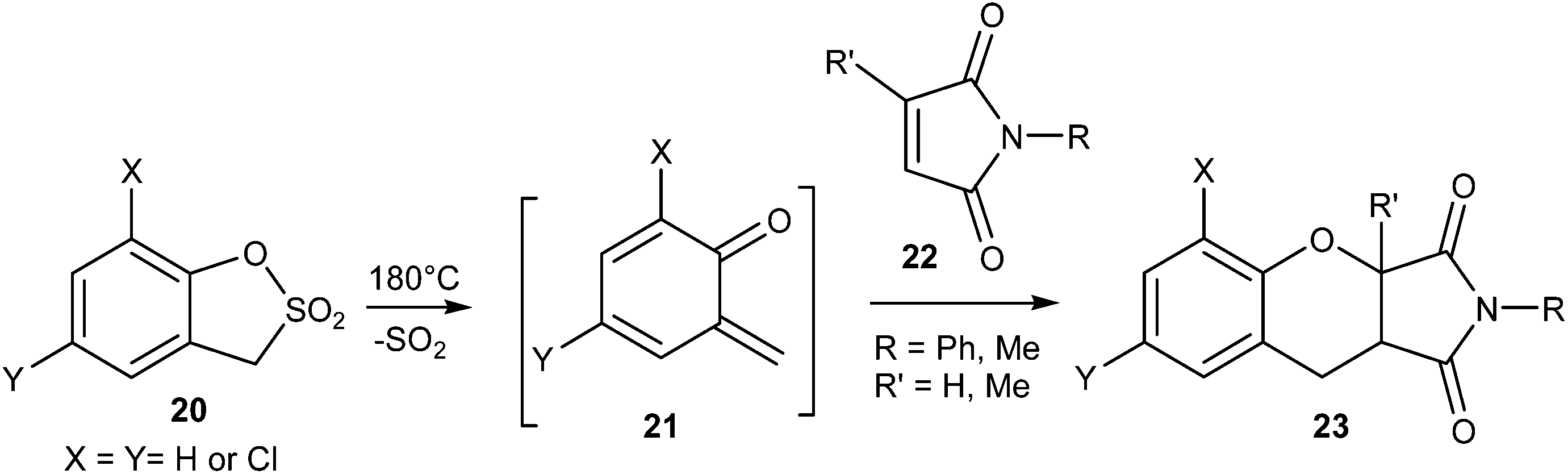 | ||
| Scheme 6 Generation of o-QM from benzosultones. | ||
Ohwada and co-workers16 reported a retro-Diels–Alder reaction of 4H-1,2-benzoxazines 24 to generate o-QMs 25 under the thermal conditions, which undergoes the Diels–Alder reaction with vinyloxy cyclohexane 26 to afford the chroman derivative 27 (Scheme 7). They also presented a mechanistic study of the retro Diels–Alder reaction of benzoxazines 24 with various substituted benzene rings. In general, o-QMs are too unstable to be isolated and observed spectroscopically, unless they are trapped with metal complexes such as Cp*Ir. In all the previously reported methods, o-QMs were generated in situ, and underwent 1,4-conjugate addition with nucleophiles or cycloaddition reaction with suitable dienophiles.
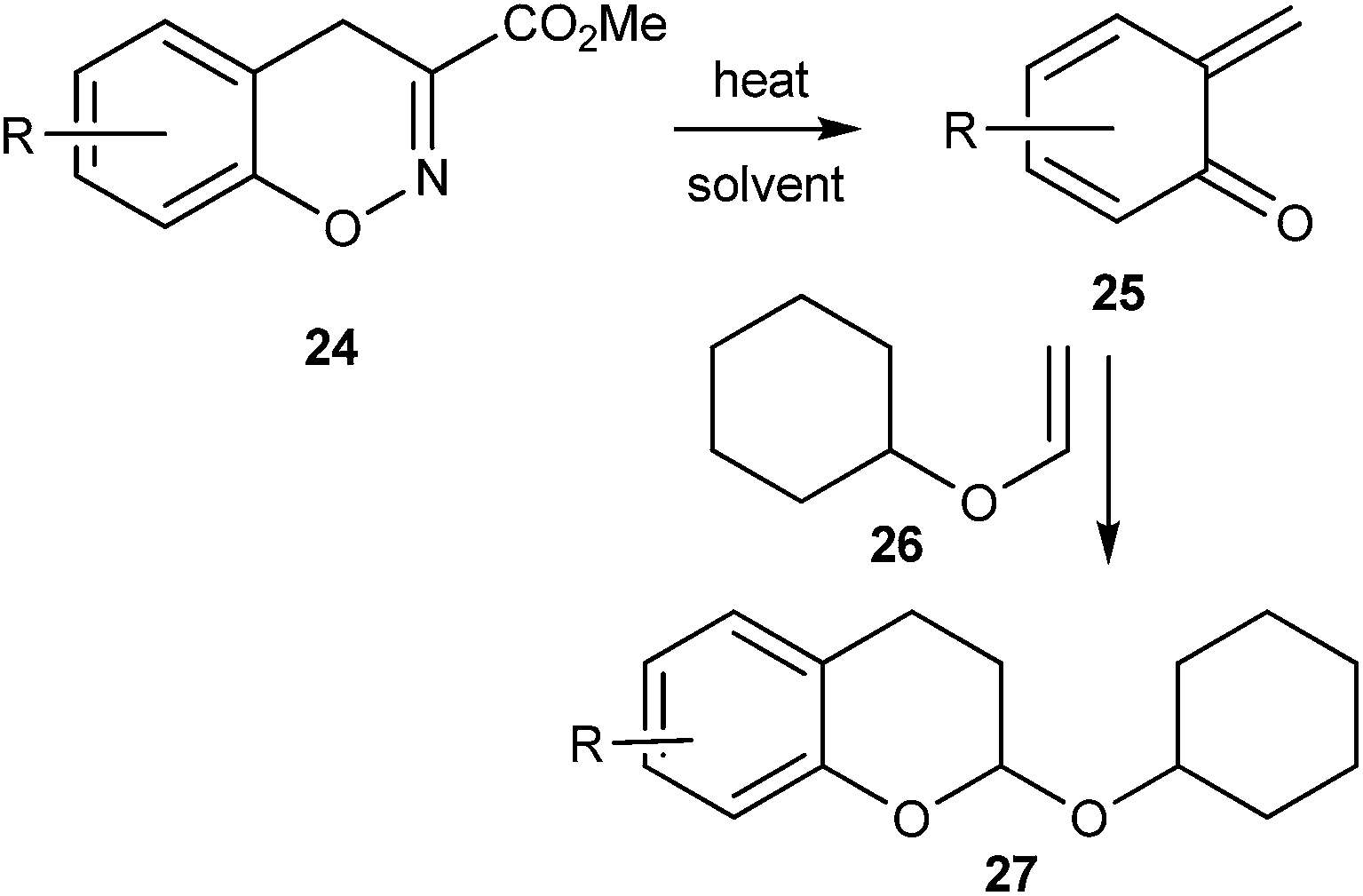 | ||
| Scheme 7 Formation of o-QM via retro-Diels–Alder reaction of 4H-1,2-benzoxazines 24. | ||
o-QMs may be generated through a diverse range of initiation methodologies (Scheme 8). Perhaps the most commonly encountered method for the generation of o-QM is the elimination of a stable molecule, typically a benzylic substituent, to generate the methylene and α-carbonyl functionalities with concomitant dearomatisation. Various precursors have been utilized for the thermolytic generation of ortho-quinone methides. It should be noted that all thermal generation techniques preclude the application of nucleophiles that are thermally unstable. With any given precursor, there is a substantial temperature range for initiation that depends upon the substituents. In general, if the process involves significant non-bonded interactions, then the temperature requirements are higher, while extended conjugation or other stabilizing factors lower the overall temperature requirements (Scheme 8).
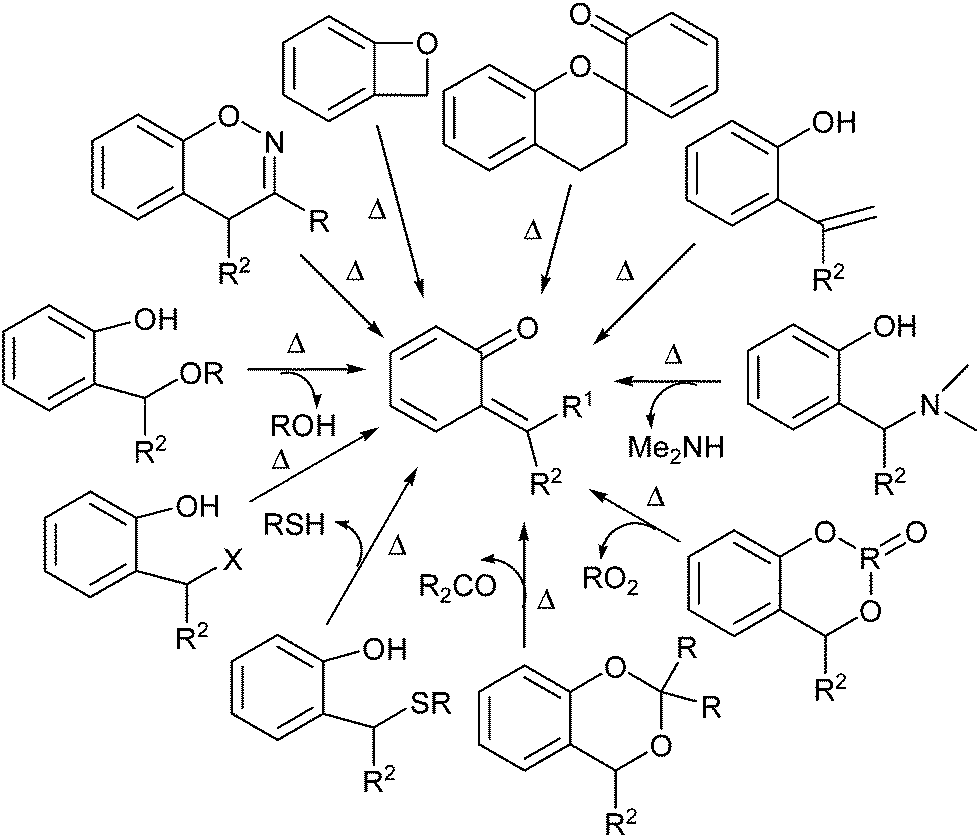 | ||
| Scheme 8 Thermal generation of o-QMs. | ||
2.2. Photochemical generation
Apart from the thermal generation of o-QM intermediates in the presence of different basic or acidic reagents, photochemical energy sources have also been extensively used to generate o-QMs. In general, o-QM intermediate 29 is generated by light-induced water elimination from ortho-hydroxybenzyl alcohols 28 or by excited state intramolecular proton transfer (ESIPT) in ortho-hydroxystyrenes 30 (Scheme 9).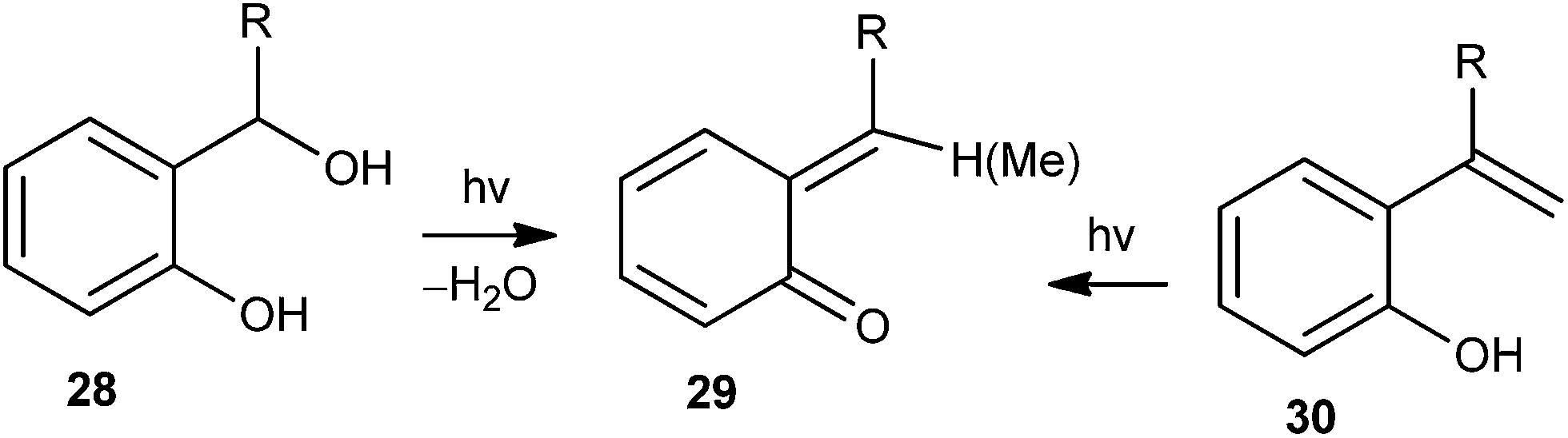 | ||
| Scheme 9 Photochemical generation of o-QMs. | ||
The majority of β-cyclodextrin inclusion complexes undergo photohydration of the olefin, preventing the formation of o-QM by insertion of a methylene group between the phenol and the olefin moieties. Miranda et al.17 reported the formation of an o-QM via C–C fragmentation of a zwitterion formed by excited state intramolecular proton transfer (ESIPT) from an ortho-allylphenol derivative 31, for the first time (Scheme 10). The mechanism has been explained in terms of the formation of the zwitterionic intermediate 32A by the initial ESIPT from the phenolic subunit to the double bond. Thus, subsequent C–C bond formation with concomitant ring opening elucidates the generation of the o-QM 32B, which would be trapped by solvent methanol to give compound 33. The direct involvement of an o-QM intermediate has been confirmed by laser flash photolysis. Use of 266 nm excitation wavelengths in acetonitrile solvent gave a well defined signal with two maxima around 300 and 400 nm.
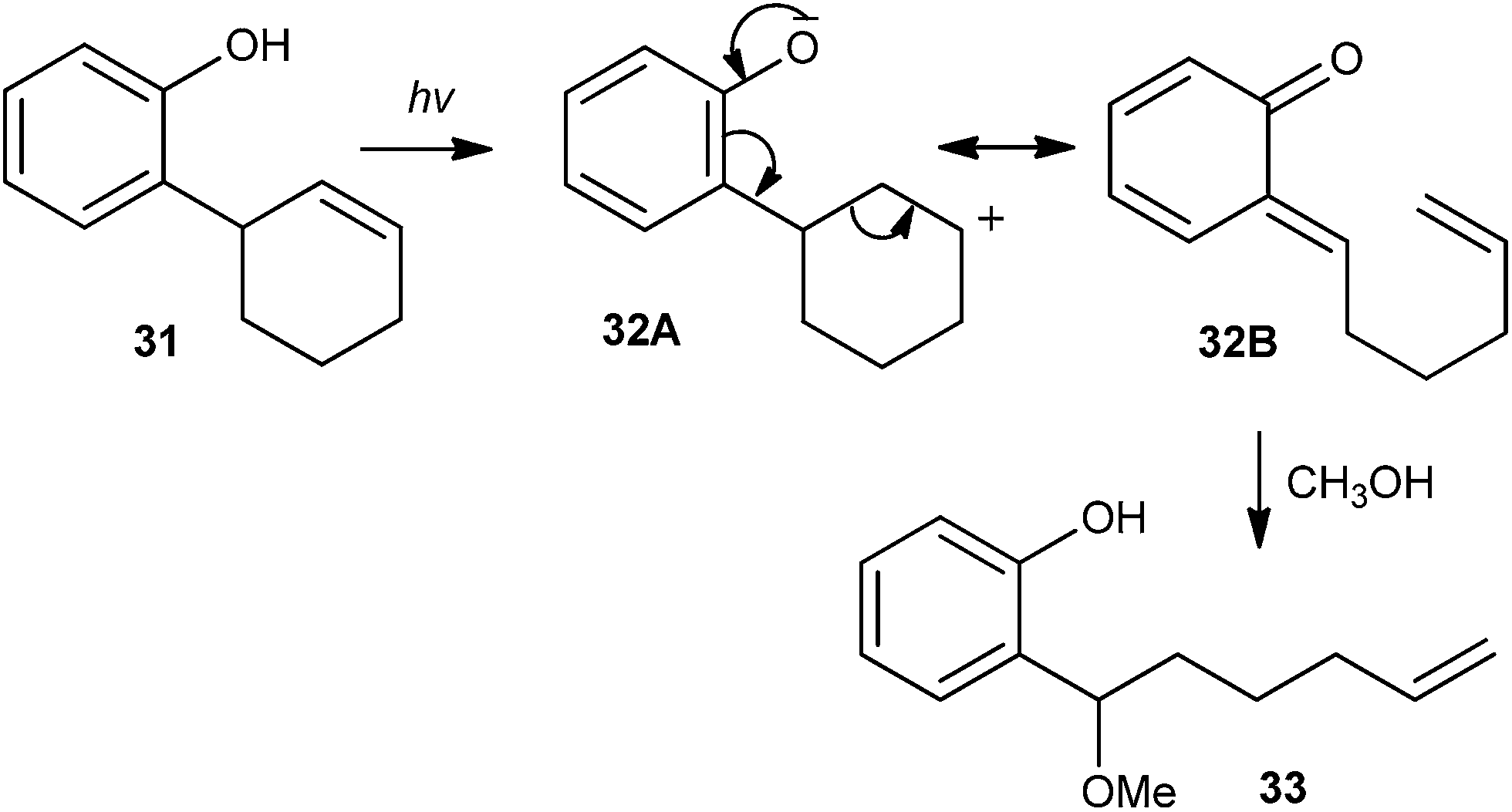 | ||
| Scheme 10 Generation of o-QM via C–C fragmentation of zwitterions. | ||
Generation of o-QM by flash photolysis of ortho-hydroxybenzyl substrates 34 in aqueous solution has also been reported (Scheme 11). Hydrogen-ion catalyzed hydration of o-QM furnished back to the ortho-hydroxybenzyl alcohol, which was anticipated to ensue through the initial fast equilibrium protonation of the carbonyl oxygen followed by trapping with water. The slower uncatalyzed hydration proceeds through nucleophilic attack of water on the QM methylene group.18
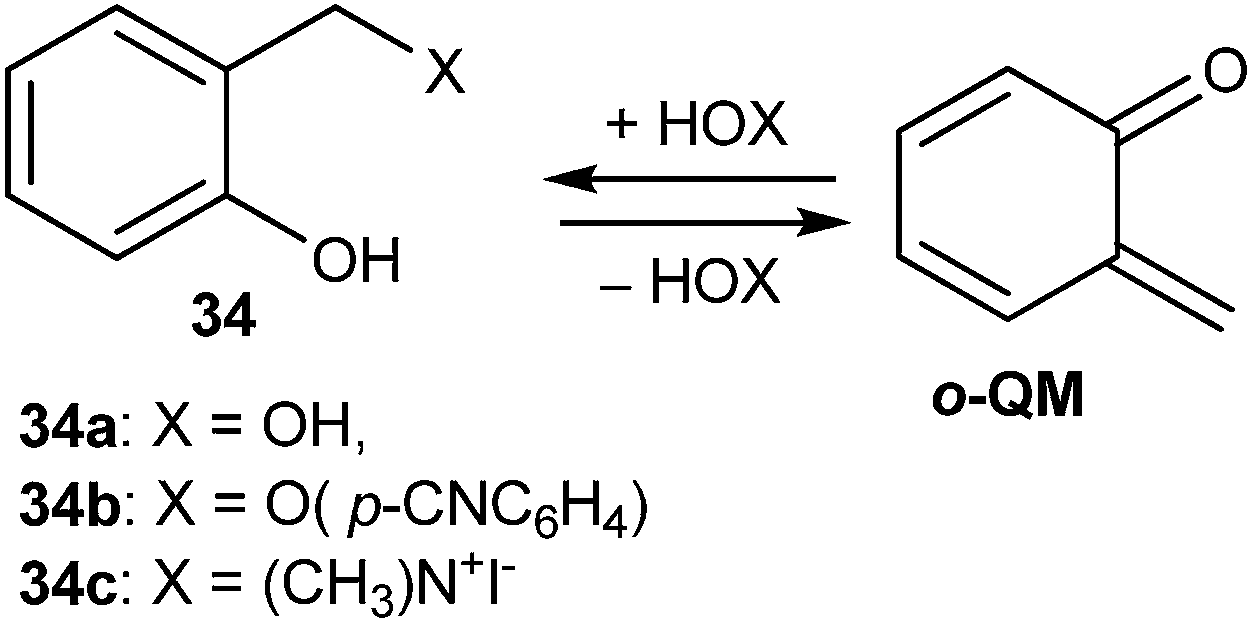 | ||
| Scheme 11 Flash photolysis of o-hydroxybenzyl substrate 34 to give o-QM. | ||
o-Quinone α-phenylmethide was produced as a short-lived transient species in aqueous solution by flash photolysis of ortho-hydroxy-α-phenylbenzyl alcohol, and its rate of decay was measured in HClO4 and NaOH solutions as well as in CH3CO2H, H2PO4−, and HCO3− buffers. Results showed that the hydration of quinone methide back to its benzyl alcohol precursor occurs by acid-, base-, and un-catalyzed routes. Kresge and co-workers19 reported the generation of o-QMs 35 and 36 by photodehydration of the corresponding ortho-hydroxybenzyl alcohols 37 and 38. ortho-Hydroxybenzyl-p-cyanophenyl ether 39 after photo-cleavage gave o-QMs 36 (Fig. 4). Flash photolytic techniques have been used to monitor the progress of the reactions because the reactions of quinone methides are very fast.
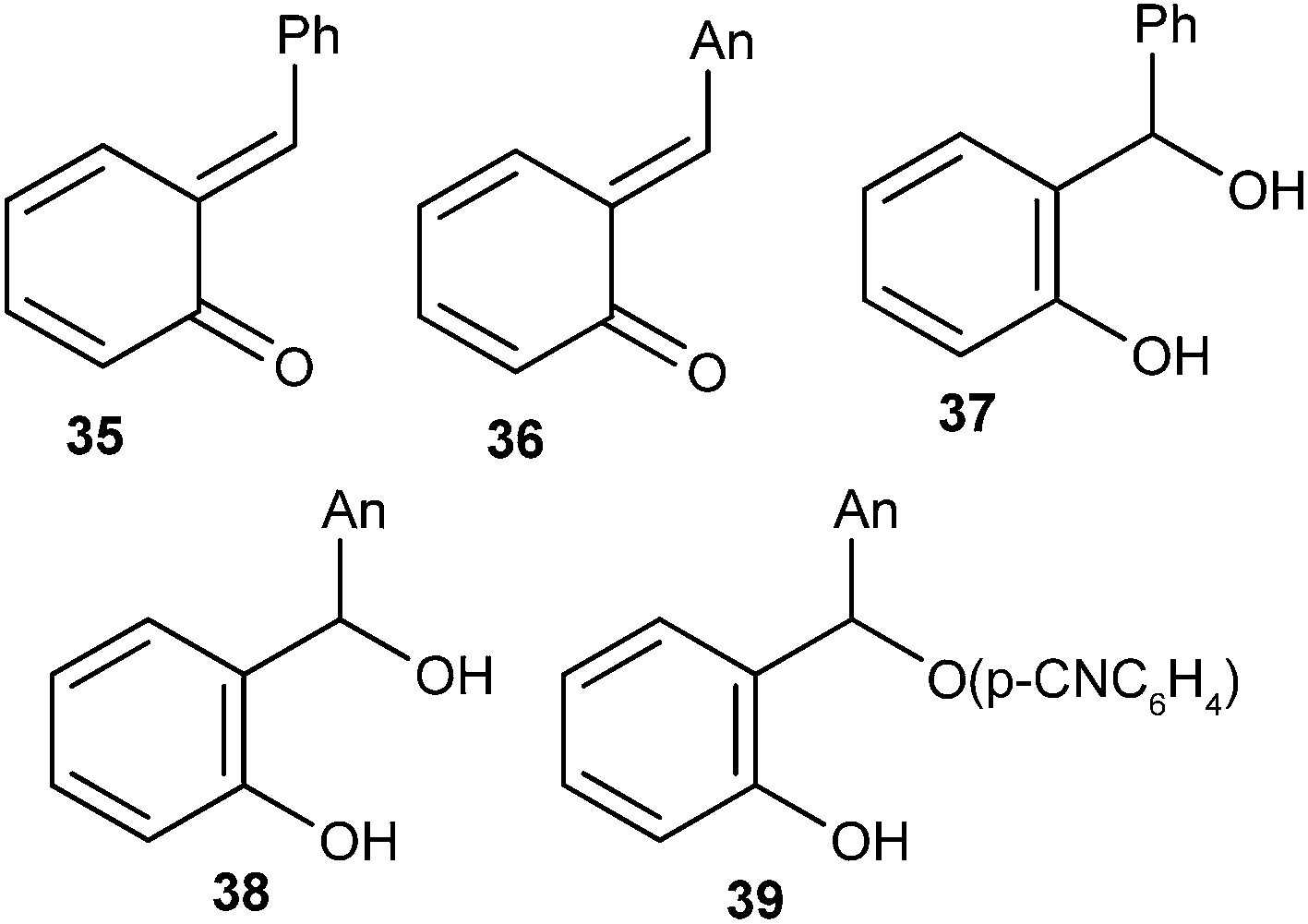 | ||
| Fig. 4 Structures of o-hydroxybenzyl alcohols and their photodehydrated o-QMs. | ||
Lukeman and Wan20 observed predominate D2O exchange at the 2′-position irrespective of D2O (MeOD) content during the study of the photochemical incorporation of deuterium on phenylphenol 40 in D2O (CH3OD)–CH3CN solutions with varying D2O (CH3OD) content. Exchange at the 2′-position (but not at the 4′-position) was observed when crystalline samples of compound 40 (deuterated OH i.e. OD) were irradiated (Scheme 12).
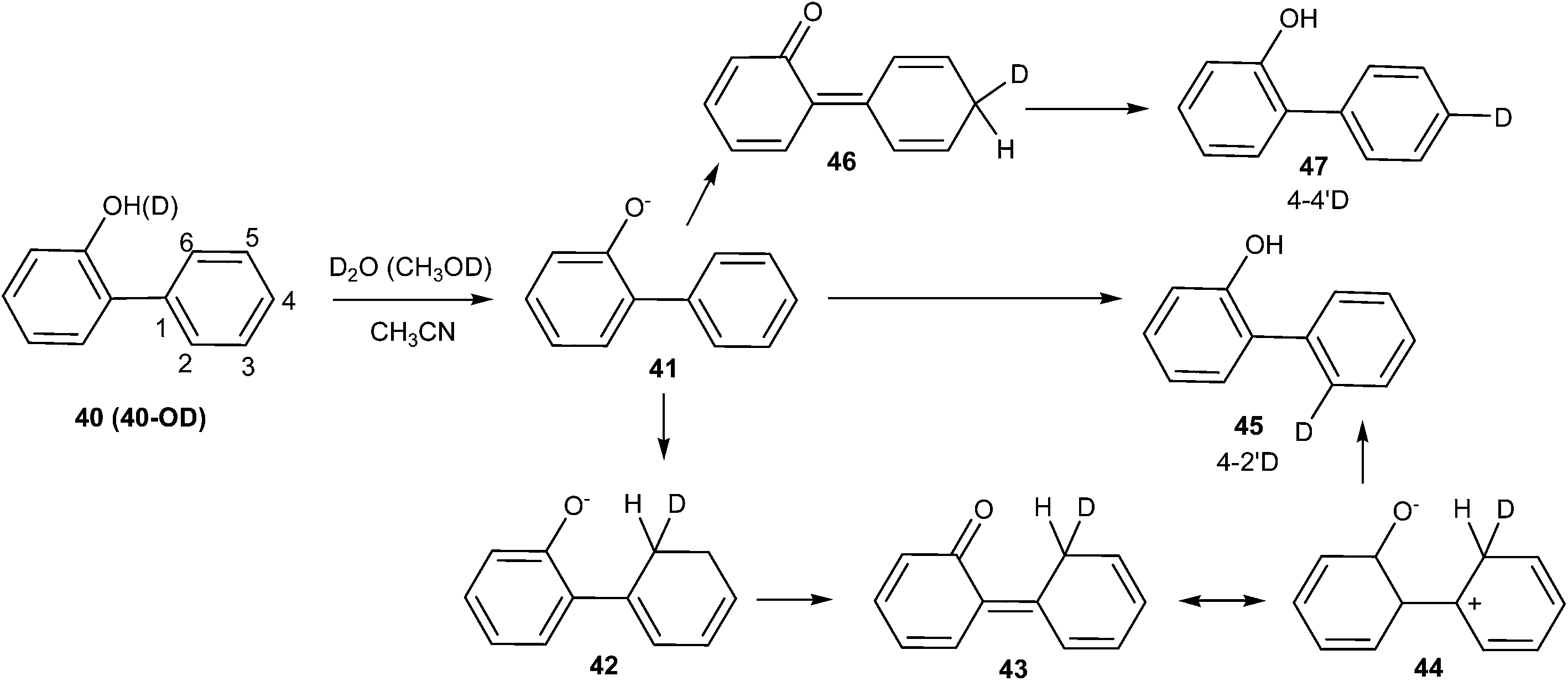 | ||
| Scheme 12 Photochemical incorporation of deuterium on phenylphenol. | ||
No deuterium exchange was observed in case of compounds 49–52, whereas 2,2′-biphenol 48 underwent similar photoexchange reaction as compound 40 (Fig. 5).
 | ||
| Fig. 5 Structure of compounds which do not undergo deuterium exchange. | ||
Wan and co-workers21 reported the generation of quinone methide 54 by ESIPT, in which naphthyl group acts as the proton acceptor (Scheme 13). The reaction has been carried out in a variety of solvents, as well as in the solid state, illustrating that the proton transfer is intrinsic (proton transfer does not require water).
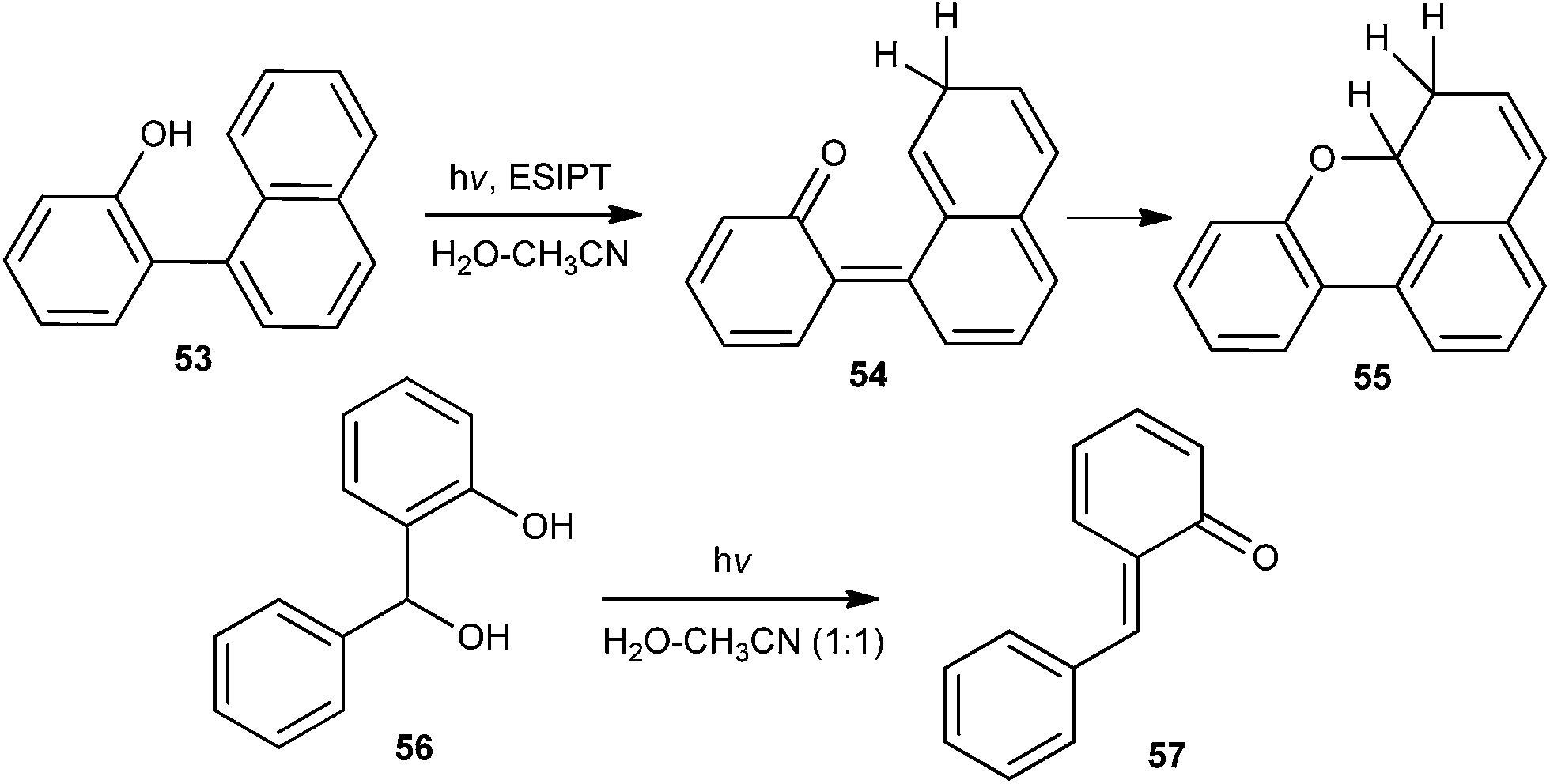 | ||
| Scheme 13 Generation of o-QM by ESIPT. | ||
Zhan and co-workers22 have reported the generation of o-thioquinone methide 59 by flash photolysis of benzothiete 58 in aqueous medium (Scheme 14). The hydration rates of the quinone methide 59 to ortho-mercaptobenzyl alcohol 60 were measured in perchloric acid solution using H2O and D2O as the solvent in acetic acid in the presence of tris(hydroxymethyl)methylammonium ion buffers.
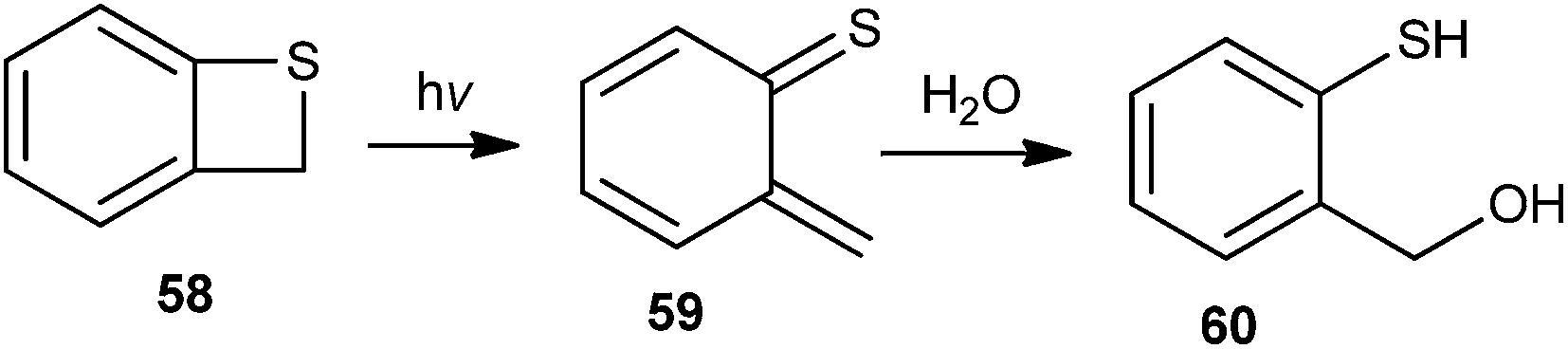 | ||
| Scheme 14 Generation of o-thioquinone methide 59. | ||
The solvent isotope effects on hydronium-ion catalysis of hydration for the two substrates are different. Solvent isotopic effect for the oxygen quinone methide 61 is kH/kD = 0.42, whereas for the sulfur substrate 59 is kH/kD = 1.66. The inverse isotope effect (kH/kD < 1) in the oxygen containing system specifies pre-equilibrium proton-transfer reaction mechanism by which protonation of the substrate on its oxygen atom being fast and reversible, and capture of the benzyl-type carbocationic intermediate is observed. On the other hand, the normal isotope effect (kH/kD > 1) in the sulfur system indicates that the protonation of the substrate on its sulfur atom is rate-determining and carbocation capture is fast (Scheme 15).
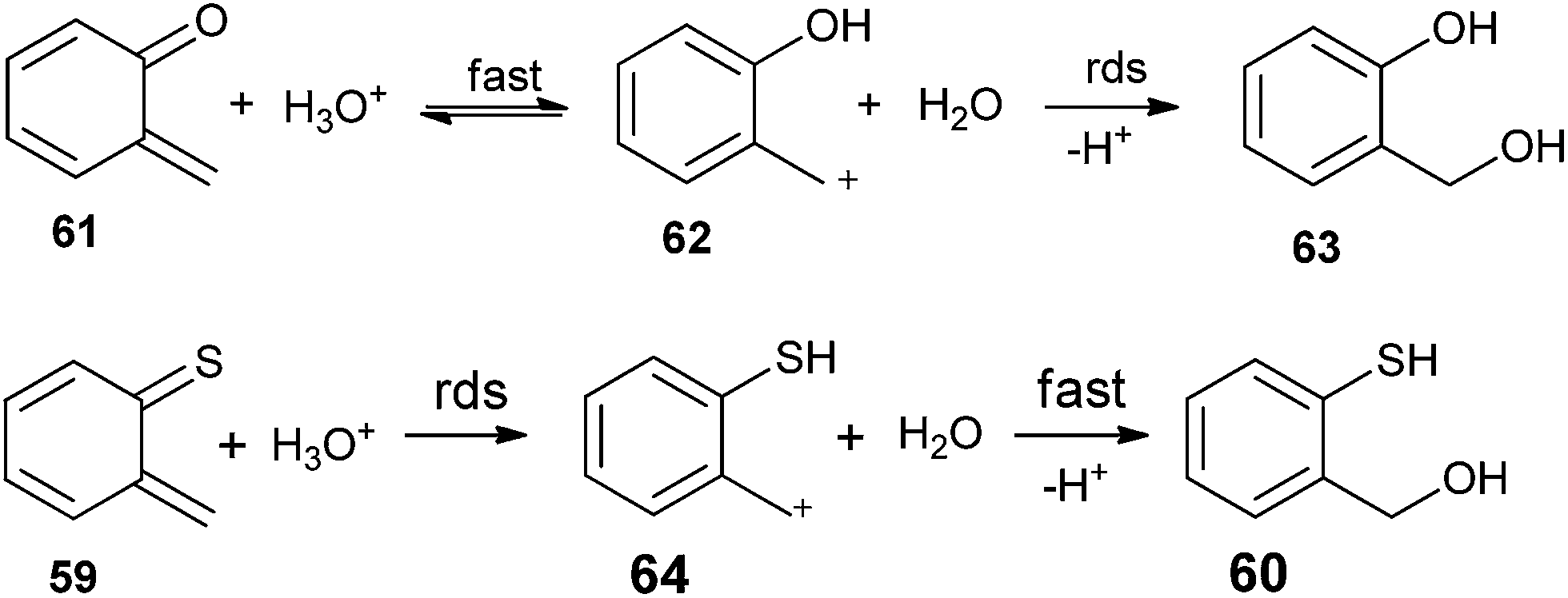 | ||
| Scheme 15 Solvent isotope effects on o-QM and o-thioquinone methide. | ||
Matsumoto and co-workers23 reported that the thermal and photochemical generation of o-QM was accelerated by the presence of water molecules in the reaction solvents via the formation of anionic micelles and vesicles. According to their report, amino phenanthrol 65 and their naphthalene analogues act as precursors and transforms to QM 68 in the presence of light (Scheme 16).
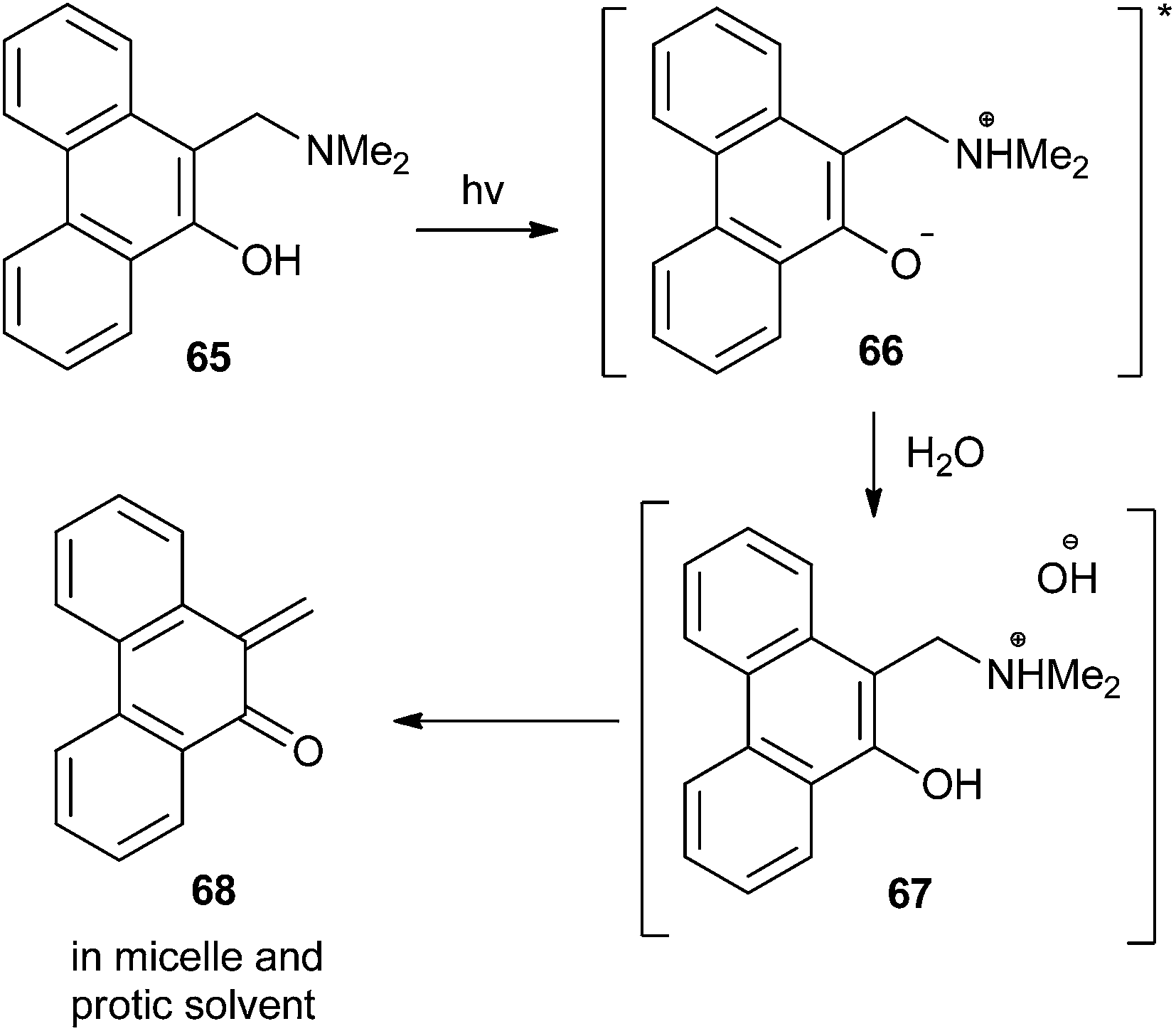 | ||
| Scheme 16 Generation of QMs by the formation of anionic micelle and vesicle. | ||
Popik and Arumugam24 reported the efficient generation of quinone methide from 2-hydroxymethyl phenol 69. Isomeric 3-hydroxy-2-naphthalenemethanol 70 and 2-hydroxy-1-naphthalenemethanol 72 resulted the respective naphthaquinone methides, 2,3-naphthoquinone-3-methide 71 and 1,2-naphthoquinone-1-methide 73 (Scheme 17). During the generation of ortho-naphthaquinone methides (o-NQMs) 71 and 73 in the flash photolysis of ammonium precursors 70c and 72c ensues within the duration of a laser pulse, irradiation of 70a and 72a as well as their ethoxy derivatives 70b and 71b provides evidence for the detection of a kinetic precursor to o-NQMs. The rate of formation of o-NQMs does not depend on the pH of the solution. Furthermore, the generation of cyclohexadienone 75 and 76 by the photochemical irradiation of three different precursors 74a–c and 76a–c have been reported (Scheme 17).
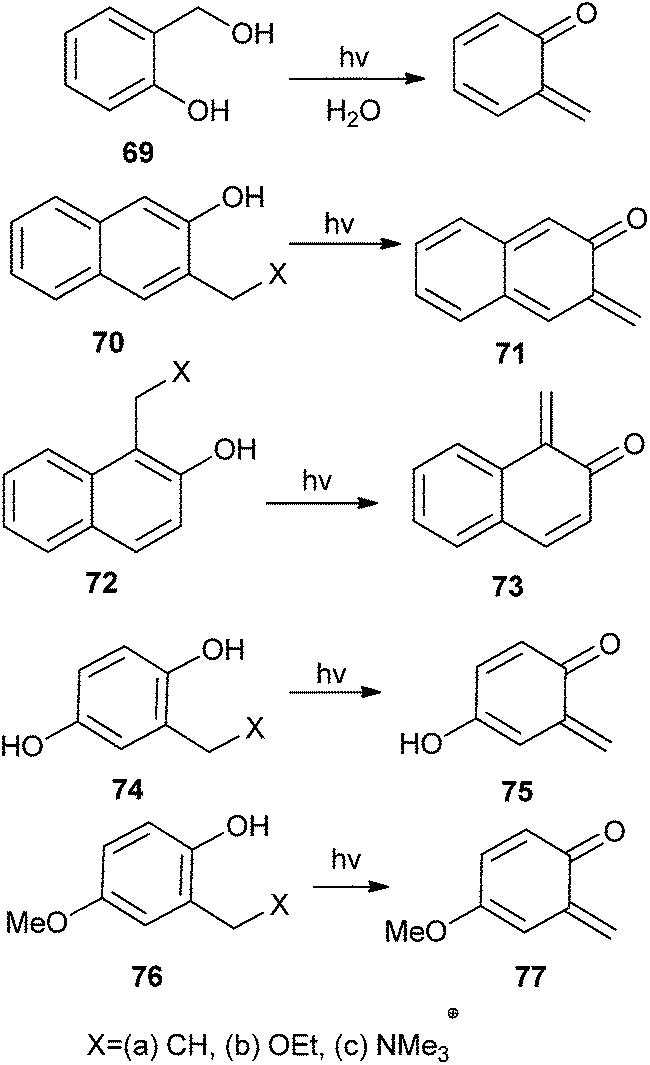 | ||
| Scheme 17 Generation of o-QM by using phenolic derivatives. | ||
Wan and co-workers25 tactically substituted the hydroxymethyl phenols with 2-hydroxy-2-adamantyl moiety and explored their photochemical reactivity. Upon the excitation of AdPh derivative 78 to its singlet excited state, it undergoes a intramolecular proton transfer at first, followed by the loss of H2O to give quinone methide 80, which has been detected by laser flash photolysis (CH3CN–H2O 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, τ = 0.55 s) and UV-vis spectroscopy (Scheme 18). The quantum yield of QM formation has been increased by three fold, and the lifetime is enhanced by the introduction of the adamantyl substituent on the ortho-hydroxymethyl phenol moiety.
:1, τ = 0.55 s) and UV-vis spectroscopy (Scheme 18). The quantum yield of QM formation has been increased by three fold, and the lifetime is enhanced by the introduction of the adamantyl substituent on the ortho-hydroxymethyl phenol moiety.
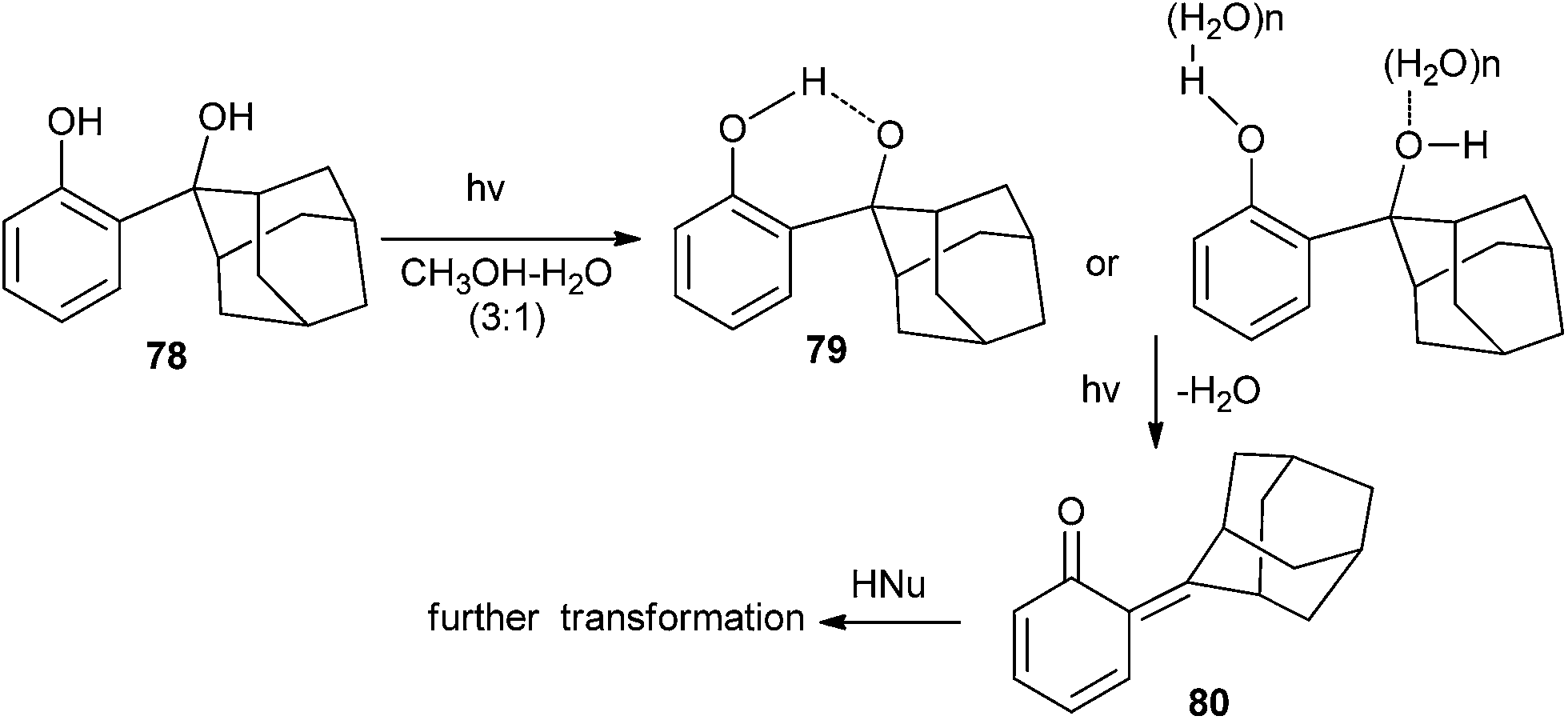 | ||
| Scheme 18 Formation of adamantyl substituted o-QM 80. | ||
Freccero and co-workers26 reported the photogeneration of new Binol quinone methides 83 and 84. They also explored mono- and bisalkylation of free nucleophiles by product distribution analysis and laser flash photolysis in water solution using Binol quaternary ammonium derivatives 81 and 82 as photoactivated precursors (Scheme 19).
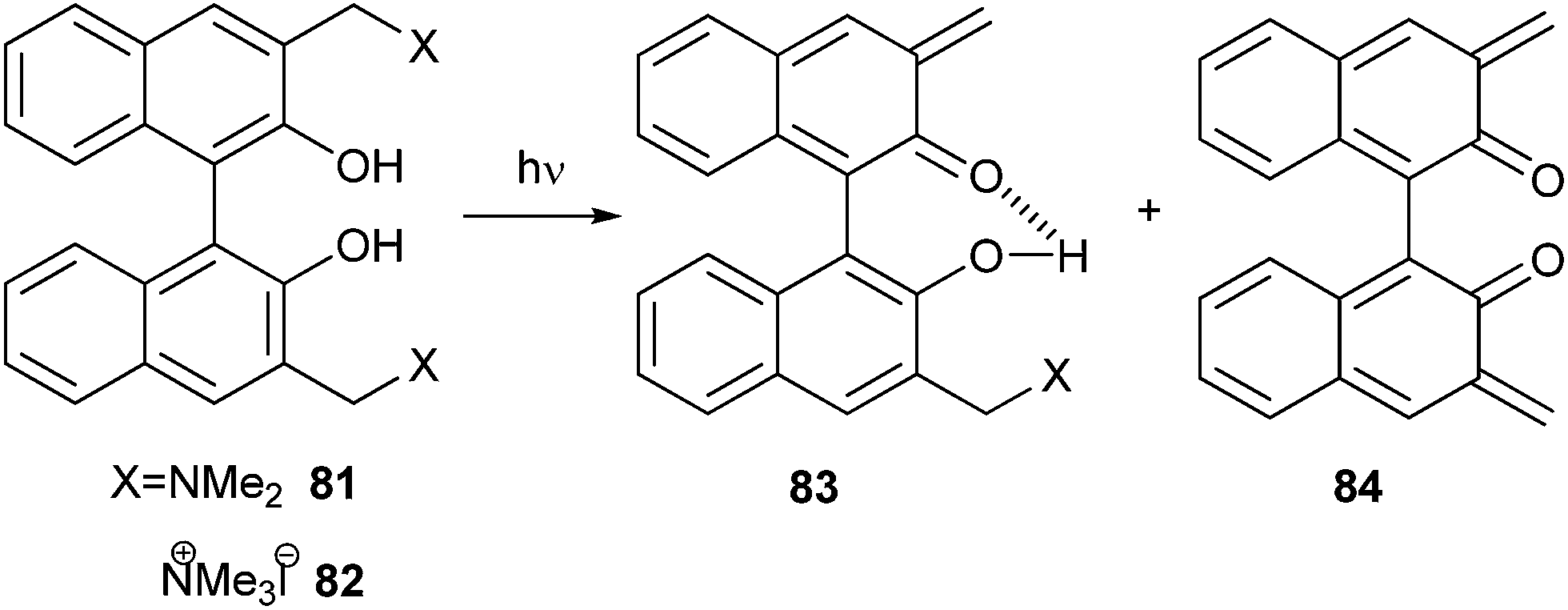 | ||
| Scheme 19 Photochemical generation of binol quinone methide. | ||
Excitation of anthracene derivative 85 to S1 initiates photodehydration giving the corresponding quinone methide 86 that was detected by laser flash photolysis (LFP) in 2,2,2-trifluoroethanol.27 The QM decays by protonation giving a cation 87, which subsequently reacts with nucleophiles to give compound 88 (Scheme 20). QMs of the anthrol series is important for its potential use in biological systems because the chromophore absorbs at wavelengths > 400 nm. Anti-proliferative investigations conducted with 2-anthrol derivative 85 on three human cancer cell lines showed higher activity for irradiated cells.
 | ||
| Scheme 20 Formation of QM by photodehydration of hydroxylated anthracene. | ||
The vinylidene-quinone methides 92 and 93 were generated by irradiation of 2-alkynylphenols 89 and 90, respectively, which were detected by laser flash photolysis in organic solvents and aqueous acetonitrile (Scheme 21).28 It was also shown that the UV-vis properties and the electrophilicity are enhanced by a β-silicon effect and solvent polarity.
 | ||
| Scheme 21 Photochemical generation of vinylidene-QMs. | ||
3. Reactions of ortho-quinone methides
The transient nature of ortho-quinone methides is due to their propensity to undergo rapid rearomatisation, either by the Michael addition of nucleophiles or often more usefully by the cycloaddition with 2π partners or via oxa-6π-electrocyclisation. Surprisingly, this rapid reactivity was historically seen as a deterent to synthetic development in the field, with the potential for dimerisation and trimerisation sometimes being major stumbling block. To avert this, it is common to employ an excess of the nucleophilic or dieneophilic components and the localized o-QM concentration is often kept low.o-QMs, being labile intermediates, have been stabilized by metal coordination. The examples of simple isolated quinone methides are rare. In fact, in condensed phases, the parent compound has only been characterized spectroscopically at lower temperatures because it is extremely reactive. Spectroscopic evidence of the zwitterionic form of the o-QM derived from vitamin E has only been possible at −78 °C, through the stabilization by interaction with N-methylmorpholine N-oxide. These results show how difficult it is to isolate quinone methides. para-Quinone methides have been discussed as intermediates in the chemistry of lignins and have been used in organic synthesis as electrophiles or electron acceptors. Because the parent o-QM molecule is unstable, NMR spectroscopic data are not available. Therefore, the metalated parent ortho-quinone methide complex was examined using two-dimensional NMR spectroscopic techniques to understand its structure and reactivity. o-QMs are also believed to be key intermediates in the action of several antitumor and antibiotic drugs.
3.1. Electrocyclisation and cycloadditions
 | ||
| Scheme 22 Synthesis of chromenes via electrocyclization of o-QMs. | ||
ortho-Quinone methides are versatile intermediates involving a minimum of seven carbon atoms, which are mainly involved in 1,4-Michael type additions as well as aza-Michael reactions with various nucleophiles. o-QMs also give Diels–Alder and hetero-Diels–Alder cycloaddition products (Scheme 23). o-QM especially derived from 4-hydroxycoumarin undergo [4 + 2] cycloaddition reaction with penta-fulvenes to afford pyranocoumarin and pyranopyrone.30
 | ||
| Scheme 23 Reactivity of ortho-quinone methide with alkenes. | ||
Ploypradith et al.31 reported the generation of o-QMs by the reaction of compound 103 with p-TsOH on silica. Hetero Diels–Alder (HDA) reaction of in situ generated o-QMs 104 with styrenes 105 and 106 gave the corresponding arylchromans 107 and 108 in good yields (Scheme 24).
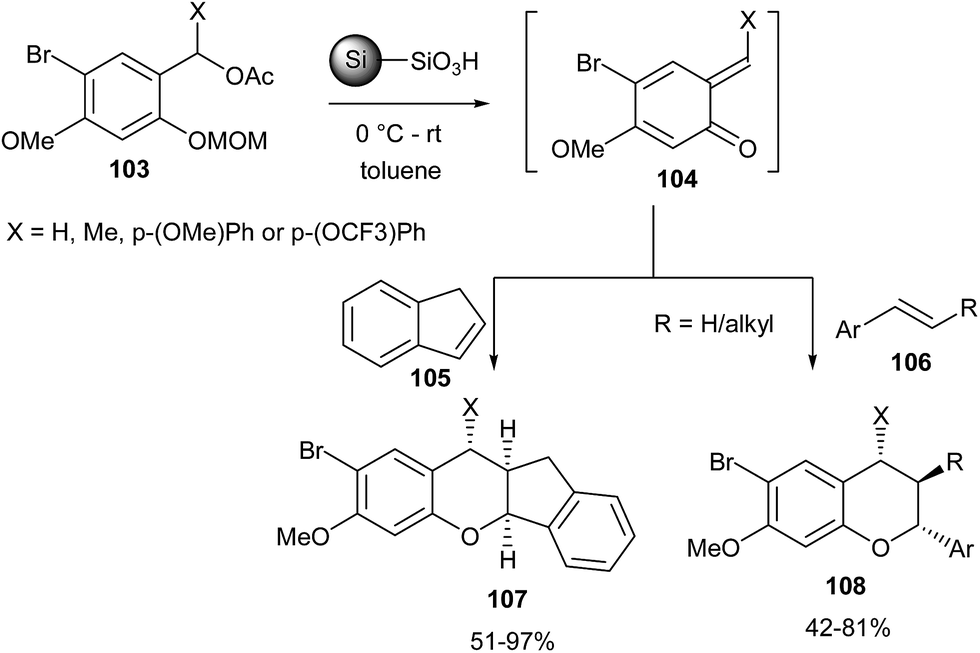 | ||
| Scheme 24 Acid-catalysed generation of o-QMs and their HDA reaction with styrenes. | ||
Sajiki and co-workers32 established a FeCl3-catalyzed method for the synthesis of 1-naphthoquinone-2-methides 112a from dihydronaphthalenes 109 and further transformation of the products in an annulation reaction with various allyl silanes to afford biologically useful dihydronaphthopyran derivatives. The annulation can proceed via an o-NQM intermediate. The site-selective cleavage of one C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond is promoted by the coordination of the two oxygen atoms of 109 to the Lewis acid to give a five-member ring, which is then cleaved to give the zwitterionic intermediate 110. Subsequent migration of the siloxymethyl group (–CH2OTBS) and aromatization provides the 2-siloxymethyl-1-naphthol 112. Further, Lewis acid induced elimination of the silanol (TBSOH) leads to an o-NQM 112a, which undergoes annulation with allyl-TMS through a hetero-Diels–Alder reaction to furnish 113 (Scheme 25).
O bond is promoted by the coordination of the two oxygen atoms of 109 to the Lewis acid to give a five-member ring, which is then cleaved to give the zwitterionic intermediate 110. Subsequent migration of the siloxymethyl group (–CH2OTBS) and aromatization provides the 2-siloxymethyl-1-naphthol 112. Further, Lewis acid induced elimination of the silanol (TBSOH) leads to an o-NQM 112a, which undergoes annulation with allyl-TMS through a hetero-Diels–Alder reaction to furnish 113 (Scheme 25).
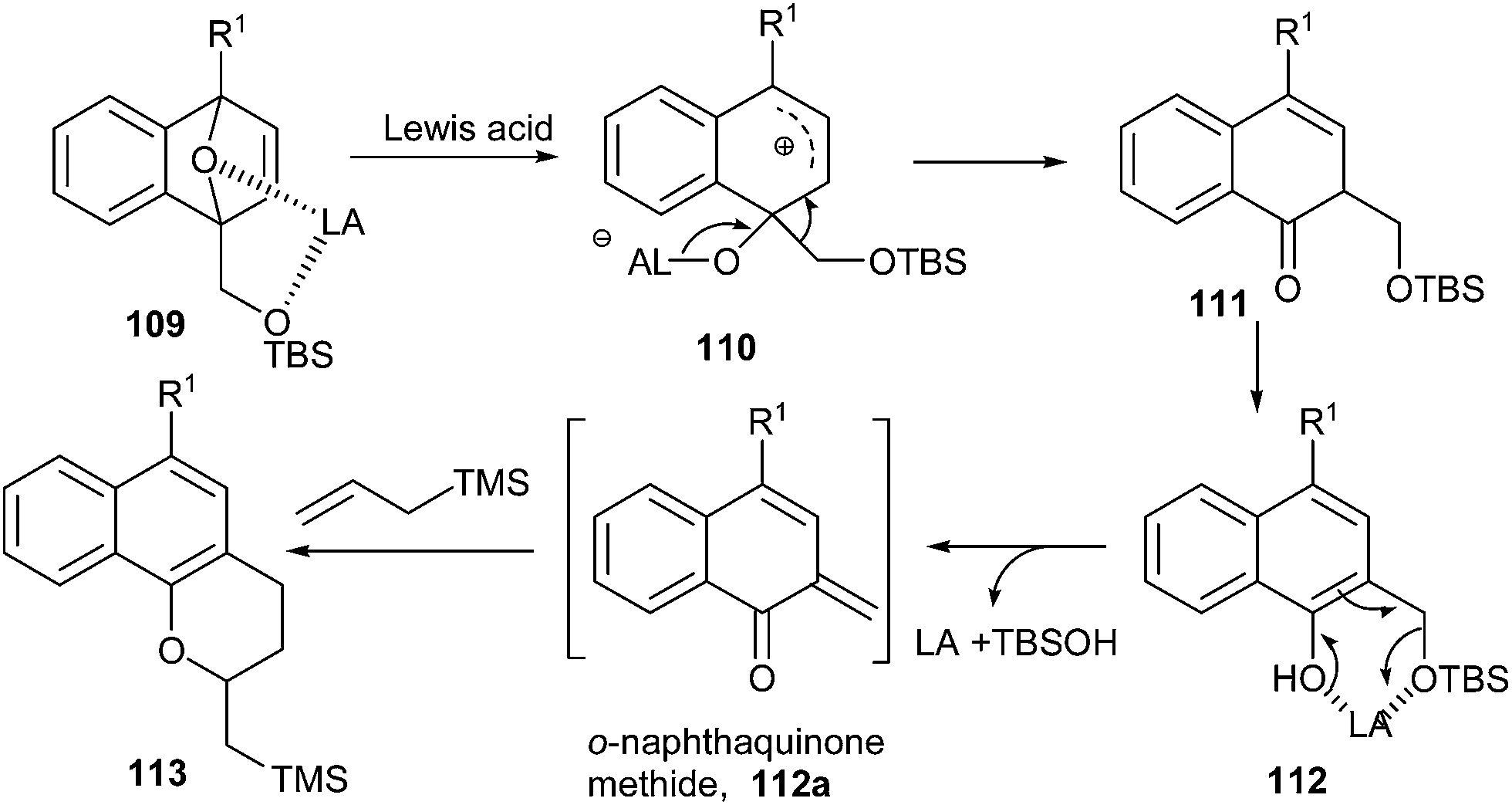 | ||
| Scheme 25 FeCl3-catalyzed synthesis of 1-naphthoquinone-2-methides. | ||
Lindsley et al.33 reported the total synthesis of polemannones B 114 and C 115, which are highly oxygenated benzoxanthenones derived from Polemannia montana, by employing a catalytic Cu(II)/sparteine oxidant system for β,β-phenolic couplings and cascade Diels–Alder reaction by tandem inverse-electron demand, producing 75–90% yield (Fig. 6).
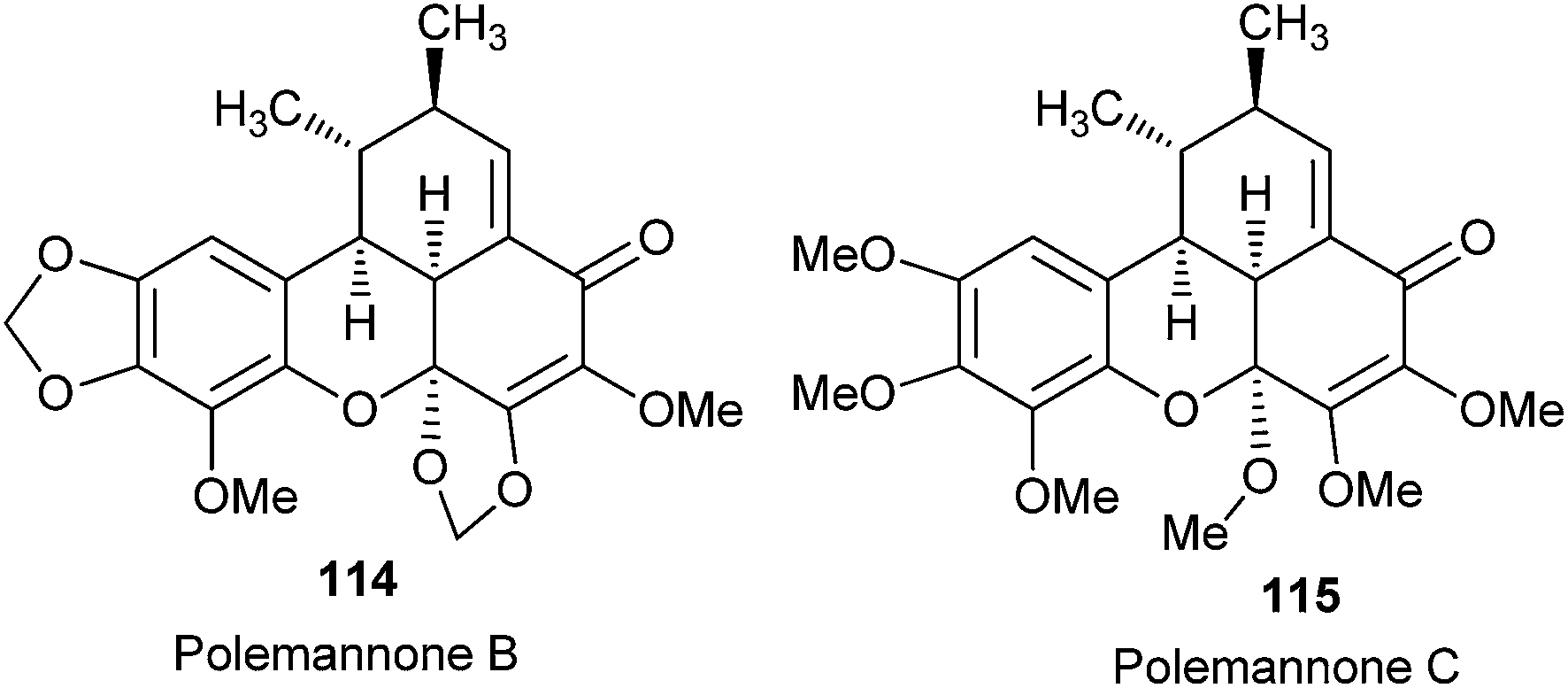 | ||
| Fig. 6 Structure of polemannones. | ||
The polemannones are unique because there is an extra electron donating ether moiety. Compounds containing electron-donating ether moieties para to the phenol, such as 116, stabilize the o-QM intermediate and provide the ‘push’ in the inverse-electron demand in the Diels–Alder reaction during the synthesis of 118 via o-QM 117 (Scheme 26). If two electron-donating ether moieties are present, the second one is always located at meta to the phenol.
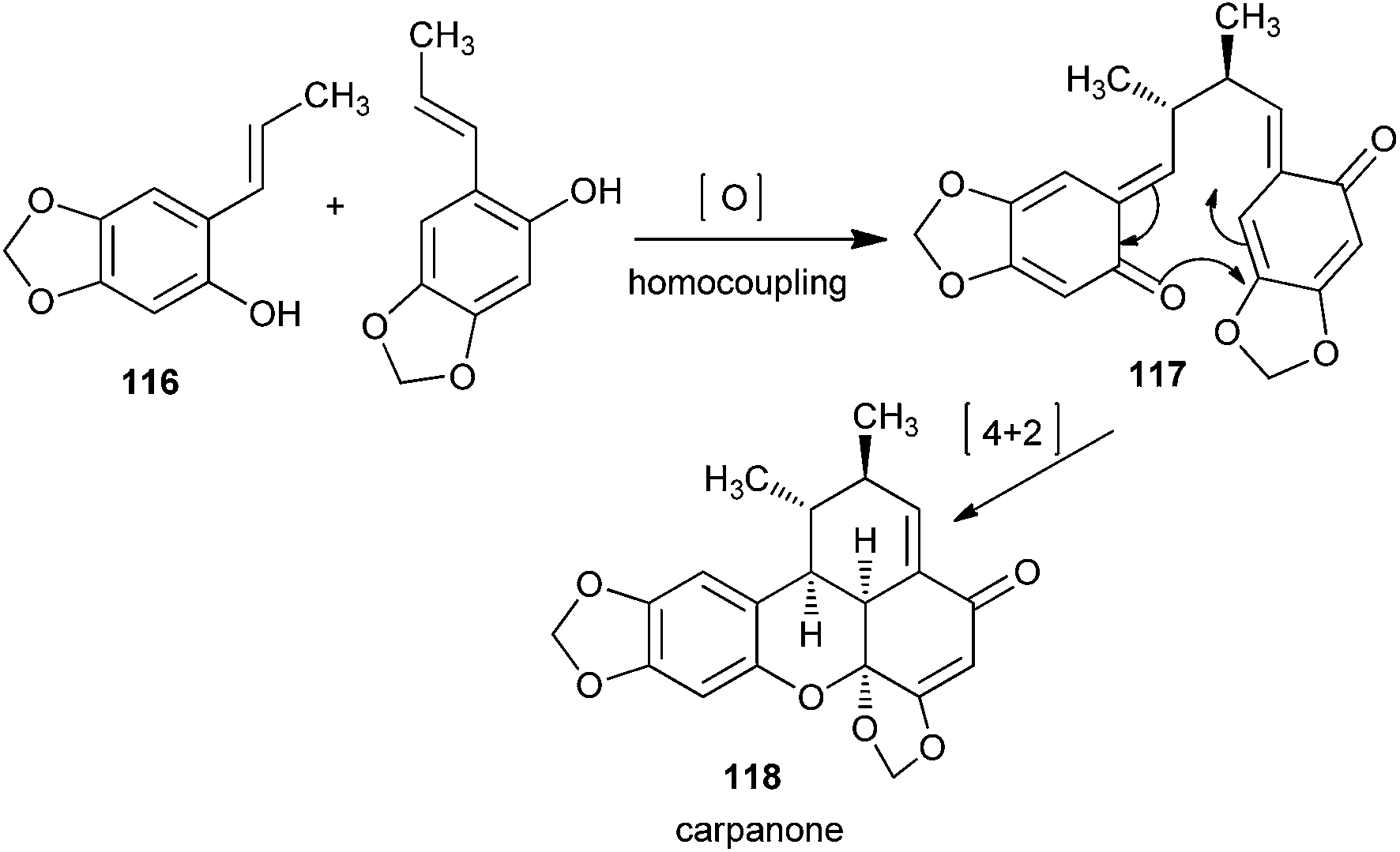 | ||
| Scheme 26 Synthesis of carpanone via electrocyclization of o-QM 117. | ||
But exceptionally, it is interesting that the o-methoxy group, as in 119, equally stabilizes the o-QM intermediate and provides the ‘push’ in the inverse electron demand in the Diels–Alder reaction to provide the unnatural benzoxanthenone 121 (Scheme 27).
 | ||
| Scheme 27 Synthesis of benzoxanthenone 121. | ||
Jha and co-workers34 reported the synthesis of naphthopyrans 133 from 2-naphthol 129, a secondary amine 124, and 3-hydroxy-2,2-dialkylpropanal 122 in the presence of a catalytic amount of p-toluenesulfonic acid. This reaction is a one-pot retro–aldol disintegration of 3-hydroxy-2,2-dialkylpropanal followed by formation of a Mannich base intermediate 130 from 2-naphthol, a secondary amine, and formaldehyde (retro–aldol product). The Mannich base undergoes disproportionation to give a QM intermediate 132 and a secondary amine. Compound 125 reacts with amine 124 to generate enamine 128. QM intermediate undergoes electrocyclization with enamines to generate the final product 133 (Scheme 28).
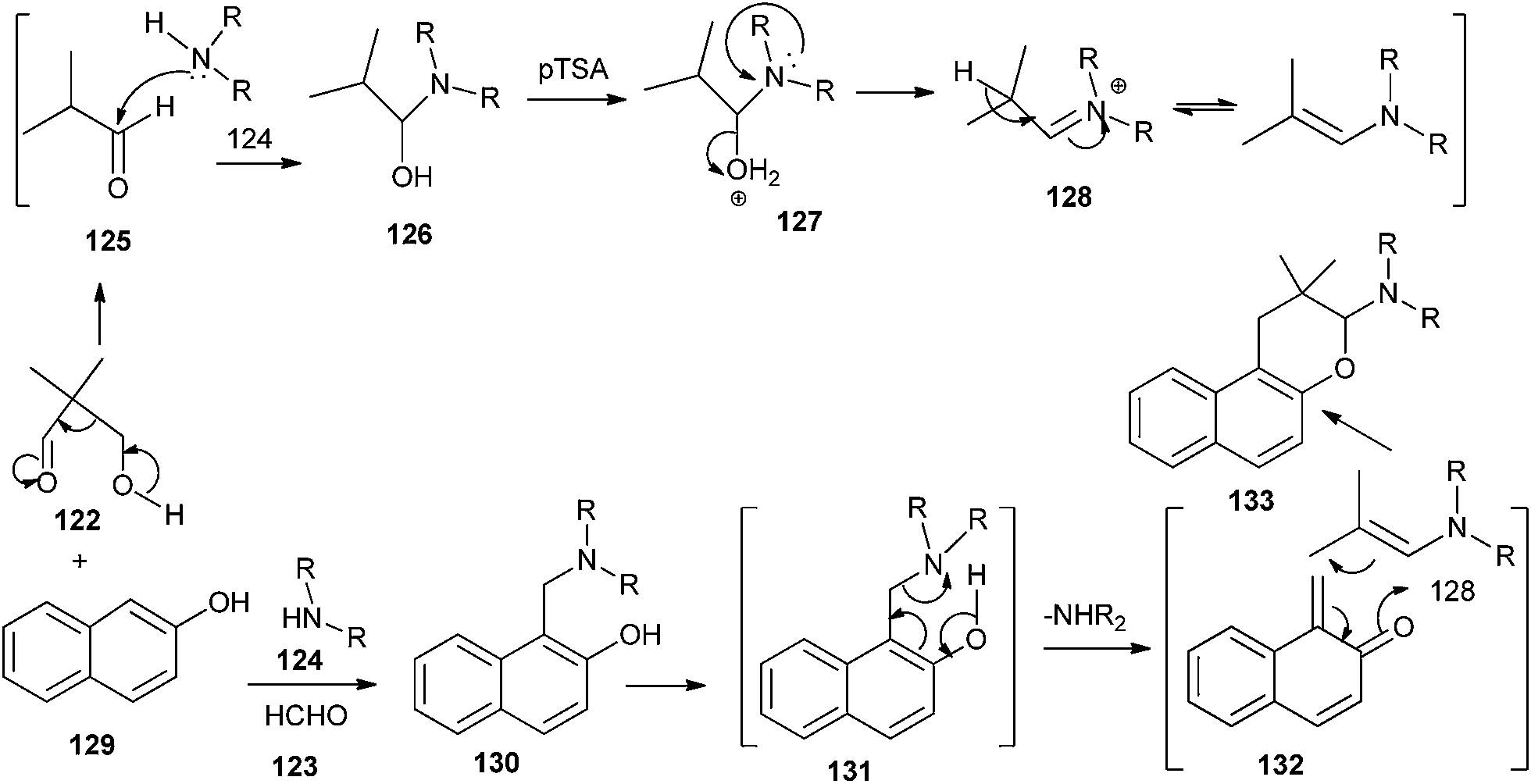 | ||
| Scheme 28 Synthesis of naphtha pyrans 133 via electrocyclization of o-QM with enamines. | ||
3,3-Dialkyl-2-morpholino-3,4-dihydro-2H-naphtho[1,2-b]pyrans 137 were synthesized by the reaction of 1-naphthol 134, morpholine 135 and compound 136 in the presence of a catalytic amount of p-TSA (Scheme 29).
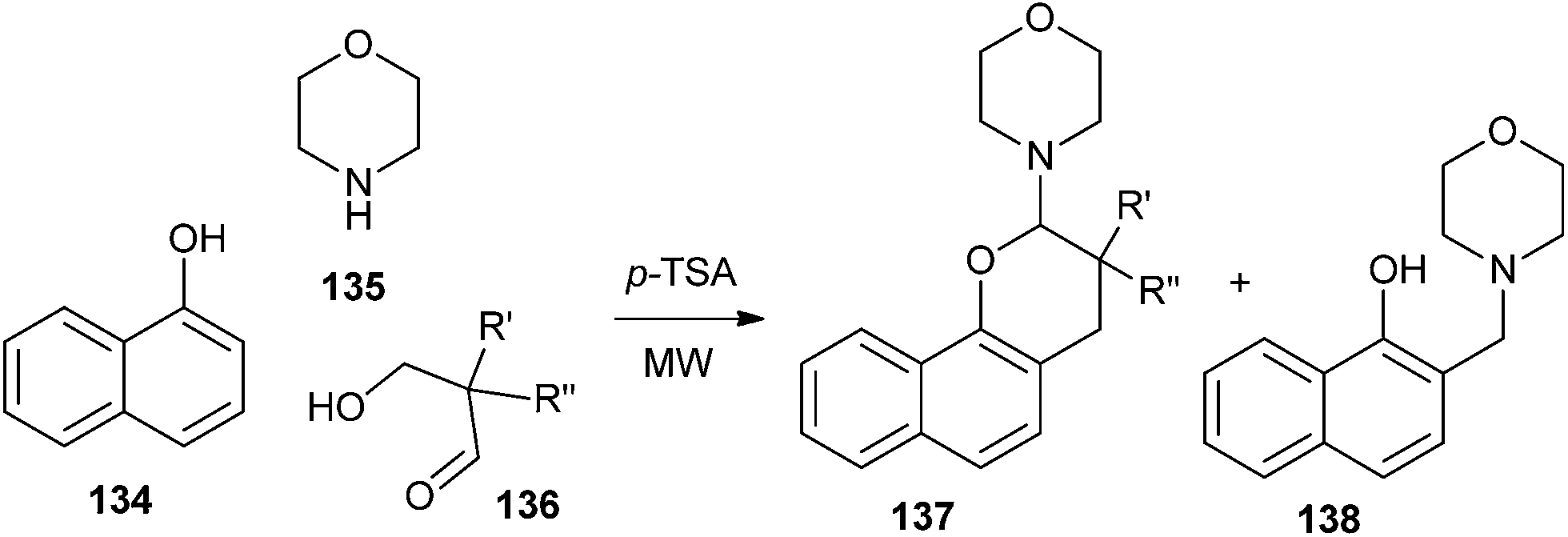 | ||
| Scheme 29 Synthesis of naphtho fused pyrans via o-QM. | ||
Lee et al.35 have reported an efficient one-pot approach for the synthesis of benzopyranobenzo pyrans and naphthopyranobenzo pyrans. Resorcinol and naphthols undergo a domino aldol type reaction followed by a hetero Diels–Alder reaction with the benzaldehyde. The reaction of compound 139 with resorcinol 140 in the presence of ethylenediamine diacetate (EDDA)/TEA first gives intermediate 141 as an aldol-type product. TEA promoted the dehydration of intermediate 141 to form quinone methides 142a and 142b. Because of a sp2-geminal effect, the endo-transition structure 142b may be more favourable than the exo-transition structure 142a due to the 1,3-allylic strain. The regiospecificity is attributed to the products 143a and 143b due to possibility of hydrogen bonding between the hydroxyl group and carbonyl group (Scheme 30).
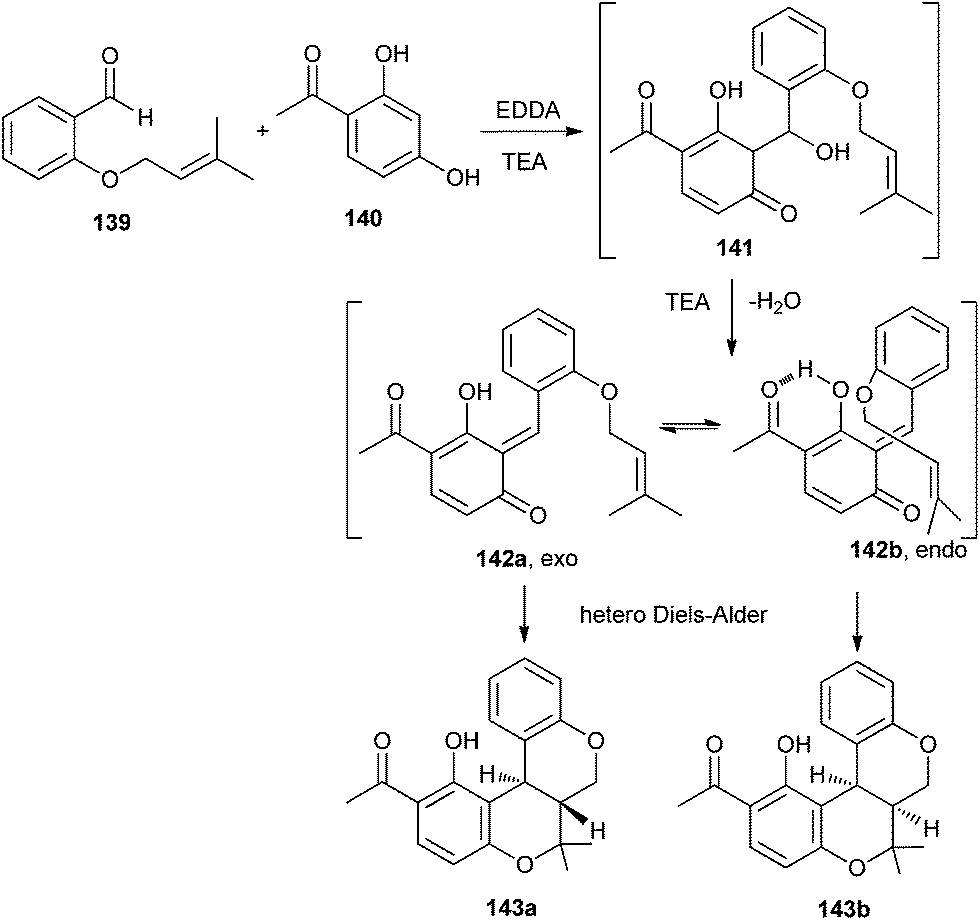 | ||
| Scheme 30 Synthesis of benzopyranobenzo pyrans and naphthopyranobenzo pyrans. | ||
ortho-Quinone methide 147 arising from a formal [2 + 2] cycloaddition between aryne 144 and DMF was found to undergo a [4 + 2] cycloaddition with ester enolate 148 or ketenimine anion to produce diverse coumarins 150 (Scheme 31).36
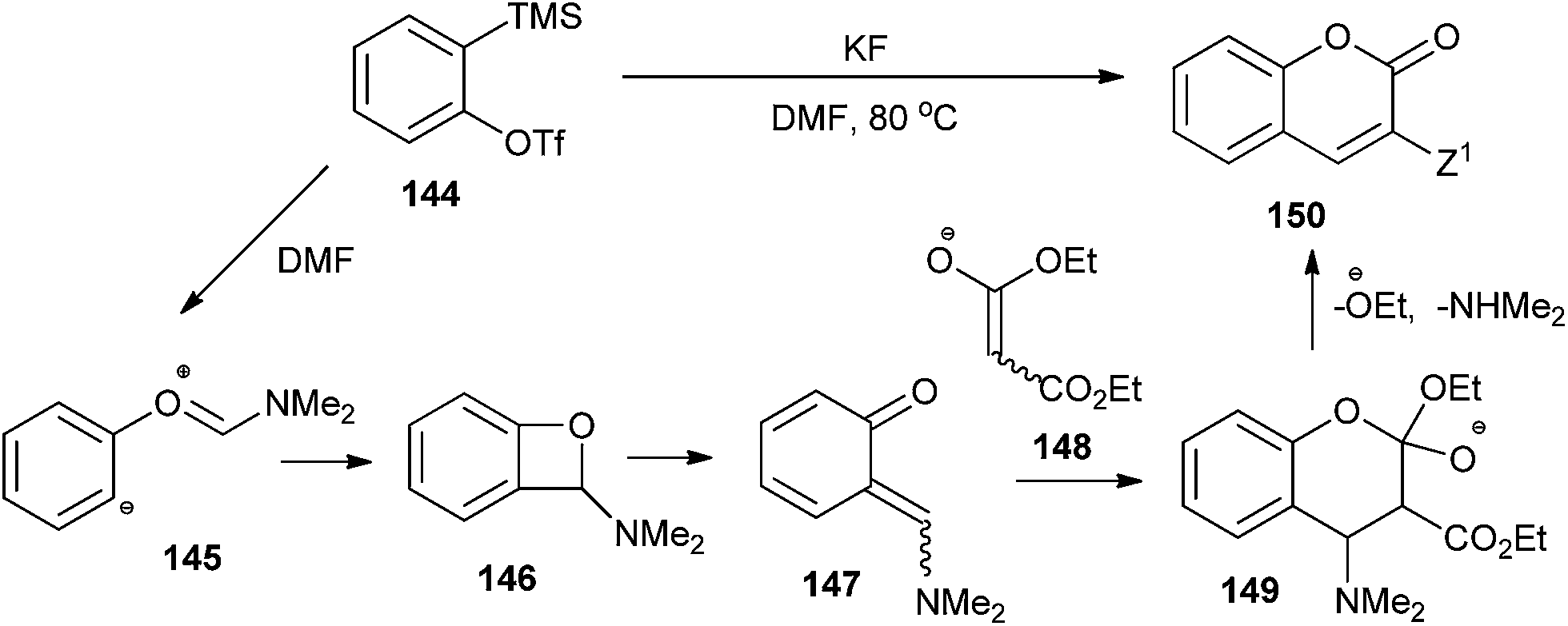 | ||
| Scheme 31 [4 + 2] Cycloaddition of o-QM with ester enolate. | ||
Silver oxide-mediated oxidation of phenol substrates 151 for the synthesis of o-QMs 152 has been developed by Lei et al.37 for the biomimetic syntheses of novel trimeric natural products, (±)-schefflone and tocopherol trimers 153 (Scheme 32).
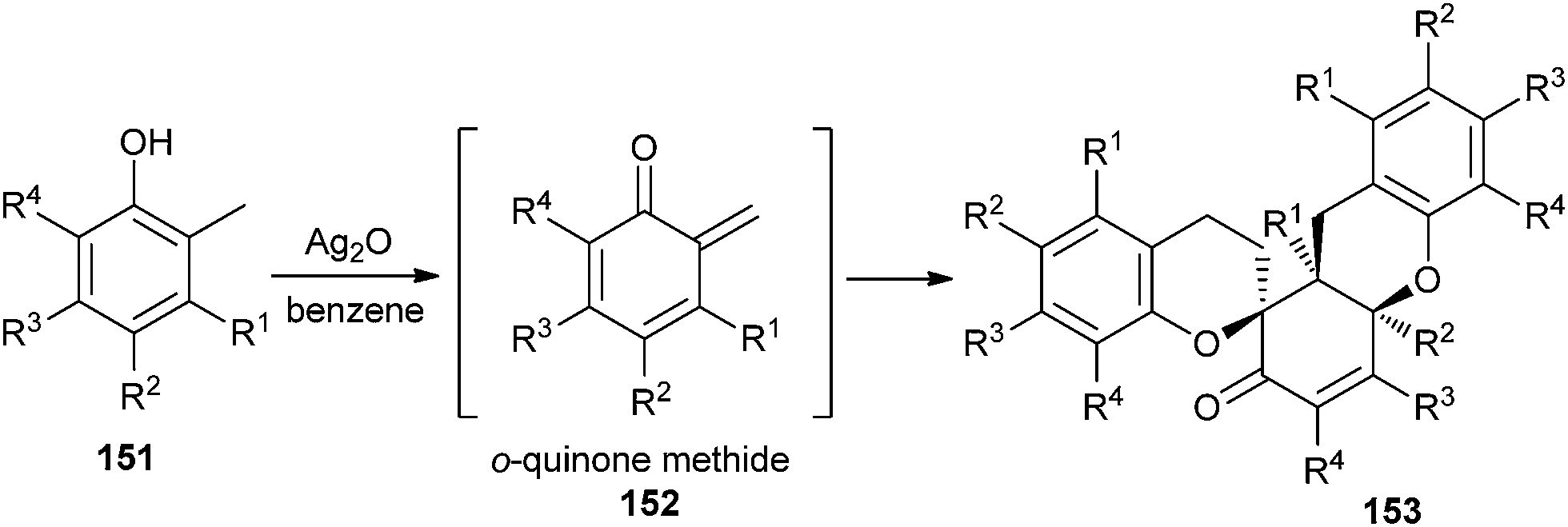 | ||
| Scheme 32 Silver oxide-mediated generation of o-QM for the synthesis of tocopherol trimers. | ||
Bharate and Singh38 have reported the synthesis of anti-malarial robustadials A and B 159 starting from the commercially accessible phloroglucinol. A biomimetic three-component reaction that involves the in situ generation of an o-QM via the Knoevenagel condensation followed by the Diels–Alder cycloaddition with (−)-β-pinene 157 gave robustadials A and B. A retrosynthetic pathway is shown in Scheme 33.
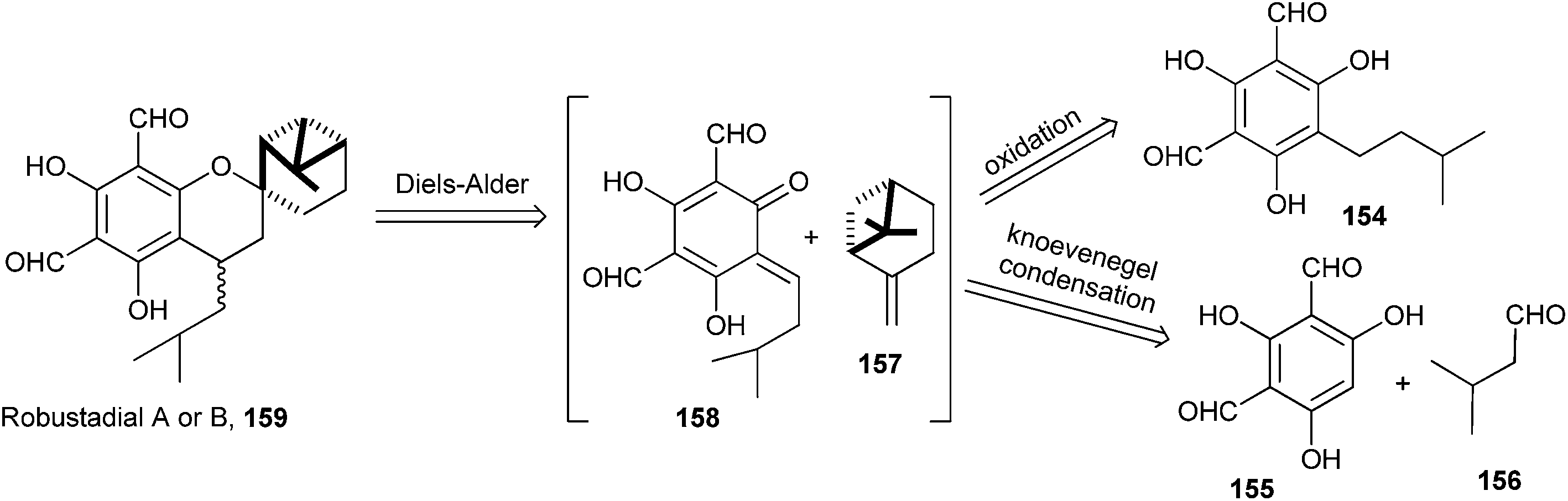 | ||
| Scheme 33 Retro-synthesis of Robustadials A and B. | ||
Robustadials A and B have been anticipated to be formed biogenetically from the Diels–Alder cycloaddition of o-QM 158 with (−)-β-pinene 157. The key intermediate o-QM 158 could be generated by the oxidation of diformylated synthon 154 or by the coupling of phenol 155 and aldehyde 156 via a Knoevenagel condensation (Scheme 34).
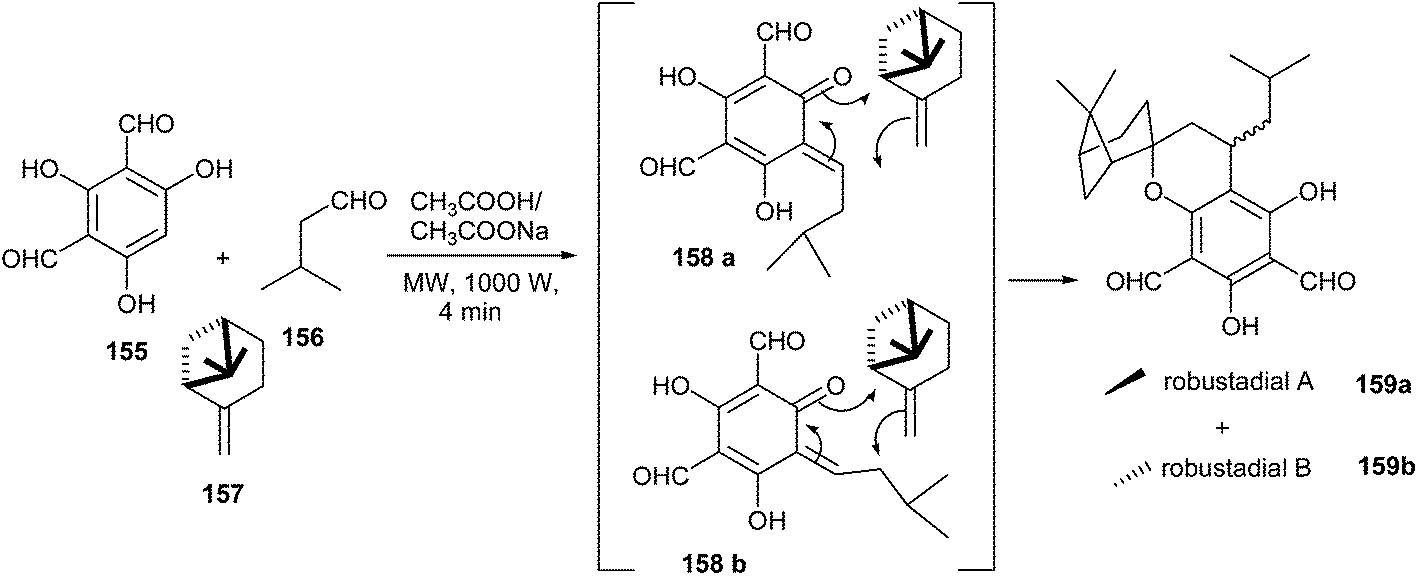 | ||
| Scheme 34 Synthesis of Robustadials A and B. | ||
The treatment of 155 with 156 and (−)-β-pinene 157 in the presence of sodium acetate in acetic acid under microwave irradiation (1000 W) for 4 min resulted in the formation of the desired products in a combined yield of 68% (Scheme 34). Similar results were obtained when the reaction was carried out under conventional heating at 80 °C for 2 h. The o-QM 158 is produced in situ by the addition of phloroglucinol 155 with isovaleraldehyde 156 followed by dehydration (Knoevenagel-like condensation). Then 157 can undergo the cycloaddition reaction with o-QM 158a and 158b via two different orientations, and each can lead to two pairs of diastereoisomers 159a and 159b. The formation of two isomers is attributed to the attack of the oxygen of the QM only to the more substituted terminus of the pinene double bond (Scheme 34).
The biosynthesis of the meroterpenoid guajadial 163 has been achieved via the hetero-Diels–Alder reaction between caryophyllene 160 and an o-QM.39 Biomimetic synthesis of guajadial 163 and psidial A 164 involved a three-component coupling reaction between caryophyllene 160, benzaldehyde 161, and diformylphloroglucinol 162 in an aqueous medium (Scheme 35). It has been envisaged that the relative stereochemistry of guajadial 163 would be partly controlled by the conformation of caryophyllene 160 during the hetero-Diels–Alder reaction. The essential o-QM 165 could be derived retrosynthetically from a simple Knoevenagel condensation between diformylphloroglucinol 162 and benzaldehyde 161. Consequently, a domino three-component one-pot reaction between diformylphloroglucinol 162, benzaldehyde 161, and caryophyllene 160 constituted a simple biomimetic synthesis of guajadial 163.
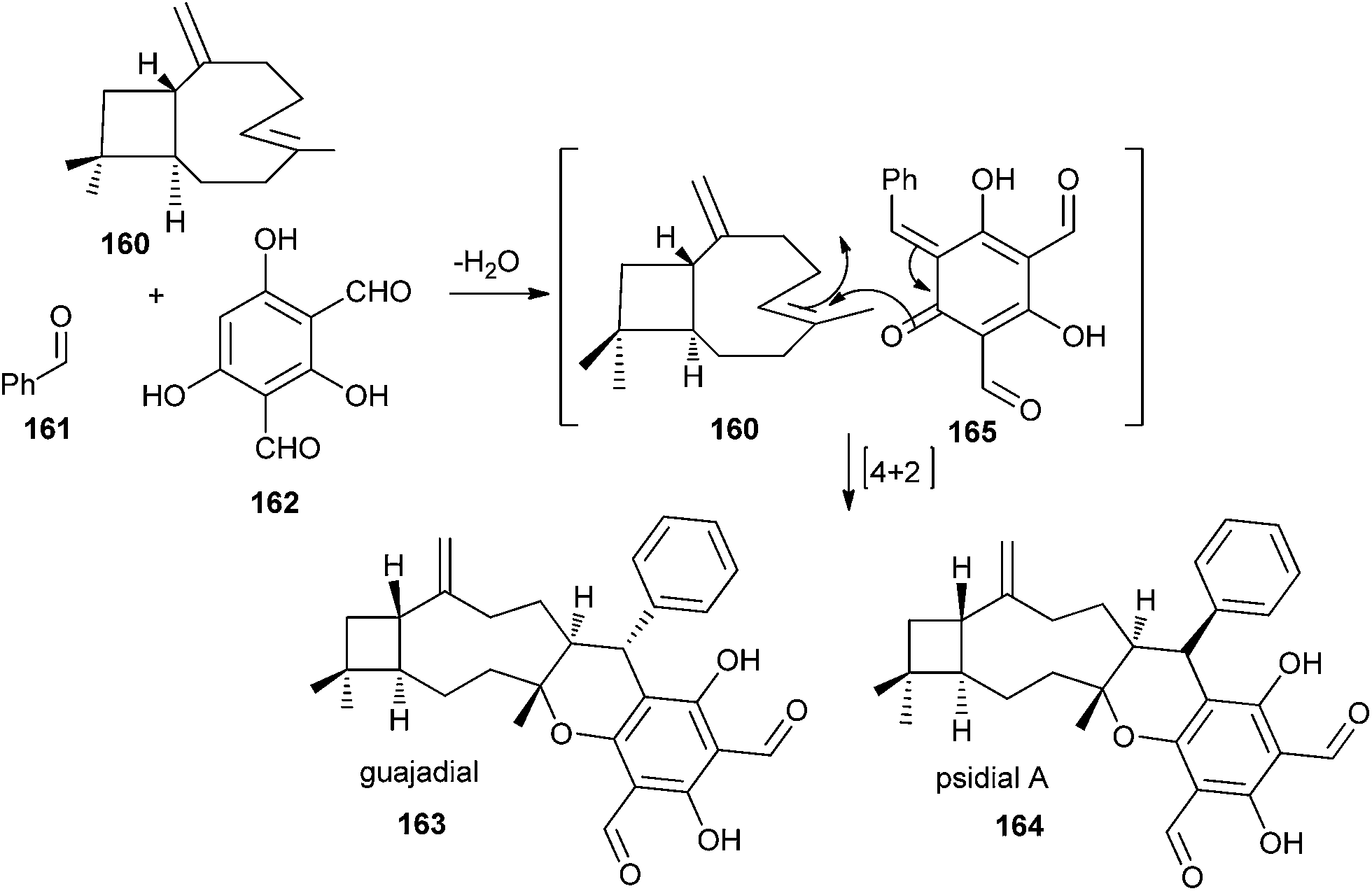 | ||
| Scheme 35 Synthesis of the guajadial and psidial A. | ||
The intermolecular Diels–Alder reactions between o-QMs and double bonds that are not electron-rich are known to be difficult to achieve due to the easy dimerization of the o-QMs, which in turn accounts for the novelty and efficiency of the protocol. Caryophyllene 160 possesses a certain degree of conformational mobility; therefore, it was not certain how this flexibility would influence the facial bias in the in vitro hetero-Diels–Alder reaction. And from 13C NMR studies and molecular mechanics calculations, we came to know that caryophyllene has four possible conformations (ααo αβo βα, ββ), which vary according to the relative nature of the exocyclic methylene and olefinic methyl groups. It has been considered that the proposed biogenetic hetero-Diels–Alder reaction must involve the βα conformer (the most stable conformer) of caryophyllene 160 with proposed transition states (Scheme 36). The stereo chemical upshot of the hetero-Diels–Alder reaction was mainly controlled by the local conformation and intrinsic chirality of caryophyllene 160.
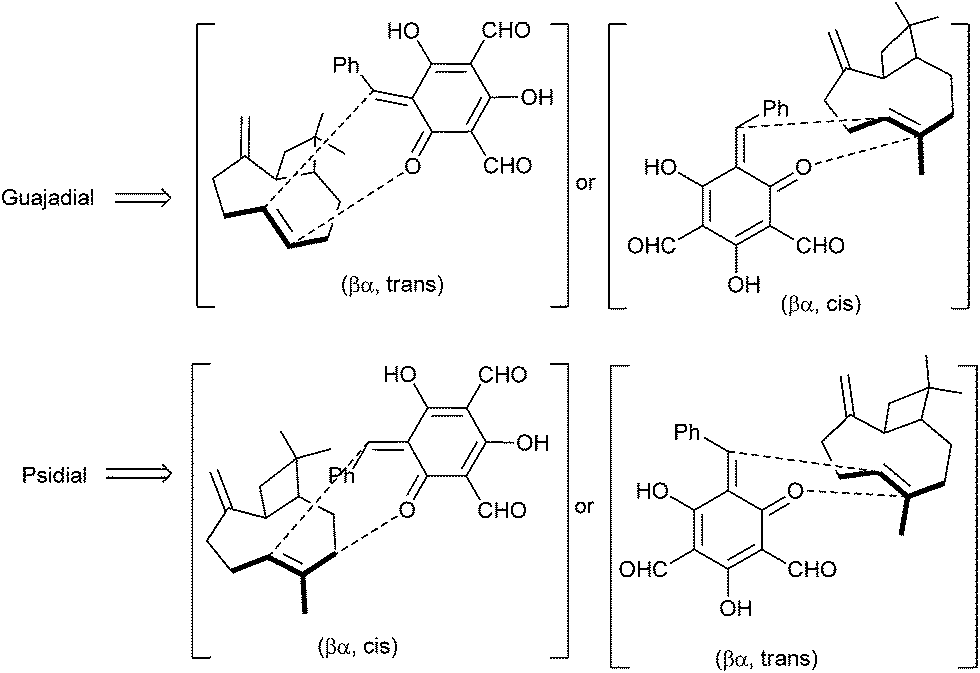 | ||
| Scheme 36 Possible conformations of guajadial and psidial A. | ||
An enantioselective cycloaddition of an o-QM 167 with a chiral enol ether 168 was reported for the first time by Selenski and Pettus,40 and it has been portrayed along with the formal synthesis of (+)-tolterodine 171 and the total synthesis of (+)-mimosifoliol 172, as shown in Scheme 37. These syntheses demonstrate a three-component one-pot benzopyran approach for the construction of chiral benzylic junctions. A low-temperature anionic method for o-QM generation was achieved in high endo/exo ratios for ensuing cycloadditions.41
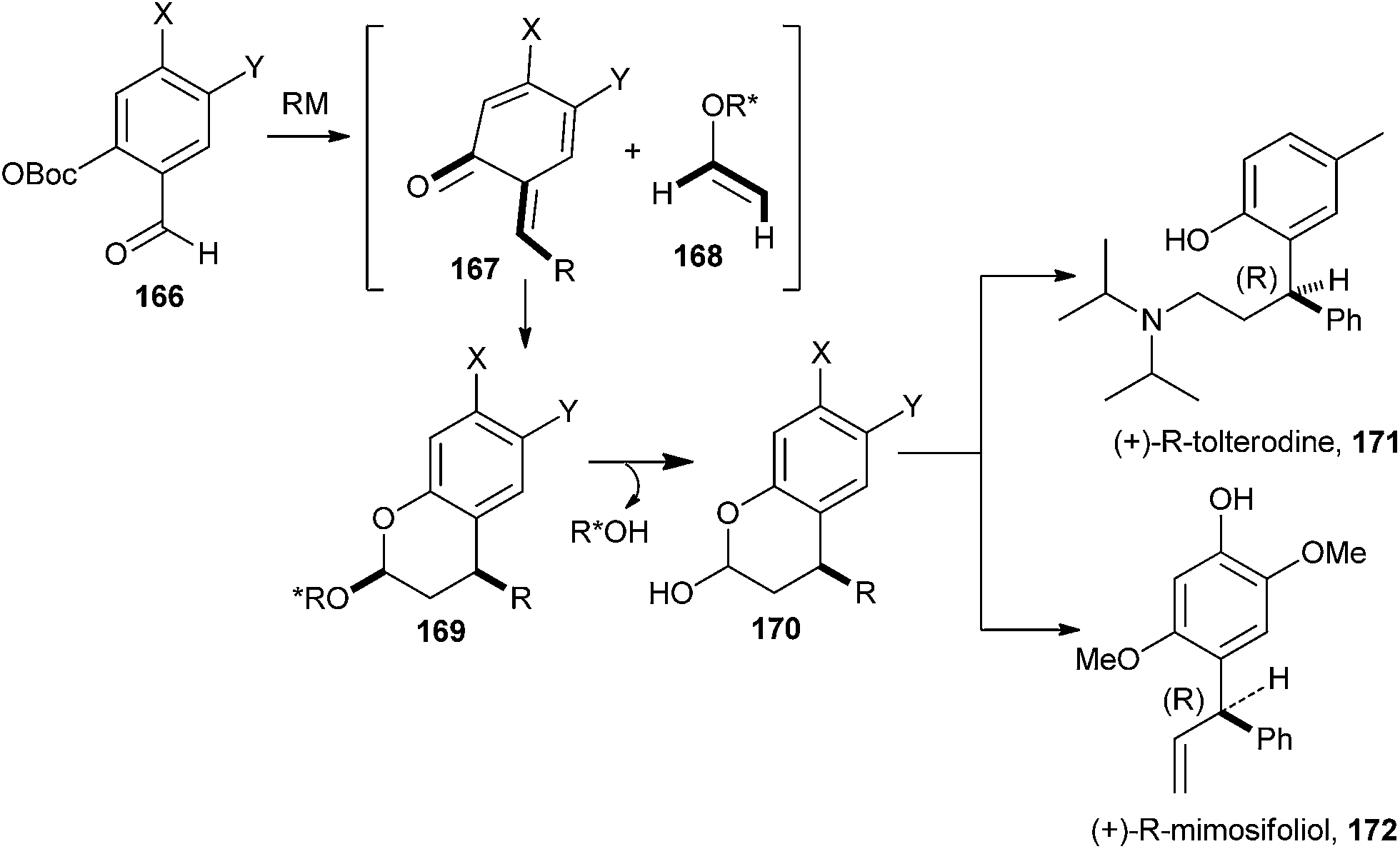 | ||
| Scheme 37 Synthesis of (+)-mimosifoliol. | ||
o-QM generation results from a series of events called the o-QM cascade. The cascade began with the nucleophilic addition of an organometallic reagent to an OBoc salicylaldehyde 166a. The consequential benzyloxy anion 166b attacks the OBoc carbonate to form a cyclic intermediate 166c, which then disintegrates to form a more stable phenoxide 166d. Within particular conditions, the β-elimination of the OBoc residue generates an o-QM 166e, as shown in Scheme 38. The cascade invasion is both metal and temperature dependent and presumably represents the strength of the oxygen-metal bond and the electrophilicity of the metal cation, like an aluminium reagent proceeds to intermediate 166b, whereas a lithium reagent leads to intermediate 166d and a Mg reagent assists the ultimate conversion to 166e. The consequential geometry of the o-QM olefin is E, except for a large group that is situated at the 6-position of the salicylaldehyde.
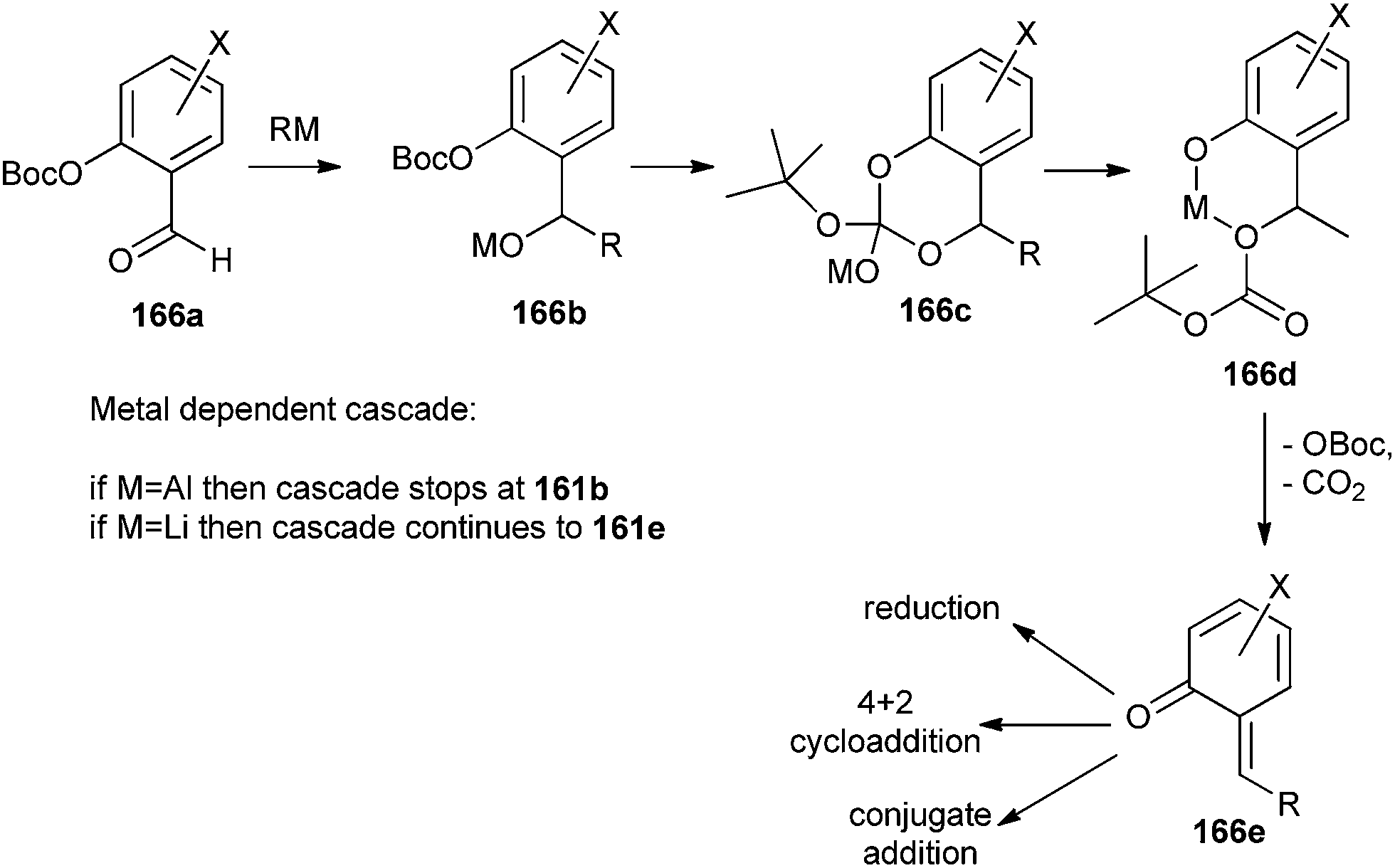 | ||
| Scheme 38 Generation of o-QM using anionic method. | ||
o-QM in the presence of chiral enol ether addicted in a diastereoselective [4 + 2] cycloaddition. The facial selectivities of enol ethers 173 and 174 through cycloaddition are shown in Scheme 39.
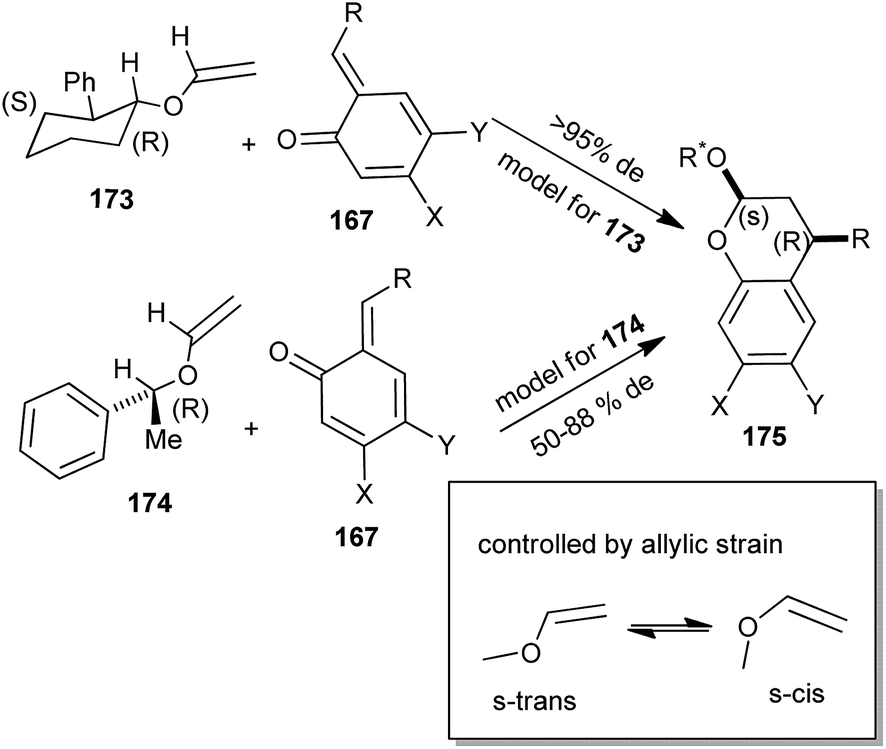 | ||
| Scheme 39 Cycloaddition reaction of o-QM with chiral enol ether. | ||
The syn affiliation flanked by the phenyl residue of the benzopyran and the oxygen substituent of the acetal results from an endo transition state. By taking pseudoallylic strain and Houk's calculation into vindication, the enol ether reacts in the s-trans conformation. The o-QM endures reaction opposite the methyl residue of enol ether 173 and opposite the phenyl residue of enol ether 174. The S-stereocenter of 173 and the 2R-stereocenter of 174 induce the S-configuration of the resulting benzyl junction of compound 175 (Scheme 39). The absolute configuration of the chiral enol ether determines the absolute configuration of the resulting benzopyran. The diastereoselectivity of trans-2S-phenyl-1R-cyclohexanol vinyl ether 174 was outstanding for all the o-QMs that ultimately created R-mimosifoliol and R-tolterodine.40
The substituted enol ether 177 should also undergo a diastereoselective cycloaddition. Addition of phenyl Grignard reagent to aldehyde 176 in the presence of enol ether 177 affords benzopyran 178 creating three chiral centers with a 60% yield and 95% de (Scheme 40).
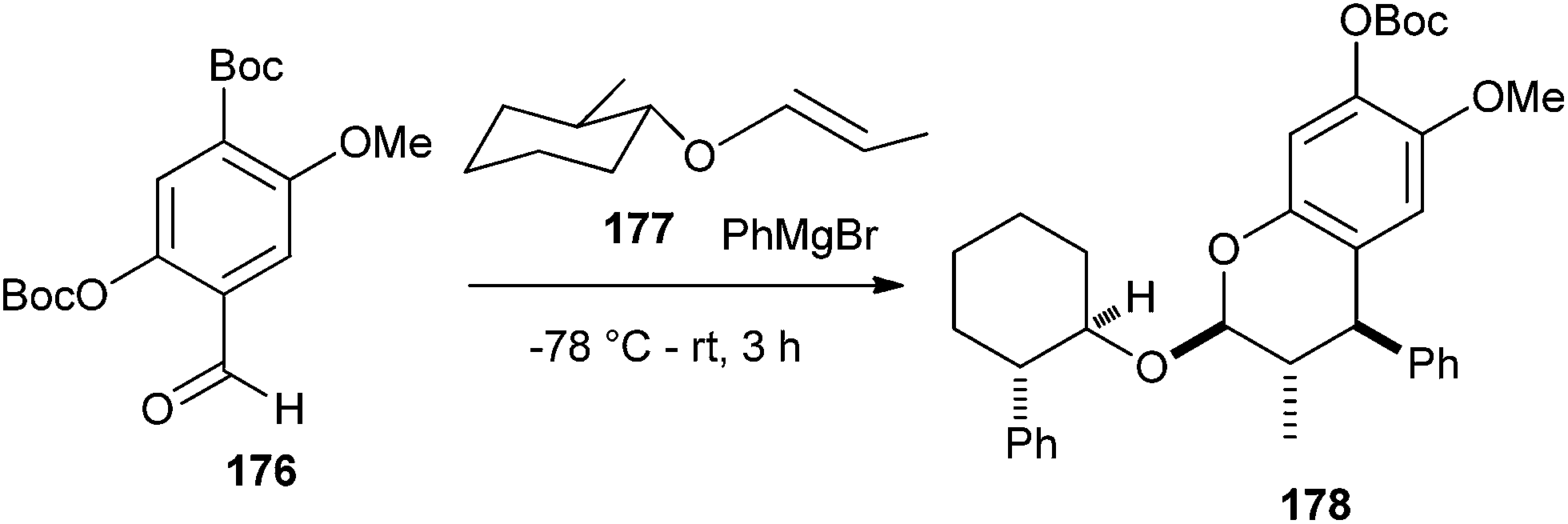 | ||
| Scheme 40 Diastereoselective synthesis of compound 178. | ||
Singh and co-workers42 have reported the biomimetic synthesis of S-Euglobals and their antileishmanial, antimalarial, and antimicrobial activities. Compound 179 was treated with formaldehyde and different terpenes like (+)-2-carene (182), (1R)-(−)-myrtenol (183), and (1R)-(−)-nopol (184) in the presence of NaOAc in CH3COOH, resulted in the development of respective pairs of regioisomers, 185 and 186 from 2-carene, 187 and 188 from myrtenol, and 189 and 190 from nopol in good yields. Similarly, treatment of 180 and 181 with formaldehyde and different terpenes in the presence of sodium acetate in acetic acid resulted in the formation of the desired cycloadducts, 191–193 and 194–196, respectively (Scheme 41).
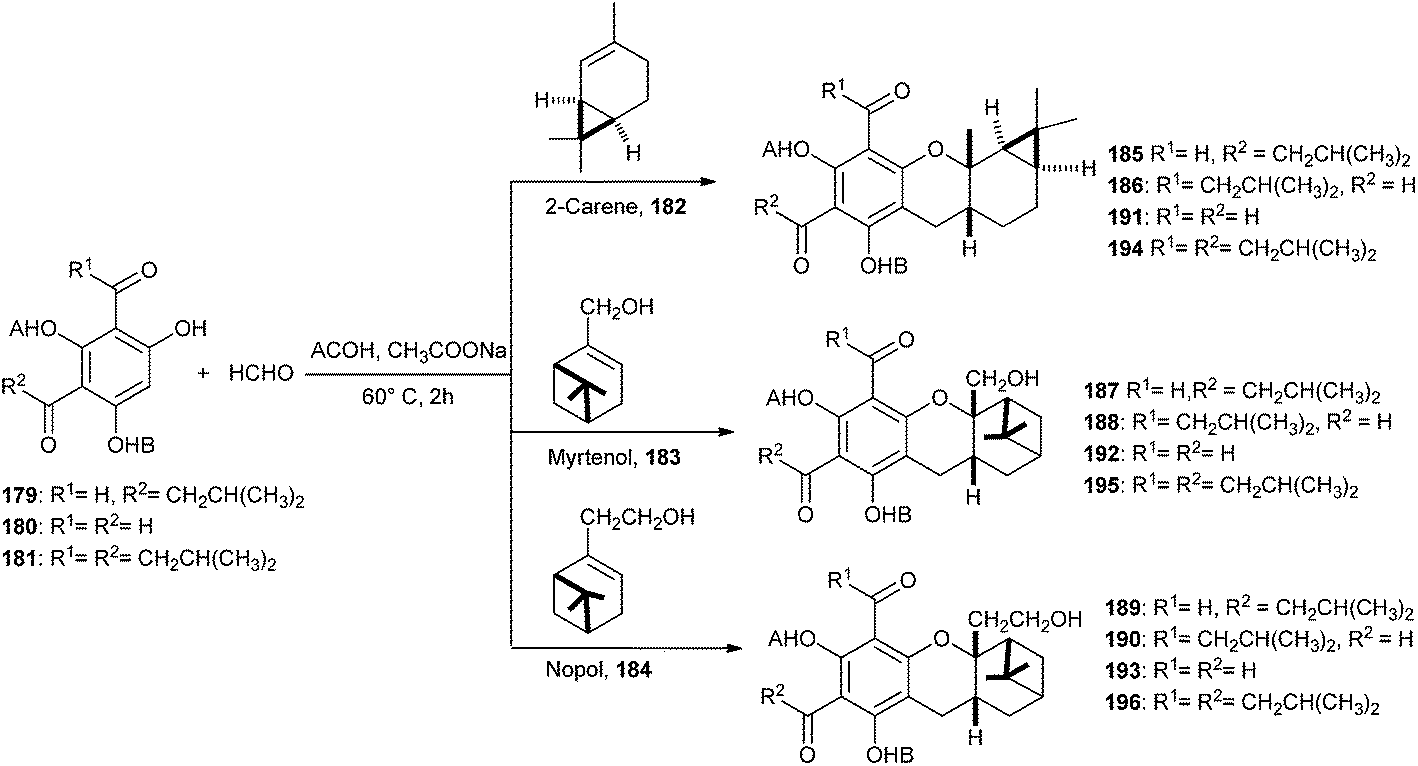 | ||
| Scheme 41 Biomimetic synthesis of S-euglobals. | ||
Singh and co-workers43 also described the synthesis of new analogues of euglobals 200–203 by a biomimetic approach (Scheme 42). These synthetic compounds differ from natural euglobals in the nature of monoterpene and acyl functionality. Oxidative addition of phloroglucinol-terpene with DDQ (2,3-dichloro-5,6-dicyano-1,4-benzoquinone) in nitromethane gave the corresponding o-QM, which further undergoes intermolecular cycloadditions with monoterpenes such as β-pinene 157, α-pinene 197, (+)-3-carene 198 and (−)-camphene 199.
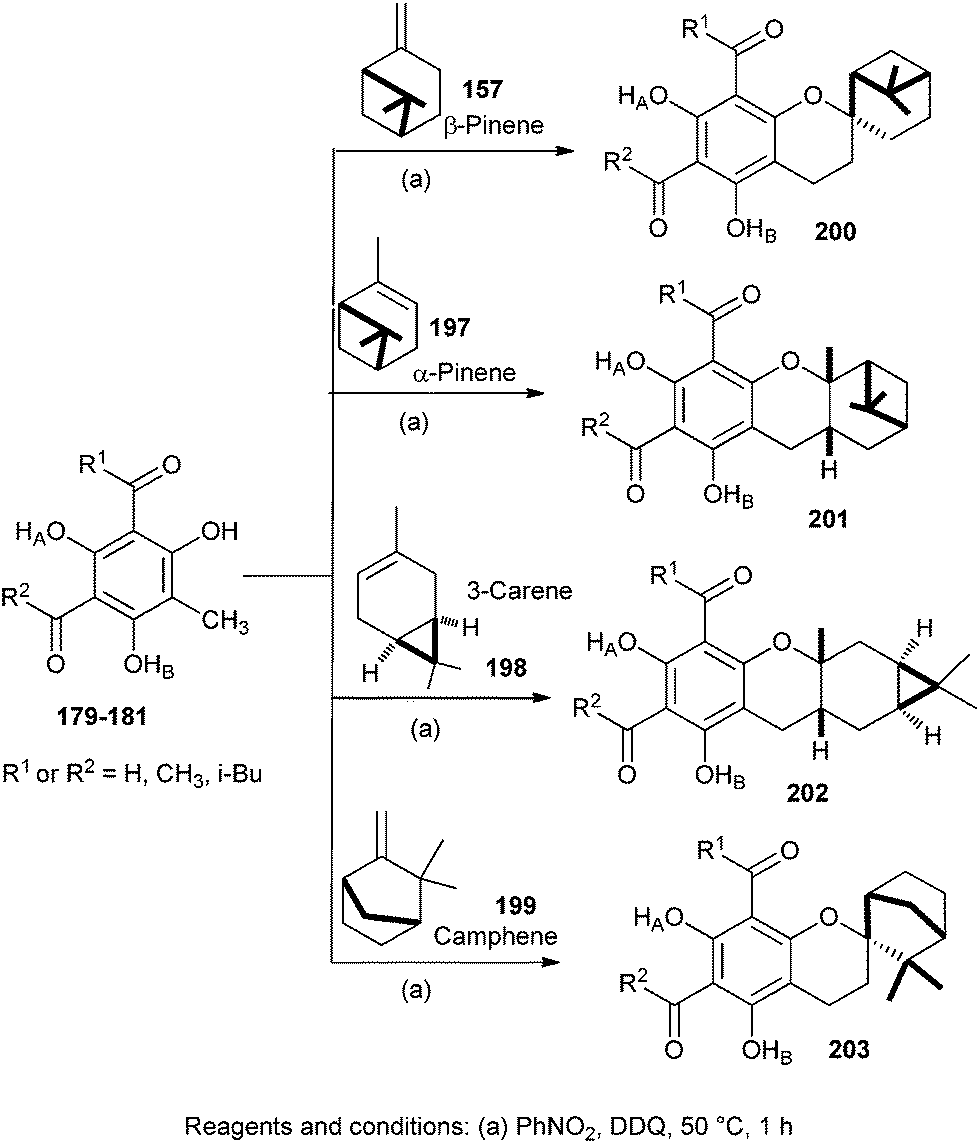 | ||
| Scheme 42 Biomimetic synthesis of S-euglobals analogues. | ||
Kucklaendera et al.44 reported 1,4,9,10-anthradiquinone as a precursor for antitumor compounds, according to which the naphtho-condensed indoles 206 were obtained by the reactions of unsubstituted 1,4-anthraquinone 204 with enamines 205 via the normal Nenitzescu method. Indoles 206 were converted to Mannich bases followed by dimerization through the Diels–Alder reaction of o-QM intermediate 207 to give the compound 208 (Scheme 43). These synthesized heterocyclic compounds were appraised for their anticancer properties in the NCI's human disease oriented in vitro anticancer screen.
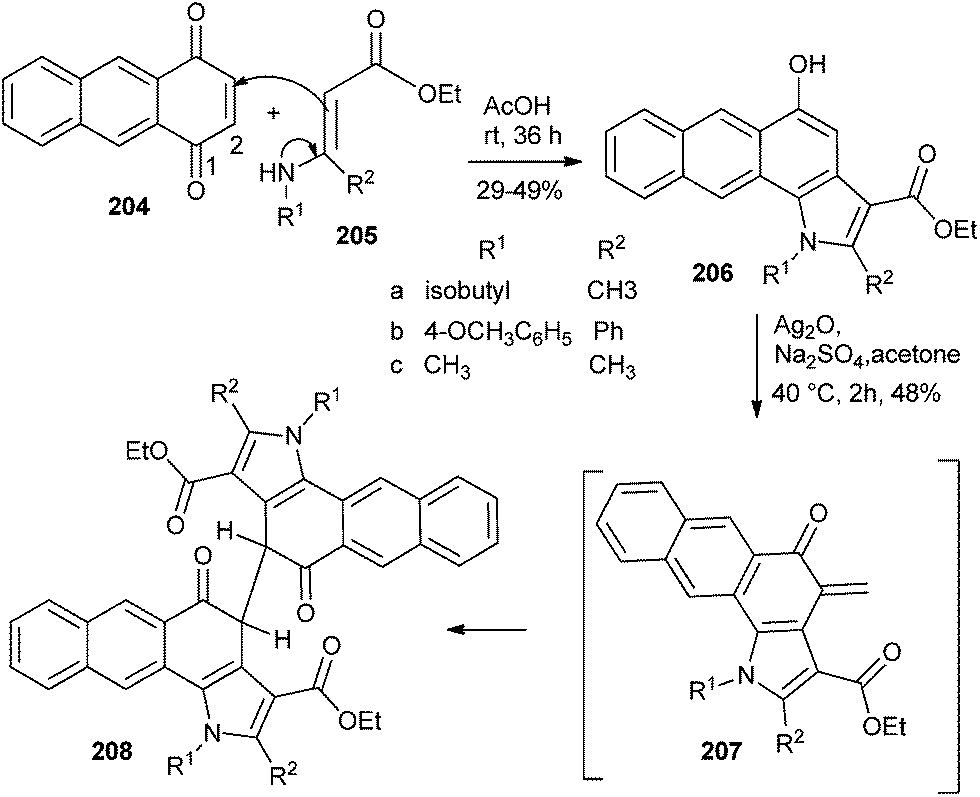 | ||
| Scheme 43 Synthesis of naphtho-condensed indoles. | ||
Generation of o-QMs by the photolysis of carboxylate derivatives 209 in aqueous acetonitrile was reported by Steinmetz and Chen.45 The carboxylate leaving groups are swiftly eliminated by photolysis of the 1,4-benzoquinone 209 to produce an o-QM intermediate 211 that is trapped by [4 + 2] cycloaddition with unreacted starting material 209 and generated photoadduct 212. The carboxylate leaving groups are easily expelled upon photocyclisation to benzoxazolines 210 under aqueous conditions (Scheme 44).
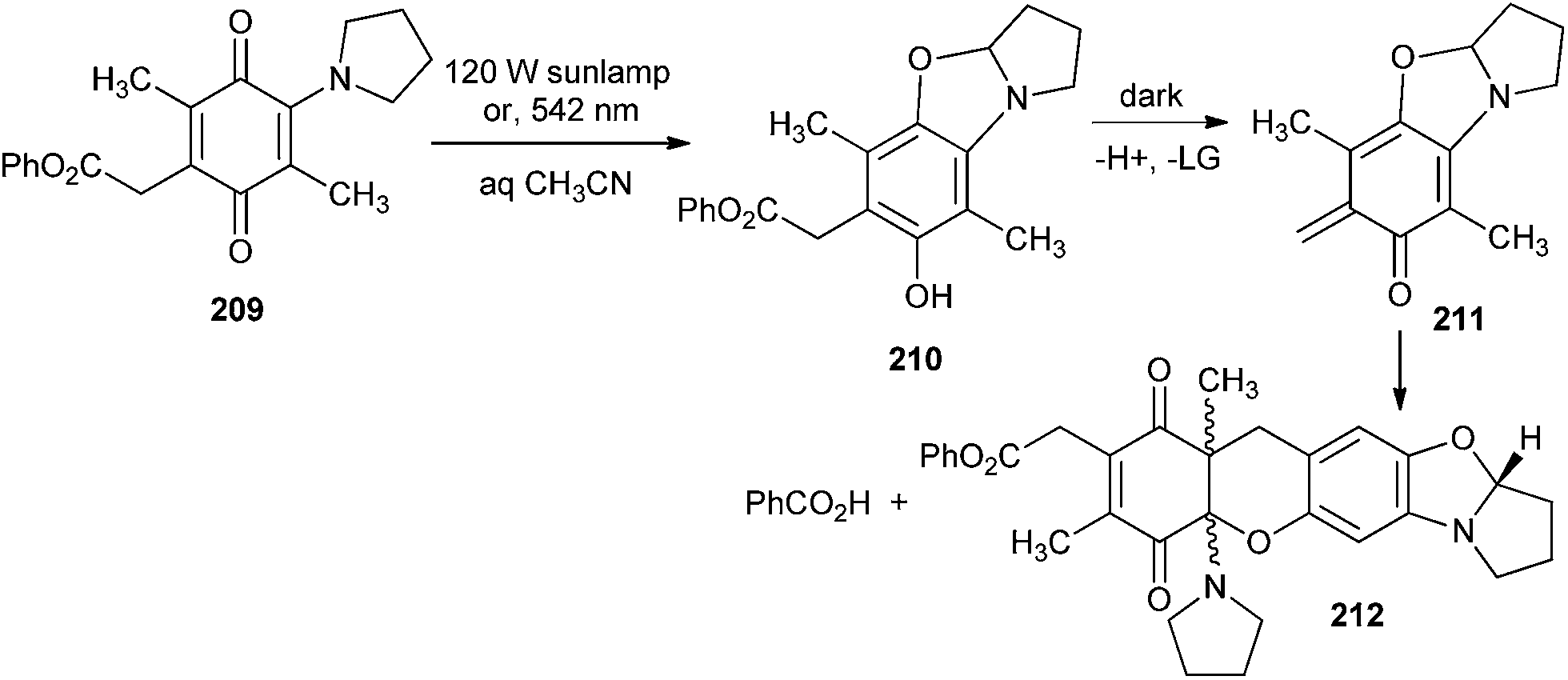 | ||
| Scheme 44 Synthesis of compound 212 via photogenerated o-QM. | ||
Photolysis of 0.02 M solution of compound 209 in a mixture of 30% D2O and 70% CD3CN with a 120 W sunlamp formed benzoic acid in 58% yield with a 100% conversion rate, according to 1H NMR spectroscopy. The other photoproduct 212 was formed in 36% yield, which was isolated by chromatography and identified as a 1:1 mixture of diastereomers by 1H and 13C NMR spectroscopy.
The o-QM intermediate 211 has been photolysed with various alkenes. Irradiation of compound 210 (10−2 M) with compound 213 (0.1 M) in 30% aqueous CH3CN produced the cycloadduct 214. The cycloadduct 214 was produced in 79–87% yields at 100% conversion rates of the reactants (Scheme 45). Other alkene trapping reagents, such as dihydropyran, ethyl vinyl ether, methyl trimethylsilyl dimethylketene acetal, and diethyl fumarate, were inadequately reactive in trapping the o-QM, which was deactivated by multiple electron-releasing substituents.
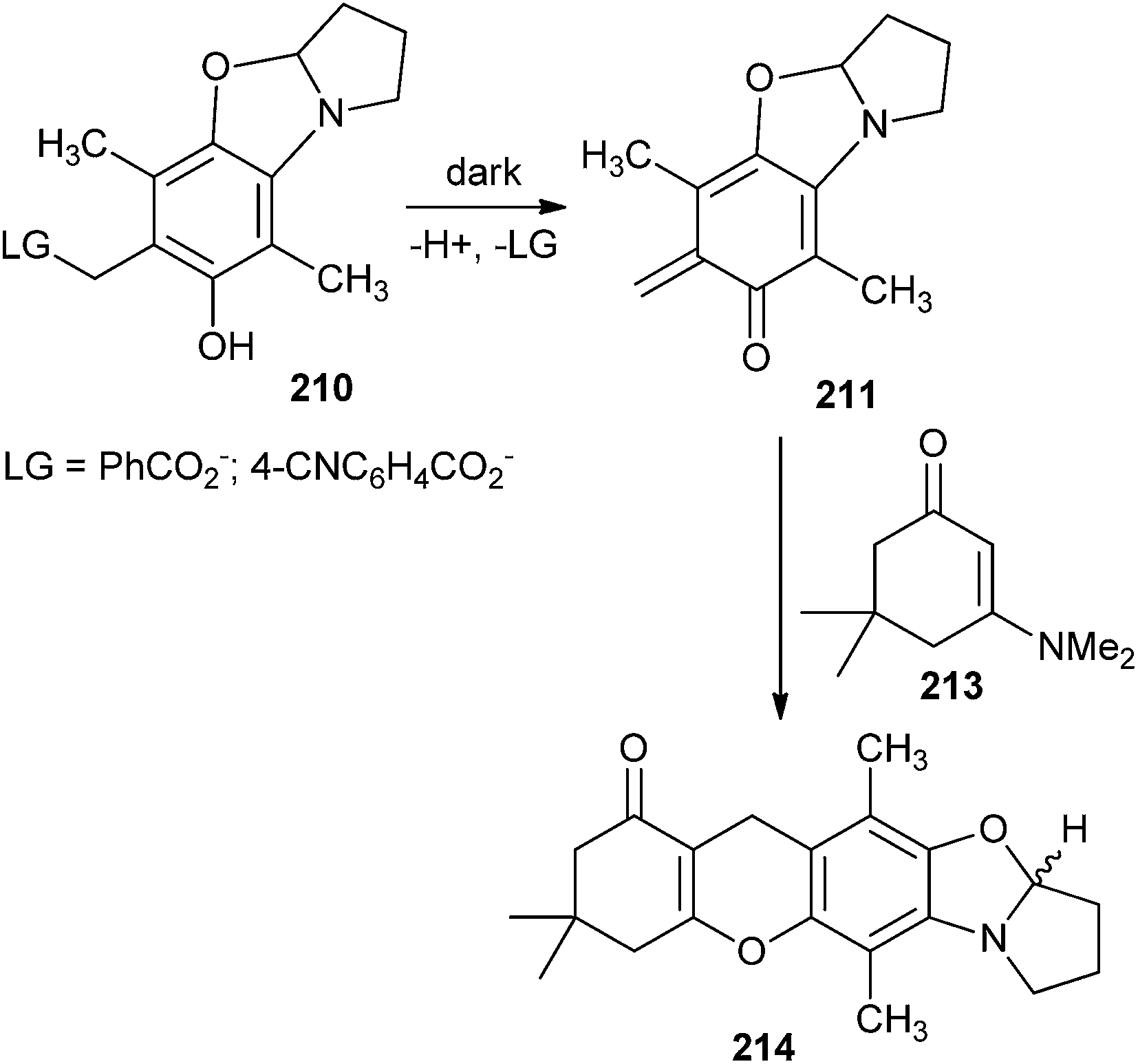 | ||
| Scheme 45 Photolysis of o-QM 211 with alkene. | ||
The immediate precursor in the photocyclization to benzoxazoline 210 is zwitterionic in character in ground state as with structure 216. From photochemistry of α-keto amides it has been shown that intermediates analogous to 216 can cyclize as well as expel phenolate or carboxylate leaving groups (Scheme 46). The mechanism for elimination of carboxylate leaving groups to form o-QM 211 is uncertain, because the elimination is possible either from 216 or 210. No evidence could be obtained to support the direct elimination of carboxylate leaving groups from 216 using nanosecond laser flash photolysis methods.
 | ||
| Scheme 46 Mechanistic analysis for the generation of o-QM 211. | ||
Barrero and co-workers46 reported the synthesis of bioactive pupehedione derivatives involving a hetero Diels–Alder cycloaddition of o-QM, which was generated by the fluoride-induced desilylation of silyl derivatives of o-hydroxybenzyl iodides. In this protocol, two variations were investigated. At first, the compound 218 was reacted with dienophiles 219 and 221, and it was found that the reaction proceeded via the formation of 220 and 222. It has also been revealed that o-QMs undergo [4 + 2] cycloadditions with ethers 219 and 221, acting as dienophiles during the course of the reaction (Scheme 47).
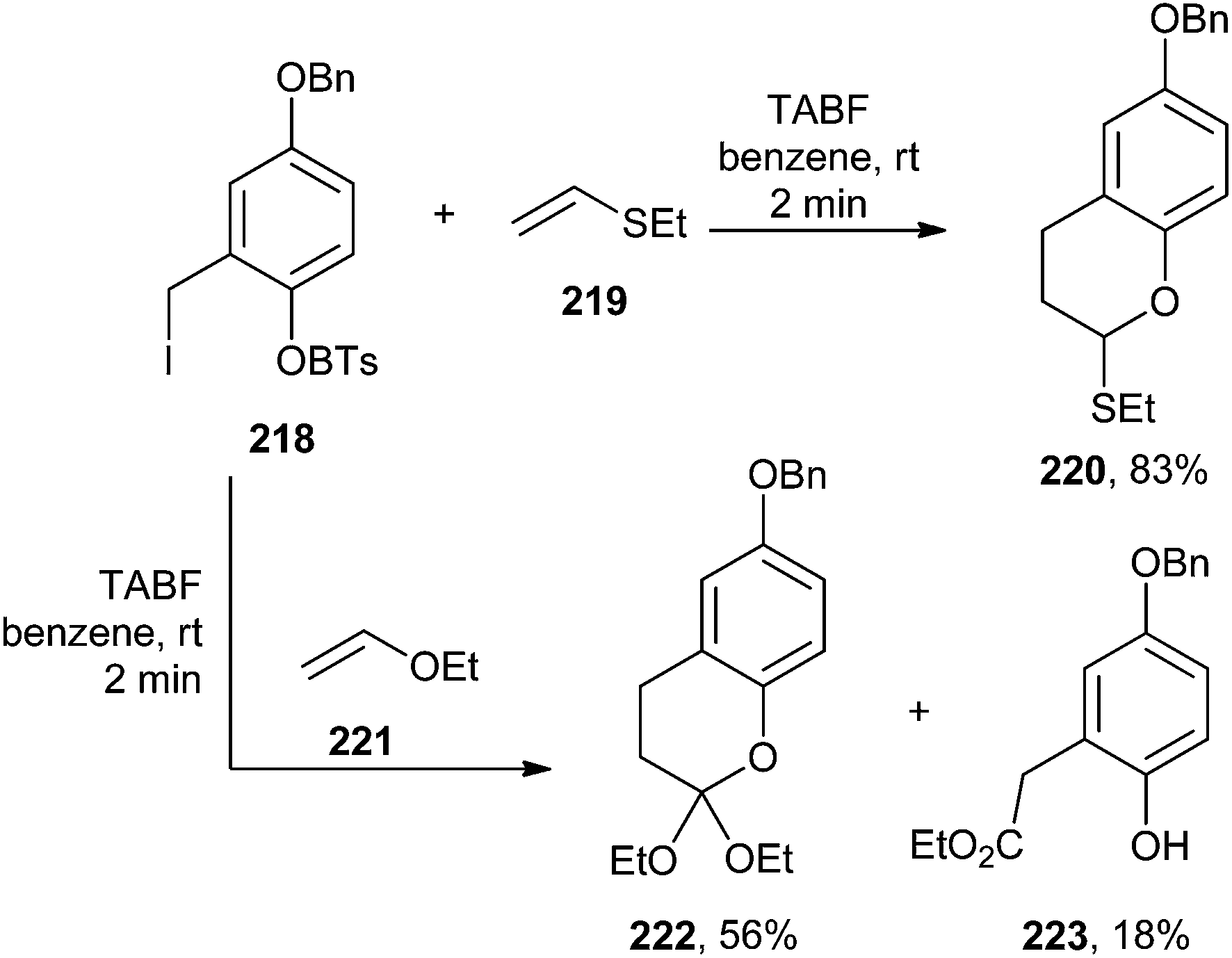 | ||
| Scheme 47 [4 + 2] Cycloadditions of o-QM with ethers. | ||
In the second attempt, variations were introduced to explore the scope of this procedure by using different oxadienic moieties. Thus, ortho-silyloxybenzyl iodide, 226 was obtained from commercial 2-hydroxybenzaldehyde 224. When this compound was treated with ethyl vinyl ether, the reaction proceeded in a similar fashion to give chroman 227 exclusively (Scheme 48).
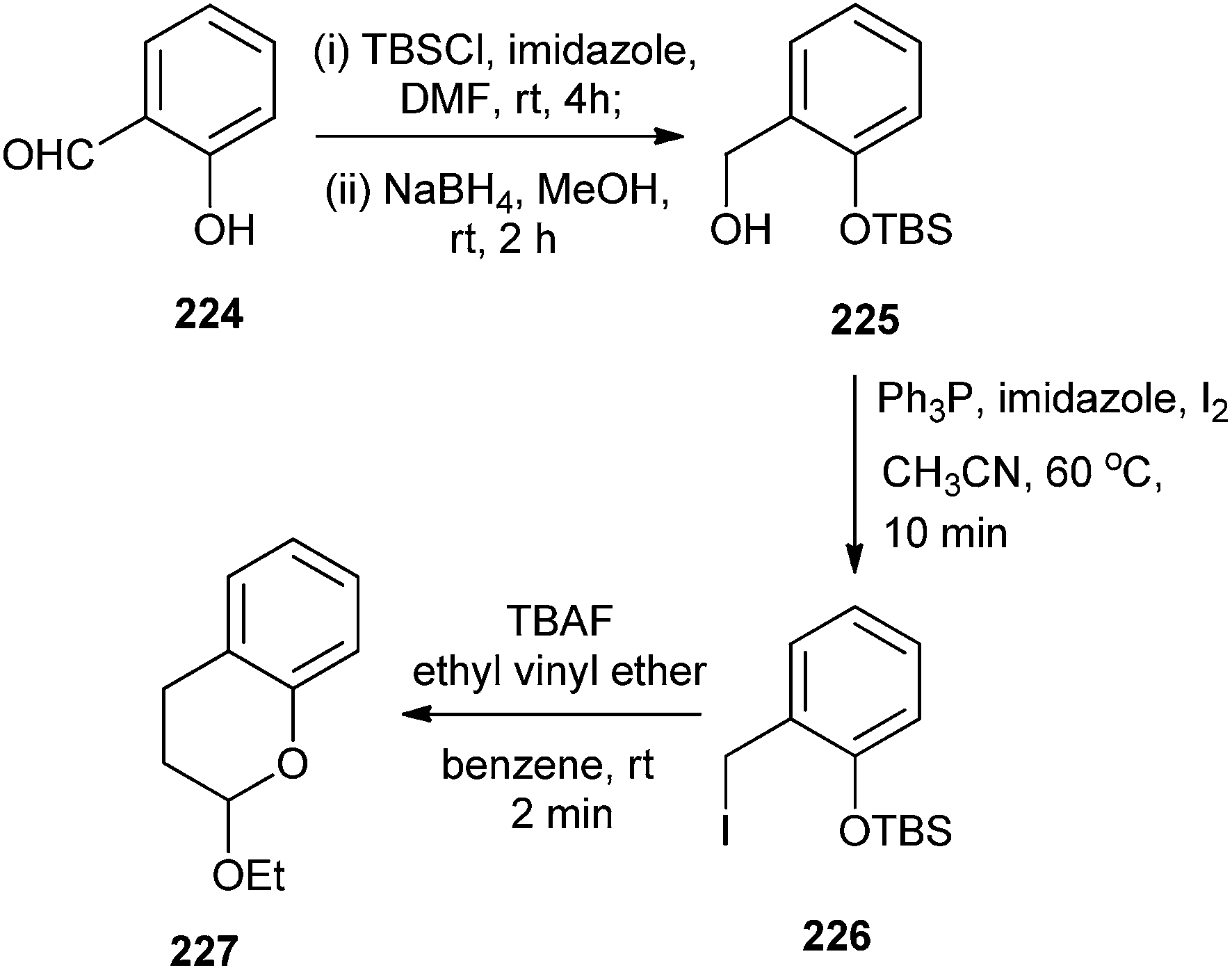 | ||
| Scheme 48 Synthesis of chroman by the reaction of o-QM and ethyl vinyl ether. | ||
The unstable 3-methylenepyridin-4-one intermediate 233 has been developed by Baldwin and co-workers47 by heating the di-Boc-pyridin-4-one derivative 232. The intermediate 233 undergoes rearomatization followed by the Diels–Alder cycloaddition with activated alkenes to give substituted pyrano[3,2-c]pyridines 234. The precursor 231 for the synthesis of 234 was obtained from commercially available 4-methoxypyridine 228. Formylation of 228 gives 229, which after reduction furnished compound 230 (Scheme 49).
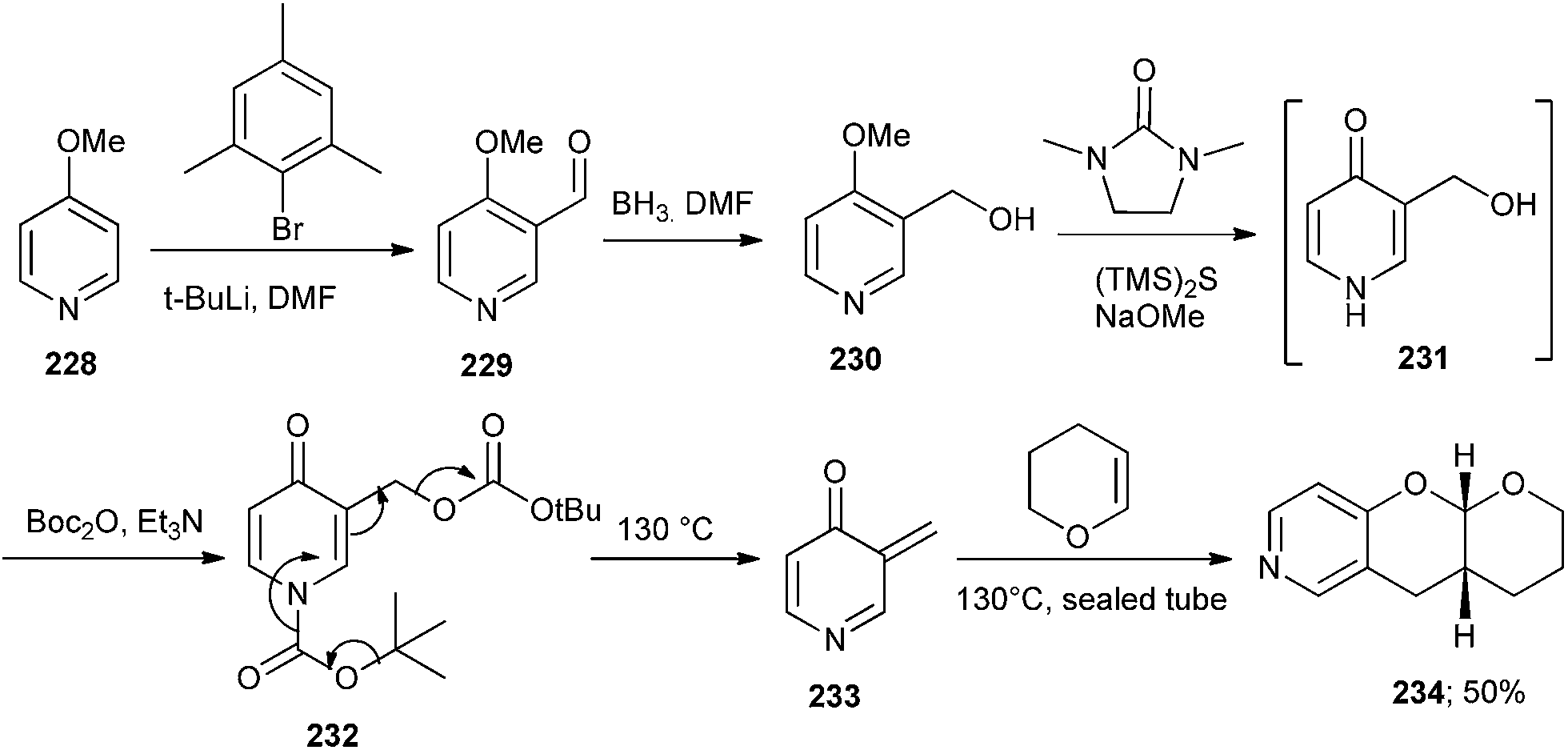 | ||
| Scheme 49 Diels–Alder cycloaddition of o-QM with activated alkenes. | ||
Unusual hetero Diels–Alder reactions between various electron-rich oxazoles and o-QMs have been achieved by Lindsey and Pettus.48 In this case, the o-QM intermediate has been generated by deprotonation of either of the ortho-OBoc hydroxymethyl aromatic compounds (240 or 241, 0.1 M in Et2O or THF) with tert-butyl magnesium bromide, which led to the migration of -OBoc and subsequent β-elimination (Scheme 50). Even though these o-QM species are extremely reactive, 4-methyl-oxazole 235 proved futile as a nucleophile. Instead, the o-QM intermediate undergoes self-destruction via known manifolds such as Diels–Alder dimerization and trimerization. The generation of o-QM 243 from compound 241 in the presence of oxazole 237 led to two adducts in a 62% combined yield. In this case, the 1,4-addition product prevails in the mixture and the [4 + 2] cycloadduct 245 arises in small amounts (1:10, 245:246), as shown in Scheme 50.
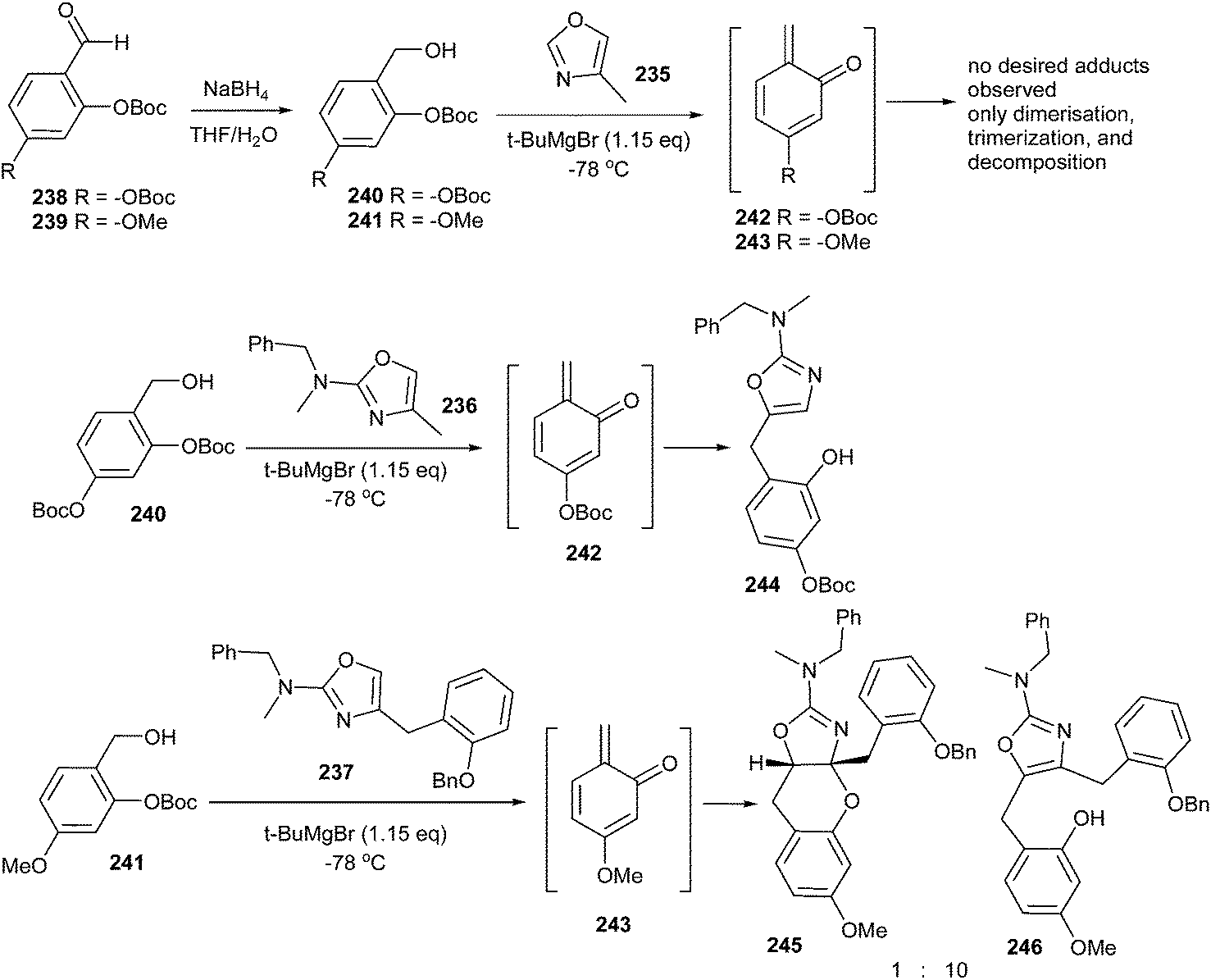 | ||
| Scheme 50 Hetero-Diels–Alder reactions between various electron-rich oxazoles and o-QMs. | ||
Surprisingly, silylated 4-hydroxymethyl oxazole 247 undergoes reaction with o-QM 242 to afford a 66% combined yield with a 3:1 mixture, favouring the [4 + 2] adduct 249 over the 1,4-conjugate addition product 250. In addition, the carbonate protected 4-hydroxymethyl oxazole 248 undergoes cycloaddition with o-QM 243, affording benzopyran 251 in a 46% yield as the only identifiable product (Scheme 51).
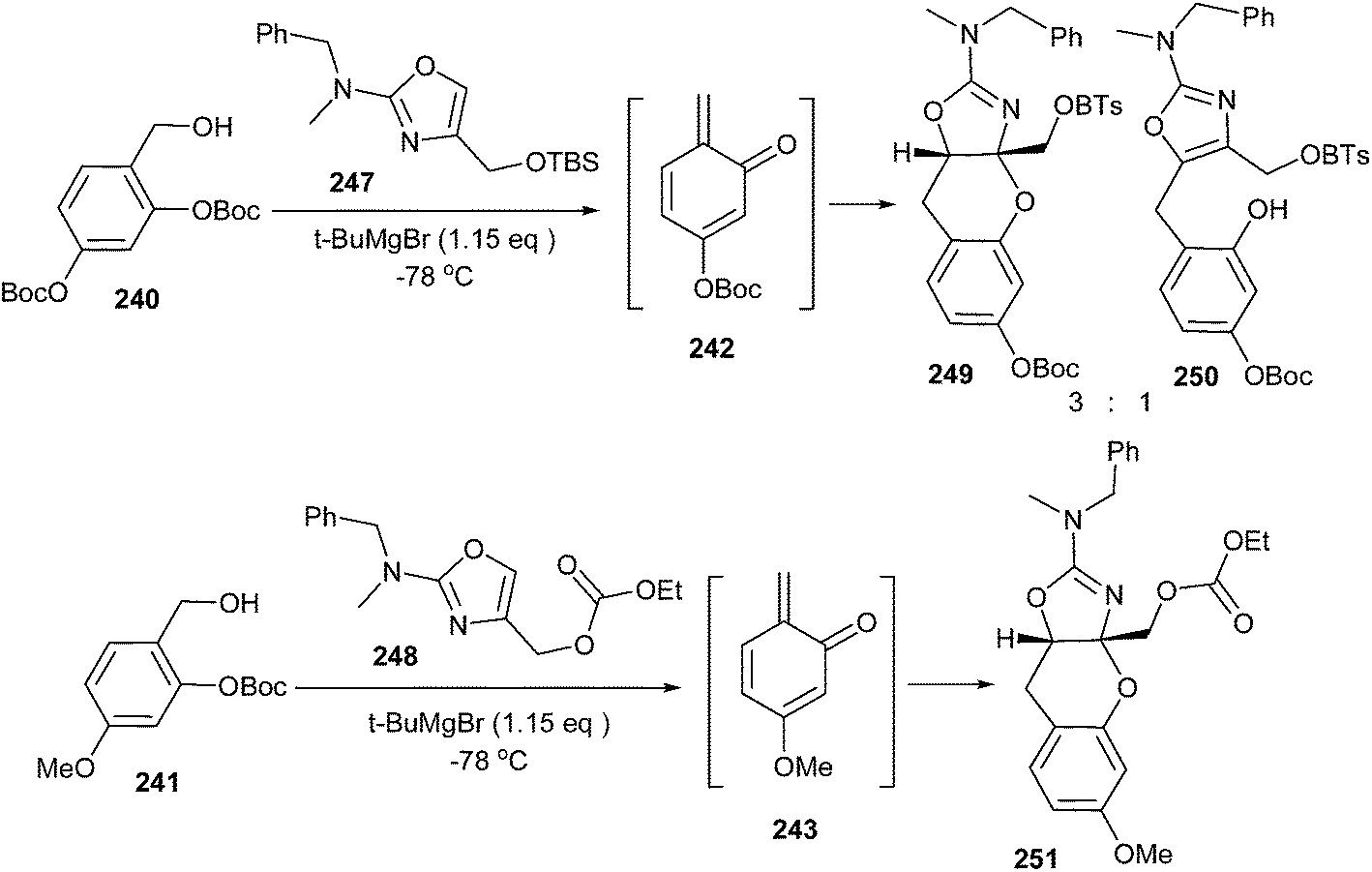 | ||
| Scheme 51 Reaction of substituted oxazoles with o-QM. | ||
The 2-amino oxazoles displaying an electron-rich substituent at the 4-position (R = H, Bn) underwent highly asynchronous reactions, leading to 1,4-conjugate addition adducts, whereas the oxazoles displaying an electron-withdrawing substituent at the 4-position (R = –OTBS, –OCO2Et) underwent a synchronous reaction, affording the [4 + 2]cycloadduct (Scheme 52).
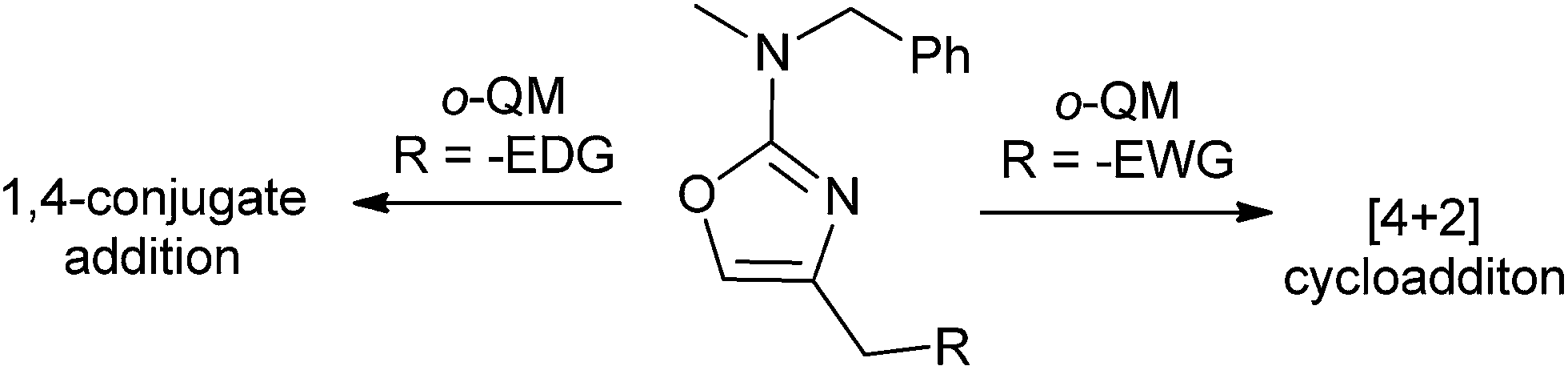 | ||
| Scheme 52 [4 + 2] Cycloadduct of oxazole derivatives. | ||
Moreover, the o-QM derived from tocopherol was trapped with DNMM (4-methyl-3,4-dihydro-2H-[1,4]oxazine) 256 via the hetero-Diels–Alder reaction with inverse electron demand, and confirmed that compound 256 occurs as a degradation product of N-methylmorpholine-N-oxide.49 As dehydro-NMM is an electron-rich olefin and o-QM is electron deficient, the hetero-Diels–Alder reaction of o-QM with ethyl vinyl ether proceeds regioselectively in a particular way, so that the α-carbon of the vinyl reacts with the oxygen and the vinylic β-carbon reacts with the methylene carbon of the QM.49b But in the case of DNMM 256, the situation was less unequivocal, as the compound contains both a vinyl ether and an enamine; hence, two directions of addition with o-QMs 254 and 255 took place, leading to tetracycles 257–260. Computations on the DFT level was performed on the shortened model compounds 255, 258, and 260, possessing a methyl group instead of the isoprenoid side chain that predicted 260 to be 12.6 kJ mol−1 more stable than 258 (Scheme 53).
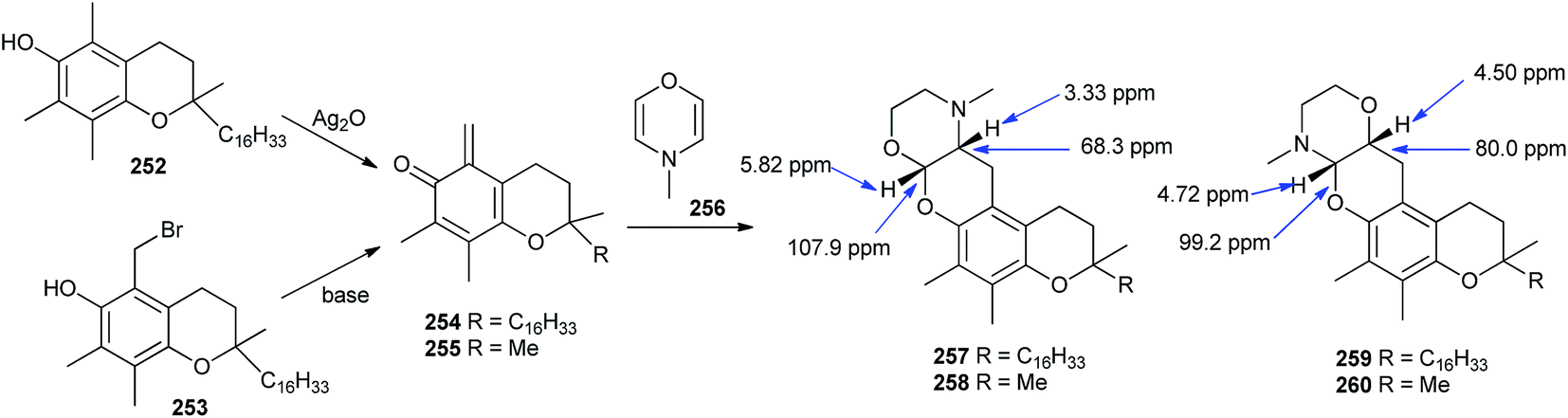 | ||
| Scheme 53 Reaction of o-QM (derived from tocopherol) with oxazine. | ||
A mechanistically interesting enantioselective [4 + 2] cycloaddition of o-QM with silyl ketene acetals by catalytic pathway is well described in a previously reported method,50 and was initiated by a chiral cinchona alkaloid-derived ammonium fluoride “precatalyst” complex to afford a variety of alkyl- and aryl-substituted 3,4-dihydrocoumarin products in excellent yields with good enantioselectivity. The electron-rich o-QM 261 was first generated by Jurd in 1977 by the reaction of sesamol and 4-methoxybenzyl alcohol under the mild Friedel–Crafts conditions followed by Ag2O oxidation. Then, the newly generated o-QM 261 was treated with butyryl chloride, thermodynamic Hunig's base, which generates stable enolate and kinetic base/chiral catalyst benzoylquinidine (BQd) 262. It has been found that the high electron density on o-QM was not compatible with the mild nucleophilic zwitterionic ketene enolate 262; therefore, silyl ketene acetal 263 was used as the ketene enolate precursor to synthesize the compound 267 (Schemes 54 and 55).
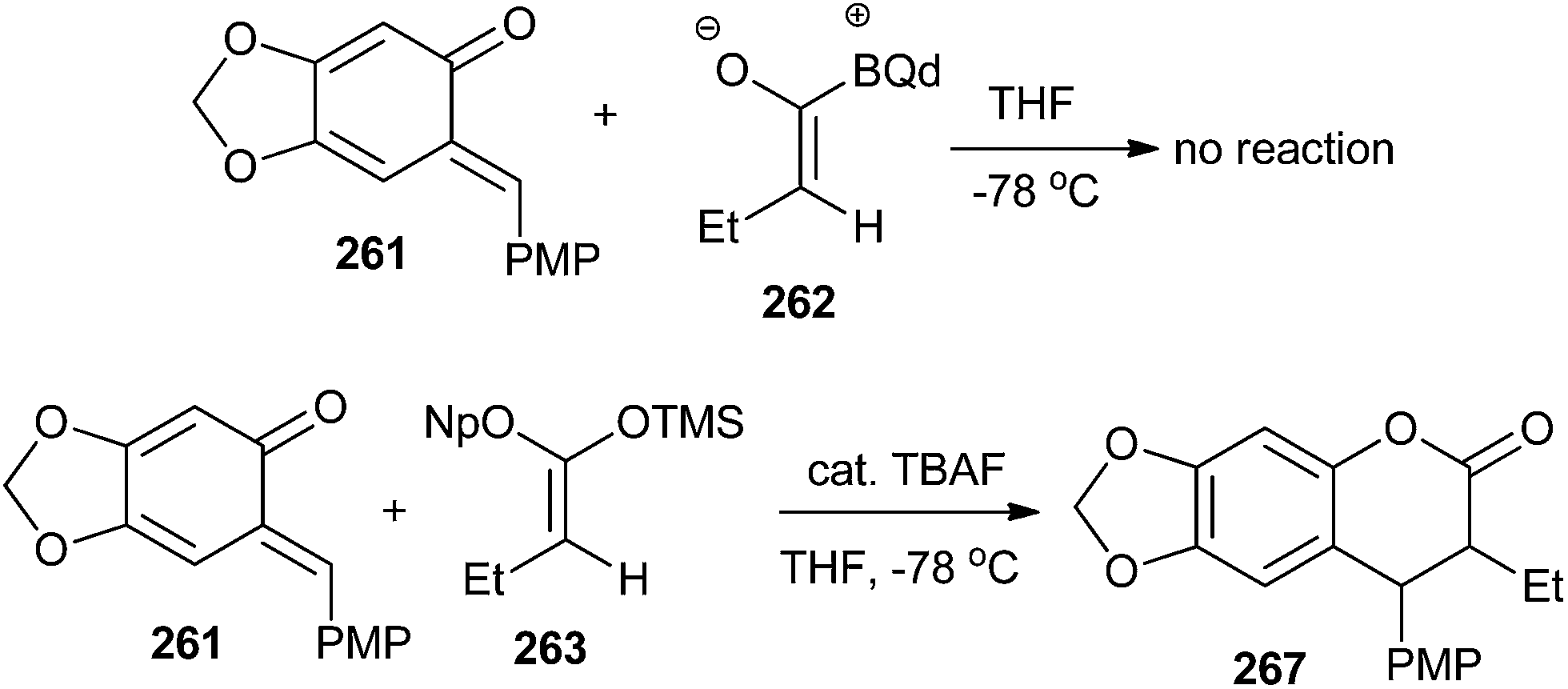 | ||
| Scheme 54 Synthesis of compound 267 utilizing silyl ketene acetal 263 and o-QM 261. | ||
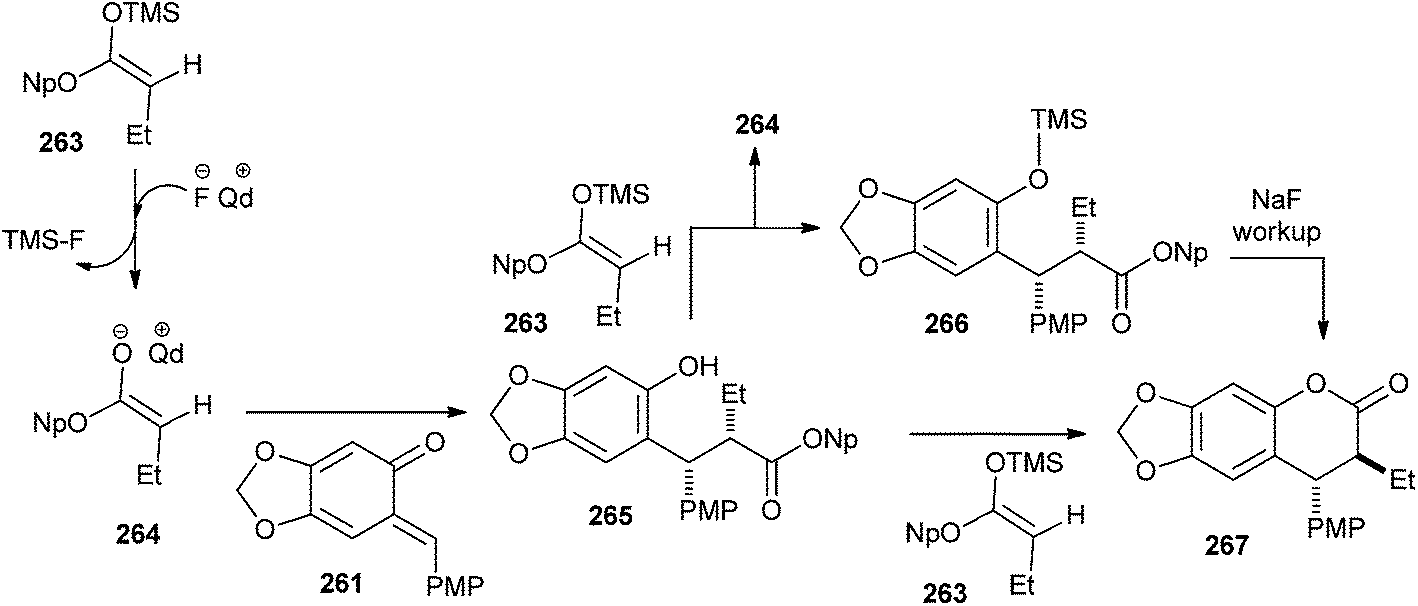 | ||
| Scheme 55 Enantioselective synthesis of compound 267. | ||
Next, reaction of o-QM 261 with 263 in the presence of catalytic tetrabutylammonium fluoride (TBAF) in THF afforded the cycloaddition product 267 with 80% ee. Thus, it shows that the precatalyst's effect on the enantioselectivity of the reaction was not dictated by steric bulk, but rather by electronic effects (Scheme 55).
A variety of chroman spiroketals have been synthesized by Pettus and co-workers51 via inverse-demand [4 + 2] cycloaddition of enol ethers with o-QMs. At low temperature, in situ generation of o-QM allows the kinetic diastereoselective construction of these motifs, providing entry to a number of unusual chroman spiroketals. When benzyl alcohol 269 (0.1 M in toluene, −78 °C) and 2–4 equivalents of the 1,3-dihydroisobenzofuran enol ether 268 were treated with t-BuMgCl, an endo-selective cycloaddition took place as the reaction mixture slowly increases to room temperature (Scheme 56). The reaction cleanly produced the chroman spiroketal 270 in 60% isolated yield (>8:1 dr).
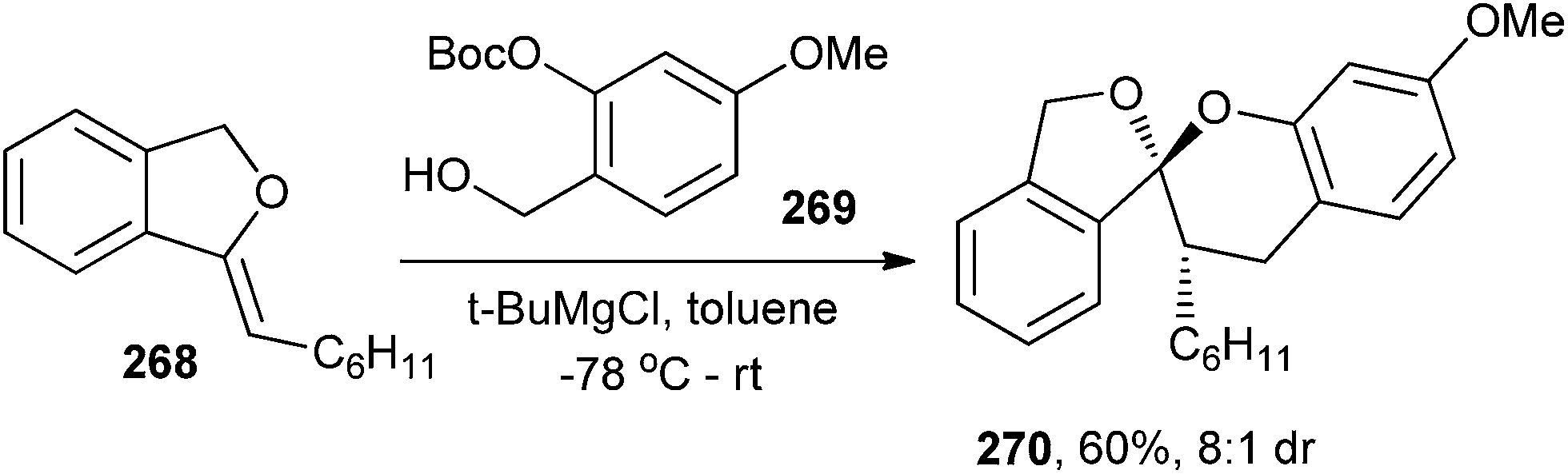 | ||
| Scheme 56 Synthesis of chroman spiroketal. | ||
The simple exo-enol ethers readily equilibrate with their endo-isomers under acidic and sometimes also thermal conditions. The major product was the expected benzopyran 273, and the mono-benzannulated spiroketal 274 (as a mixture of diastereomers) was observed in a 25% yield during the reaction of compound 271 with endo-enol ether 272. Apparently, this was the result of the cycloaddition of the intermediate o-QM with 4,5-dihydro-2,4-dimethylfuran (Scheme 57).12,13
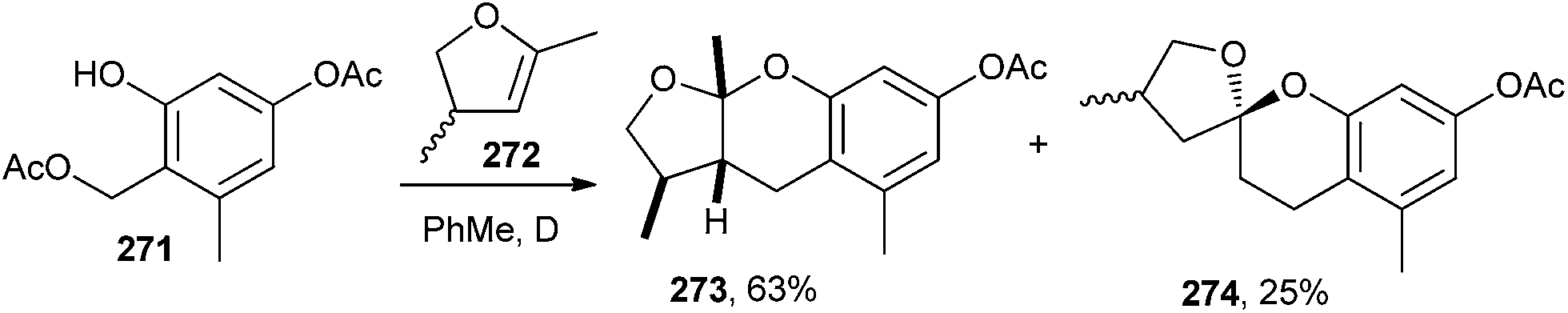 | ||
| Scheme 57 Synthesis of benzopyran 273 and mono-benzannulated spiroketal 274. | ||
The intermediacy of o-QM has been employed to develop a concise strategy for the synthesis of isoflavans via the Diels–Alder reaction between o-QM and aryl-substituted enol ethers 278 followed by the reductive cleavage of the acetal group.52 The method is extended toward the total syntheses of equol, 3′-hydroxyequol, and vestitol. A reductive removal of the methoxy group from acetal 279 was achieved by using BF3·OEt2 and triethylsilane to give isoflavan 280 (Scheme 58).
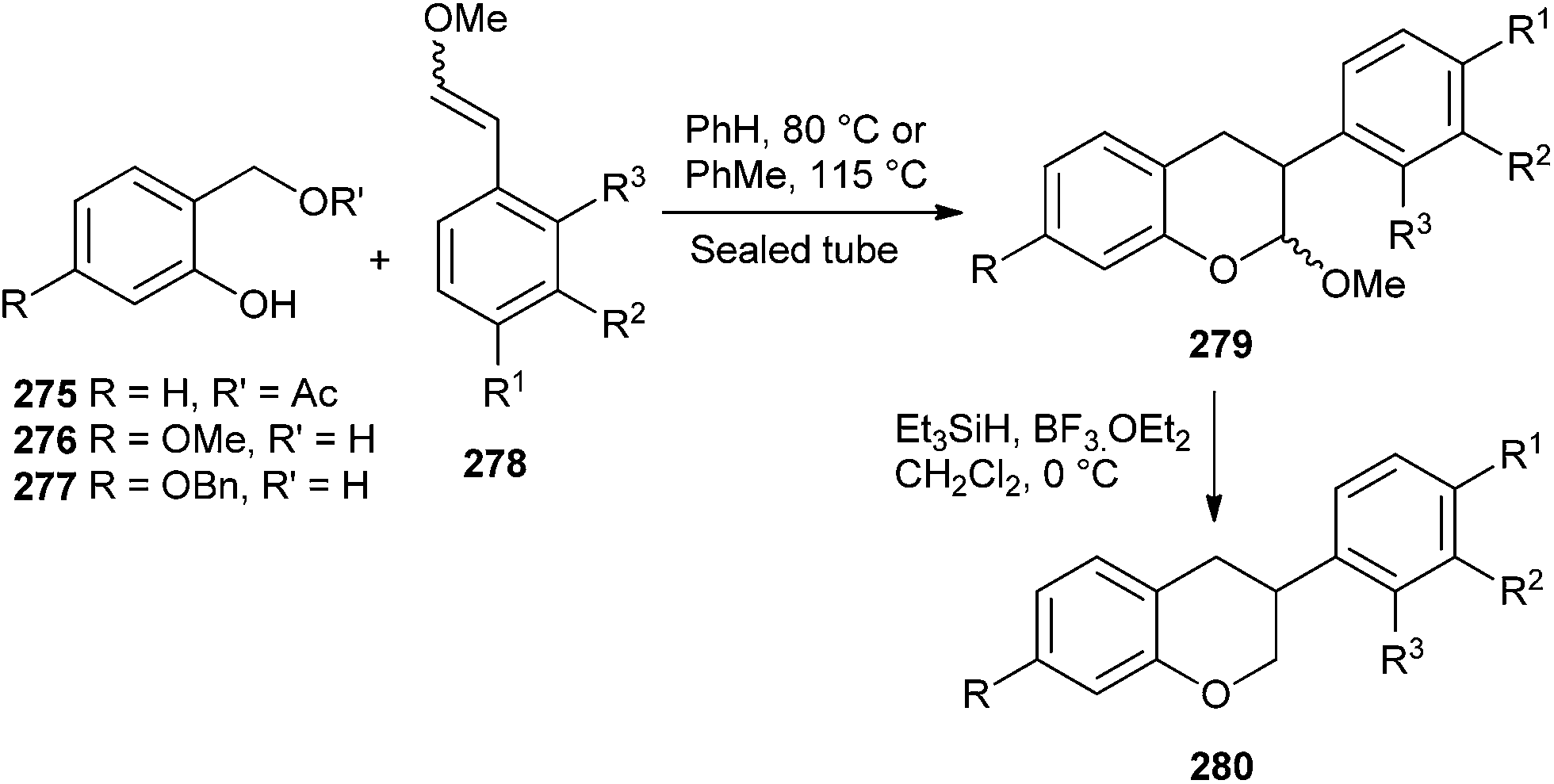 | ||
| Scheme 58 Synthesis of isoflavan 280 via intermediacy of o-QM. | ||
It has been envisioned that the Lewis acid should be able to catalyze both the [4 + 2] cycloaddition and the reductive removal of the methoxy group of the acetal moiety in a one-pot transformation. In this course, the reaction of phenol 277 and enol ether 278a with BF3·OEt2 in CH2Cl2 at 0 °C in the presence of triethylsilane in the same pot gave isoflavan 280a in a 34% yield (Scheme 59).
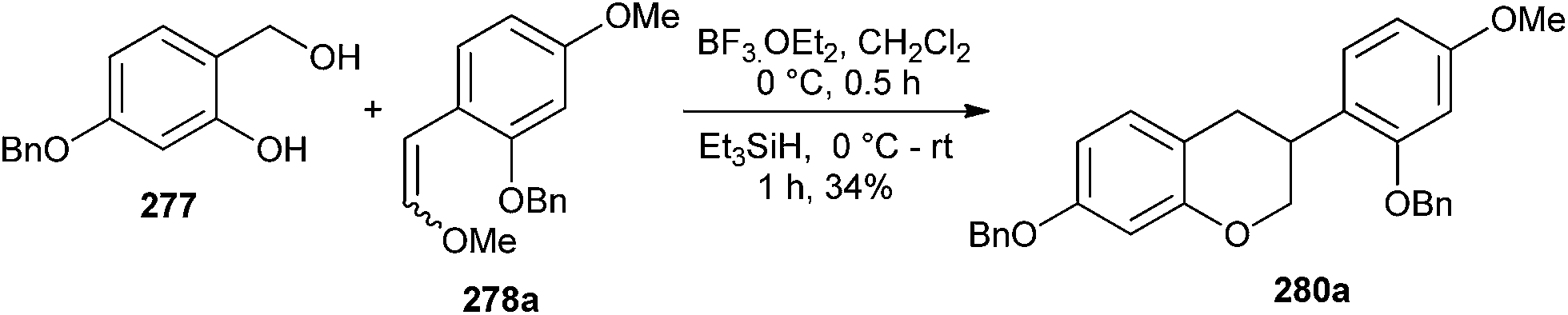 | ||
| Scheme 59 [4 + 2] Cycloaddition reaction of substituted phenol with enol ether 278a. | ||
 | ||
| Scheme 60 Synthesis of 5,2,1-dibenzooxathiazocine-2,2-dioxides by hetero [4 + 4] cyclization. | ||
 | ||
| Scheme 61 Intramolecular [5 + 2] cycloaddition of phenoxonium intermediates. | ||
3.2. Addition of nucleophiles
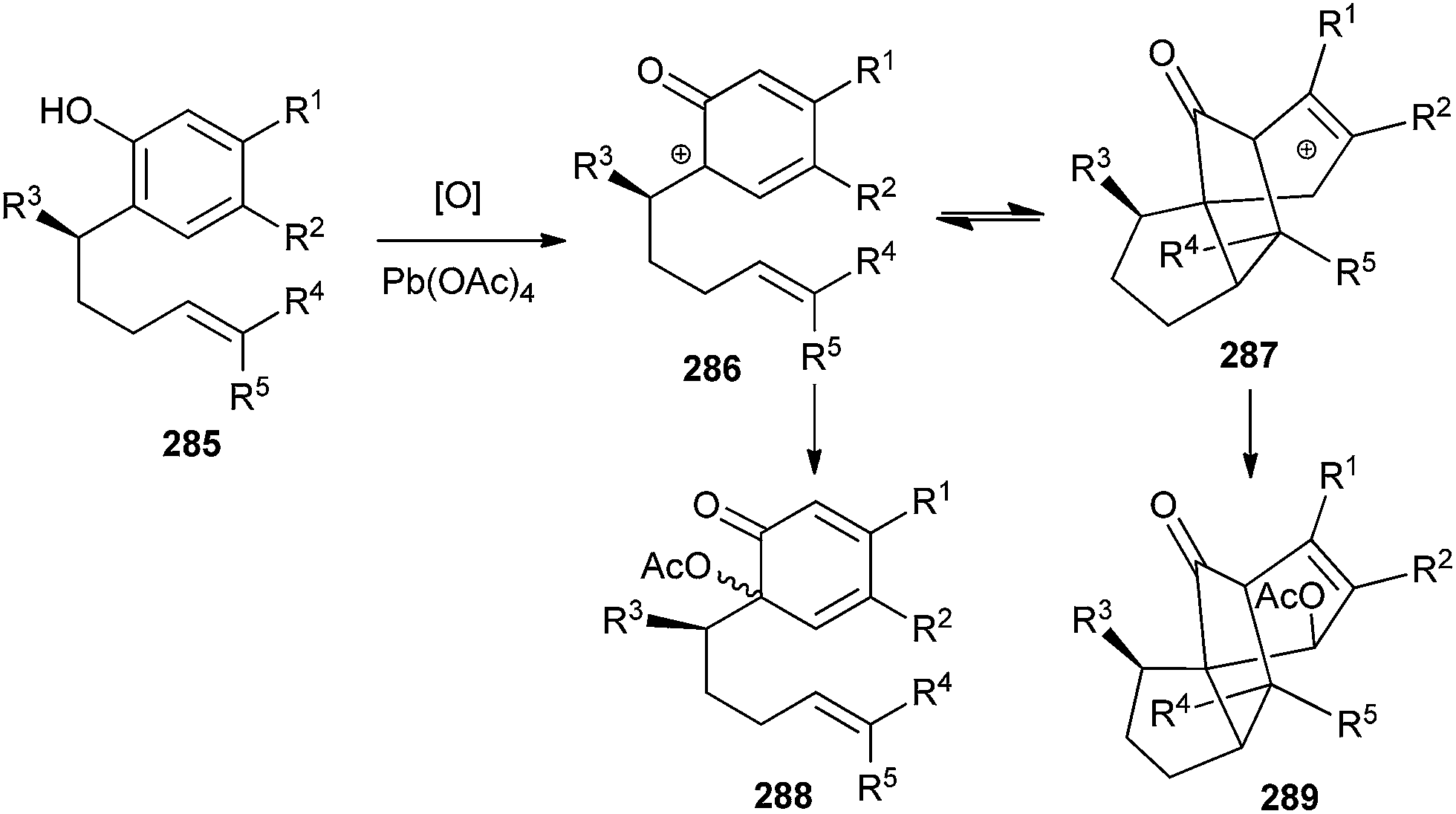
Bromination of α-tocopherol 290 for the formation of 294 is not possible by simple bromination because an ethano-dimer of α-tocopherol 290 after bromination affords pyrano-spirodimer of α-tocopherol 291 in > 95% yields. Compound 290 and 291 constitute a largely reversible redox pair.55 Bromine oxidizes one α-tocopherol 290 to an ortho-quinone methide (o-QM) intermediate 292, which is immediately attacked by the phenolic OH group of the tocopherol. It is evidently favoured over the 1,4-addition of evolving HBr by a pre-organizational effect, as the ethano-bridge keeps the two reacting moieties in close distance. This acid facilitates the opening of 291 to regenerate the o-QM 292.56 Thus, the OH group was protected by TMS, which continuously traps a small percentage of the o-QM intermediate present and cannot react back to give compound 291. In this way, 291 is eventually completely converted into 293 (Scheme 62). Further reaction of 293 with elemental bromine followed by desilylation provided the target bisbromide 294.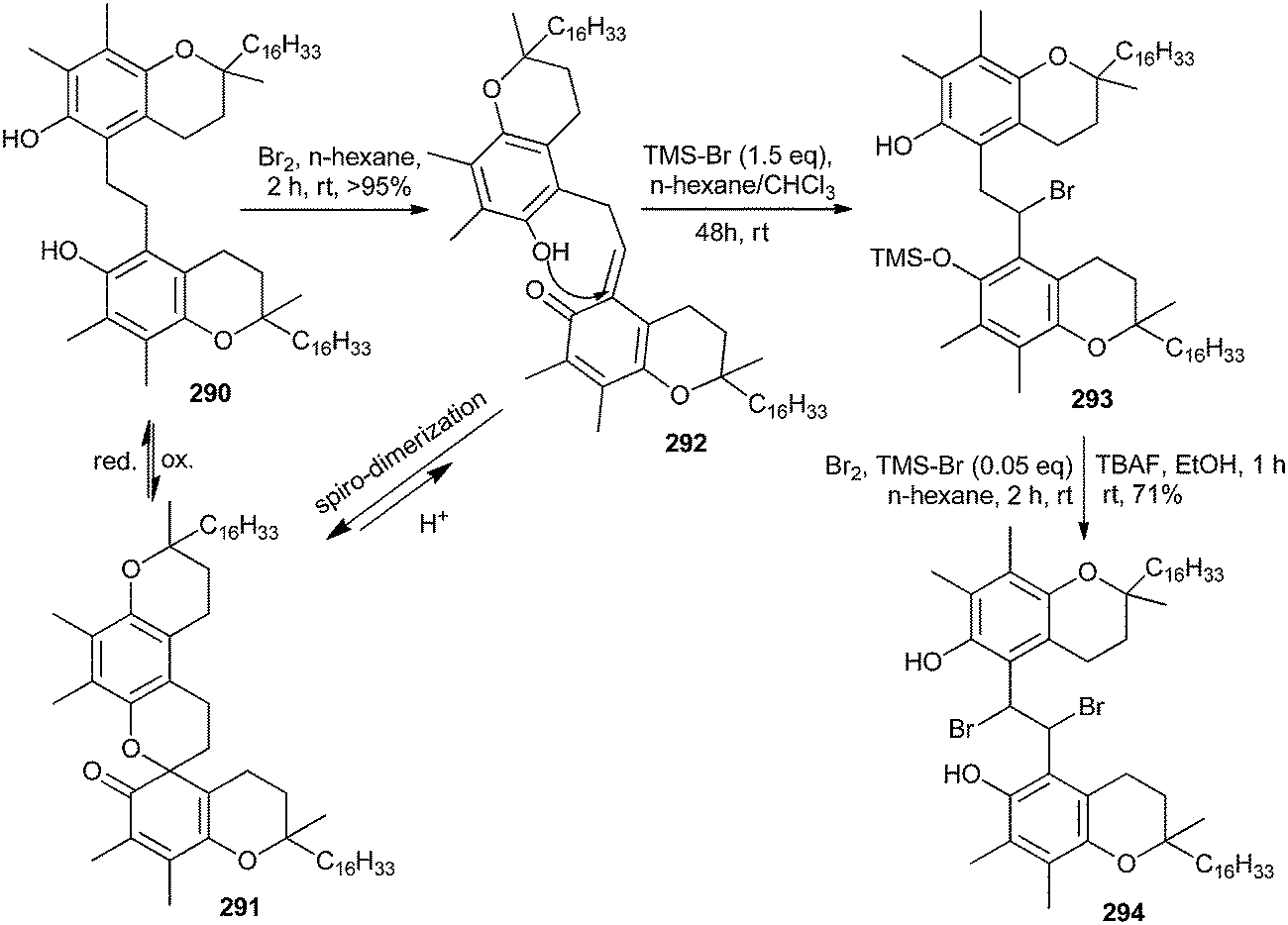 | ||
| Scheme 62 Bis-bromination of ethano-dimer of α-tocopherol. | ||
The pseudo three-component condensation reaction of 2-naphthol with aldehydes in the presence of various catalysts to form xanthenes has been widely studied. The reaction proceeds through the in situ formation of o-QMs with 2-naphthol acting as a nucleophile. However, the three-component condensation reactions of 2-naphthol and aldehydes with other nucleophiles are rarely reported in previously studied reports. Singh and co-workers57 generated substituted xanthenes 297 by one-pot condensation of 2-naphthol with aliphatic/aromatic aldehydes 295 in the presence of P2O5 or InCl3 as catalysts under solvent-free conditions. They also reported a one-pot synthesis of tetrahydrobenzoxanthenone 299 and diazabenzoanthracenedione 300 in the presence of an Indium(III) chloride catalyst by a three-component cyclocondensation of aldehydes 295, β-naphthol 296 and cyclic 1,3-dicarbonyl compounds 298 under solvent-free conditions in high yields. This transformation has also been effectively carried out by using P2O5 as a catalyst. The use of aldehyde 295, β-naphthol 296, and 3-oxo-3-phenyldithiopropionic acid methyl ester 301 in equimolar amounts in the presence of InCl3 produces benzo[f]chromene 302 in good yields (Scheme 63).
 | ||
| Scheme 63 Cyclocondensation reaction via o-QMs intermediates. | ||
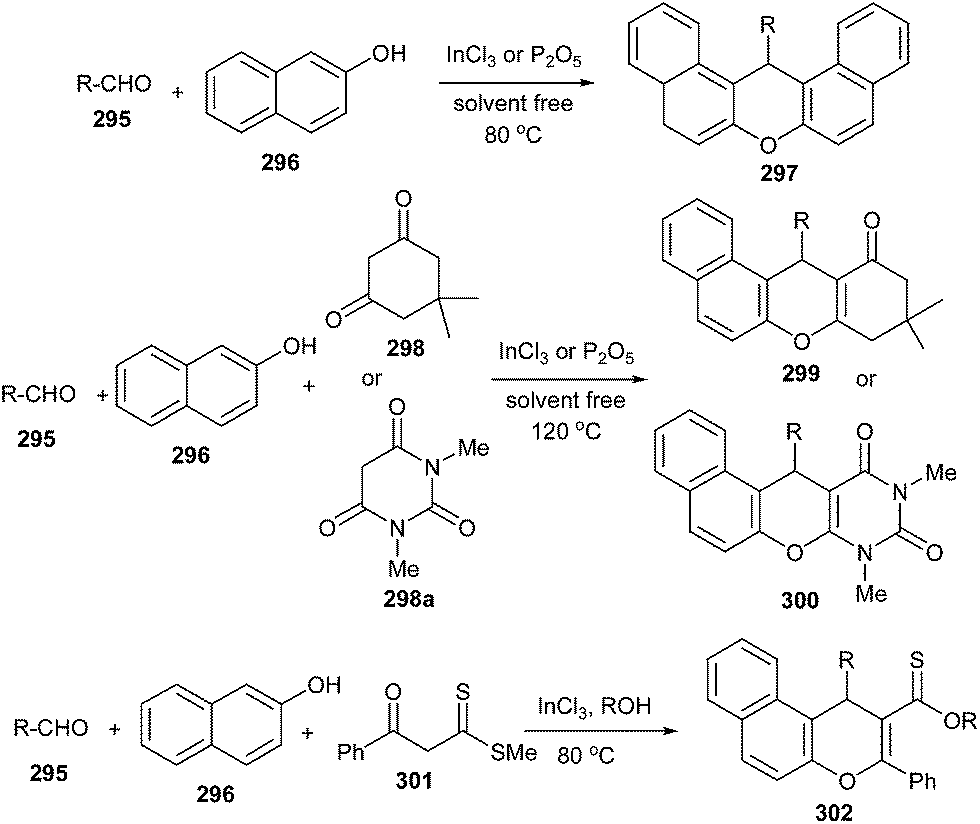
The nucleophilic addition of β-naphthol 296 to aldehyde 295 in the presence of InCl3 gives naphthoquinone methide intermediate 303, which then reacts with the nucleophile like 1,3-dicarbonyl compound 298 followed by the addition of the phenolic hydroxyl moiety to the carbonyl of ketone, providing cyclic hemiketal, which on dehydration afforded 299 (Scheme 64).
 | ||
| Scheme 64 Nucleophilic addition of β-naphthol to aldehyde via o-QM intermediacy. | ||
The in situ formation of o-QM intermediates in the presence of solid silica chloride under sonication followed by the nucleophilic attack of amide has been reported to lead to the formation of amidoalkyl-2-naphthol.58 Due to the collapse of the cavitation bubbles near the surface of the catalyst, the oxygen of the carbonyl group might have easily replaced the chlorine atom present on the catalyst silica chloride to give the activated aldehyde, which could be attacked by β-naphthol to give o-QM 307. Thus, the in situ generated o-QM 307 upon reaction with amide 308 furnished the envisioned amidoalkyl naphthol 309 (Scheme 65). This reaction was previously reported under the influence of P2O5, which acts as a catalyst as well as a water scavenger.59
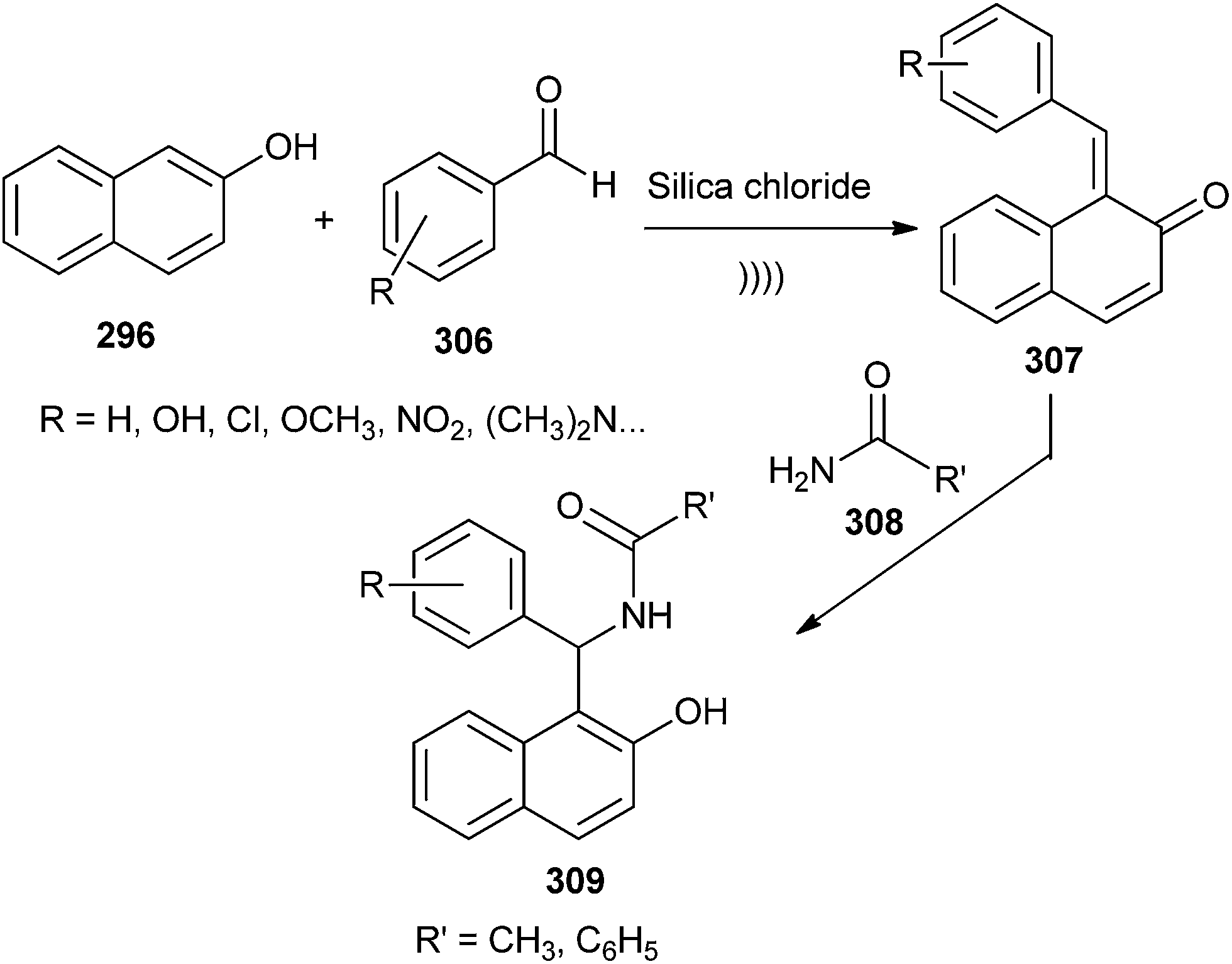 | ||
| Scheme 65 Synthesis of amidoalkyl naphthols. | ||
Nagawade and Shinde60 reported the rapid synthesis of biologically significant amidoalkyl naphthols using a catalytic amount of iodine. Chlorinated solvents (dichloroethane > dichloromethane > chloroform) gave the highest yields. The yields of the end products depend on the polarity of the solvent used, and after a through screening, 1,2-dichloroethane was found to be the best solvent for the protocol. o-QM has also been generated by the treatment of β-naphthol, aryl aldehyde and ureas or amides in the presence of catalytic amounts of p-TSA at 125 °C.61 In addition to that hafnium(IV) bis(perfluorooctanesulfonyl)imide complex,62 (Hf(NPf2)4), NaHSO4·H2O63 and H3PW12O40 have also been shown to be effective catalysts for the same reaction, with respect to reaction times and yields in the molten salt media.64
2′-Aminobenzothiazolomethyl naphthols or 5-(2′-aminobenzothiazolomethyl)-6-hydroxyquinolines 313 was prepared by the reaction of o-QM 312, and 2-aminobenzothiazole. o-QM was generated in situ from the reaction of β-naphthol or 6-hydroxyquinoline and aldehyde 311 (Scheme 66).65
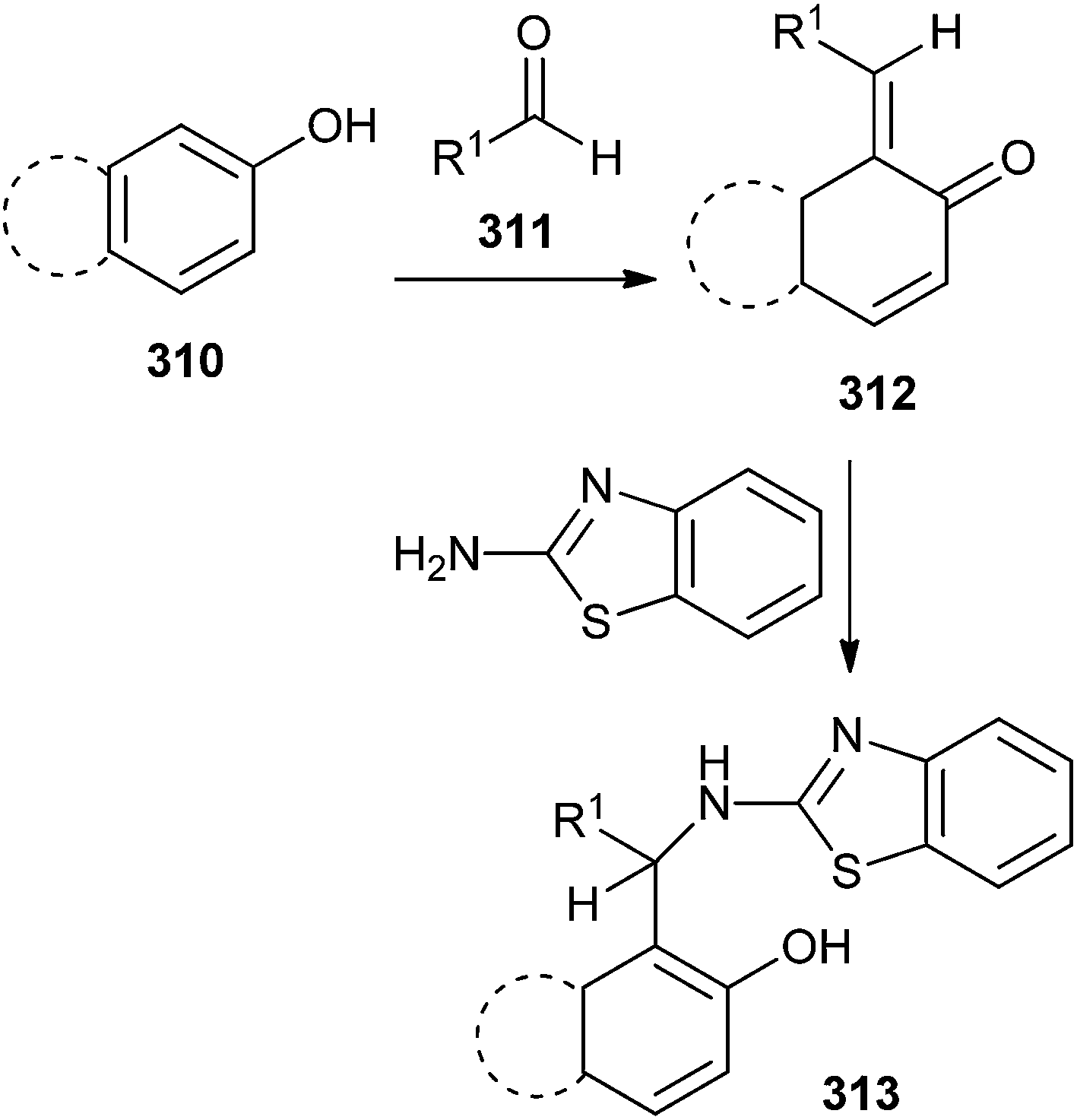 | ||
| Scheme 66 Synthesis of 2′-aminobenzothiazolomethyl naphthols. | ||
1,2-Dihydronaphtho[2,1-b]furans and 2,3-dihydrobenzofurans 316 substituted at C-2 by an acyl or aryl group were prepared in a diastereoselective manner. The reaction goes through the formation of the o-quinone methide intermediate followed by a Michael-type addition of the ylide 315 to the o-QM with a consequent intramolecular nucleophilic substitution. The reaction is applicable to a range of Mannich bases and pyridinium salts with a variety of versatile functional groups (Scheme 67).66
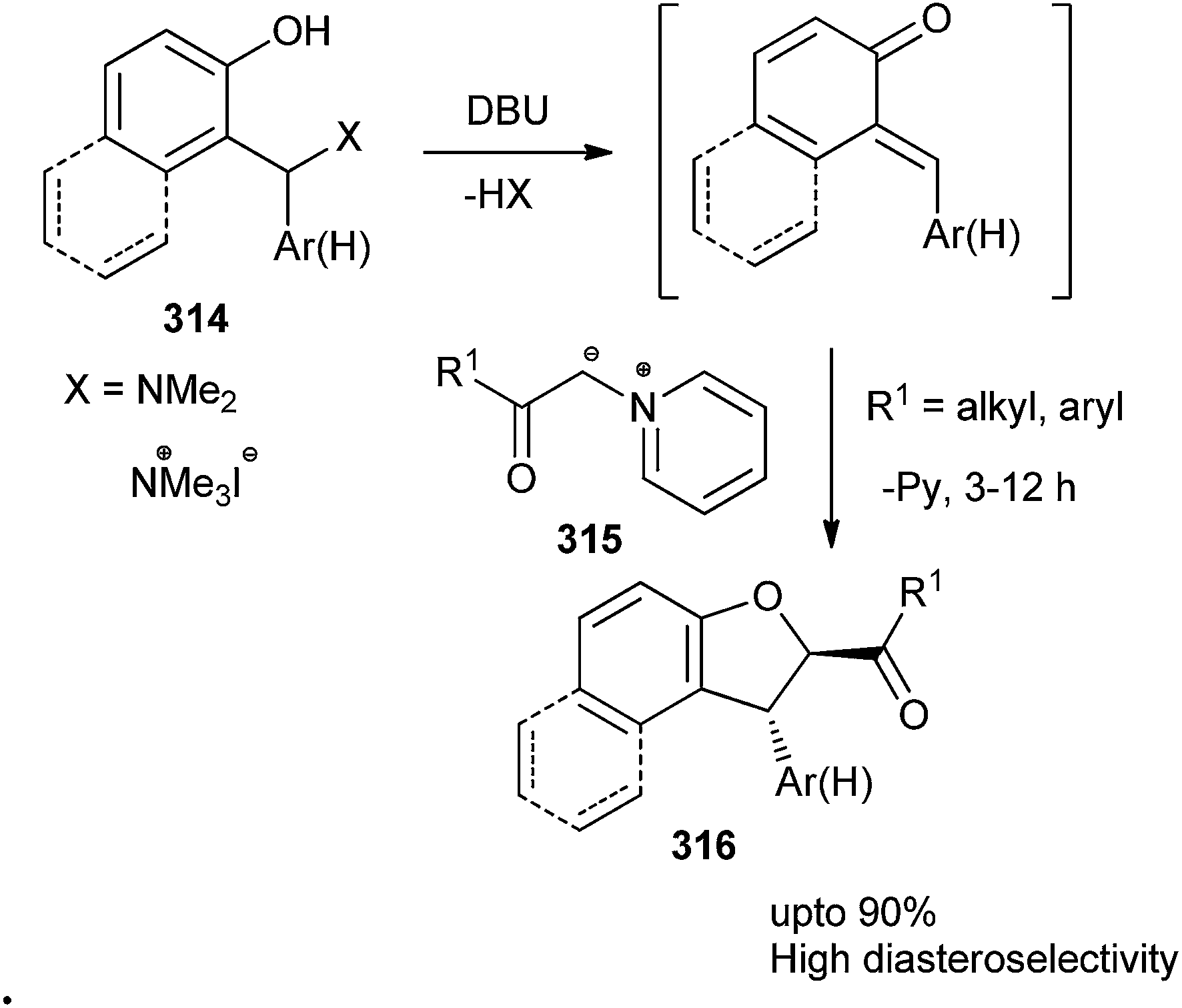 | ||
| Scheme 67 Diastereoselective synthesis of naphthofurans and benzofurans. | ||
Salvadoria and co-workers67 reported the oxidation of α-tocopherol 317 leading to o-QM species 318, which when reacted with bromide as a nucleophile from hydrogen bromide, affords the benzylic brominated product 319 (Scheme 68).
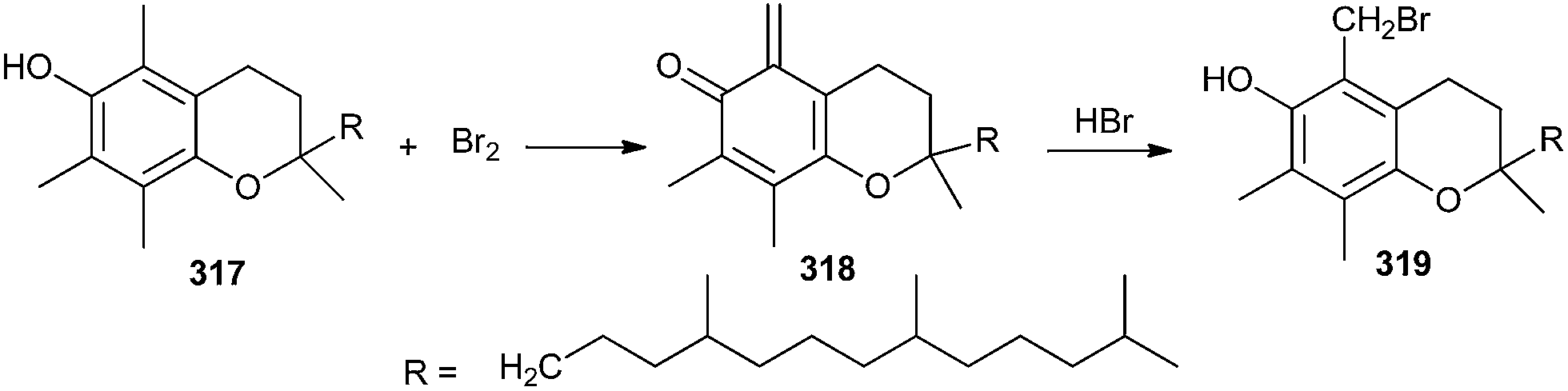 | ||
| Scheme 68 Bromination of α-tocopherol. | ||
The bromo-α-tocopherols 317 and 320 after treating with nucleophilic triphenyl phosphene followed by acylation gives the furotocooherols 325 & 326 (Scheme 69).68
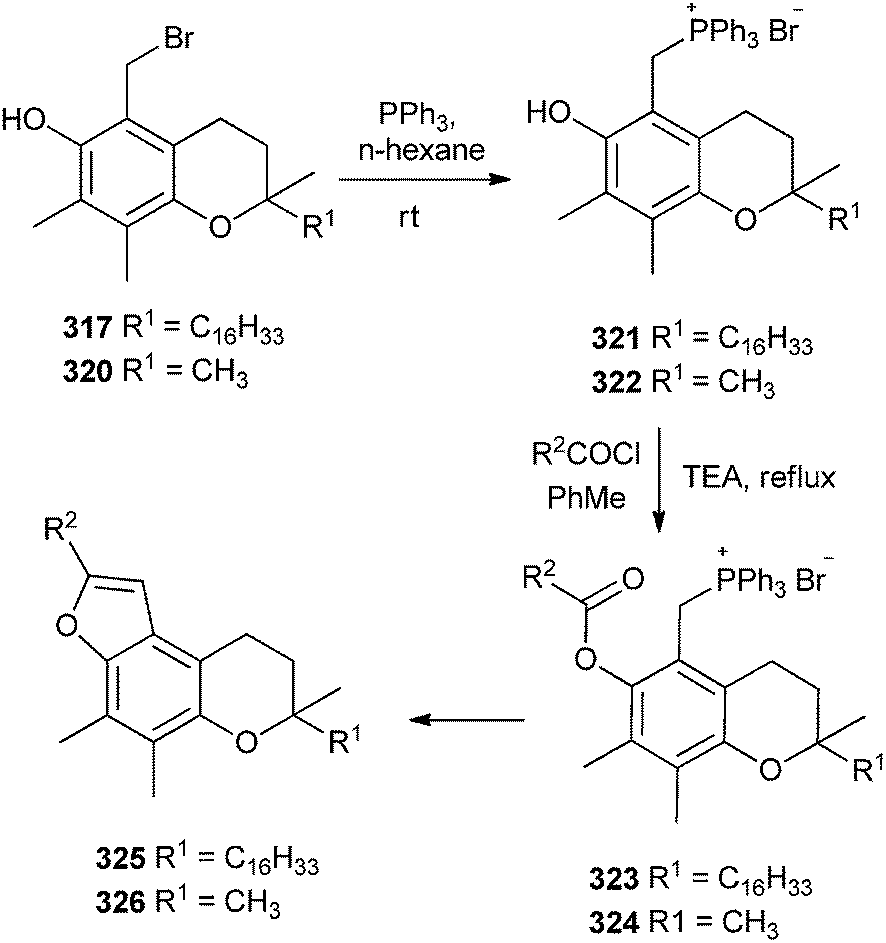 | ||
| Scheme 69 Synthesis of urotocooherols from bromo-α-tocopherols. | ||
Chen and co-workers69 reported the stereoselective synthesis of trans-dihydrobenzofurans 332 by the reaction of o-QM 328 and sulfur ylides 329. This gave intermediate 330, which undergoes trans-elimination–cyclization to afford 332. o-QM intermediates have been generated from 2-tosylalkylphenols 327 under mild basic conditions (Scheme 70).
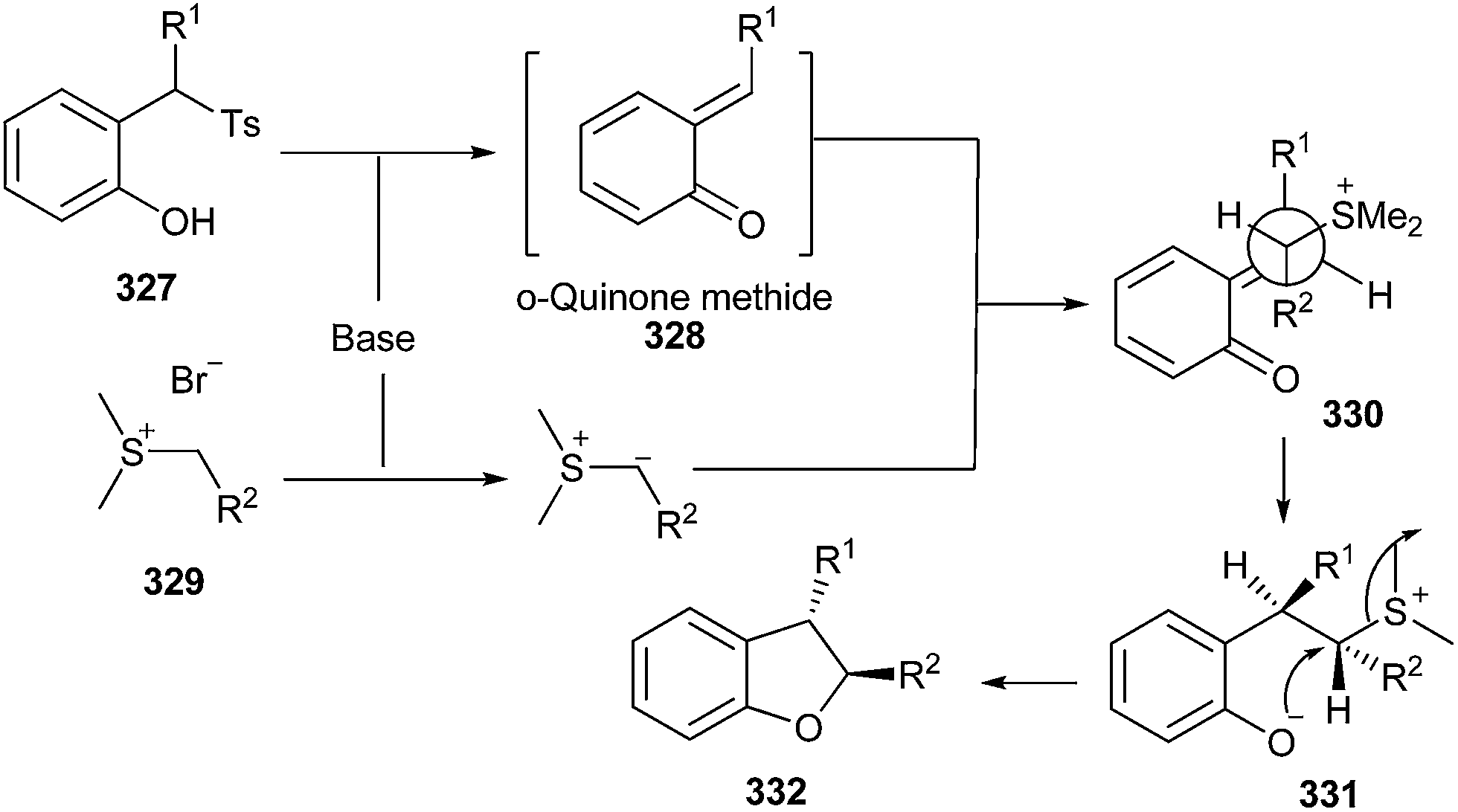 | ||
| Scheme 70 Stereoselective synthesis of trans-dihydrobenzofurans. | ||
Chiral biphenols were found to catalyze the enantioselective asymmetric addition of aryl- or alkenylboronates to o-QMs 333. Thus, substituted 2-styrylphenols 336 were obtained in high yields (up to 95%) with high enantiomeric ratios (up to 98:2) in the presence of 10 mol% of 3,3′-Br2-BINOL 335 (Scheme 71).70
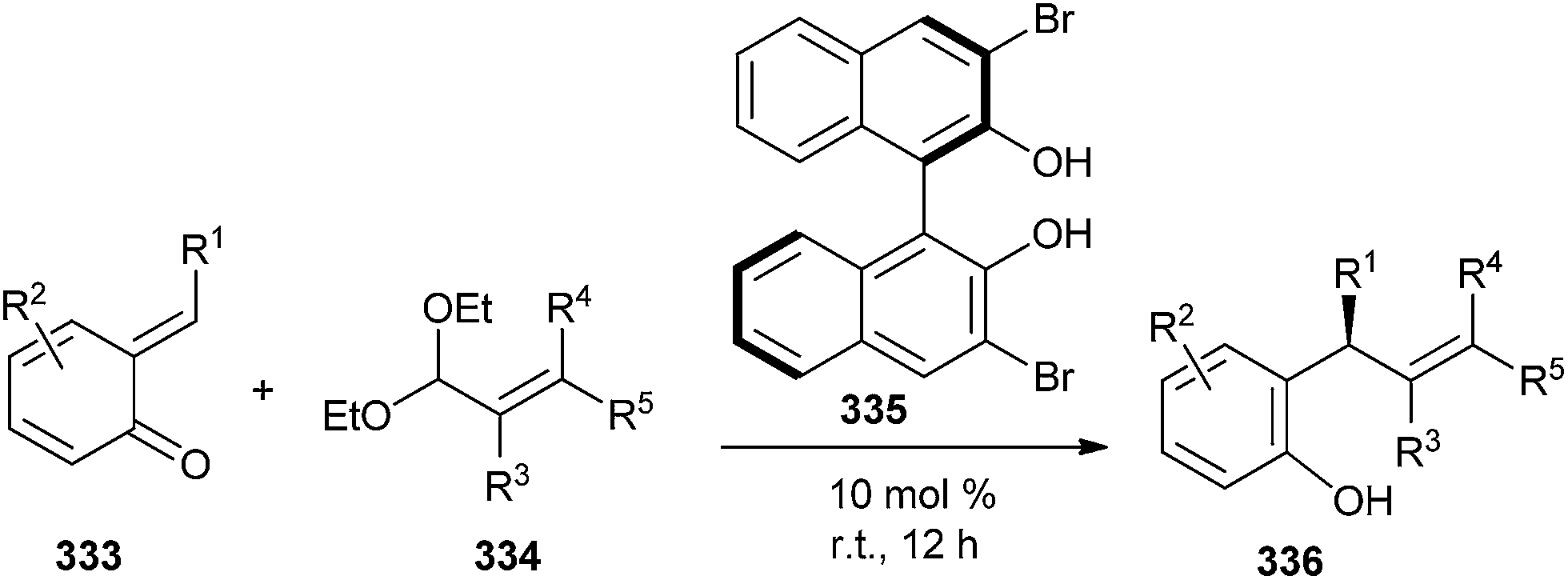 | ||
| Scheme 71 Enantioselective addition of aryl- or alkenylboronates to o-QMs. | ||
Kumar and co-workers2,71 have developed an efficient and catalyst-free approach for C–H hydroarylation of in situ generated o-QM 339 with electron-rich arenes (tert-aryl amines 340) in a glycerol medium. A number of 3-substituted 4-hydroxycoumarin 341 and 4-hydroxypyrone derivatives were obtained in good to excellent yields. The reaction takes place in water without any catalyst, and is highly regioselective (Scheme 72). They have used coumarins, indole, and 4-hydroxypyrone derivatives to generate corresponding o-QMs. It is noteworthy that in all the cases, para substituted products were exclusively obtained. The reason of regioselectivity is the steric hindrance of N-alkyl group and ortho hydrogen of N,N-dialkylanilines, which provides the para selectivity.
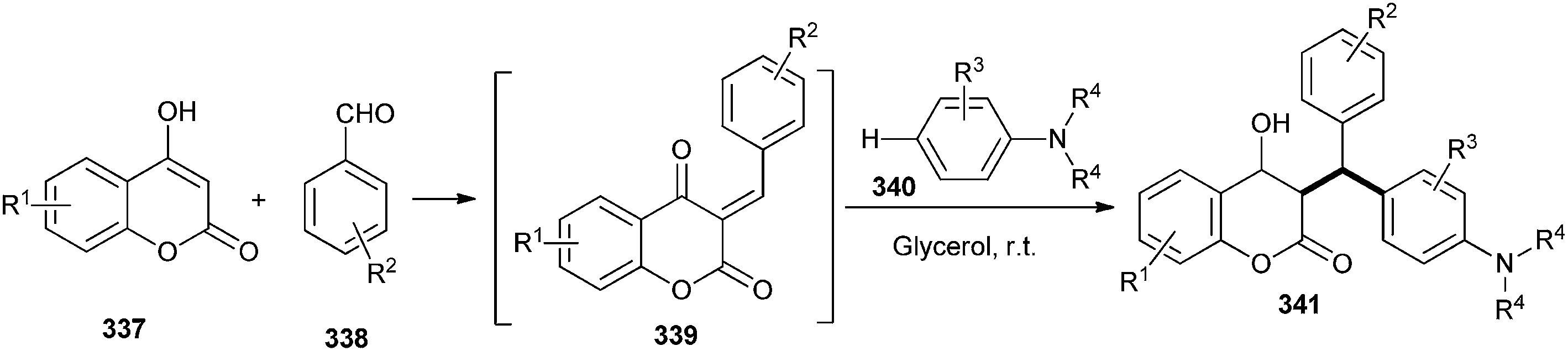 | ||
| Scheme 72 C–H hydroarylation of in situ generated o-QM. | ||
Antonio and Freccero72 have reported the synthesis and the unexpected photochemical reactivity of a new class of naphthalimide derivatives (NIs) 342 bearing quinone methide precursor (QMPs) at the imide moiety by irradiation of the NI core (Scheme 73). Photogeneration of the transient QM 343 through a PET mechanism between the phenolic precursors and the NI core was rationalized. Such reactivity represents the first case of non-direct phototriggering of QMs via photoinduced electron transfer (PET) involving a tethered peripheral moiety.
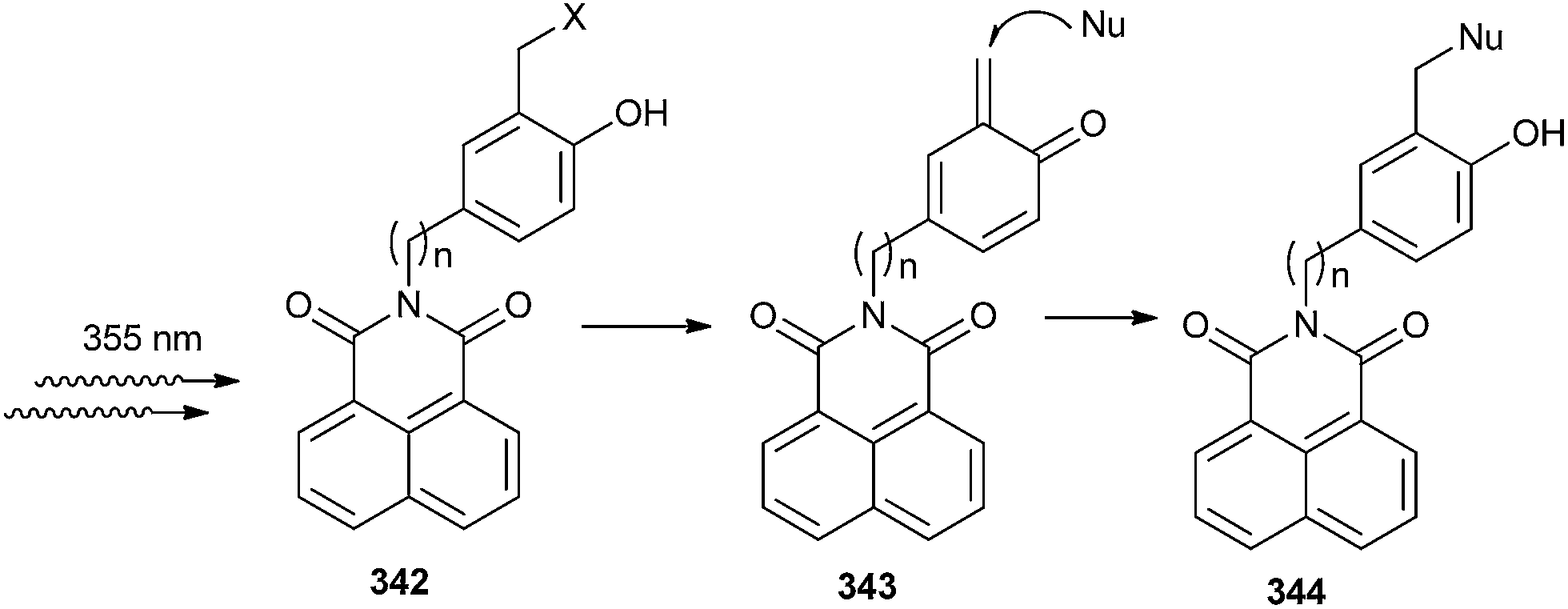 | ||
| Scheme 73 Phototriggering of QMs via photoinduced electron transfer (PET). | ||
Freccero and co-workers26 have reported the photoinduced generation of new binol quinone methides 349, which undergo mono- and bisalkylation of free nucleophiles by product distribution analysis and laser flash photolysis in water solution using binol quaternary ammonium derivatives 346 and 352 as photoactivated precursors (Schemes 74 and 75). N and S nucleophiles are powerfully competitive in alkylation with the hydration reaction.
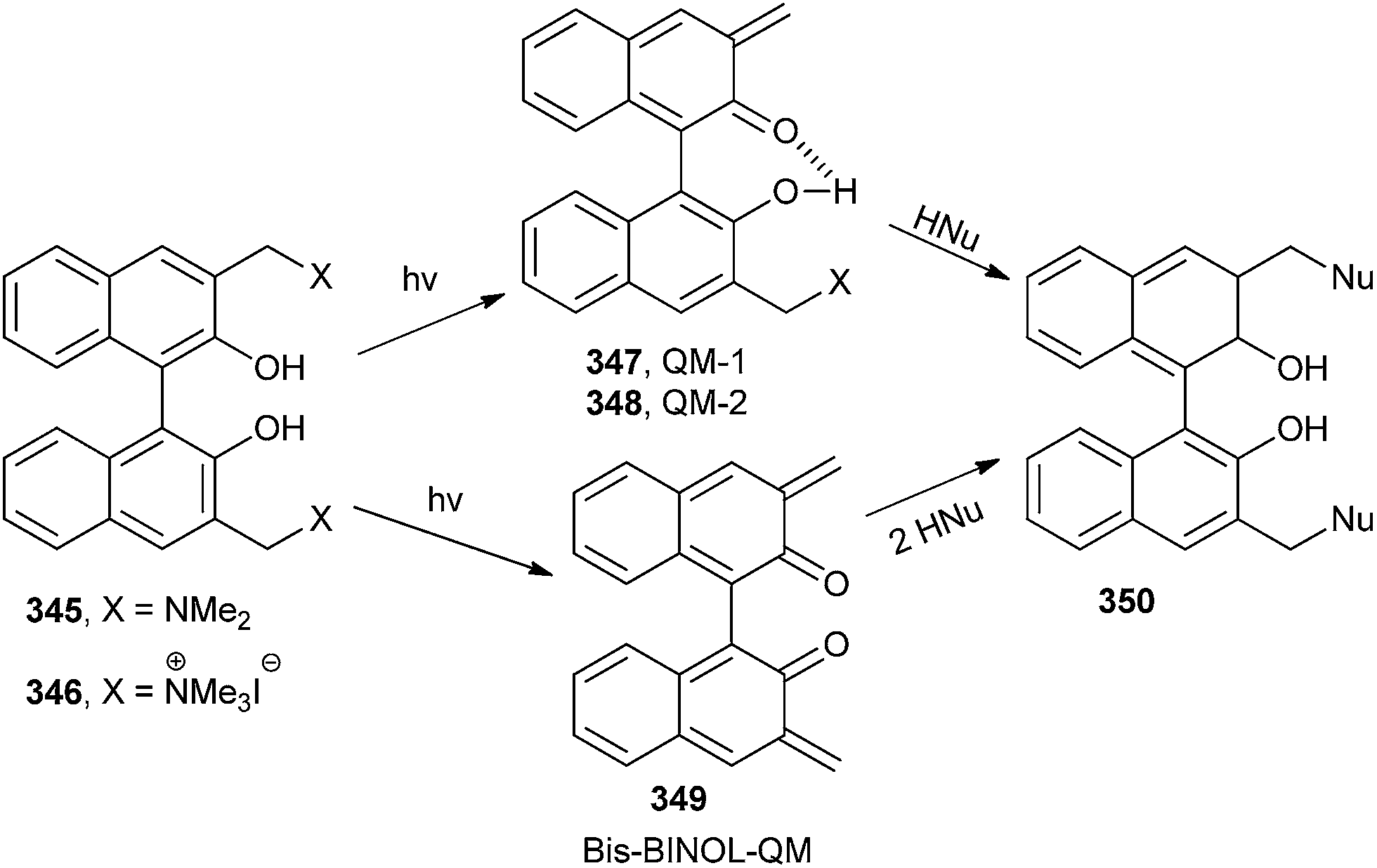 | ||
| Scheme 74 Addition of nucleophiles on binol quinone methides. | ||
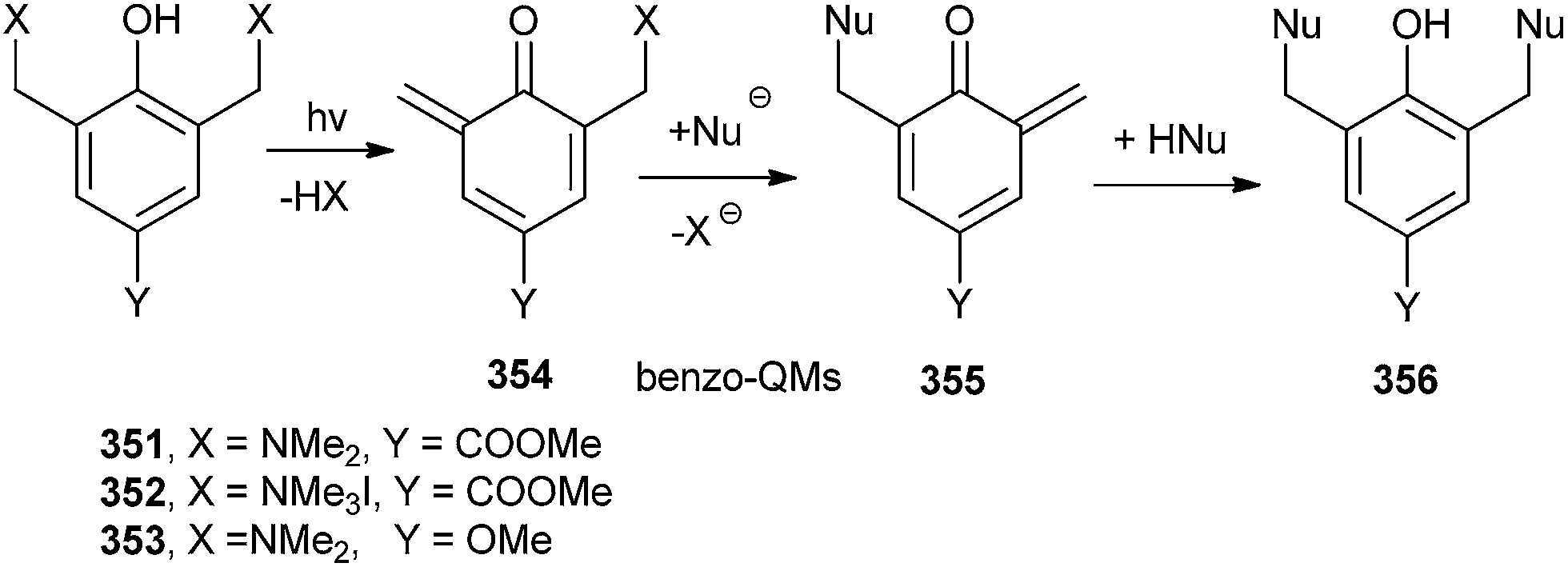 | ||
| Scheme 75 Bis-alkylation of photoinduced o-QM 354 and 355. | ||
The DNA cross-linking potency of the water-soluble binol quaternary ammonium salt 346 was investigated as a pH function and compared to that of other quaternary ammonium salts proficient for photogenerated benzo-QM by gel electrophoresis. DFT calculations in the gas phase and in water bulk on the binol and benzo quaternary ammonium salts 346 and 352 evidence structural and electrostatic features of the binol derivative, which might offer a validation of its promising high photo-cross-linking efficiency.
o-QM 358 (NDI-QM) has been generated by activation of suitable quinone methide precursors (QMPs) 357 under biocompatible conditions such as mild thermal digestion (40 °C), UV-Vis irradiation and mild mono-electronic reduction (Scheme 76).73 The conjugated NDI-QMPs might have been acting as a hybrid ligand-alkylating structures selectively targeting G-4 folding oligonucleotides.
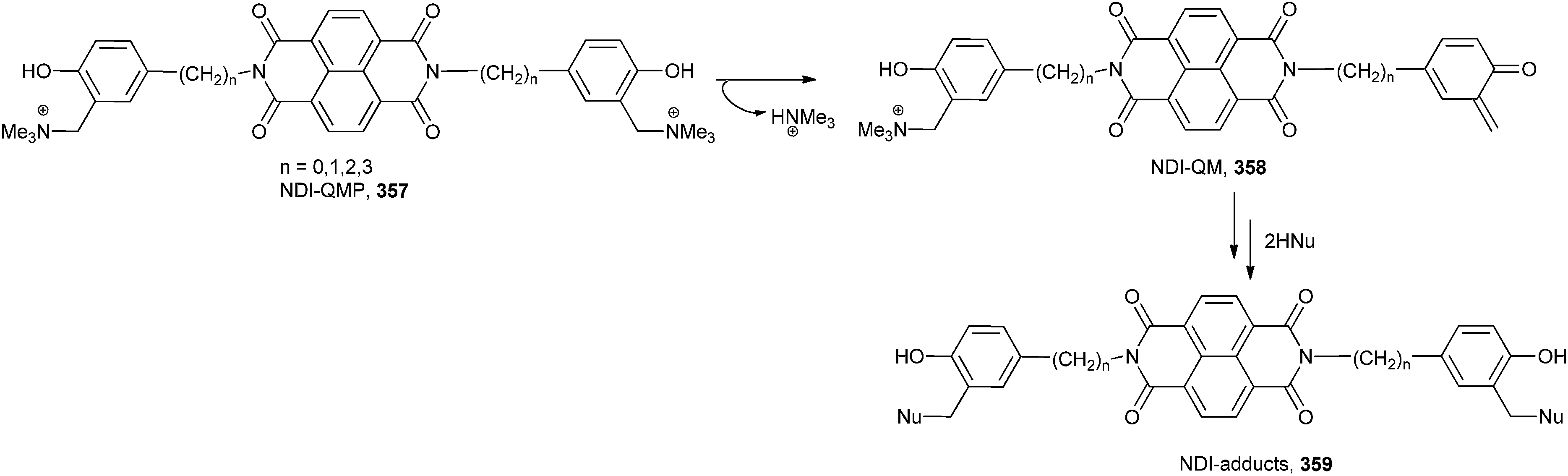 | ||
| Scheme 76 Addition of nucleophile on NDI-QM 358. | ||
3.3. Alkylation on o-QM: DNA alkylation
The formation of QMs depends on the strength of the leaving group attached to the benzylic position of o-QM 360 in the presence of biological nucleophiles (dN).74,75 The formation and reaction of QMs are also highly responsive to the presence of electron-withdrawing and electron-donating groups. The observed trends of reactivity are consistent with the electron-deficient nature of the QM intermediate. Electron-donating groups greatly facilitate initial QM generation and its subsequent regeneration from adducts formed by the nucleophiles of deoxynucleosides (dN) that also function as good leaving groups. Conversely, electron-withdrawing groups greatly suppress initial formation of QM and its regeneration from the reversible deoxynucleoside adducts 362 (Scheme 77). The stability of o-QM is mainly influenced by the electronic perturbation over it, and the kinetics of the product formation is also altered by the reaction with deoxynucleosides.76 Electron-rich QMs react considerably more slowly but selectively with nucleophiles than the electron-poor QMs. These fundamental characteristics provide the basis for explaining the product profiles observed after DNA has been exposed to various QMs.77 These characteristics are used to fine-tune the stability and reactivity of QM conjugates for target-promoted and gene-specific alkylation.78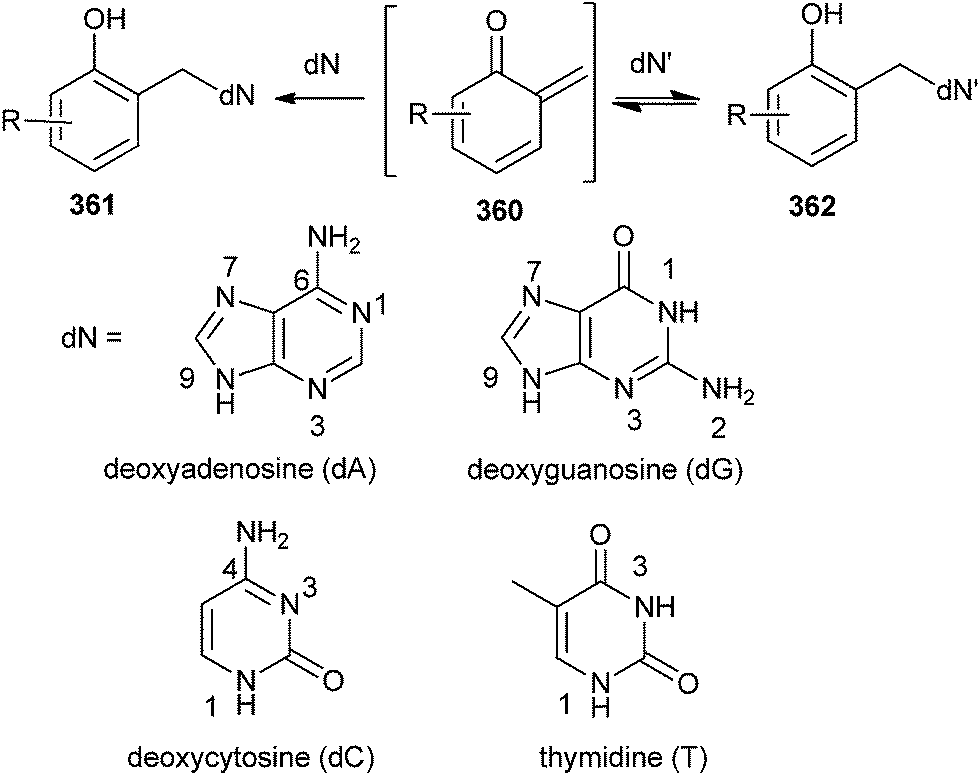 | ||
| Scheme 77 Generation of deoxynucleoside and o-QM adducts. | ||
Rokita and co-workers79 reported the formation of the reversible quenching adducts 366 under physiological conditions between o-QM 364 and deoxycytidine 363 by selective oxidation using bis[(trifluoroacetoxy)iodo]benzene (BTI), which induces a surprising rearrangement driven by over oxidation of a derivative lacking an alkyl substituent at the 4-position of the QM (Scheme 78).
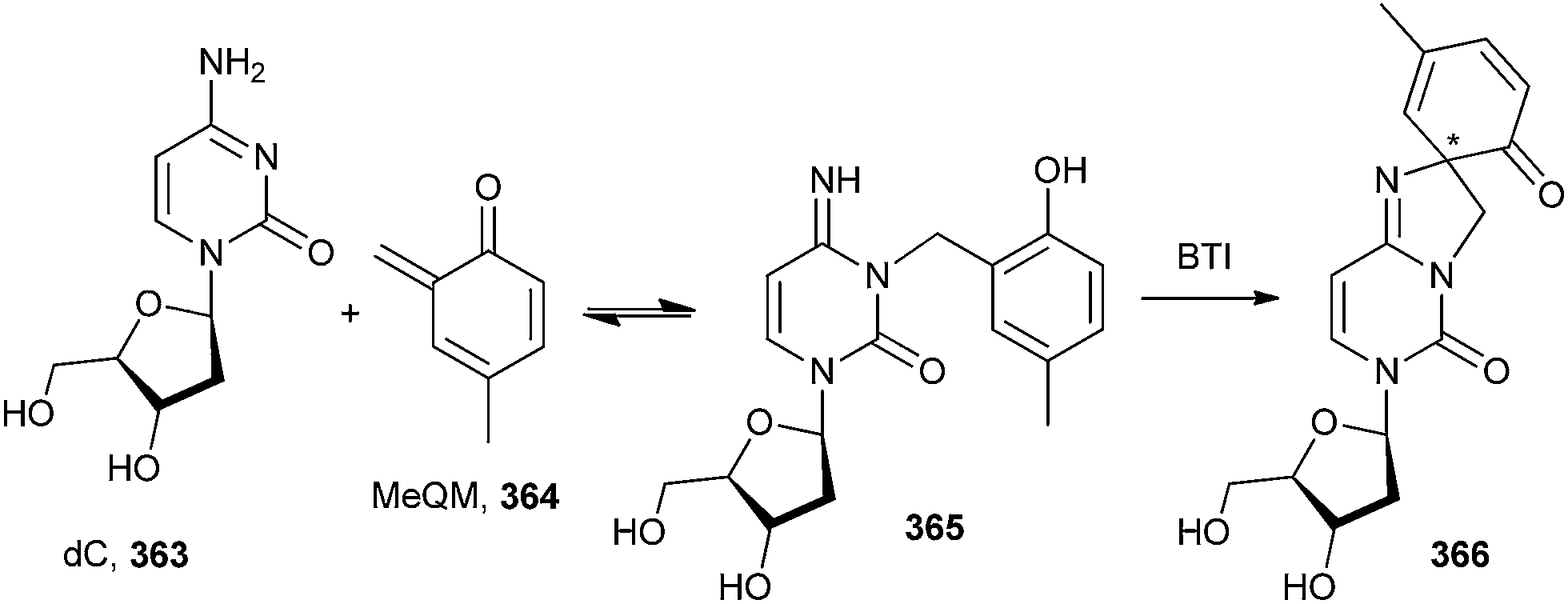 | ||
| Scheme 78 Reaction of o-QM with deoxycytidine 363. | ||
The rates of alkylation of 2′-deoxyguanosine 369 and guanosine 370 with o-quinone α-(p-anisyl)methide 368 (Scheme 79) were studied by flash photolysis in a series of aqueous sodium hydroxide solutions and bicarbonate ion, t-butylhydrogenphosphonate ion as well as in biphosphate ion buffers.80 Rate constants showed the reaction of o-quinone α-(p-anisyl)-methide 368 with guanosine and deoxyguanosine to be quite a fast process. Even it was considerably faster than the biologically wasteful reaction of the quinone methide with water, which is the ubiquitous medium in biological systems, and hence makes the quinone methide a potent guanosine and deoxyguanosine alkylator.
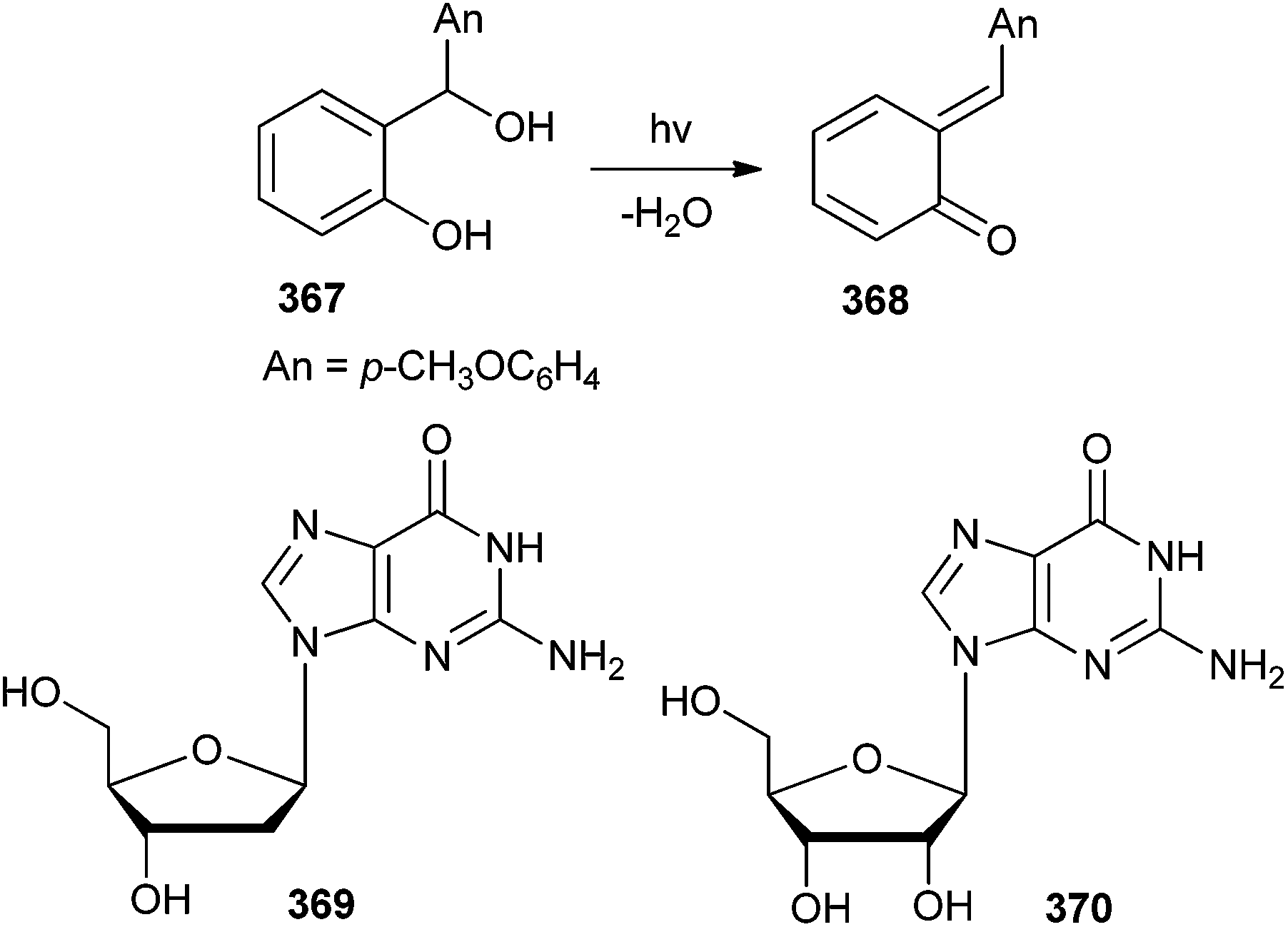 | ||
| Scheme 79 Alkylation of 2′-deoxyguanosine and guanosine on o-quinone α-(p-anisyl)methide. | ||
The selenide derivatives 371 can generate covalent interstrand cross-linking with its complementary strand through the formation of o-QM intermediate 373, induced by periodate oxidation.81 It has been evidenced by hydroxyl radical footprinting experiments that the quinone appendage specifically alkylated the cytosine base (nuc) extending the duplex formed between the conjugate and the target strand (Scheme 80).
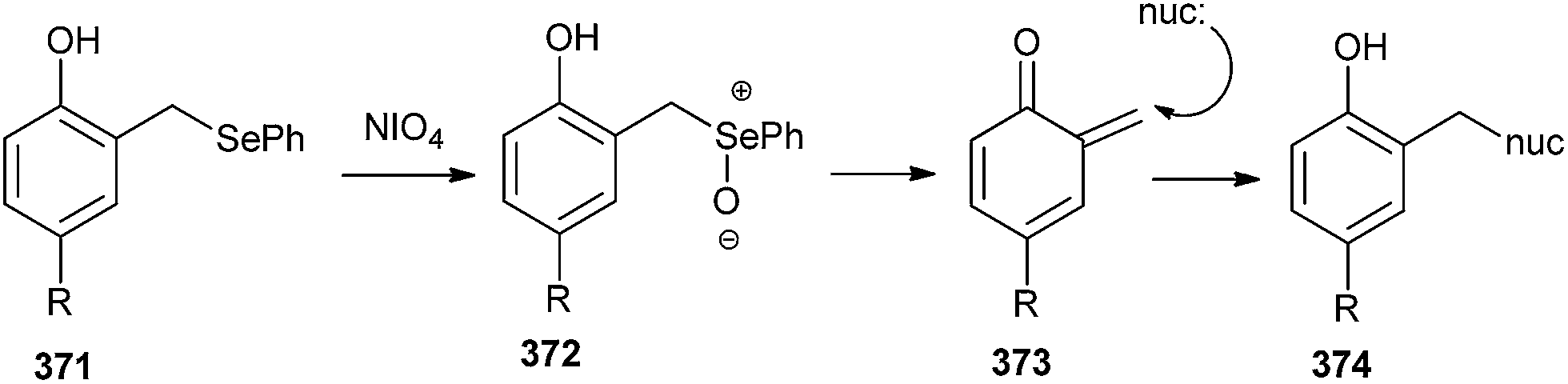 | ||
| Scheme 80 Alkylation of o-QM by using cytosine base (nuc). | ||
It is well known that cation on porphyrin can increase both selective binding affinity to nucleic acid or protein and solubility in aqueous conditions. Hence, with photoinducing, o-QM intermediate and singlet oxygen could be formed and they may play an important role for both alkylation and oxidative damage to nucleic acids or proteins. In all probability, porphyrin 377 was photochemically activated and induced to form both singlet oxygen and o-QM intermediate 378, which led to both oxidative damage and the alkylation of the organism in the bacterial system (Scheme 81).82
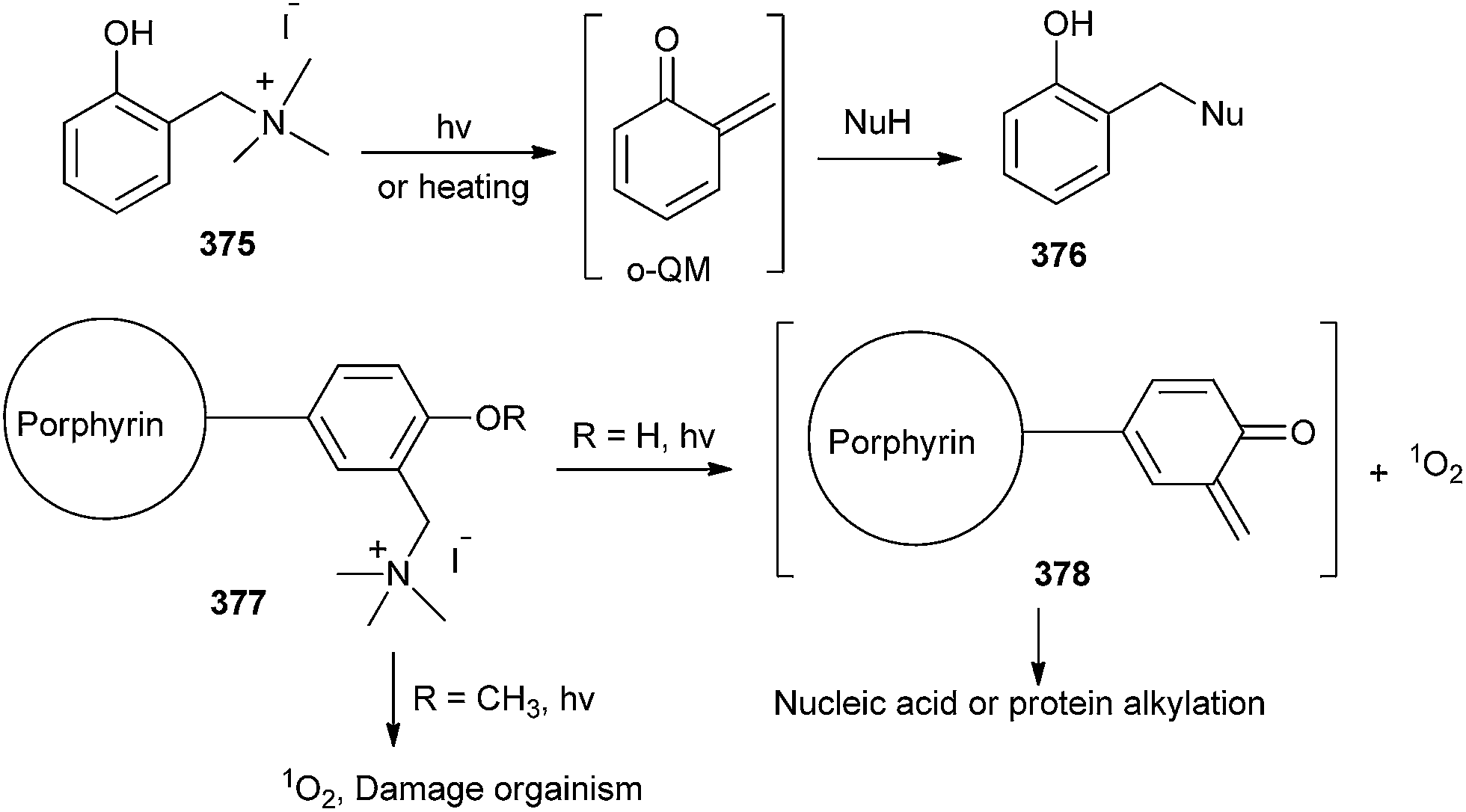 | ||
| Scheme 81 Binding of nucleic acid or protein on o-QM having porphyrin unit. | ||
Song and co-workers83 reported three water-soluble DNA cross-linking phenol quaternary ammonium derivatives, which could inhibit the transcription in vitro by photo-activation. The cross-linking agent 379 could be a significant reason for the late apoptosis of tumour cells. The twisted form and two quaternary ammonium groups could anchor DNA strands and facilitate the binding of DNA strands and the cross-linking between strands can then proceed through a bis(quinone methide) intermediate 380. The in situ formed o-QM has also been trapped by t-BuNH2 (Scheme 82).
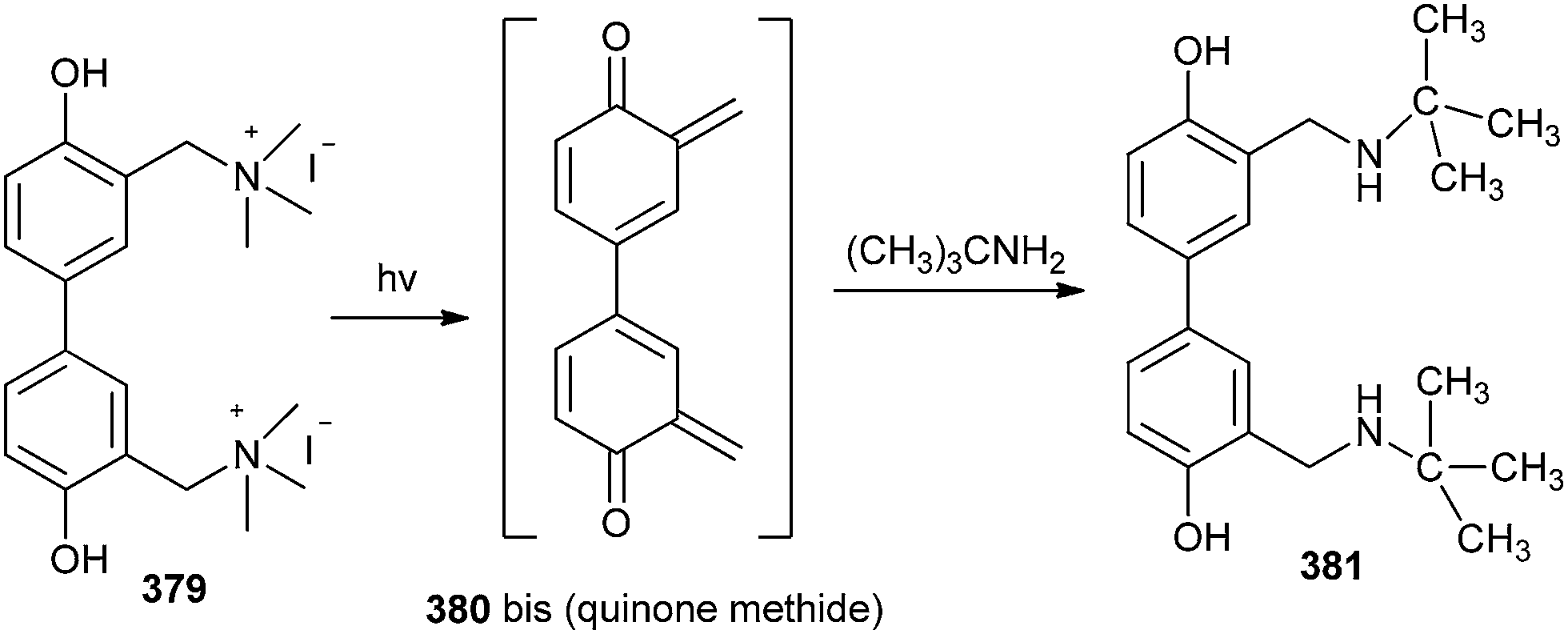 | ||
| Scheme 82 Trapping of bis(quinone methide) intermediate by t-BuNH2. | ||
Quinolinyl QM 387 was designed through the fluoride-induced QM generation by desilylation and elimination of acetate group from compound 386 (Scheme 83). The incorporation of the quinolinyl substitute was designed to enhance the potential interactions with DNA through possible partial intercalation and charges of the quinoline moiety and also for the investigation of the steric effect on the reactivity of QMs. Authors have reported that the N1-dG adduct 388 was selectively formed with quinolinyl QM as a favored dG adduct in 30% aqueous DMF and 10 mM phosphate buffer (pH 7.0).84
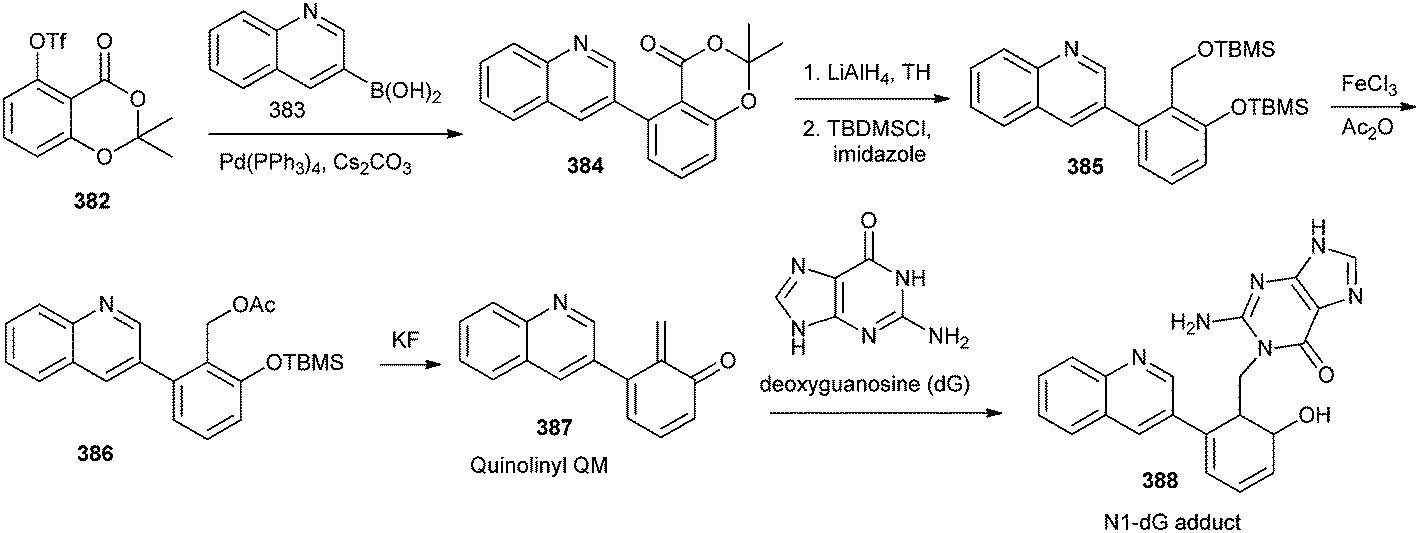 | ||
| Scheme 83 Synthesis of N1-dG adduct using quinolinyl QM and deoxyguanosine. | ||
A novel G-quadruplex (G-4), ligand/alkylating hybrid structures 391 tethering the naphthalene diimide moieties to quaternary ammonium salts of Mannich bases, as quinone-methide precursors 389, was activated by mild thermal digestion (40 °C).85 The bis-substituted naphthalene diimides were capably synthesized and their reactivity as activatable bis-alkylating agents was explored in the presence of thiols and amines in aqueous buffered solutions (Scheme 84). During alkylation, it has been found that the reversible process is a precondition to DNA alkylation, which in turn strengthens the G-quadruplex structural reorganization.
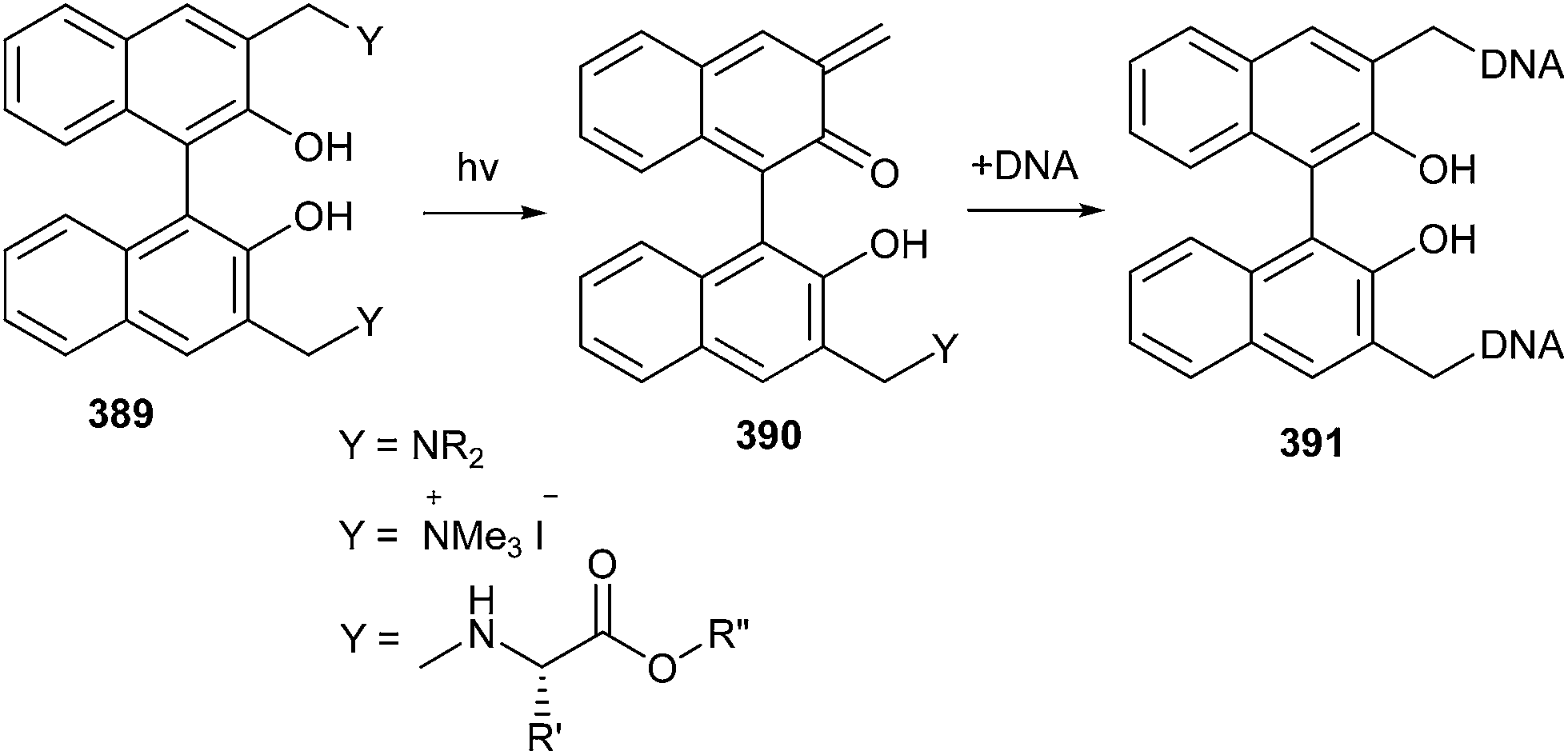 | ||
| Scheme 84 DNA alkylation o-QM precursors to generate G-quadruplex. | ||
Freccero and co-workers86 also reported a photoactivation of new BINOL-amino acid and BINOL-amino ester conjugates acquiesced alkylating and DNA cross-linking agents with high photoefficiency and superior cytotoxicity. While using nucleosides as nucleophiles, only the conjugate adducts with deoxycytidine at the N3 (dC-N3), deoxyadenosine at both N1 and NH2 (dA-N1 and dA-NH2), and deoxyguanosine at NH2 (dG-NH2) are stable in water solution. Conversely, the alkylation adducts at N7 of both dG and dA are thermodynamically unstable, because the reversal of the alkylation becomes a competitive process.
The alkylation reaction of 9-methyladenine and 9-methylguanine (as prototype substrates of deoxyadenosine and deoxyguanosine) with the parent o-QM have been studied by using DFT at the B3LYP/6-311+G(d,p) level.87 The guanine oxygen atom (O6) (ΔGq gas, 5.6 kcal mol−1) followed by the adenine N1 (ΔGq gas, 10.3 kcal mol−1) are more nucleophilic, whereas other centres have comparatively lower nucleophilicity. But water as a solvent diminishes the nucleophilicity of both 9-methyladenine N1 (DGq solv, 14.5 kcal mol−1) and 9-methylguanine O6 (ΔGq solv, 17.0 kcal mol−1), and as a consequence it reverts the purine base nucleophilicity. Regarding the product stability, calculations predict that only two adducts of o-QM with 9-methyladenine at NH2 and N1 positions, are in lower energy than reactants, in the gas and water phases. However, adduct at N1 can easily be dissociated in water. In particular, the oxygen alkylation adduct becomes slightly unstable in water (¢Gsolv, +1.4 kcal mol−1), and the N7 alkylation product remains only moderately more stable than free reactants (¢Gsolv, −2.8 kcal mol−1).
Pettus et al.88 have reported the total synthesis of all of the known chiral Cleroindicins from the known substituted benzaldehyde 392, which has been treated with [(2-(trimethyl-silyl)ethoxy)methyl]lithium 393 resulted in Boc migration, and afforded an intermediate phenoxide. This phenoxide generated the o-QM by in situ elimination, which then underwent succeeding in situ 1,4-reduction with sodium borohydride to produce the phenol 394 in a 68% yield. Mitsunobu coupling of compound 394 with amide 395 afforded the inverted amide 396 in an 85% yield and >99% ee (Scheme 85).
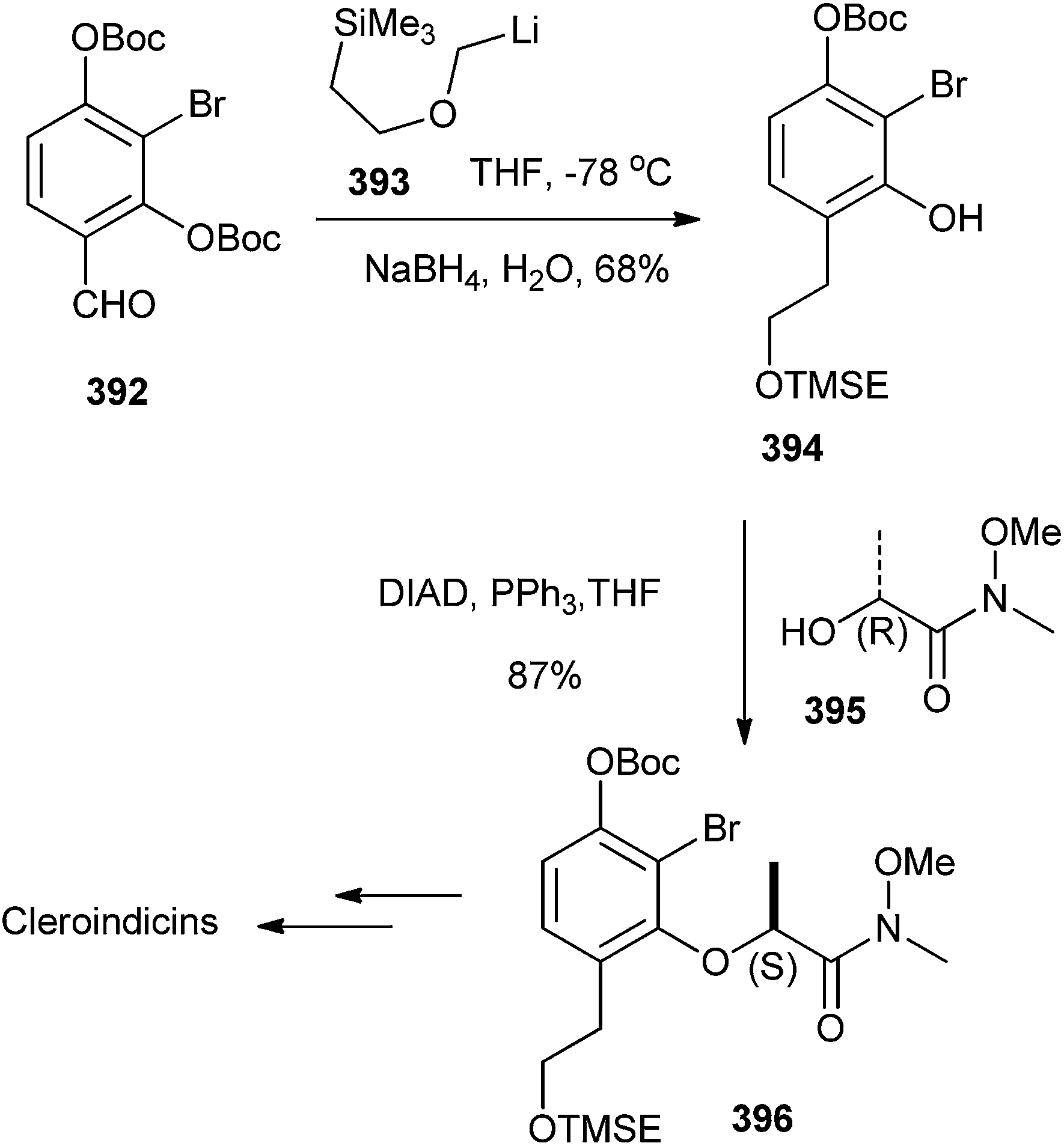 | ||
| Scheme 85 Synthesis of chiral Cleroindicins from the substituted benzaldehyde 392. | ||
3.4. Coupling reactions
The coupling of o-QM has been done by redox reactions of vitamin E in 1,2-dichloroethane with oxidants in water/1,2-dichloroethane interface.89 Here, the oxidation reaction of α-tocopherol (TOH) with MnO4− in DCE has been explained by a series of reactions (eqn (1)–(3)). After the formation of α-TD in hexane and o-QM of α-TOH produced during the oxidation of α-TOH dimerizes easily to α-TD, as shown in Scheme 86.| 3α-TOHDCE + 2MnO4DCE− + 4H2ODCE → 2MnO2DCE + 3α-TODCE + 5OHDCE− | (1) |
| α-TODCE+ + OHDCE− → α-QMDCE + H2ODCE | (2) |
| 2α-QMDCE → α-TDDCE | (3) |
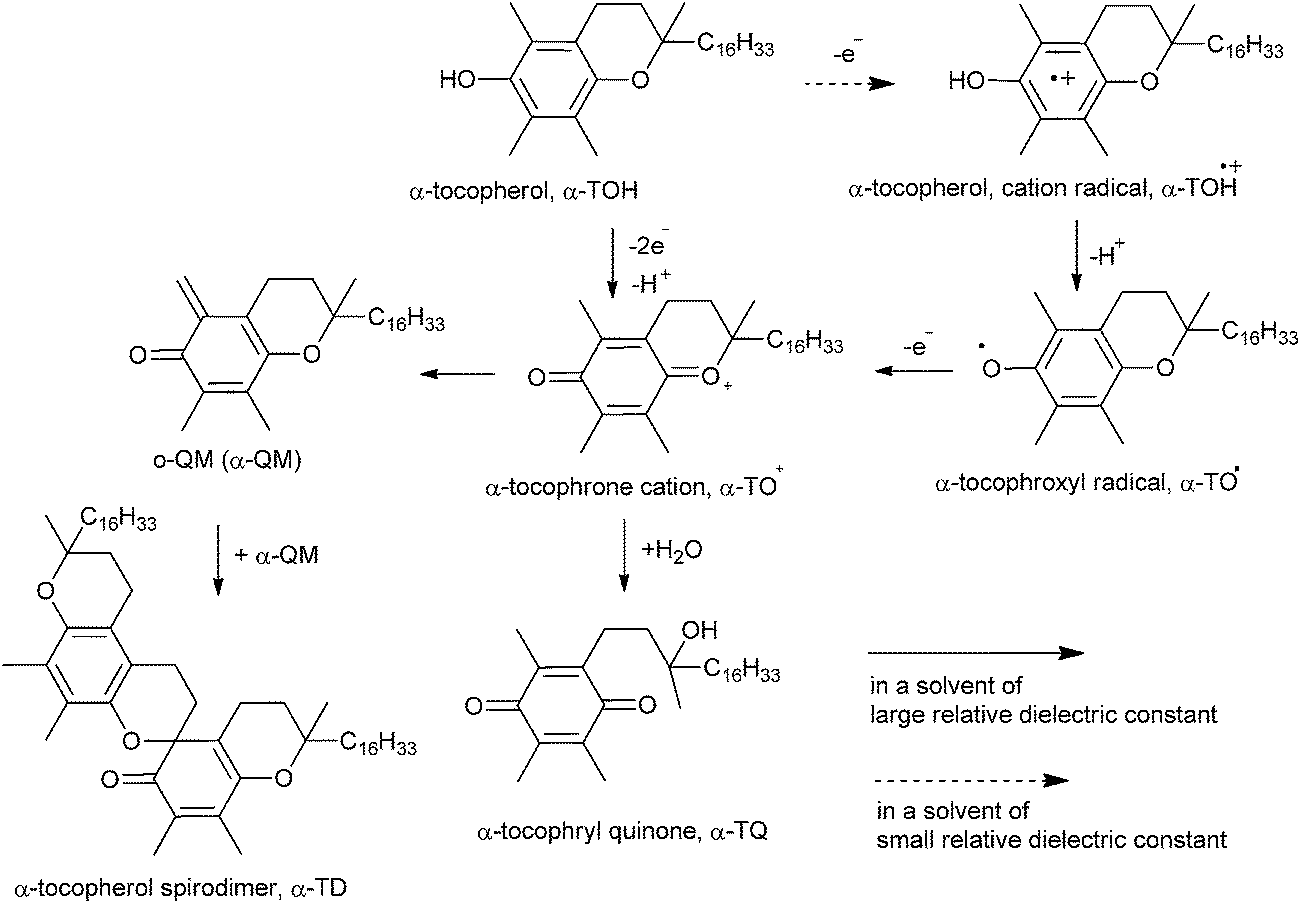 | ||
| Scheme 86 Formation and dimerization of α-TD. | ||
3.5. Complex formation with metals
Korthals and Wulff90 reported that the reactions of Chromium carbene complexes of type 398 with enynyl propargyl ethers 397 in the presence of a base would generate hexahydrodibenzopyrans 399 after an oxidative workup. The involvement of a [4 + 2] cycloaddition of a Cr complexed o-QM intermediate was inferred from the observed high asymmetric induction (Scheme 87).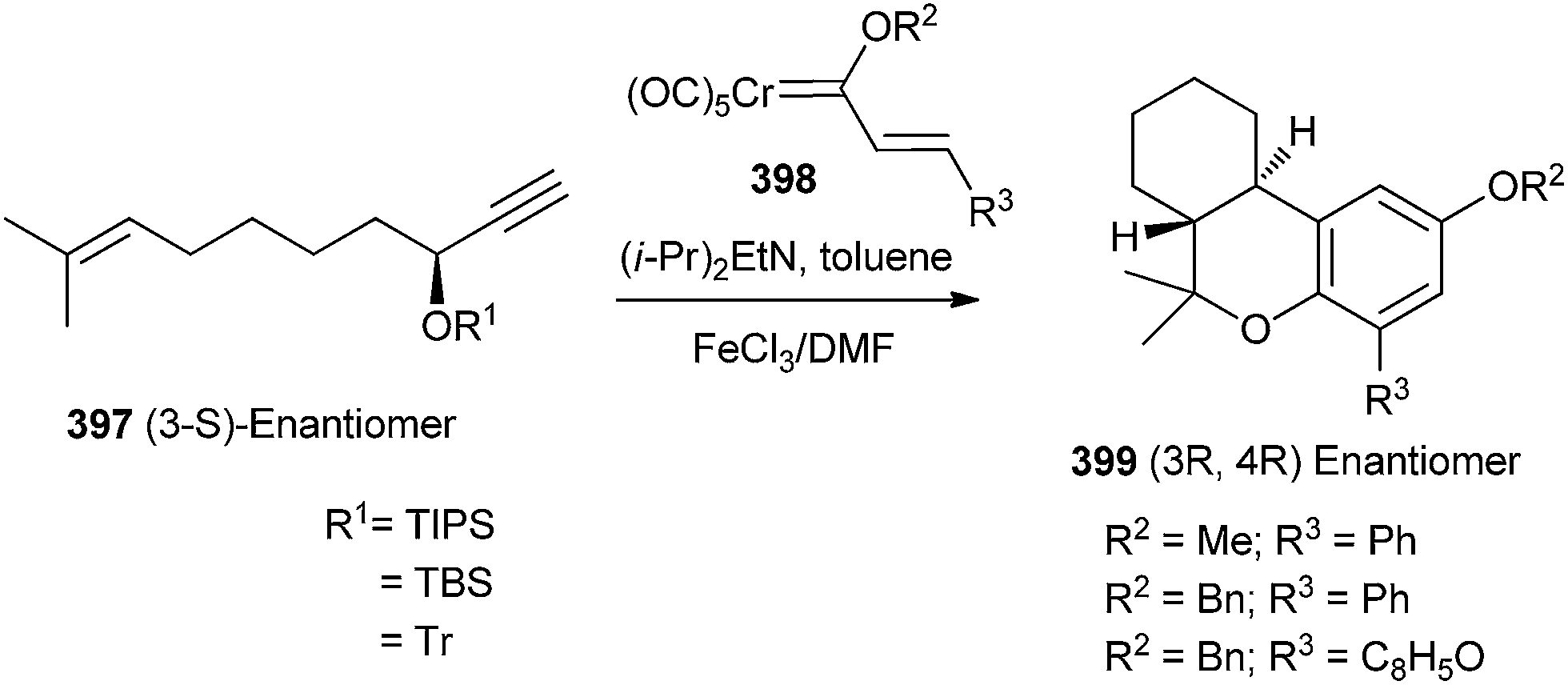 | ||
| Scheme 87 [4 + 2] Cycloaddition of a Cr complexed o-QM with enynyl propargyl ethers. | ||
Compound 401 was prepared through the Diels–Alder reaction of the o-QM intermediate 406. The methyl group at the C-9 position of o-QM 406 puts into effect the formation of a single diastereomer from the Diels–Alder reaction possessing a chair transition state in an exo-cycloaddition where the CH3 is in an equatorial position. Such a chair transition state is possible in 406, where the cycloaddition occurs anti to the chromium. But in the case of diastereomer 407, the Cr is on the opposite face of the o-QM, where the chair transition state for the exo-cycloaddition anti to the metal has an axial methyl. The (3S,5S)-enyne 403 is an incompatible case and gives a mixture of diastereomers, the two most predominate of which are formed via an exo cycloaddition with a chair transition state with an axial methyl 407 and an endo-cycloaddition with a chair transition state with an equatorial methyl 408 (Scheme 88).
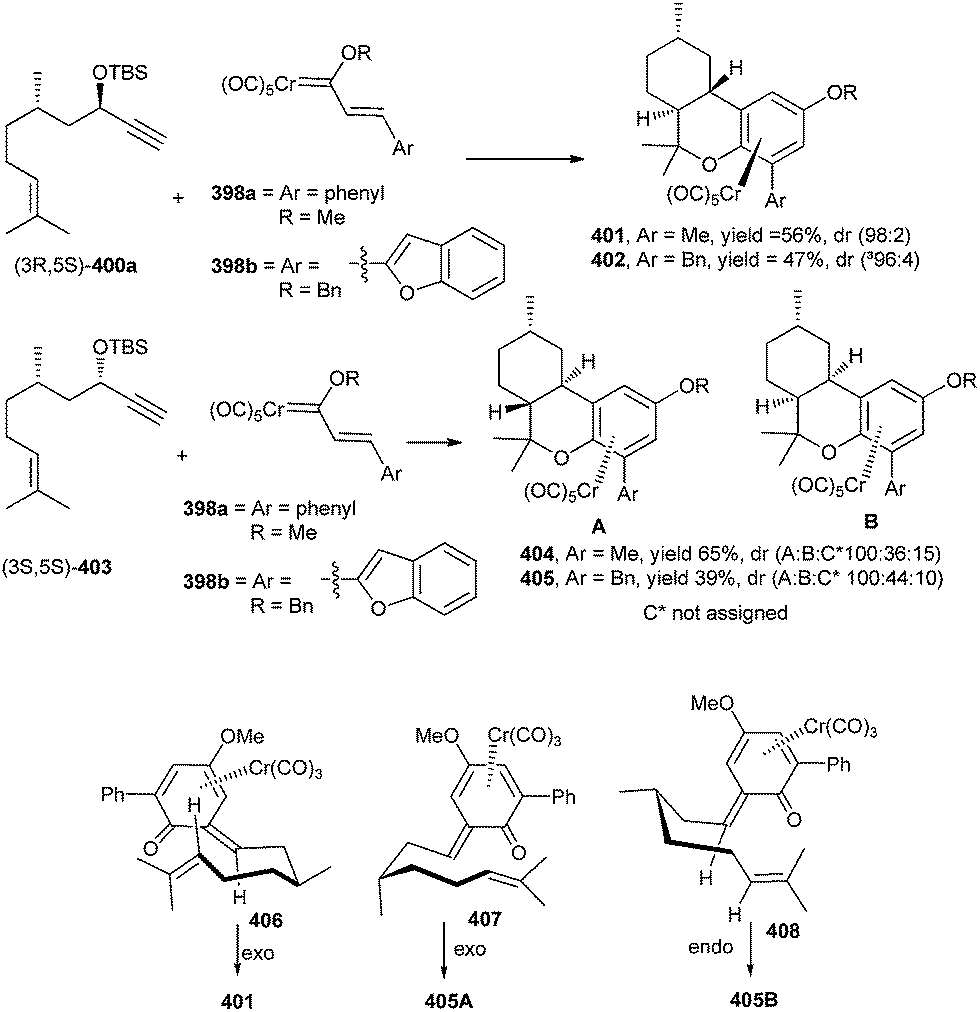 | ||
| Scheme 88 Diastereoselactive synthesis of hexahydrodibenzopyrans via a chair transition state of a Cr complexed o-QM. | ||
Wulff and co-workers91 also disclosed the synthesis of allocolchicinoids that involves the benzannulation reaction of the Fischer chromium carbene complexes and alkynes with high to moderate diastereoselectivity. One of the diastereomer 409 was recemized, but not the other 413. The intermediacy of an o-quinone methide chromium tricarbonyl complexes 411 & 412 and the intramolecular hydrogen bonding assist the reversible dissociation of methanol in compound (aS, 7S) 409 to give (aR, 7R) 410. Due to the intramolecular H-bonding, it only becomes possible in diastereomer 409, whereas the opposite diastereomer 413 would not participate, and thus retains its enantiomeric purity (Scheme 89).
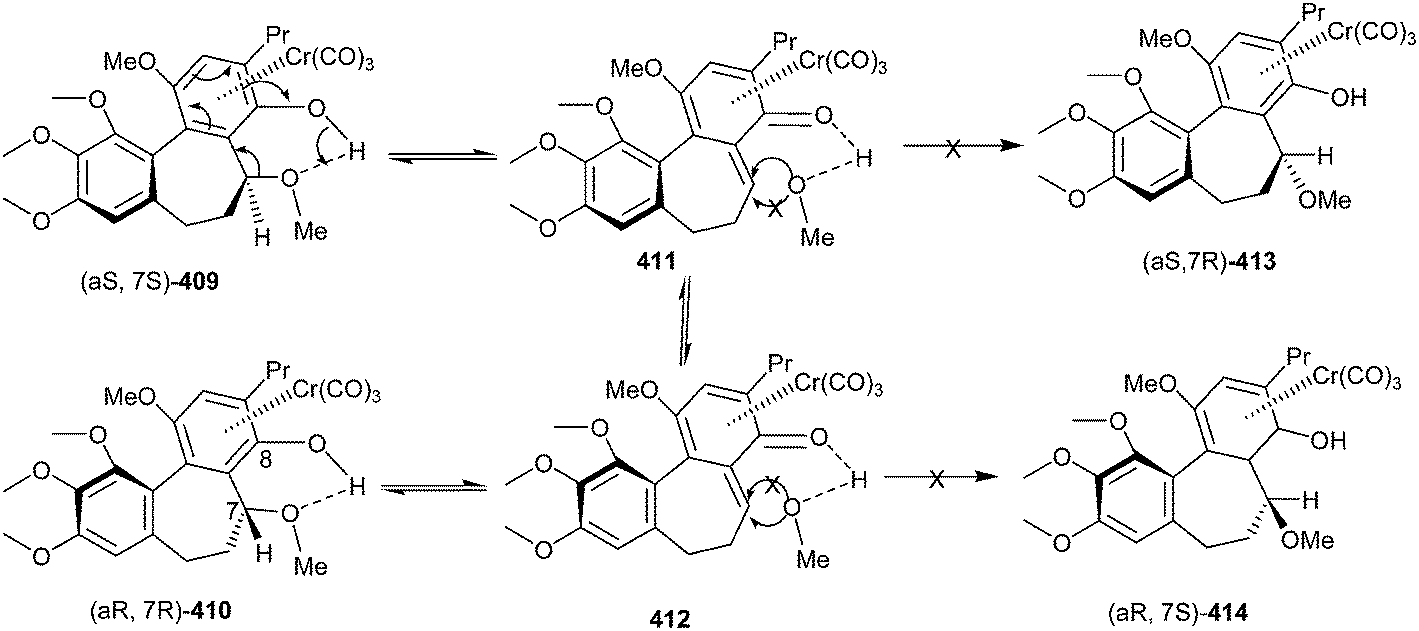 | ||
| Scheme 89 Synthesis of allocolchicinoids involving Fischer chromium carbene complex and alkynes. | ||
QMs could be stabilized by the steric bulk or by co-ordination to electron-rich transition metal complexes. The mechanism of an enantioselective palladium-catalyzed alkene difunctionalization reaction gives evidence of rapid ligand exchange between palladium and copper, as well as a correlation between ligand electronic nature and enantioselectivity.92 A substrate having an alkene with an adjacent ortho-phenol and a linked nucleophile 415 was used. ortho-Phenol not only coordinates the Pd(II) intermediate 416, preventing hydride elimination, but it also allows for the in situ formation of an electrophilic quinone methide 418, necessary for the second functionalization (Scheme 90). So this approach contrasts with others in the oxidation of the substrate, because the Pd centre does not promote the addition of the second nucleophile.
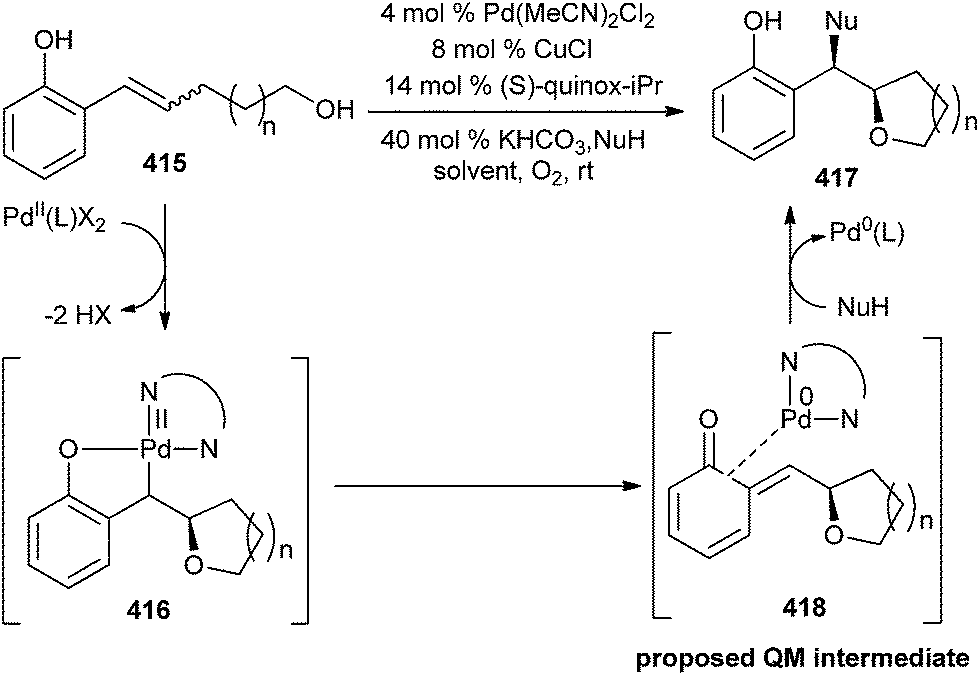 | ||
| Scheme 90 Enantioselective palladium-catalyzed difunctionalization of alkene. | ||
Pd(II) catalyzes the aerobic reductive coupling reactions of organometallic reagents and styrenes through o-QM intermediates.93 Compound A initially oxidizes the alcoholic solvent to generate the Pd-hydride intermediate B, which coordinates with alkene 419 to give cationic complex C. The alkene inserts into the Pd-hydride yielding D and D′. As the consequence of formation of the benzylic addition product only, it has been emphasized that intermediate D′ proceeds to product via formation of o-QM E, with concomitant reduction of Pd(II) to Pd(0) and generation of 420. Subsequently, ethanol reacts with the o-QM E to liberate the ether product 420 and the Pd(0) F, which has been reoxidized by CuCl2 or O2 (Scheme 91).
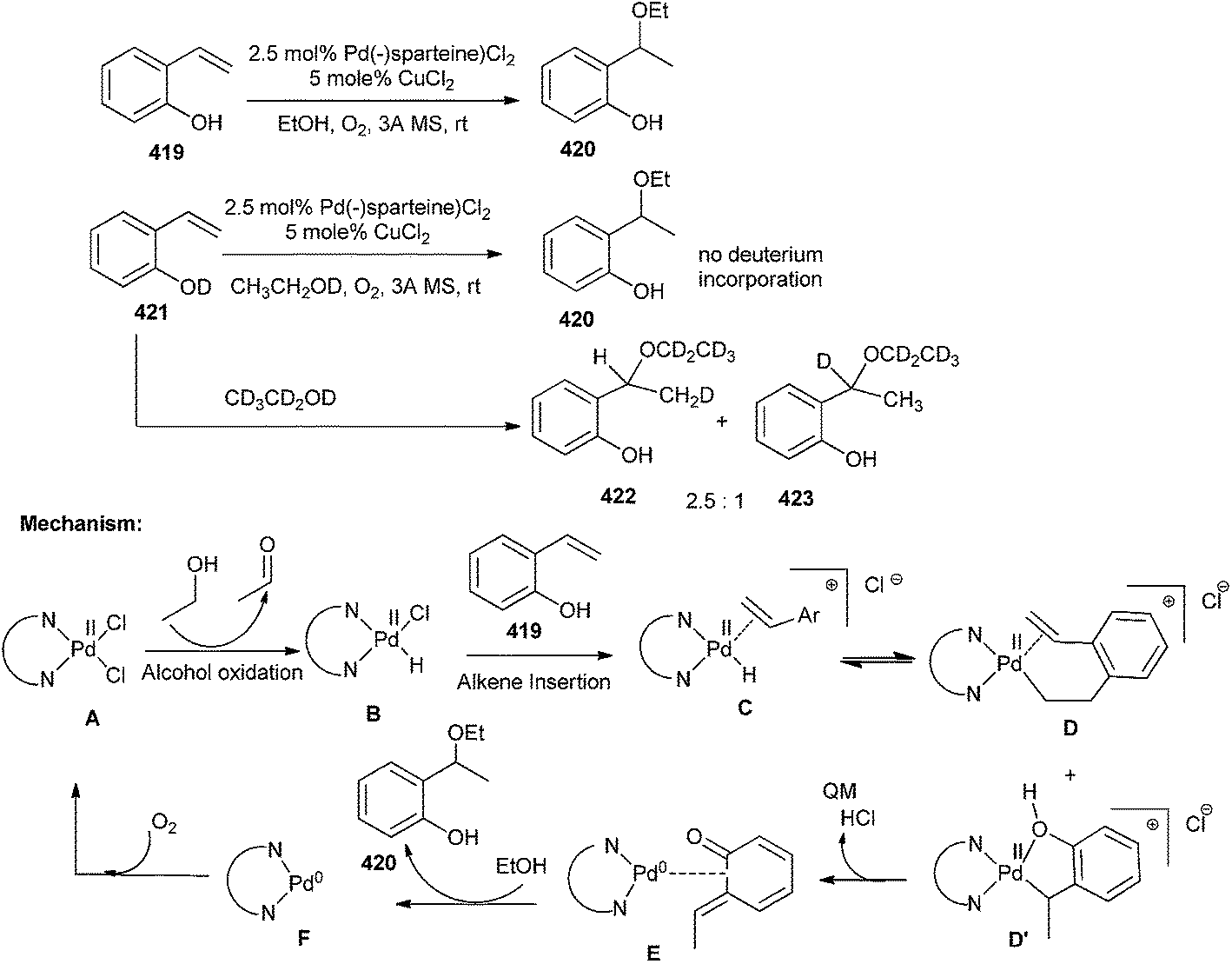 | ||
| Scheme 91 Pd(II) catalyzed reductive coupling of organometallic reagents and styrenes via o-QM intermediate. | ||
Phenols when bound to pentaammine osmium(II) 424, readily react with aldehydes 425 to generate the complexes of o-QMs 427. While these complexes are remarkably stable, when exposed to air or dissolved in water, treatment with CAN oxidizes the metal and releases the intact QM ligand, which can be trapped by a suitable dienophile 428 to give the compound 429 (Scheme 92).94
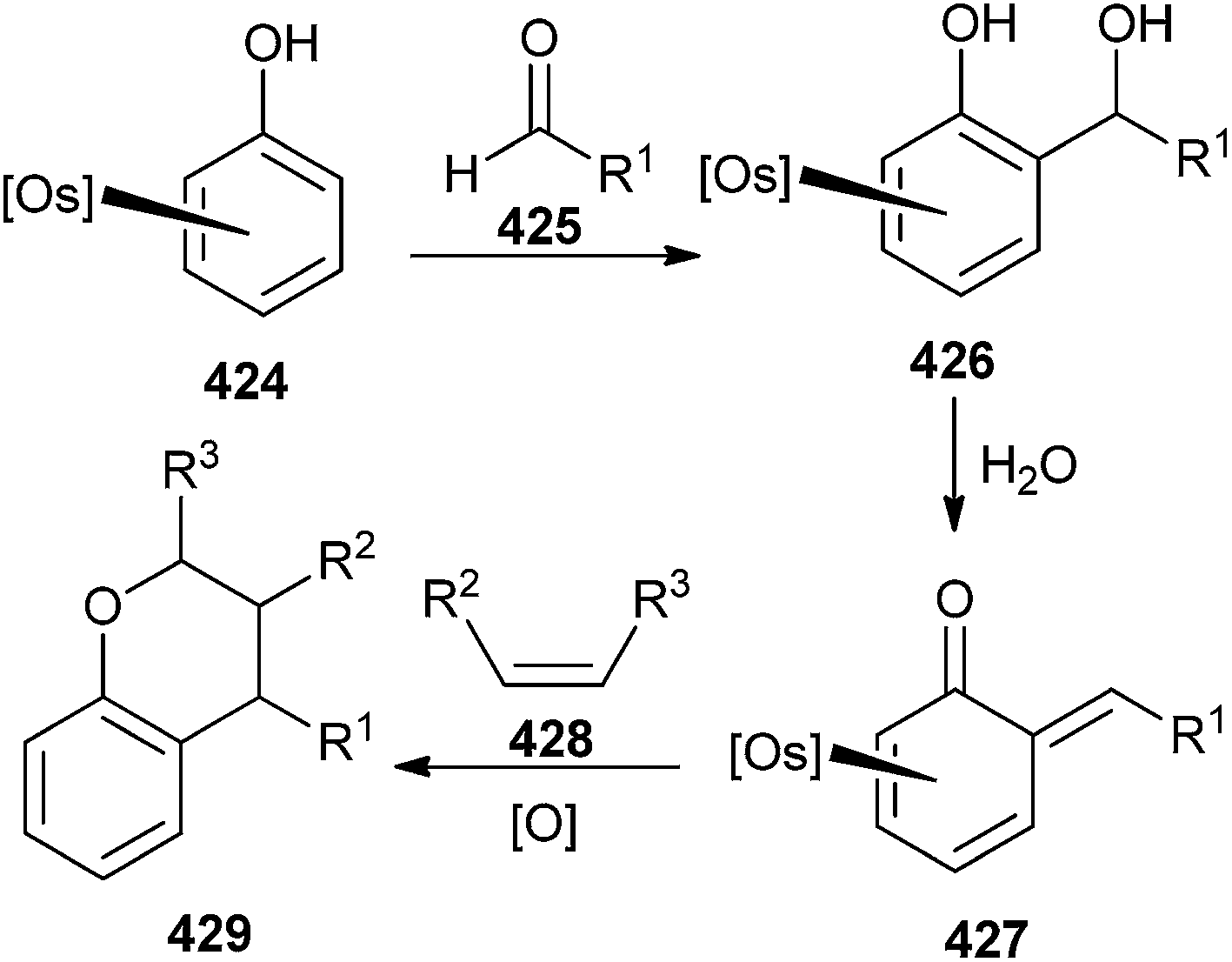 | ||
| Scheme 92 Trapping of Os-complexed o-QM by different alkenes. | ||
Whenever the phenol complex was treated with acetaldehyde, it gave the o-QM 430 with small amounts of acetaldehyde complex [Os(NH3)5(acetaldehyde)]2+. The reaction of complex 430 with an excess of 3,4-dihydropyran and 1.5 equiv of CAN in acetonitrile provided tetrahydropyranochromene 431 (Scheme 93).
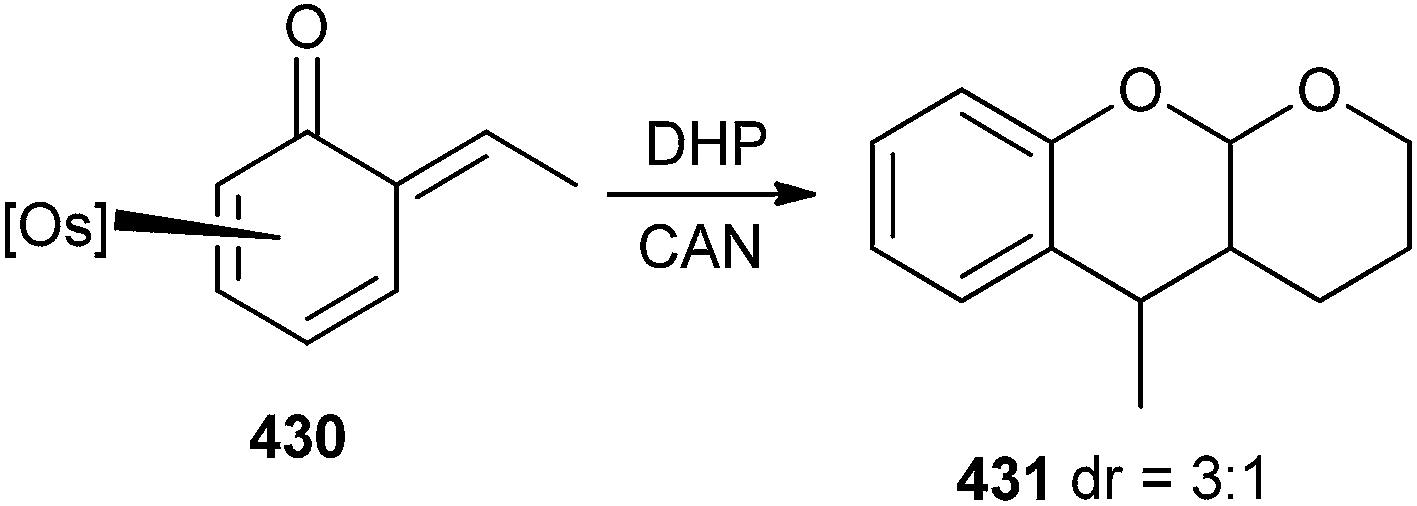 | ||
| Scheme 93 Reaction of Os-complexed o-QM with 3,4-dihydropyran. | ||
The intramolecular Diels–Alder reaction was accomplished by using the o-QM 432, which was formed by treatment of the phenol complex 424 with (R)-citronellal and pyridine. Compound 432, after treatment with CAN, leads to the demetallation and cycloaddition to form the benzo[c]chromene 433 in 52% yield as a single diastereomer (Scheme 94).
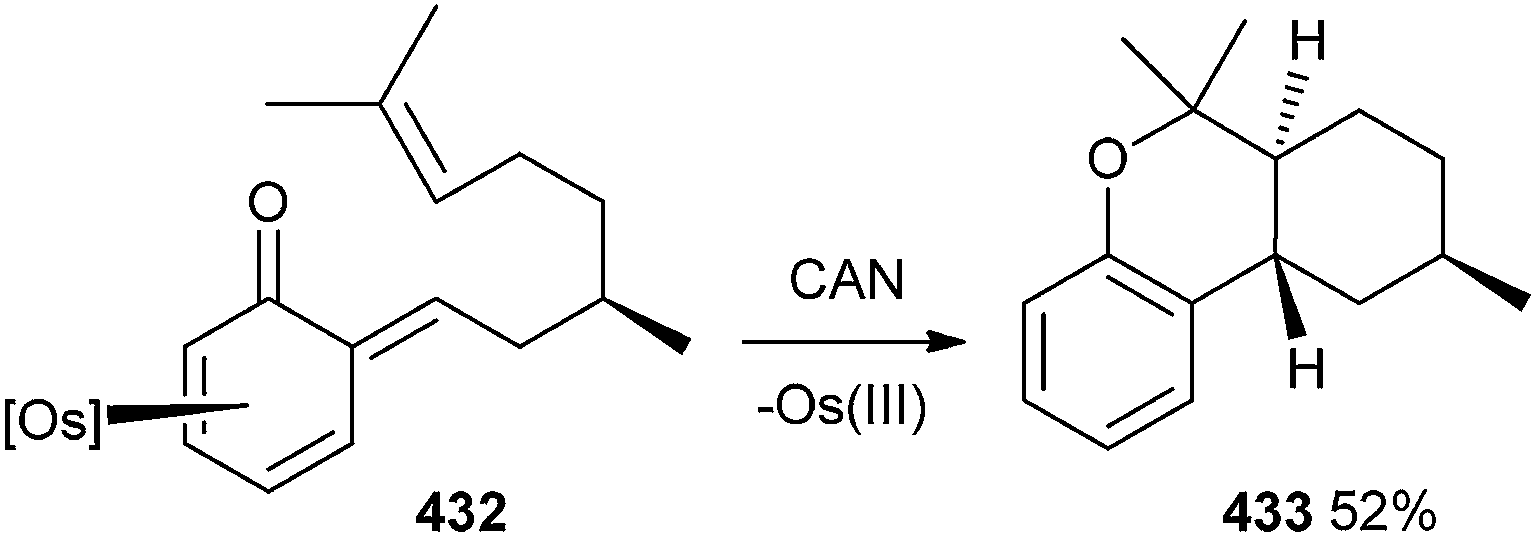 | ||
| Scheme 94 Diastereoselective synthesis of benzo[c]chromene. | ||
Cycloaddition reactions of activated olefins and styrenes 435 with o-quinine methides 436 in the presence of PtCl4 and AuCl3 gave a wide range of 2-alkyl or 2-aryl chromans 437. Good diastereoselectivity (up to >99:1) of the chromans was obtained (Scheme 95).95
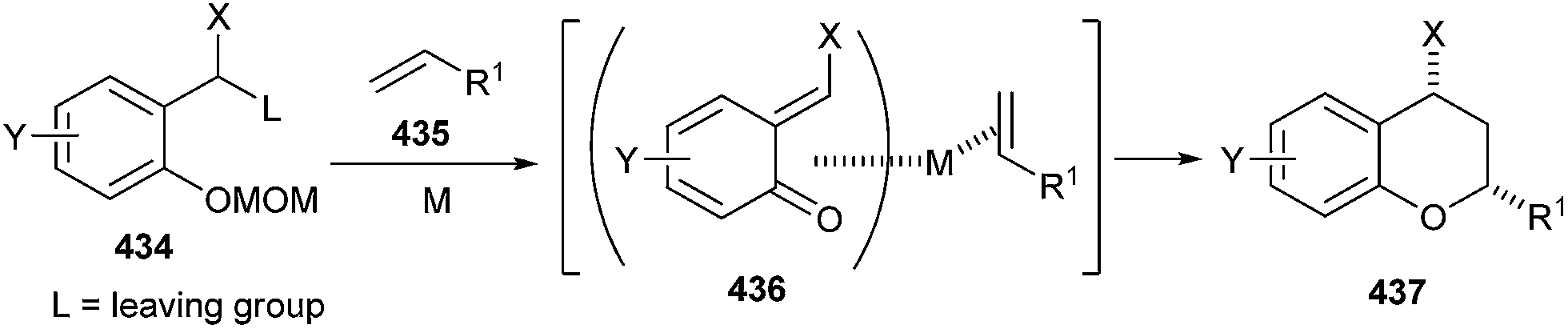 | ||
| Scheme 95 Cycloaddition reactions of activated olefins with o-QM. | ||
The reactivity96 of an o-QM can be understood from MIII resonance form in which the carbon can be viewed as bearing a negative charge (Scheme 96). It has been emphasized that the stabilisation of o-QM could be correlated to the degree of deviation of the carbonyl/alkene segment of the o-QM from planarity with the coordinated diene segment, which is a consequence of metal back-bonding into the aromatic system.
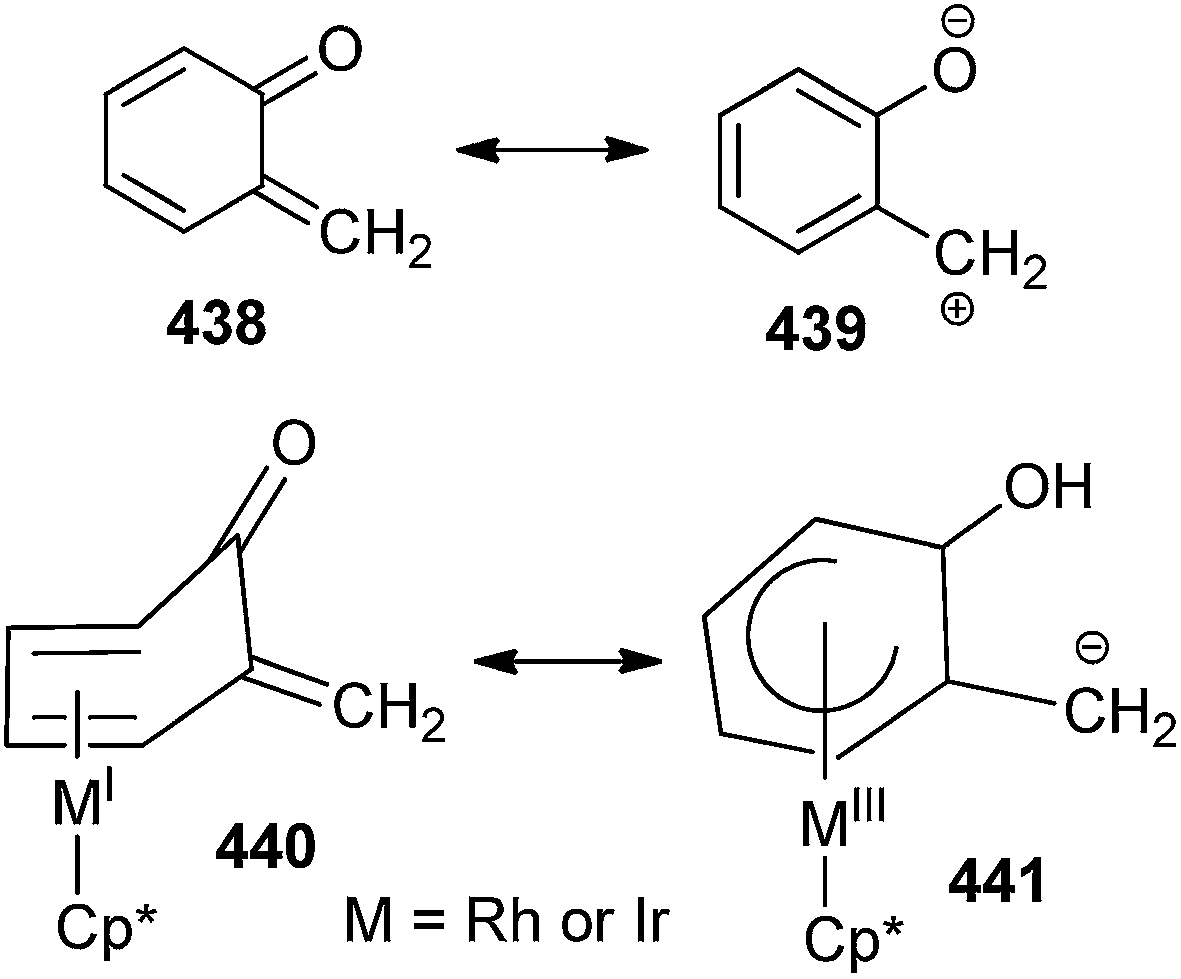 | ||
| Scheme 96 Resonating forms of metal o-QM complex. | ||
Metal stabilised o-QM showed a complete reverse effect; the methylene carbon became nucleophilic upon transition metal coordination. For example, in Cp*Ir(o-QM) 442 the exocyclic carbon undergoes nucleophilic substitution reactions, e.g. with iodine or methyl propiolate, and [3 + 2] dipolar cycloadditions, as in the case of the electron-poor dienophile N-methylmaleimide 443 (Scheme 97). The partial charge and hinge angle follow the order Ru < Co < Rh < Ir. The Ru and Co complexes predicted high reactivity. Mono substituted nucleophilic metal complexes were isolated to establish the importance to the application of this chemistry for the stereoselective synthesis.
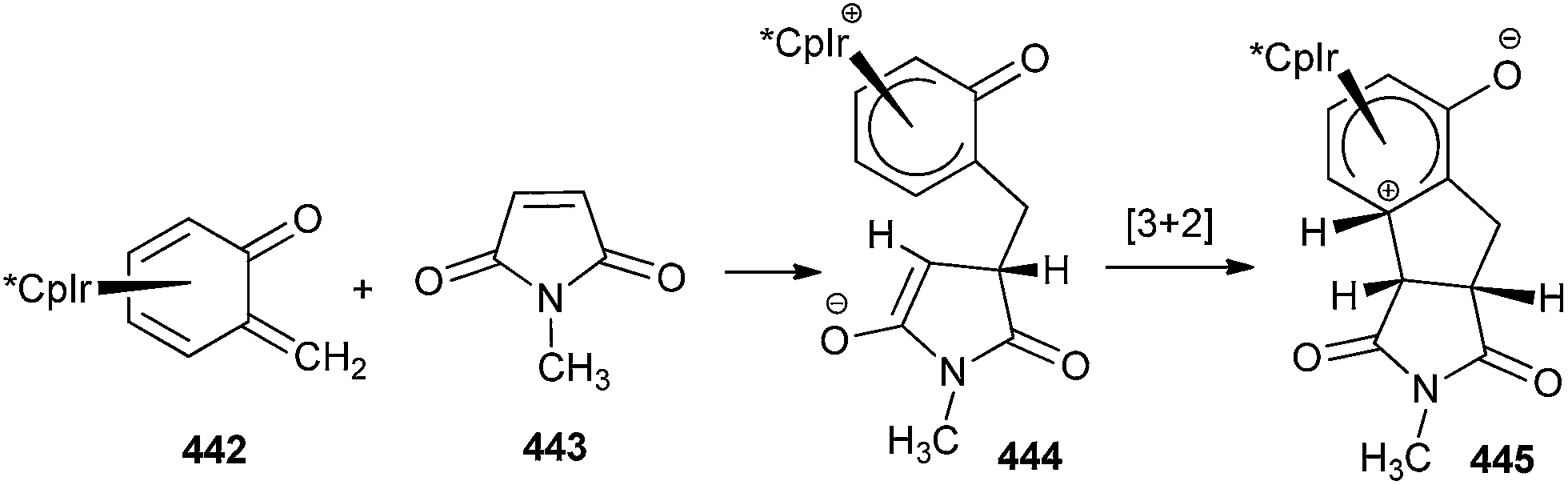 | ||
| Scheme 97 Cycloaddition reaction of Cp*Ir(o-QM) with N-methylmaleimide. | ||
Knoevenagel reaction of 2-hydroxy-1,4-naphthoquinone, 4-hydroxycoumarin, and 4-hydroxy-6-methylcoumarin with paraformaldehyde generated respective quinone methides 447, 448 and 449. They applied these in situ generated intermediate in the hetero-Diels–Alder reaction with β-vinyl-meso tetraphenylporphyrinatozinc(II) 446, leading to macrocycles containing 5,10-dioxobenzo[g]chromene, 5,6-dioxobenzo[h]-chromene 450, pyrano[3,2-c]coumarin 451, and benzopyran 452 motifs at the β-position (Scheme 98).97
 | ||
| Scheme 98 Hetero-Diels–Alder reaction of o-OMs with β-vinyl-meso tetraphenylporphyrinatozinc(II). | ||
3.6. Rearrangement reactions
The chemical transformation of fluorogenic BCC triggered by thiols proceeds through a tandem benzoquinone reduction and QM type rearrangement reactions, which are spontaneous and irreversible at physiological temperatures in aqueous media.98 In general, the intracellular reduction of the quinone moiety 453 produces the corresponding hydroquinone 454 that instinctively releases the deactivated drug and gives compound 455 via a QM-type rearrangement (Scheme 99). | ||
| Scheme 99 Generation of deactivated drug via QM-type rearrangement. | ||
QM-type rearrangement reaction sequence has been employed as part of the fluorescence releasing mechanism for latent fluorophores or the drugs release mechanism of the prodrugs, as shown in Scheme 100.
 | ||
| Scheme 100 Drugs release mechanism of the prodrugs. | ||
Silva and Bozzelli99 have reported that o-QM, a 6-methylene-2,4-cyclohexadiene-1-one is an important intermediate in lignin and alkyl benzene combustion, and so the thermal decomposition of o-QM is pertinent to the combustion of transportation fuels (containing toluene) and of biomass and low-rank coals (containing lignin). A potential pathway leading to o-QM 463 formation has been demonstrated by the reaction of the benzyl radical 464 with O2, which would also be important in toluene combustion (Scheme 101).
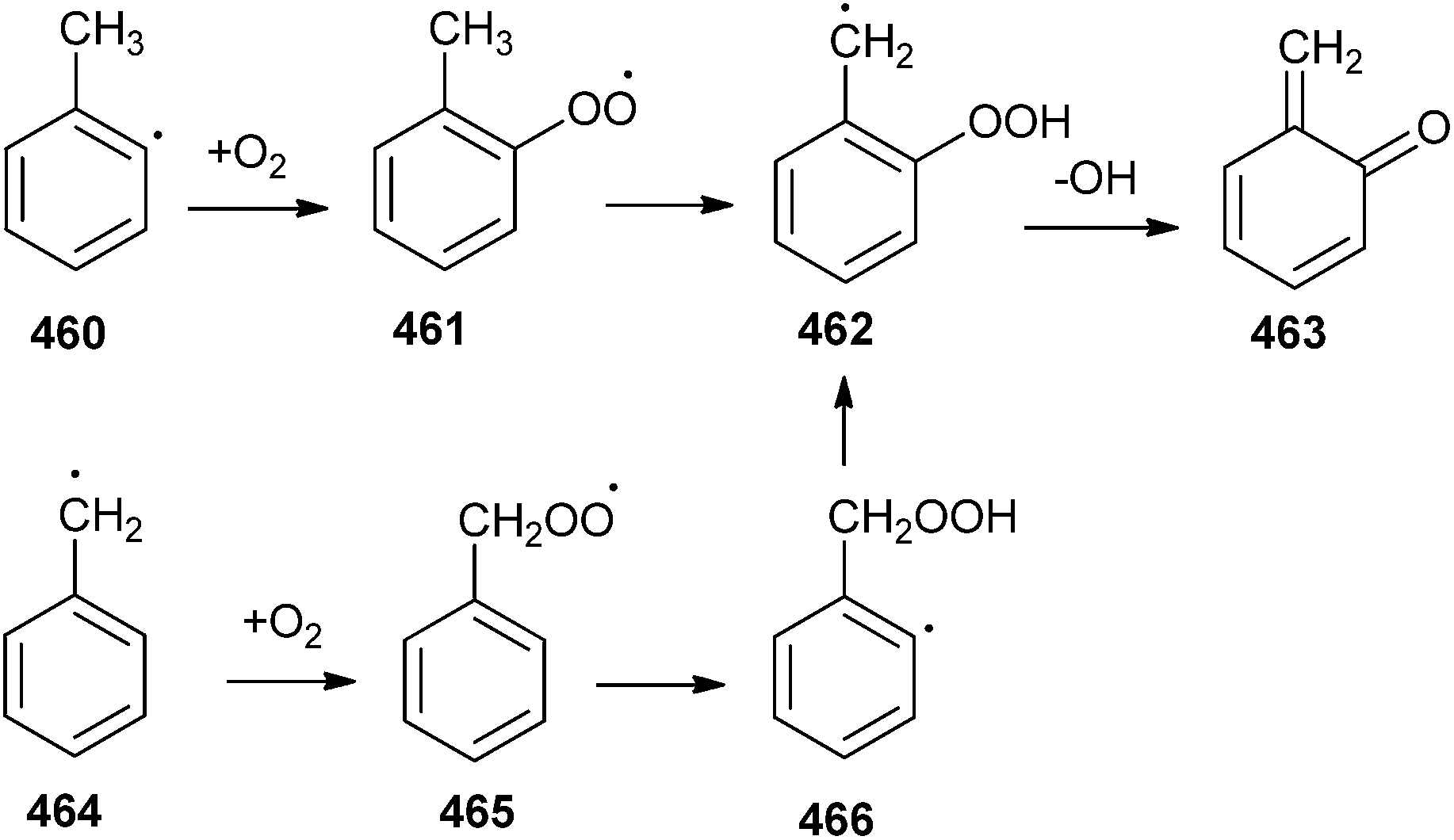 | ||
| Scheme 101 Combustion of toluene. | ||
The formation of tropone via a hydroxyphenylcarbene intermediate 467 from o-QM undergoes an intramolecular hydrogen shift to produce 2-hydroxphenylycarbene 467 in a concerted process (Scheme 102).
 | ||
| Scheme 102 Synthesis of tropone from o-QM. | ||
Eventually, the mechanism for fulvene (+CO) formation has been considered. The ring opened product 470 cyclises to the bicyclic intermediate bicyclo[3.2.0]hepta-3,5-dien-1-one 471, which rearranges to yield spiro[2.4]hepta-4,6-dien-1-one (472). This intermediate subsequently dissociates to fulvene 473 and CO (Scheme 103).
 | ||
| Scheme 103 Synthesis of fulvene. | ||
It has been assumed100 that the structure obtained after proton transfer is a resonance hybrid of two possible forms, zwitterionic (ortho-quinoid, 474B) and keto enes (ortho-quinoid, 474C), as shown in Scheme 104. The wave function of the real structure is a linear combination of the wave functions of two resonance forms. The results demonstrate that not only resonance enhances the strength of hydrogen bonds, but also that the formation of intramolecular hydrogen bonds leads to more efficient resonance. The value of this stabilization can be estimated as a difference in energy between the proton transfer state and the open one, which is about 5 kcal mol−1 in N-methyl-2-hydroxybenzylideneamine.
 | ||
| Scheme 104 Resonating structures of zwitterions (ortho-quinoid, and keto enes). | ||
Reaction of guaiazulene101 475 with 2-methoxybenzaldehyde 476 in methanol in the presence of hexafluorophosphoric acid at 25 °C gives (3-guaiazulenyl)(2-methoxyphenyl)methylium hexafluorophosphate 477 with 93% yield. Similarly, reaction of 475 with 3-methoxybenzaldehyde under the same reaction conditions affords (3-guaiazulenyl)(3-methoxyphenyl)methylium hexafluorophosphate 478 (91% yield) or with 4-methoxybenzaldehyde gives (3-guaiazulenyl)(4-methoxyphenyl)methylium hexafluorophosphate 479 (97% yield). The crystal structures of these monocarbenium-ion compounds, possessing interesting resonance forms, are stabilized by the 3-guaiazulenyl and anisyl (2-, 3-, or 4-methoxyphenyl) groups (Scheme 105).
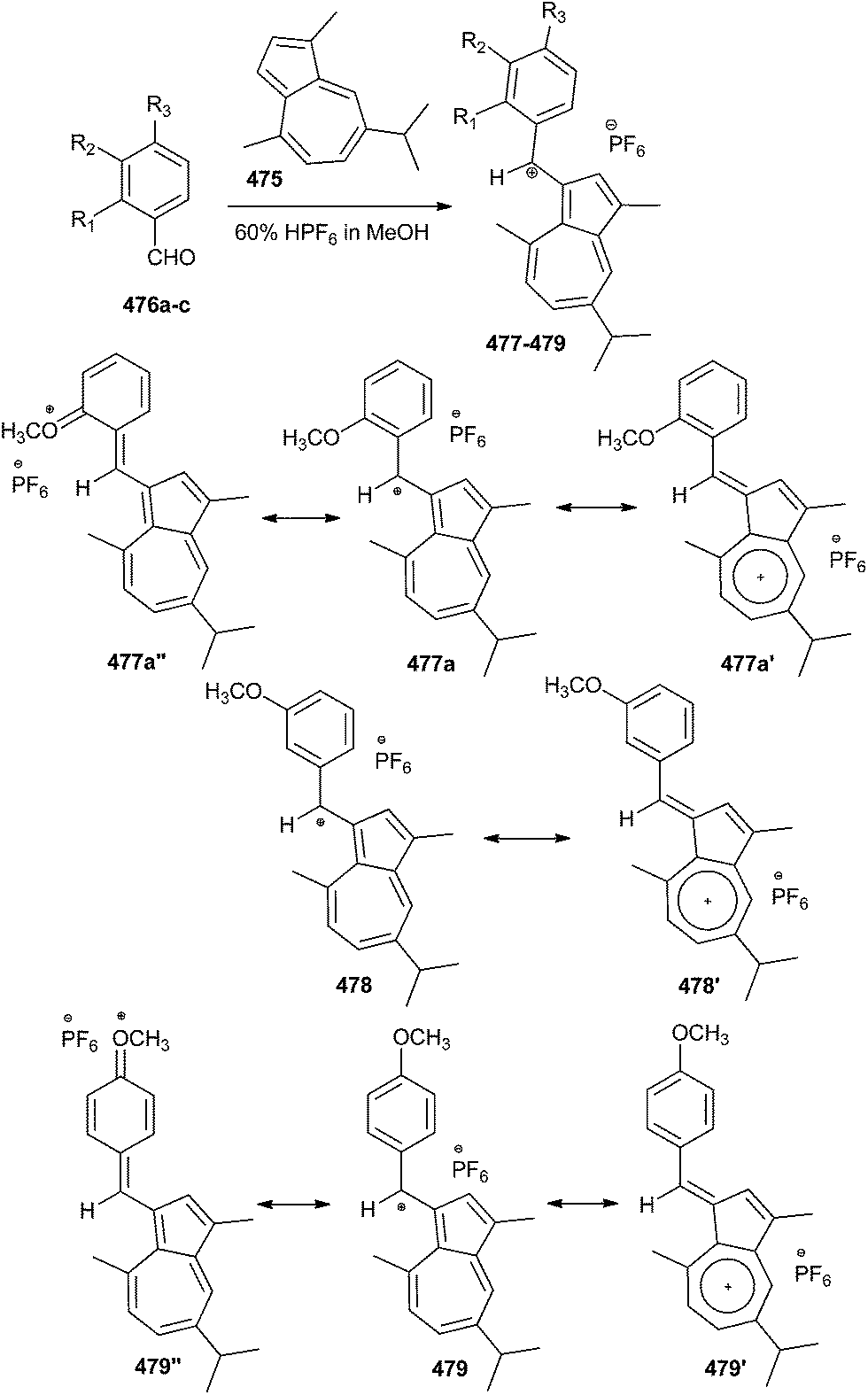 | ||
| Scheme 105 Reaction of guaiazulene with methoxybenzaldehyde and resonating structure of products 477–479. | ||
The regiochemical product of Pictet–Spengler cyclization reactions, focussed toward the preparation of the pentacyclic core of the ecteinascidin class of antitumor antibiotics, has been explored on two different phenolic substrates.102 The reduction of the β-lactam unit in compound 480 proceeds through the coordinated lithium complex 481, which also prevents the over-reduction of the corresponding aldehyde group. Following aqueous quench with NH4Cl, condensation of the secondary amine group onto the hemi-aminal unit 481 led to the key iminium ion intermediate 482. The expected hydrogen bonding between the C-4 benzylic amine and the E-ring phenolic residue directs the Pictet–Spengler cyclization ortho to the phenol. Finally, spontaneous elimination of the C-4 benzylamine group occurs through the intermediacy of o-QM 483 to afford the C3–C4 unsaturated pentacyclic compound 484 as the major product (Scheme 106).
 | ||
| Scheme 106 Pictet–Spengler cyclization reaction for the synthesis of C3–C4 unsaturated pentacyclic compound 484. | ||
3.7. Tautomerism
After oxidation, quercetin 485 exists in the four tautomeric forms 486–489, one o-quinone isomer 486 and three quinone methide isomers 487, 488 and 489.103 Due to the relatively high abundance of the quinone methide isomers, glutathionyl adduct formation occurs at positions C6 and C8 of the quercetin A ring to give 6-GSQ 490 and 8-GSQ 491 (Scheme 107). | ||
| Scheme 107 Glutathionyl adduct formation at the quercetin ring. | ||
Depending on the orientation of the indole rings around the single bond, it has been predicted that the pairs of syn-periplanar (sp) and anti-periplanar (ap) rotamers for the o-quinone, quinone methide, and quinonimine tautomers of dimers 493–495, two geometrical isomers (E and Z) are possible for the extended forms.104 In order to resolve a reasonable entrant among these structures for the observed two-electron oxidation products of the dimers, all tautomeric quinones of dimers 493–495 were geometry-optimized in vacuum at the PBE 0/6-31+G(d,p) level of theory. The pentahydroxybiindolyl 498 was isolated from unprecedented hydroxylated indole derivative by oxidation of dimer 494 (Scheme 108).
 | ||
| Scheme 108 Structures of dimers of indole and hydroxybiindolyl. | ||
Pestacin occurs naturally as a racemic mixture of (S) 499 and (R) 501 enantiomers.105 This racemization probably occurs post-biosynthetically as enzyme mediated reactions with stereospecificity. A racemization mechanism proceeds through a cationic intermediate 500, which has the desirable feature of being stabilized by different resonating structures. These intermediates are further stabilized by their ability to tautomerize into the three neutral o-QMs 501–503 (Scheme 109).
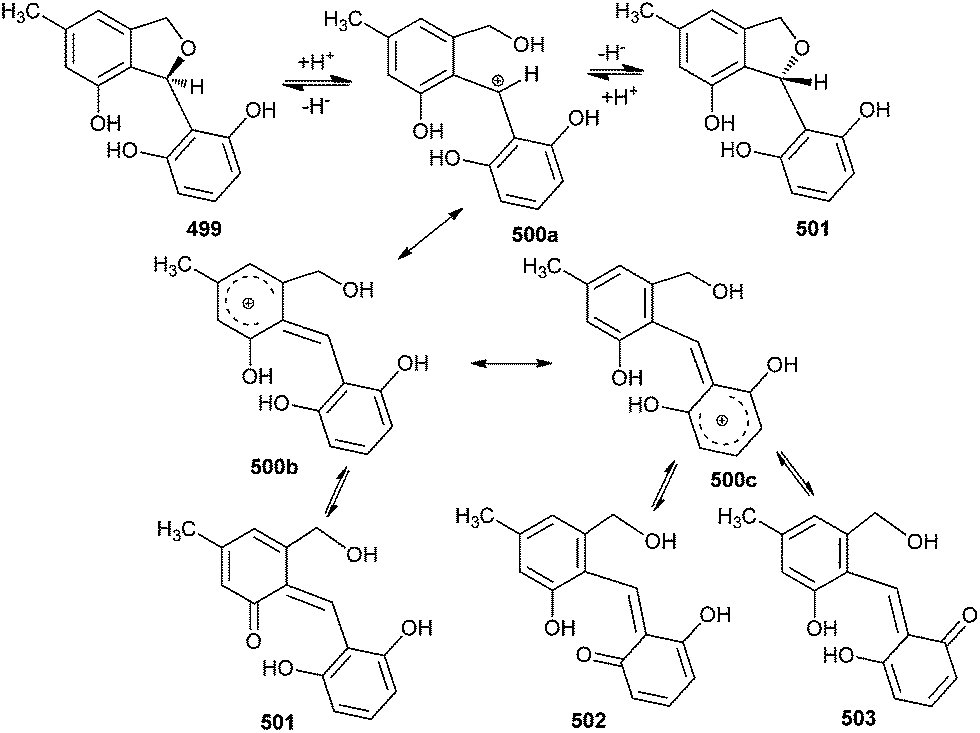 | ||
| Scheme 109 Resonating structures of Pestacin. | ||
It has been identified that certain aryl quinones experience a tautomeric isomerization to an intermediate QM because of the low barriers of rotation about central aryl-quinone C–C bond, which brings significant implications to consider in the design of synthetic routes leading to natural aryl quinones such as hibarimicin B.106 The rotational barrier of phenol is significantly lowered due to the stabilization of the transition state structure, leading to the interconversion of 504 and 506 by tautomerization to intermediate QM 505 (Scheme 110).
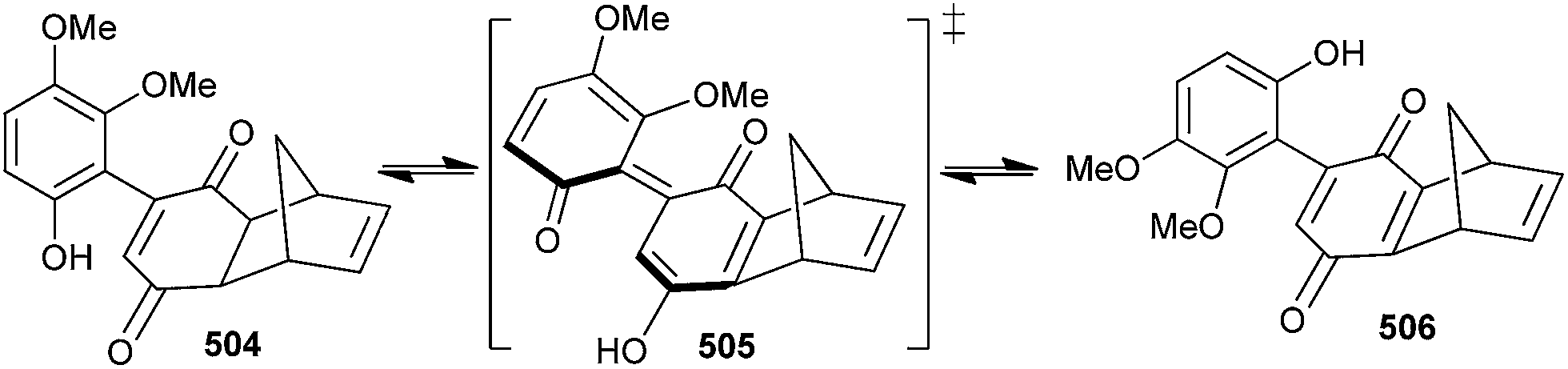 | ||
| Scheme 110 Tautomerization in quinone methide. | ||
3.8. Photochemical reactions
The photochemistry of a number of p-biphenyls and terphenyls substituted with hydroxyl and hydroxymethyl groups in aqueous solutions has been studied in detail by Wan and co-workers.107 They reported that the simple hydroxyl group can strongly and appropriately activate substituted benzenes or biphenyls towards dehydroxylation due to its strongly electron-donating nature, as well as for being carrier of an acidic proton. The intermediates formed are the corresponding quinone methides (QMs) or biphenyl quinone methides (BQM, 508 & 511), as shown in Scheme 111. | ||
| Scheme 111 Photochemical synthesis of compounds 509 and 512. | ||
There was no pH effect on the photosolvolysis quantum yield of the hydroxyterphenyls 513 in the pH 4–12 region, so the most probable mechanism for photosolvolysis is the formation of excited state phenolate followed by dehydroxylation to give terphenyl quinone methides (TQM, 514), which after reaction with methanol gives compound 515 (Scheme 112).107a
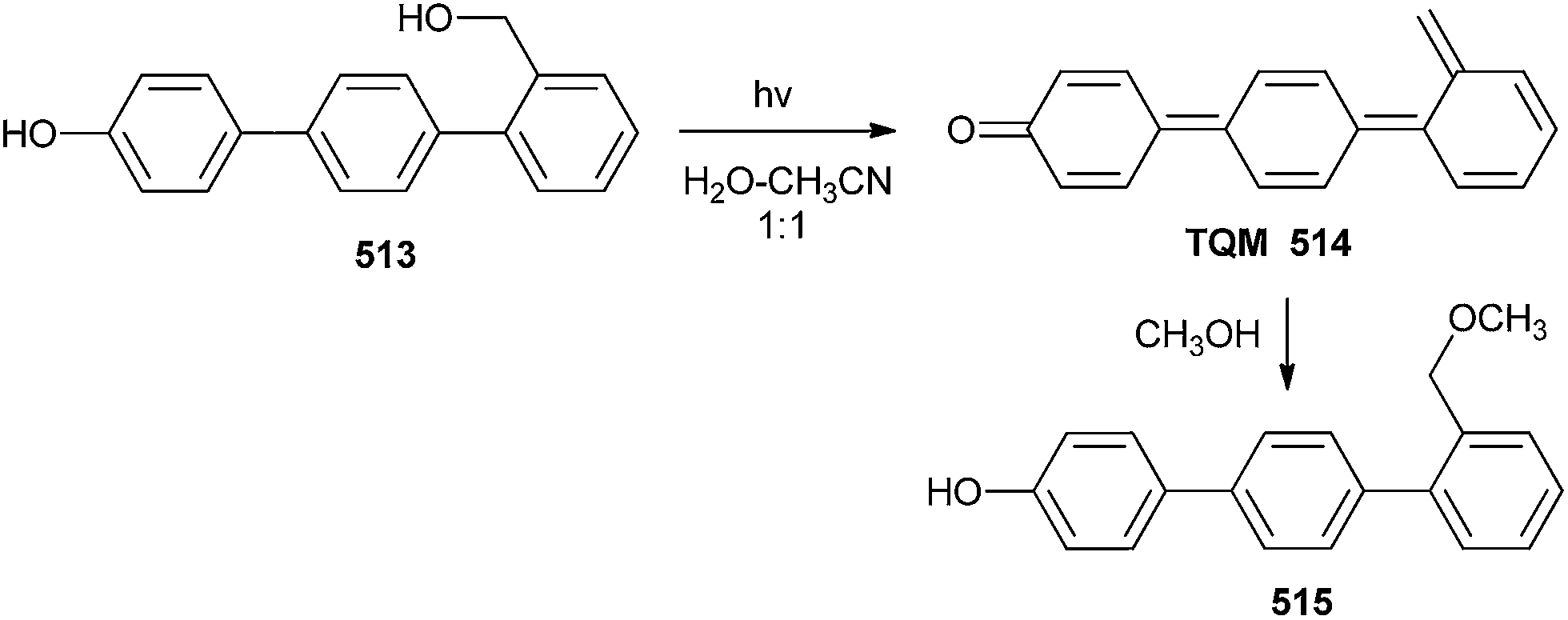 | ||
| Scheme 112 Reaction of methanol with terphenyl quinone methide 514. | ||
Compound 516 with competing photosolvolytic reaction generated compound 517 by the ESIPT process, with or without water.108 At higher water contents, an intermediate 518 (with one or more water molecules) underwent the loss of hydroxide ions from the benzylic position with the assistance of the hydronium ion to generate quinone methide 519, and finally over all solvolysis (attack by CH3OH at the terminal quinone methide position) gave compound 520 (Scheme 113).
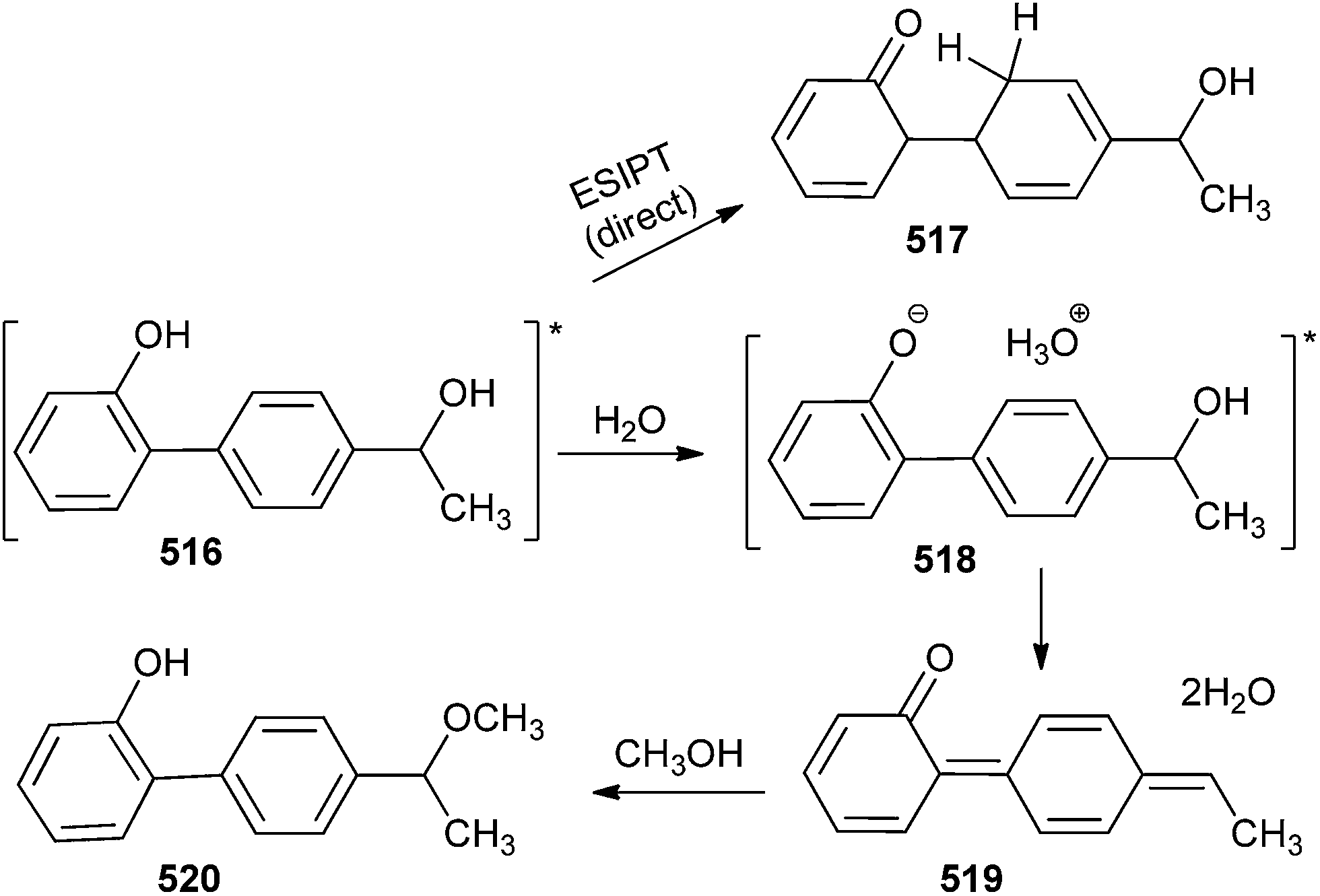 | ||
| Scheme 113 Photosolvolytic reaction of quinone methide 519. | ||
The compound 9-(2′-hydroxyphenyl) anthracene 521 undergoes proficient photoaddition of water and alcohols at the 9- and 10-positions of the anthracene moiety to give isolable triphenylmethanol or triphenylmethyl ether type of products.21 The reaction is supposed to ensue via water-mediated formal excited state an intramolecular proton transfer (ESIPT) from the phenolic OH to the 10-position of the anthracene ring, generating an o-QM intermediate that is observable by nanosecond laser flash photolysis, and is trappable with nucleophiles. Irradiation in deuterated solvents led to incorporation of one deuterium atom at the methylene position in the photoaddition product, and partial deuterium exchange of the 10-position of recovered starting material, which is reliable with the proposed formal excited state proton transfer mechanism (Scheme 114). The deuterium exchange and the photoaddition have got maximum quantum efficiency in 5 M water (in CH3CN or CH3OH), and no reaction was observed in the absence of a hydroxylic solvent, thus representing the sensitivity of this type of ESIPT to solvent composition. Once formed, QM 522 is transformed to 523 (hydration product) or returns to 521 via enolization. These two reaction pathways from 522 are not really astonishing apart from the isolation of the stable anthracene hydrates and alcohol adducts (Scheme 114).
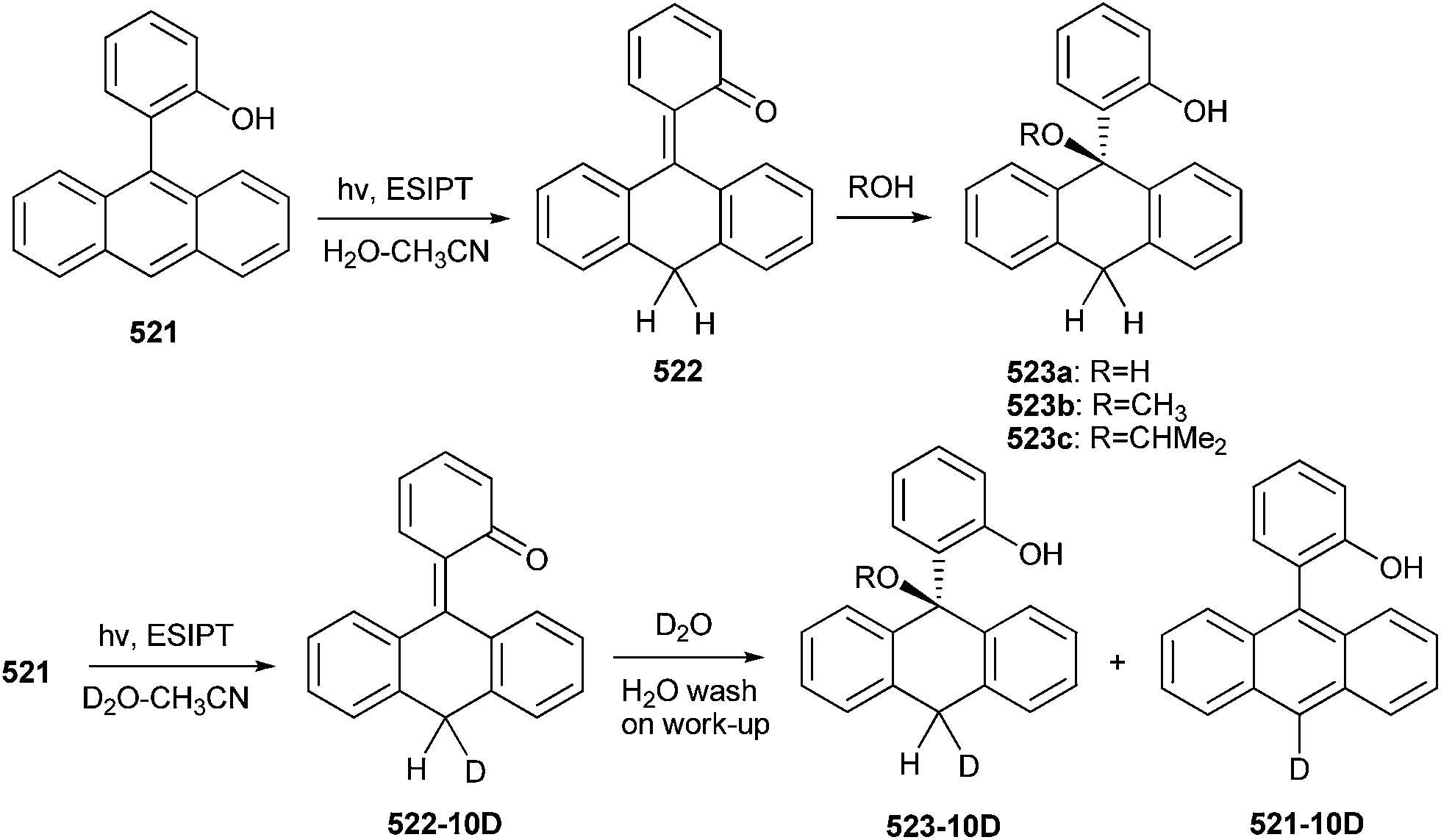 | ||
| Scheme 114 Photoaddition of hydroxylic solvents at 10-position of the anthracene. | ||
In general, caged carbonyl compounds109 are prepared by their acetalization with (2,5-dimethoxyphenyl)ethylene glycol followed by oxidative demethylation to produce the corresponding (1,3-dioxolane-4-yl)-1,4-benzoquinones. (2,5-Dihydroxyphenyl) ethylene glycol acetals of aldehydes and ketones 524a–e upon irradiation (at 300 nm) resulted in their efficient cleavage and in the regeneration of the carbonyl compound. This proceeds via photoinduced transfer of the phenolic proton to the dioxolane oxygen in the 3-position accompanied by the C–O bond cleavage and the formation of an intermediate o-QM 525. o-QM is very reactive and could undergo rapid hydration or tautomerization to form corresponding unstable hemiacetals 526 and/or 527. The hydrolysis of 526 and 527 liberates the carbonyl compound 528 (Scheme 115).
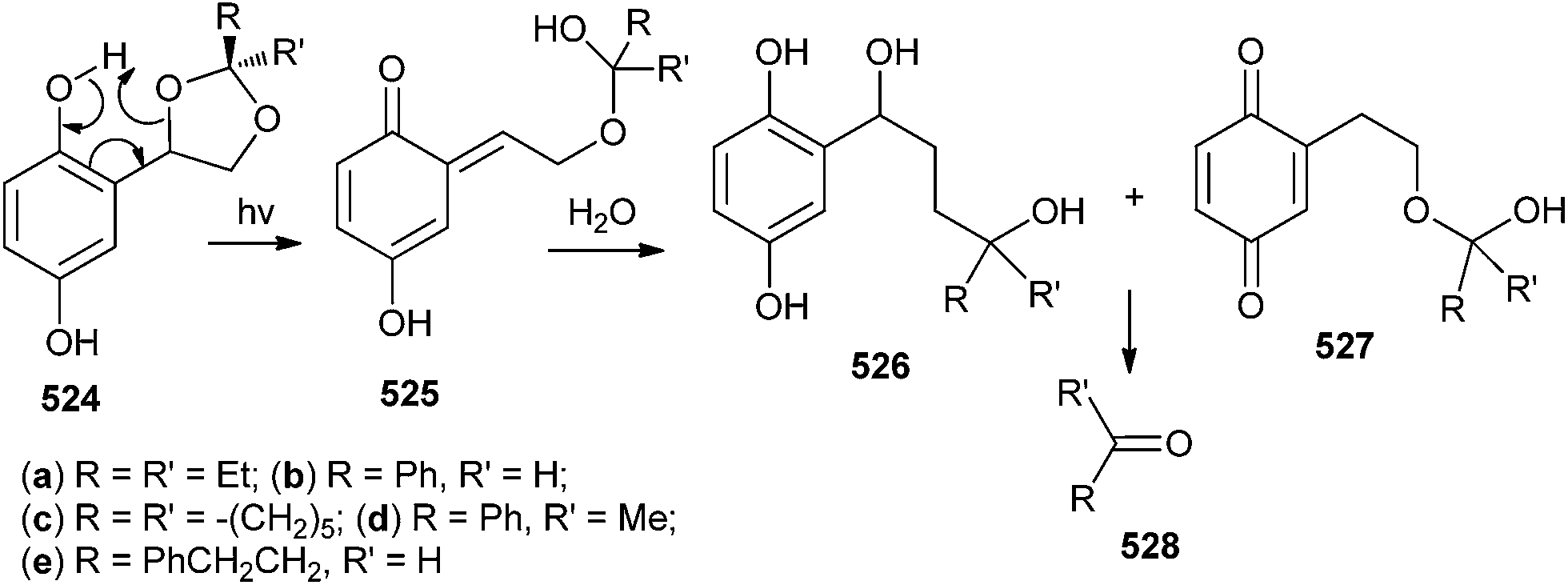 | ||
| Scheme 115 Regeneration of the carbonyl compound by irradiation of ethylene glycol acetals. | ||
The dibenzo[1,4]dioxins 529 underwent a photochemical aryl–ether bond homolysis to generate reactive 2-spiro-6-cyclohexa-2,4-dienone 531 and subsequent biphenylquinone intermediates.110 Under steady-state irradiation, the 2,2′-biphenylquinones 532 undergo an excited state hydrogen abstraction from the organic solvent to give the corresponding 2,2-dihydroxybiphenyls 533 (Scheme 116).
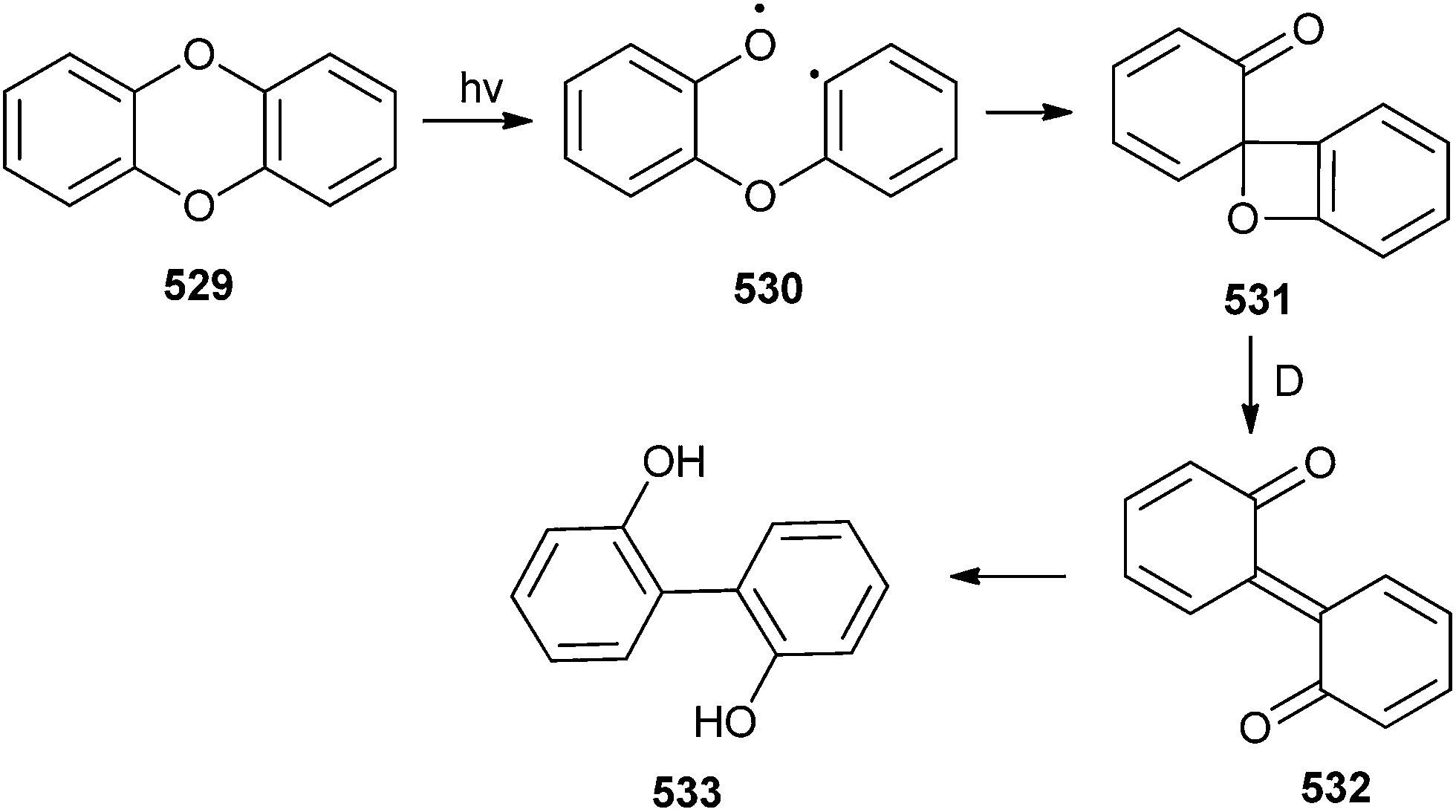 | ||
| Scheme 116 Photochemical aryl-ether bond homolysis of dibenzo[1,4]dioxin. | ||
The flash photolysis of methyl 2-hydroxyphenyldiazoacetate 534 in dilute aqueous perchloric acid solution and acetic acid and biphosphate ion buffers generated a transient species that was identified as o-quinone-α-carbomethoxymethide 535 (Scheme 117).111 This structural assignment is based on the solvent isotope effects, the form of buffer catalysis, UV absorption maxima, and the identity of decay rate constants with those determined for the transient, obtained by flash photolysis of other more conventional quinone methide precursors such as the benzyl alcohol methyl 2-hydroxy mandelate 536 and its acetate, 2-acetoxy-2-hydroxyphenylacetate 537.
 | ||
| Scheme 117 Photolysis of methyl 2-hydroxyphenyl diazoacetate. | ||
New (2-adamantyl)naphthol derivatives 536–540, quinone methide precursors (QMP, Scheme 118) were synthesized and their photochemical reactivity was investigated by preparative photolyses, fluorescence spectroscopy, and laser flash photolysis (LFP). All derivatives undergo photosolvolysis via QM intermediates. Presence of adamantly moiety results in different reactivity pattern compared to the non-substituted structure. They also investigated the anti-proliferative activity of the QMPs on three human cancer cell lines: HCT 116 (colon), MCF-7 (breast), and H 460 (lung). Exposure of cells treated with 5 to 300 nm of irradiation led to enhanced anti-proliferative effect on MCF-7 cell line, classifying 5 (or QM5) as a potential lead for further anti-proliferative studies.112
 | ||
| Scheme 118 Quinone methide precursors of (2-adamantyl)naphthol derivatives. | ||
4. Concluding remarks
In this review, we present the power and diversity of applications of ortho-quinone methides (o-QMs), which have become powerful, fascinating, and highly efficient intermediates in organic synthesis. Recent advances toward the generation, reactivity and application of o-QMs in organic synthesis offer a bright future for the development of novel methodologies for the synthesis of natural and man-made compounds. The cases cited in this review have been selected to highlight the most promising applications of o-QMs in organic synthesis. It can be seen that many well-established different methodologies were successfully applied to o-QMs by worldwide laboratories to more environmentally benign approaches. This area is clearly expanding in several aspects and will reveal spectacular applications in the near future, ranging from organic synthesis, material chemistry, fine chemicals, and pharmaceuticals to the synthesis of natural products. Definitely, much more remains to be done in this field, and in the next few years, we will see many new, exciting findings in o-QMs chemistry. Moreover, it will lead to the serendipitous discovery of many more new reactions, for as much as we may know, chemistry always has new and intriguing surprises in store. Therefore, we sincerely hope that this overview will be of interest for the synthetic community as well as for the general readers. Despite the flurry of recent interest in this species, it is unlikely that we have seen a peak, and it is probable that in future years o-QMs will be viewed in the same light as radicals, carbenes, benzynes or similar reactive intermediates.Acknowledgements
We sincerely thank all co-workers and collaborators whose names appear in the related references for their great contribution to the project. The financial support from the Science and Engineering Research Board (SERB) and the Council of Scientific and Industrial Research (CSIR), New Delhi is gratefully acknowledged. A.N., S.C. and N.A. thank University Grants Commission, New Delhi for fellowship.Notes and references
- For reviews see: (a) R. W. V. D. Water and T. R. R. Pettus, Tetrahedron, 2002, 58, 5367–5405 CrossRef; (b) H. Amouri and J. L. Bras, Acc. Chem. Res., 2002, 35, 501–510 CrossRef CAS PubMed; (c) S. B. Ferreira, F. D. C. da Silva, A. C. Pinto, D. T. G. Gonzaga and V. F. Ferreira, J. Heterocycl. Chem., 2009, 46, 1080–1097 CrossRef CAS.
- (a) R. Li, X. Wang, Z. Wei, C. Wu and F. Shi, Org. Lett., 2013, 15, 4366–4369 CrossRef CAS PubMed; (b) A. Kumar, M. Kumar and M. K. Gupta, Green Chem., 2012, 14, 2677–2681 RSC.
- N. J. Willis and C. D. Bray, Chem.–Eur. J., 2012, 18, 9160–9173 CrossRef CAS PubMed.
- (a) Quinone Methides, ed. S. E. Rokita, Wiley, Hoboken, NJ, 2009 Search PubMed; (b) H. Wang and S. E. Rokita, Angew. Chem., Int. Ed., 2010, 49, 5957–5960 CrossRef CAS PubMed.
- E. Modica, R. Zanaletti, M. Freccero and M. Mella, J. Org. Chem., 2001, 66, 41–52 CrossRef CAS PubMed.
- D. Zhang, M. Ogan, R. Gedamke, V. Roongta, R. Dai, M. Zhu, J. K. Rinehart, L. Klunk and J. Mitroka, Drug Metab. Dispos., 2003, 7, 837–845 CrossRef PubMed.
- (a) S. E. Rokita, J. Yang, P. Pande and W. A. Greenberg, J. Org. Chem., 1997, 62, 3010–3012 CrossRef CAS PubMed; (b) W. F. Veldhuyzen, A. J. Shallop, R. A. Jones and S. E. Rokita, J. Am. Chem. Soc., 2001, 123, 11126–11132 CrossRef CAS PubMed.
- (a) P. Pande, J. Shearer, J. Yang, W. A. Greenberg and S. E. Rokita, J. Am. Chem. Soc., 1999, 121, 6773–6779 CrossRef CAS; (b) P. Wang, R. Liu, X. Wu, H. Ma, X. Cao, P. Zhou, J. Zhang, X. Weng, X. L. Zhang, X. Zhou and L. Weng, J. Am. Chem. Soc., 2003, 125, 1116–1117 CrossRef CAS PubMed; (c) D. Verga, S. N. Richter, M. Palumbo, R. Gandolfi and M. Freccero, Org. Biomol. Chem., 2007, 5, 233–235 RSC; (d) S. E. Wolkenberg and D. L. Boger, Chem. Rev., 2002, 102, 2477–2495 CrossRef CAS PubMed.
- T. H. Koch, B. L. Barthel, B. T. Kalet, D. L. Rudnicki, G. C. Post and D. J. Burkhart, Top. Curr. Chem., 2008, 283, 141–170 CrossRef CAS PubMed.
- (a) S. R. Angle and W. Yang, J. Am. Chem. Soc., 1990, 112, 4524–4528 CrossRef CAS; (b) G. Gaudiano, M. Frigerio, P. Bravo and T. H. Koch, J. Am. Chem. Soc., 1990, 112, 6704–6709 CrossRef CAS; (c) G. Gaudiano and T. H. Koch, Chem. Res. Toxicol., 1991, 4, 2–16 CrossRef CAS PubMed; (d) S. R. Angle and W. Yang, J. Org. Chem., 1992, 57, 1092–1097 CrossRef CAS; (e) S. R. Angle, J. D. Rainer and C. Woytowicz, J. Org. Chem., 1997, 62, 5884–5892 CrossRef CAS.
- P. Wang, Y. Song, L. Zhang, H. He and X. Zhou, Curr. Med. Chem., 2005, 12, 2893–2913 CrossRef CAS PubMed.
- R. Rodriguez, R. M. Adlington, J. E. Moses, A. Cowley and J. E. Baldwin, Org. Lett., 2004, 6, 3617–3619 CrossRef CAS PubMed.
- C. D. Bray, Org. Biomol. Chem., 2008, 6, 2815–2819 CAS.
- A. F. Barrero, J. F. Q. del Moral, M. M. Herrador, P. Arteaga, M. Cortes, J. Benites and A. Rosellon, Tetrahedron, 2006, 62, 6012–6017 CrossRef CAS.
- K. Wojciechowski and K. Dolatowska, Tetrahedron, 2005, 61, 8419–8422 CrossRef CAS.
- H. Sugimoto, S. Nakamura and T. Ohwada, J. Org. Chem., 2007, 72, 10088–10095 CrossRef CAS PubMed.
- J. Delgado, A. Espinos, M. C. Jimenez and M. A. Miranda, Chem. Commun., 2002, 2636–2637 RSC.
- M. K. Boyd and G. M. Zopp, Annu. Rep. Prog. Chem., Sect. B: Org. Chem., 2002, 98, 543–579 RSC.
- (a) Y. Chiang, A. J. Kresge and Y. Zhu, J. Am. Chem. Soc., 2002, 124, 717–722 CrossRef CAS PubMed; (b) Y. Chiang, A. J. Kresge and Y. Zhu, J. Am. Chem. Soc., 2001, 123, 8089–8094 CrossRef CAS PubMed.
- M. Lukeman and P. Wan, J. Am. Chem. Soc., 2002, 124, 9458–9464 CrossRef CAS PubMed.
- M. Flegel, M. Lukeman, L. Huck and P. Wan, J. Am. Chem. Soc., 2004, 126, 7890–7897 CrossRef CAS PubMed.
- Y. Chiang, A. J. Kresge, O. Sadovski and H.-Q. Zhan, J. Org. Chem., 2005, 70, 1643–1646 CrossRef CAS PubMed.
- J. Matsumoto, M. Ishizu, R. Kawano, D. Hesaka, T. Shiragami, Y. Hayashi, T. Yamashita and M. Yasuda, Tetrahedron, 2005, 61, 5735–5740 CrossRef CAS.
- (a) S. Arumugam and V. V. Popik, J. Am. Chem. Soc., 2009, 131, 11892–11899 CrossRef CAS PubMed; (b) A. Kulikov, S. Arumugam and V. V. Popik, J. Org. Chem., 2008, 73, 7611–7615 CrossRef CAS PubMed; (c) S. Arumugam and V. V. Popik, J. Org. Chem., 2010, 75, 7338–7346 CrossRef CAS PubMed.
- N. Basaric, I. Zabcic, K. M. Majerski and P. Wan, J. Org. Chem., 2010, 75, 102–116 CrossRef CAS PubMed.
- S. N. Richter, S. Maggi, S. C. Mels, M. Palumbo and M. Freccero, J. Am. Chem. Soc., 2004, 126, 13973–13979 CrossRef CAS PubMed.
- D. Skalamera, K. M. Majerski, I. M. Kleiner, M. Kralj, P. Wan and N. Basaric, J. Org. Chem., 2014, 79, 4390–4397 CrossRef CAS PubMed.
- F. Doria, C. Percivalle and M. Freccero, J. Org. Chem., 2012, 77, 3615–3619 CrossRef CAS PubMed.
- M. J. Adler and S. W. Baldwin, Tetrahedron Lett., 2009, 50, 5075–5079 CrossRef CAS.
- (a) K. Nakatani, N. Higashida and I. Saito, Tetrahedron Lett., 1997, 38, 5005–5008 CrossRef CAS; (b) K. Chiba, T. Hirano, Y. Kitano and M. Tada, Chem. Commun., 1999, 691–692 RSC.
- P. Batsomboon, W. Phakhodee, S. Ruchirawat and P. Ploypradith, J. Org. Chem., 2009, 74, 4009–4012 CrossRef CAS PubMed.
- Y. Sawama, Y. Shishido, T. Yanase, K. Kawamoto, R. Goto, Y. Monguchi, Y. Kita and H. Sajiki, Angew. Chem., Int. Ed., 2013, 52, 1515–1519 CrossRef CAS PubMed.
- O. O. Fadeyi, R. N. Daniels, S. M. DeGuire and C. W. Lindsley, Tetrahedron Lett., 2009, 50, 3084–3087 CrossRef CAS.
- P.-J. J. Huang, T. S. Cameron and A. Jha, Tetrahedron Lett., 2009, 50, 51–54 CrossRef CAS.
- Y. R. Lee, Y. M. Kim and S. H. Kim, Tetrahedron, 2009, 65, 101–108 CrossRef CAS.
- H. Yoshida, Y. Ito and J. Ohshita, Chem. Commun., 2011, 47, 8512–8514 RSC.
- D. Liao, H. Li and X. Lei, Org. Lett., 2012, 14, 18–21 CrossRef CAS PubMed.
- S. B. Bharate and I. P. Singh, Tetrahedron Lett., 2006, 47, 7021–7024 CrossRef CAS.
- (a) A. L. Lawrence, R. M. Adlington, J. E. Baldwin, V. Lee, J. A. Kershaw and A. L. Thompson, Org. Lett., 2010, 12, 1676–1679 CrossRef CAS PubMed; (b) X.-L. Yang, K.-L. Hsieh and J.-K. Liu, Org. Lett., 2007, 9, 5135–5138 CrossRef CAS PubMed.
- C. Selenski and T. R. R. Pettus, J. Org. Chem., 2004, 69, 9196–9203 CrossRef CAS PubMed.
- (a) R. V. De Water, D. Magdziak, J. Chau and T. R. R. J. Pettus, J. Am. Chem. Soc., 2000, 122, 6502–6503 CrossRef; (b) A. Arduini, A. Bosi, A. Pochini and R. Ungaro, Tetrahedron, 1985, 41, 3095–3103 CrossRef CAS; (c) C. Selenski, L. Mejorado and T. R. R. Pettus, Synlett, 2004, 1101–1103 CAS.
- S. B. Bharate, S. I. Khan, B. L. Tekwani, M. Jacob, I. A. Khan and I. P. Singh, Bioorg. Med. Chem., 2008, 16, 1328–1336 CrossRef CAS PubMed.
- S. B. Bharate, K. K. Bhutani, S. I. Khan, B. L. Tekwani, M. R. Jacob, I. A. Khan and I. P. Singh, Bioorg. Med. Chem., 2006, 14, 1750–1760 CrossRef CAS PubMed.
- L. W. Schenck, K. Kuna, W. Frank, A. Albert, C. Asche and U. Kucklaender, Bioorg. Med. Chem., 2006, 14, 3599–3614 CrossRef CAS PubMed.
- Y. Chen and M. G. Steinmetz, J. Org. Chem., 2006, 71, 6053–6060 CrossRef CAS PubMed.
- A. F. Barrero, J. F. Q. del Moral, M. M. Herrador, P. Arteaga, M. Cortes, J. Benites and A. Rosellon, Tetrahedron, 2006, 62, 6012–6017 CrossRef CAS.
- K. Tchabanenko, M. G. O. Taylor, R. M. Adlington and J. E. Baldwin, Tetrahedron Lett., 2006, 47, 39–41 CrossRef CAS.
- C. C. Lindsey and T. R. R. Pettus, Tetrahedron Lett., 2006, 47, 201–204 CrossRef CAS PubMed.
- (a) F. Liebner, P. Schmid, C. Adelwohrer and T. Rosenau, Tetrahedron, 2007, 63, 11817–11821 CrossRef CAS; (b) T. Rosenau, Encyclopedia of Vitamin E, ed. V. R. Preedy and R. R. Watson, CABI: Oxford, Cambridge, 2007, pp. 21–44 and 69–96 Search PubMed.
- E. Alden-Danforth, M. T. Scerba and T. Lectka, Org. Lett., 2008, 10, 4951–4953 CrossRef CAS PubMed.
- M. A. Marsini, Y. Huang, C. C. Lindsey, K.-L. Wu and T. R. R. Pettus, Org. Lett., 2008, 10, 1477–1480 CrossRef CAS PubMed.
- S. J. Gharpure, A. M. Sathiyanarayanan and P. Jonnalagadda, Tetrahedron Lett., 2008, 49, 2974–2978 CrossRef CAS.
- T. B. Samarakoon, M. Y. Hur, R. D. Kurtz and P. R. Hanson, Org. Lett., 2010, 12, 2182–2185 CrossRef CAS PubMed.
- J. C. Green and T. R. R. Pettus, J. Am. Chem. Soc., 2011, 133, 1603–1608 CrossRef CAS PubMed.
- T. Rosenau, C. Adelwohrer, E. Kloser, K. Mereiter and T. Netscher, Tetrahedron, 2006, 62, 1772–1776 CrossRef CAS.
- H. M. Fales, H. A. Lloyd, J. A. Ferretti, J. V. Silverton, D. G. Davis and H. J. Kon, J. Chem. Soc., Perkin Trans. 2, 1990, 1005–1010 RSC.
- (a) G. C. Nandi, S. Samai, R. Kumar and M. S. Singh, Tetrahedron, 2009, 65, 7129–7134 CrossRef CAS; (b) S. Samai, G. C. Nandi and M. S. Singh, Tetrahedron, 2012, 68, 1247–1252 CrossRef CAS; (c) R. Kumar, G. C. Nandi, R. K. Verma and M. S. Singh, Tetrahedron Lett., 2010, 51, 442–445 CrossRef CAS.
- B. Datta and M. A. Pasha, Ultrason. Sonochem., 2011, 18, 624–628 CrossRef CAS PubMed.
- G. C. Nandi, S. Samai, R. Kumar and M. S. Singh, Tetrahedron Lett., 2009, 50, 7220–7222 CrossRef CAS.
- R. R. Nagawade and D. B. Shinde, Mendeleev Commun., 2007, 17, 299–300 CrossRef CAS.
- M. M. Khodaei, A. R. Khosropour and H. Moghanian, Synlett, 2006, 916–920 CrossRef CAS.
- M. Hong, C. Cai and W. B. Yi, Chin. Chem. Lett., 2011, 22, 322–325 CrossRef CAS.
- H. R. Shaterian and H. Yarahmadi, ARKIVOC, 2008, ii, 105–114 Search PubMed.
- A. Dorehgiraee, H. Khabazzadeh and K. Saidi, ARKIVOC, 2009, vii, 303–310 Search PubMed.
- (a) A. Kumar, M. S. Rao and V. K. Rao, Aust. J. Chem., 2010, 63, 1538–1540 CrossRef CAS; (b) A. Hosseinian and H. R. Shaterian, Phosphorus, Sulfur Silicon Relat. Elem., 2012, 187, 1056–1063 CrossRef CAS; (c) A. Shaabani, A. Rahmati and E. Farhangi, Tetrahedron Lett., 2007, 48, 7291–7294 CrossRef CAS.
- V. A. Osyanin, D. V. Osipov and Y. N. Klimochkin, J. Org. Chem., 2013, 78, 5505–5520 CrossRef CAS PubMed.
- F. Mazzini, T. Netscher and P. Salvadoria, Tetrahedron, 2005, 61, 813–817 CrossRef CAS.
- C. Adelwohrer, T. Rosenau, W. H. Binder and P. Kosma, Tetrahedron, 2003, 59, 3231–3235 CrossRef CAS.
- M.-W. Chen, L.-L. Cao, Z.-S. Ye, G.-F. Jiang and Y.-G. Zhou, Chem. Commun., 2013, 49, 1660–1662 RSC.
- Y. Luan and S. E. Schaus, J. Am. Chem. Soc., 2012, 134, 19965–19968 CrossRef CAS PubMed.
- A. Kumar, M. Kumar, M. K. Gupta and L. P. Gupta, RSC Adv., 2012, 2, 8277–8280 RSC.
- C. Percivalle, A. La Rosa, D. Verga, F. Doria, M. Mella, M. Palumbo, M. Di Antonio and M. Freccero, J. Org. Chem., 2011, 76, 3096–3106 CrossRef CAS PubMed.
- M. Nadai, F. Doria, M. Di Antonio, G. Sattin, L. Germani, C. Percivalle, M. Palumbo, S. N. Richter and M. Freccero, Biochimie, 2011, 93, 1328–1340 CrossRef CAS PubMed.
- E. E. Weinert, K. N. Frankenfield and S. E. Rokita, Chem. Res. Toxicol., 2005, 18, 1364–1370 CrossRef CAS PubMed.
- E. Modica, R. Zanaletti, M. Freccero and M. Mella, J. Org. Chem., 2001, 66, 41–52 CrossRef CAS PubMed.
- E. E. Weinert, R. Dondi, S. Colloredo-Melz, K. N. Frankenfield, C. H. Mitchell, M. Freccero and S. E. Rokita, J. Am. Chem. Soc., 2006, 128, 11940–11947 CrossRef CAS PubMed.
- E. E. Weinert, K. N. Frankenfield and S. E. Rokita, Chem. Res. Toxicol., 2005, 18, 1364–1370 CrossRef CAS PubMed.
- Q. Zhou and S. E. Rokita, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 15452–15457 CrossRef CAS PubMed.
- M. P. McCrane, E. E. Weinert, Y. Lin, E. P. Mazzola, Y.-F. Lam, P. F. Scholl and S. E. Rokita, Org. Lett., 2011, 13, 1186–1189 CrossRef CAS PubMed.
- Y. Chiang and A. J. Kresge, Org. Biomol. Chem., 2004, 2, 1090–1092 CAS.
- Y. Du, X. Weng, J. Huang, D. Zhang, H. Ma, D. Chen, X. Zhou and J.-F. Constant, Bioorg. Med. Chem., 2010, 18, 4149–4153 CrossRef CAS PubMed.
- J. Zhang, X. Wu, X. Cao, F. Yang, J. Wang, X. Zhou and X.-L. Zhang, Bioorg. Med. Chem. Lett., 2003, 13, 1097–1100 CrossRef CAS PubMed.
- Y. Song, P. Wang, J. Wu, X. Zhou, X.-L. Zhang, L. Weng, X. Cao and F. Liang, Bioorg. Med. Chem. Lett., 2006, 16, 1660–1664 CrossRef CAS PubMed.
- Q. Zhou, T. Xu and J. B. Mangrum, Chem. Res. Toxicol., 2007, 20, 1069–1074 CrossRef CAS PubMed.
- M. Di Antonio, F. Doria, S. N. Richter, C. Bertipaglia, M. Mella, C. Sissi, M. Palumbo and M. Freccero, J. Am. Chem. Soc., 2009, 131, 13132–13141 CrossRef CAS PubMed.
- F. Doria, S. N. Richter, M. Nadai, S. Colloredo-Mels, M. Mella, M. Palumbo and M. Freccero, J. Med. Chem., 2007, 50, 6570–6579 CrossRef CAS PubMed.
- M. Freccero, R. Gandolfi and M. Sarzi-Amade, J. Org. Chem., 2003, 68, 6411–6423 CrossRef CAS PubMed.
- T. A. Wenderski, S. Huang and T. R. R. Pettus, J. Org. Chem., 2009, 74, 4104–4109 CrossRef CAS PubMed.
- T. Okugaki, M. Kasuno, K. Maeda and S. Kihara, J. Electroanal. Chem., 2010, 639, 67–76 CrossRef CAS.
- K. A. Korthals and W. D. Wulff, J. Am. Chem. Soc., 2008, 130, 2898–2899 CrossRef CAS PubMed.
- A. V. Vorogushin, W. D. Wulff and H.-J. Hansen, Tetrahedron, 2008, 64, 949–968 CrossRef CAS PubMed.
- K. H. Jensen, J. D. Webb and M. S. Sigman, J. Am. Chem. Soc., 2010, 132, 17471–17482 CrossRef CAS PubMed.
- K. M. Gligorich, Y. Iwai, S. A. Cummings and M. S. Sigman, Tetrahedron, 2009, 65, 5074–5083 CrossRef CAS PubMed.
- S. M. Stokes Jr, F. Ding, P. L. Smith, J. M. Keane, M. E. Kopach, R. Jervis, M. Sabat and W. D. Harman, Organometallics, 2003, 22, 4170–4171 CrossRef.
- S. Radomkit, P. Sarnpitak, J. Tummatorn, P. Batsomboon, S. Ruchirawat and P. Ploypradith, Tetrahedron, 2011, 67, 3904–3914 CrossRef CAS.
- D. A. Lev, D. B. Grotjahn and H. Amouri, Organometallics, 2005, 24, 4232–4240 CrossRef CAS.
- J. C. J. M. D. S. Menezes, A. T. P. C. Gomes, A. M. S. Silva, M. A. F. Faustino, M. G. P. M. S. Neves, A. C. Tomé, F. C. da Silva, V. F. Ferreira and J. A. S. Cavaleiro, Synlett, 2011, 1841–1844 CAS.
- S.-T. Huang, K.-N. Ting and K.-L. Wang, Anal. Chim. Acta, 2008, 620, 120–126 CrossRef CAS PubMed.
- G. da Silva and J. W. Bozzelli, J. Phys. Chem. A, 2007, 111, 7987–7994 CrossRef CAS PubMed.
- A. Koll, A. Karpfen and P. Wolschann, J. Mol. Struct., 2006, 790, 55–64 CrossRef CAS.
- S. Takekuma, M. Tamura, T. Minematsu and H. Takekuma, Tetrahedron, 2007, 63, 12058–12070 CrossRef CAS.
- G. Vincent, J. W. Lane and R. M. Williams, Tetrahedron Lett., 2007, 48, 3719–3722 CrossRef CAS PubMed.
- H. Jacobs, W. J. F. van der Vijgh, G. H. Koek, G. J. J. Draaisma, M. Moalin, G. P. F. van Strijdonck, A. Bast and G. R. M. M. Haenen, Free Radical Biol. Med., 2009, 46, 1567–1573 CrossRef CAS PubMed.
- A. Pezzella, L. Panzella, O. Crescenzi, A. Napolitano, S. Navaratman, R. Edge, E. J. Land, V. Barone and M. Ischia, J. Am. Chem. Soc., 2006, 128, 15490–15498 CrossRef CAS PubMed.
- J. K. Harper, A. M. Arif, E. J. Ford, G. A. Strobel, J. A. Porco Jr, D. P. Tomer, K. L. Oneill, E. M. Heidere and D. M. Granta, Tetrahedron, 2003, 59, 2471–2476 CrossRef CAS.
- U. S. M. Maharoof and G. A. Sulikowski, Tetrahedron Lett., 2003, 44, 9021–9023 CrossRef CAS.
- (a) M. Xu, C. Z. Chen and P. Wan, J. Photochem. Photobiol., A, 2008, 198, 26–33 CrossRef CAS; (b) C.-G. Huang, K. A. Beveridge and P. Wan, J. Am. Chem. Soc., 1991, 113, 7676–7684 CrossRef CAS; (c) Y. Shi and P. Wan, J. Chem. Soc., Chem. Commun., 1995, 1217–1218 RSC; (d) M. Lukeman and P. Wan, J. Am. Chem. Soc., 2003, 125, 1164–1165 CrossRef CAS PubMed.
- N. B. Aein and P. Wan, J. Photochem. Photobiol., A, 2009, 208, 42–49 CrossRef CAS.
- A. P. Kostikov, N. Malashikhina and V. V. Popik, J. Org. Chem., 2009, 74, 1802–1804 CrossRef CAS PubMed.
- S. Rayne, R. Sasaki and P. Wan, Photochem. Photobiol. Sci., 2005, 4, 876–886 CAS.
- Y. Chiang, A. J. Kresge and Y. Zhu, Phys. Chem. Chem. Phys., 2003, 5, 1039–1042 RSC.
- J. Veljkovic, L. Uzelac, K. Molcanov, K. M. Majerski, M. Kralj, P. Wan and N. Basaric, J. Org. Chem., 2012, 77, 4596–4610 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |