DOI:
10.1039/C2RA20490H
(Paper)
RSC Adv., 2012,
2, 5565-5570
Received
15th March 2012
, Accepted 3rd April 2012
First published on 4th April 2012
Abstract
Aminobromination of α,β-unsaturated nitro compounds with benzyl carbamate and N-bromosuccinimide as nitrogen/bromine sources was reported. This new catalytic system tolerates a wide range of aromatic substrates, as well as heterocyclic and aliphatic substrates, resulting in good chemical yields. The reaction also proceeds smoothly with water as a medium in high efficiency. This practical aminobromination method was also proved to be suitable for large-scale preparation. Furthermore, the N-carbobenzoxy protecting group could be easily cleaved to afford the free vicinal haloamines.
Introduction
The vicinal haloamine compounds are important and useful building blocks in organic synthesis and medicinal chemistry.1 These haloamines can also be easily converted into other valuable intermediates, such as β-substituted α-amino acid, aziridines and vicinal diamines.2 Aminohalogenation is a useful tool for the synthesis of these vicinal haloamines, by constructing carbon–nitrogen and carbon–halogen bonds in tandem.3–6 In the past years, we and other groups have reported many regio- and stereoselective aminohalogenation systems with readily available functionalized alkenes as starting materials, including α,β-unsaturated carboxylic esters,7 α,β-unsaturated nitriles,8 α,β-unsaturated ketones,9 methylenecyclopropanes10 and β-nitrostyrenes.11 Although great progress has been achieved on the alkene substrates, these aminohalogenation processes suffer from the limitation of the nitrogen sources, which are usually limited to sulfonamides.7–12 In addition, N,N-dichlorocarbamates have been used as nitrogen source for aminohalogenation in 1960s.13 However, these pioneering reactions show many of the characteristics of a free-radical addition reaction, which resulted in moderate regio- and stereoselectivities.
Recently, several amides and carbamates have been reported as nitrogen sources for the aminohalogenation of β-nitrostyrenes, which included, succinimide/NCS,14N-bromoacetamide,15 benzamides/NCS,16tert-butyl N,N-dichlorocarbamate17 and N,N-dibromourethane.18 Although these contributions expand the scope of nitrogen sources, most of them have the shortcomings of long reaction times, high loading amounts of catalyst and low chemical yields. Furthermore, the N-protecting groups are difficult to remove from the resulting haloamine product, which greatly limits the development and applications of such aminohalogenation reactions. In our ongoing research on this reaction,19 we found that benzyl carbamate/NBS was an efficient nitrogen/halogen source for the aminohalogenation of α,β-unsaturated nitro compounds. The advantage of such an exploration is that the N-carbobenzoxy protecting group from the resulting products can be easily cleaved to give free vicinal haloamines, as well as a lower amount of catalyst and shorter reaction time.14–19 Herein, we reported a facile and efficient aminobromination of α,β-unsaturated nitro compounds with benzyl carbamate/NBS as a new nitrogen/bromine source by using 5 mol% K3PO4 as the catalyst, giving dibrominated products with up to 97% chemical yields, which was deprotected to give free haloamine (Scheme 1). This new system could complete within 3 h, which greatly shortened the reaction time compared to the previous process.19
 |
| | Scheme 1 Aminobromination with CbzNH2/NBS. | |
Results and discussion
Initially, β-nitrostyrene 1a, benzyl carbamate 2 and NBS were mixed in acetonitrile and stirred at room temperature without the use of any catalyst. No haloamine product was obtained and most of the starting materials remained even after the reaction time was prolonged to 12 h (entry 1, Table 1). After many tries, we found that the reaction proceeded smoothly catalyzed by 20 mol% K2CO3, giving product in 80% yield in 5 h (entry 2). Then, Na2CO3, NaHCO3, KOH and NaOH were tested in the reaction, however, no improvements were obtained at all (entries 3–6). The chemical yield was further increased to 88% when 20 mol% K3PO4 was used as catalyst (entry 7). The results showed that only 5 mol% catalyst was enough for the current system, and had almost no effect on the chemical yield (entries 8–9).
| Entry |
Catalyst |
Amount (mol%) |
Time (h) |
Yield (%)b |
|
Conditions: substrate 1a (0.5 mmol), NBS (1.5 mmol), CbzNH2 (1.5 mmol), CH3CN (3 mL), at room temperature.
Isolated yields.
NR: No reaction was observed.
|
| 1 |
No |
0 |
12 |
NRc |
| 2 |
K2CO3 |
20 |
5 |
80 |
| 3 |
Na2CO3 |
20 |
5 |
55 |
| 4 |
NaHCO3 |
20 |
12 |
Trace |
| 5 |
KOH |
20 |
12 |
NRc |
| 6 |
NaOH |
20 |
12 |
NRc |
| 7 |
K3PO4 |
20 |
5 |
88 |
| 8 |
K3PO4 |
10 |
5 |
90 |
| 9 |
K3PO4 |
5 |
5 |
93 |
Then, a scan of common organic solvents was carried out (Table 2). The reaction could also proceed smoothly in dichloromethane, THF, acetone and methanol, giving the desired haloamines with good chemical yields (entries 1, 2, 4 and 7). No reaction was detected with DMSO and cyclohexane, and almost all the starting materials remained (entries 3 and 6). Acetonitrile was the most suitable solvent for this reaction. Only 3 h was needed to consume all the β-nitrostyrene and resulted in an excellent yield (92%, entry 5). The reaction time also showed effects on the chemical yields. A dramatic lower chemical yield was obtained when the reaction was stopped at 2 h (76% yield, entry 10).
Table 2 Optimization of reaction conditionsa
| Entry |
Solvent |
Time (h) |
Yield (%)b |
|
Conditions: substrate 1a (0.5 mmol), NBS (1.5 mmol), CbzNH2 (1.5 mmol), K3PO4 (5 mol%), solvent (3 mL), at room temperature.
Isolated yields.
NR: No reaction was observed.
|
| 1 |
DCM |
5 |
80 |
| 2 |
THF |
5 |
82 |
| 3 |
DMSO |
5 |
NRc |
| 4 |
Acetone |
5 |
83 |
| 5 |
MeCN |
3 |
92 |
| 6 |
Cyclohexane |
5 |
Trace |
| 7 |
MeOH |
5 |
81 |
| 8 |
Toluene |
5 |
43 |
| 9 |
MeCN |
1 |
65 |
| 10 |
MeCN |
2 |
76 |
| 11 |
MeCN |
4 |
91 |
Very recently, we developed a green aminohalogenation of β-nitrostyrenes with t-butyl N,N-dichlorocarbamate as nitrogen/halogen source.17b We then tried to investigate the potential of using water as the medium for this aminobromination system based on benzyl carbamate. We were surprised to find that the reaction could proceed smoothly with acetonitrile/water (ratio = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) as solvent and complete within 8 h, resulting in 83% yield (Scheme 2). This could serve as another example for green aminohalogenation reaction.
:3) as solvent and complete within 8 h, resulting in 83% yield (Scheme 2). This could serve as another example for green aminohalogenation reaction.
 |
| | Scheme 2 Aminobromination with water as medium. | |
Then, several α,β-unsaturated nitro compounds were subjected to this reaction to examine the scope and limitation of the current aminobromination reaction (Table 3). As shown in Table 3, a wide range of α,β-unsaturated nitro compounds worked well in the reaction, and completed within 5 h, giving 55–95% chemical yields. The electronegativity of the substituents on the aromatic rings showed almost no effects on the chemical yields. Both electron-rich (entries 7, 8 and 10–12) and electron-deficient substrates (entries 2–6, 13 and 18) could participate well in the reaction, even for methoxy (entries 10–11) and trifluoromethyl (entry 13) groups. On the contrary, the reactivity depended on the position of substituents on the aromatic ring. Especially, the substrates with the ortho-substituted aromatic ring needed longer to consume all the starting material and gave a lower yield (55%, entry 2) probably due to the steric hindrance. Notably, substrates with heterocyclic groups 1n and 1o were also well tolerated in this reaction along with good chemical yields (entries 14 and 15). Furthermore, an aliphatic substrate 1p, (E)-1-nitronon-1-ene was used in the reaction, and only 3 h reaction time was needed with 82% chemical yield (entry 16). This is really the first example that aliphatic substrates can work well in an aminohalogenation system based on α,β-unsaturated nitro compounds until now.11,15–19 Finally, the stereochemistry of these haloamino nitro compounds has been confirmed by the X-ray diffraction analysis of 3a (Fig. 1).
 |
| | Fig. 1 ORTEP diagram showing of compound 3a. | |
Table 3 Scope of the aminobrominationa
| Entry |
R
|
Product |
time |
Yield (%)b |
|
Conditions: substrate 1 (0.5 mmol), NBS (1.5 mmol), CbzNH2 (1.5 mmol), K3PO4 (5 mol%), acetonitrile (3 mL), at room temperature.
Isolated yields.
|
| 1 |
C6H5 |
3a
|
3 |
93 |
| 2 |
2-ClC6H4 |
3b
|
5 |
55 |
| 3 |
3-ClC6H4 |
3c
|
3 |
88 |
| 4 |
4-BrC6H4 |
3d
|
3 |
91 |
| 5 |
3-FC6H4 |
3e
|
3 |
86 |
| 6 |
4-FC6H4 |
3f
|
3 |
85 |
| 7 |
3-MeC6H4 |
3g
|
3 |
97 |
| 8 |
4-MeC6H4 |
3h
|
3 |
62 |
| 9 |
1-naphthyl |
3i
|
3 |
89 |
| 10 |
3-MeOC6H4 |
3j
|
3 |
83 |
| 11 |
4-MeOC6H4 |
3k
|
3 |
95 |
| 12 |
4-BnOC6H4 |
3l
|
3 |
78 |
| 13 |
4-CF3C6H4 |
3m
|
3 |
83 |
| 14 |
1-furyl |
3n
|
3 |
68 |
| 15 |
1-thienyl |
3o
|
3 |
72 |
| 16 |
n-heptyl |
3p
|
3 |
82 |
| 17 |
3-Br-4-MeOC6H3 |
3q
|
3 |
87 |
| 18 |
4-CNC6H4 |
3r
|
3 |
81 |
The current system was found to have a broad scope of substrates, excellent regioselectivities and high chemical yields. Furthermore, it was proven to be efficient for large scale preparation (ten gram-scale or greater). Good yield (81%) and excellent regioselectivity were obtained, even the amount of β-nitrostyrene 1a was increased from 0.15 g to 10.0 g in this reaction (Scheme 3). To our delight, the reaction could complete within 18 h, and only a slight decrease in the yield was detected comparing to the 0.15 g scale reaction. This result allows the large scale preparation of vicinal haloamino nitro compounds, useful organic intermediates.20
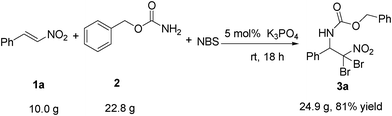 |
| | Scheme 3 Large-scale aminobromination. | |
According to the regiochemistry of the reaction, an Michael addition pathway involving predominant formation of Michael addition intermediate was proposed in Scheme 4 for this aminobromination reaction.17b In the initial step, benzyl carbamate reacts with NBS forming the intermediate A, which is activated by K3PO4 and results in intermediate B. B undergoes Michael addition to β-nitrostyrene 1a giving intermediate C, followed by the trapping of Br+ from NBS to from intermediate D, a normal monobrominated product. Then, the Br+ ion migrates from the N–Br of the amide to the α-position of nitro group, resulting in the final dibrominated product 3a.
 |
| | Scheme 4 Proposed mechanism. | |
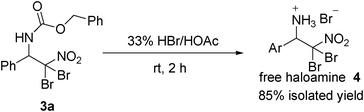
Although various nitrogen sources, including sulfonamides, acid amides and carbamates, have been reported for aminohalogenation,12–19 almost no reports described the deprotection of the resulted aminohalogenation products. The carbobenzoxy usually serves as a conventional amine protecting group, which can be easily removed under hydrogen atmosphere in the presence of Pd/C21 or direct treating with HBr.22 When the aminobromination product 3a was stirred with Pd/C as catalyst under hydrogen atmosphere, 1H NMR disclosed that a mixture was obtained after 1 h, which could not be isolated and purified. After many trials, the free haloamine 4 was obtained with high isolated yield 85% with 2 h, when 3a was stirred with 33% HBr/HOAc at room temperature (Scheme 5). This study could be looked as the first successful deprotection of aminohalogenation product under simple and mild condition.
Experimental section
General information
Unless otherwise stated, all reagents were purchased from commercial sources and used without further purification. Reaction progress was monitored by TLC using silica gel 60F-254 with detection by UV. Flash chromatography was performed using silica gel 60 (200–300 mesh). Thin layer chromatography was carried out on silica gel 60 F-254 TLC plates of 20 cm × 20 cm. Melting points are uncorrected. IR spectra were collected on Bruker Vector 22 in KBr pellets. 1H and 13C NMR (TMS used as internal standard) spectra were recorded with a Bruker ARX300 spectrometer. High resolution mass spectra for all the new compounds were done by Micro mass Q-Tof instrument (ESI). The crystal structure was recorded on a X-ray diffraction spectrometer.
Typical procedure for aminohalogenation of α,β-unsaturated nitro compounds with CbzNH2/NBS
Into a vial were added substrates 1 (0.5 mmol), NBS (1.5 mmol), CbzNH2 (1.5 mmol), K3PO4 (5 mol%). Then, 3 mL of acetonitrile was added to the vial. The solution was stirred at room temperature without the protection of inert gas and monitored by TLC. When the reaction was completed, the mixture was directly purified by TLC plate (Petroleum ether/EtOAc, 4:1).
Benzyl 2,2-dibromo-2-nitro-1-phenylethylcarbamate (3a).
White solid (93% yield). mp 86–87 °C. 1H NMR (300 MHz, CDCl3): δ 7.35–7.45 (m, 10H), 6.42 (d, J = 10.2 Hz, 1H), 5.84 (d, J = 10.2 Hz, 1H), 5.08–5.17 (m, 2H). 13C NMR (75 MHz, CDCl3): δ 154.92, 135.58, 133.38, 129.8, 129.1, 128.67, 128.63, 128.52, 128.41, 93.83, 67.99, 65.15. IR (KBr): ν = 3280, 3064, 2965, 1690, 1573, 1531, 1251 cm−1. HRMS (ESI/[M+Na]+) Calcd For C16H14N2O4Br2Na: 480.9186; found: 480.9193.
Benzyl 2,2-dibromo-1-(2-chlorophenyl)-2-nitroethylcarbamate (3b).
White solid (55% yield). mp 98–99 °C. 1H NMR (300 MHz, CDCl3): δ 7.29–7.43 (m, 9H), 6.03 (d, J = 10.3 Hz, 1H), 5.79 (d, J = 10.1 Hz, 1H), 5.02–5.21 (m, 2H). 13C NMR (75 MHz, CDCl3): δ 154.74, 135.99, 135.40, 131.90, 130.42, 128.9, 128.85, 128.71, 128.68, 128.61, 128.41, 93.10, 68.10, 64.52. IR (KBr): ν = 3339, 3035, 2949, 1693, 1567, 1536, 1352, 1286, 1260, 1028 cm−1. HRMS (ESI/[M+Na]+) Calcd. For C16H13N2O4Br2ClNa: 514.8798; found 514.8802.
Benzyl 2,2-dibromo-1-(3-chlorophenyl)-2-nitroethylcarbamate (3c).
White solid (88% yield). mp 87–89 °C. 1H NMR (300 MHz, CDCl3): δ 7.47–7.57 (m, 2H), 7.28–7.43 (m, 7H), 6.03 (d, J = 10.0 Hz, 1H), 5.94 (d, J = 10.0 Hz, 1H), 5.05–5.19 (m, 2H). 13C NMR (75 MHz, CDCl3): δ 154.75, 135.38, 132.45, 131.85, 130.69, 128.69, 128.62, 128.41, 128.13, 124.26, 92.99, 68.10, 64.59. IR (KBr): ν = 3257, 3059, 2967, 1704, 1683, 1574, 1540, 1494, 1258 cm−1. HRMS (ESI/[M+Na]+) Calcd. For C16H13N2O4Br2ClNa: 514.8797; found 514.8802.
Benzyl 2,2-dibromo-1-(4-bromophenyl)-2-nitroethylcarbamate (3d).
Colorless oil (91% yield). 1H NMR (300 MHz, CDCl3): δ 7.47–7.57 (m, 2H), 7.28–7.42 (m, 7H), 6.03 (d, J = 9.89 Hz, 1H), 5.94 (d, J = 9.8 Hz, 1H), 5.03–5.19 (m, 2H). 13C NMR (75 MHz, CDCl3): δ 154.73, 135.38, 132.45, 131.85, 130.69, 128.69, 128.62, 128.41, 124.26, 92.99, 68.10, 64.59. IR (KBr): ν = 3409, 3310, 3034, 2958, 1708, 1577, 1490, 1323, 1232 cm−1. HRMS (ESI/[M+Na]+) Calcd. For C16H13N2O4Br3Na: 558.8284; found 558.8298.
Benzyl 2,2-dibromo-1-(3-fluorophenyl)-2-nitroethylcarbamate (3e).
White solid (86% yield). mp 102–104 °C. 1H NMR (300 MHz, CDCl3): δ 7.29–7.42 (m, 6H), 7.07–7.25 (m, 3H), 6.04 (d, J = 10.3 Hz, 1H), 5.81 (d, J = 10.1 Hz, 1H), 5.06–5.2 (m, 2H). 13C NMR (75 MHz, CDCl3): δ 162.43 (d, 1JCF = 248.09 Hz), 154.82, 135.73(d, 3JCF = 6.43 Hz), 135.42, 130.23 (d, 3JCF = 7.81 Hz), 128.68, 128.6, 128.39, 125.06, 116.87 (d, 2JCF = 21.13 Hz), 116.2 (d, 2JCF = 23.12 Hz), 92.91, 68.12, 64.60. IR (KBr): ν = 3278, 3063, 2968, 1689, 1575, 1533, 1251, 1236 cm−1. HRMS (ESI/[M+Na]+) Calcd. For C16H13N2O4Br2FNa: 498.9105; found 498.9099.
Benzyl 2,2-dibromo-1-(4-fluorophenyl)-2-nitroethylcarbamate (3f).
White solid (85% yield). mp 91–93 °C. 1H NMR (300 MHz, CDCl3): δ 7.39–749 (m, 2H), 7.28–7.38 (m, 5H), 7.02–7.13 (m, 2H), 6.03 (d, J = 10.1 Hz, 1H), 5.83 (d, J = 10.2 Hz, 1H), 5.05–5.19 (m, 2H). 13C NMR (75 MHz, CDCl3): δ 163.35 (d, 1JCF = 250.46 Hz), 154.81, 135.46, 131.02 (d, 3JCF = 8.37 Hz), 129.32, 128.68, 128.59, 128.37, 115.74 (d, 2JCF = 22.04 Hz), 93.55, 68.06, 64.52. IR (KBr): ν = 3264, 3062, 3038, 1701, 1686, 1579, 1511, 1326, 1257, 1232 cm−1. HRMS (ESI/[M+Na]+) Calcd. For C16H13N2O4Br2FNa: 498.9104; found 498.9099.
Benzyl 2,2-dibromo-2-nitro-1-m-tolylethylcarbamate (3g).
White solid (97% yield). mp 109–111 °C. 1H NMR (300 MHz, CDCl3): δ 7.13–7.46 (m, 9H), 6.01 (d, J = 10.3 Hz, 1H), 5.87 (d, J = 10.4 Hz, 1H), 5.01–5.24 (m, 2H), 2.35 (s, 3H). 13C NMR (75 MHz, CDCl3): δ 154.84, 138.44, 135.53, 133.25, 130.53, 129.77, 128.65, 128.57, 128.52, 128.43, 125.98, 93.79, 67.95, 65.11, 21.48. IR (KBr): ν = 3275, 3060, 3038, 1705, 1688, 1574, 1533, 1251, 1054 cm−1. HRMS (ESI/[M+Na]+) Calcd. For C17H16N2O4Br2Na 494.9336; found 494.935.
Benzyl 2,2-dibromo-2-nitro-1-p-tolylethylcarbamate (3h).
White solid (62% yield). mp 93–95 °C. 1H NMR (300 MHz, CDCl3): δ 7.27–7.43 (m, 7H), 7.14–7.22 (m, 2H), 6.01 (d, J = 10.2 Hz, 1H), 5.83 (d, J = 10.2 Hz, 1H), 5.05–5.19 (m, 2H), 2.36 (s, 3H). 13C NMR (75 MHz, CDCl3): δ 154.89, 139.88, 135.57, 130.32, 129.34, 128.89, 128.65, 128.52, 128.40, 94.06, 67.93, 64.95, 21.23. IR (KBr): ν = 3411, 3312, 3033, 2957, 1712, 1575, 1513, 1324, 1233, 1049 cm−1. HRMS (ESI/[M+Na]+) Calcd. For C17H16N2O4Br2Na: 494.9340; found 494.9350.
Benzyl 2,2-dibromo-1-(naphthalen-1-yl)-2-nitroethylcarbamate (3i).
Yellow solid (89% yield). mp 103–105 °C. 1H NMR (300 MHz, CDCl3): δ 8.44 (d, J = 8.12 Hz, 1H), 7.45–7.98 (m, 6H), 7.27–7.43 (m, 5H), 7.1 (d, J = 10.2 Hz, 1H), 5.94 (d, J = 10.1 Hz, 1H), 5.01–5.18 (m, 2H). 13C NMR (75 MHz, CDCl3): δ 155.01, 135.44, 133.74, 132.01, 131.16, 130.56, 129.03, 128.64, 128.52, 128.39, 127.32, 126.41, 125.24, 124.92, 123.44, 93.76, 68.02, 58.63. IR (KBr): ν = 3412, 3311, 3065, 3035, 1713, 1577, 1322, 1058, 910 cm−1. HRMS (ESI/[M+Na]+) Calcd For C20H16N2O4Br2Na: 530.9350; found 530.9350.
Benzyl 2,2-dibromo-1-(3-methoxyphenyl)-2-nitroethylcarbamate (3j).
White solid (83% yield). mp 97–99 °C. 1H NMR (300 MHz, CDCl3): δ 7.28–7.45 (m, 6H), 6.89–7.05 (m, 3H), 6.01 (d, J = 10.3 Hz, 1H), 5.81 (d, J = 10.3 Hz, 1H), 5.05–5.19 (m, 2H), 3.81 (s, 3H). 13C NMR (75 MHz, CDCl3): δ 159.52, 154.82, 135.50, 134.74, 129.67, 128.64, 128.52, 128.4, 121.17, 115.16, 114.92, 93.53, 67.98, 65.06, 55.38. IR (KBr): ν = 3286, 3062, 2960, 2886, 1691, 1574, 1531, 1251, 1230, 1056 cm−1. HRMS (ESI/[M+Na]+) Calcd For C17H16N2O5Br2Na: 510.9304; found 510.9299.
Benzyl 2,2-dibromo-1-(4-methoxyphenyl)-2-nitroethylcarbamate (3k).
Yellow oil (95% yield). 1H NMR (300 MHz, CDCl3): δ 7.29–7.44 (m, 7H), 6.84–6.93 (m, 2H), 5.99 (d, J = 10.4 Hz, 1H), 5.79 (d, J = 10.2 Hz, 1H), 5.05–5.21 (m, 2H), 3.81 (s, 3H). 13C NMR (75 MHz, CDCl3): δ 160.54, 154.85, 135.57, 130.28, 128.64, 128.51, 128.38, 125.17, 113.99, 94.28, 67.92, 64.73, 55.33. IR (KBr): ν = 3411, 3317, 2959, 2839, 1713, 1574, 1513, 1250, 1030 cm−1. HRMS (ESI/[M+Na]+) Calcd For C17H16N2O5Br2Na: 510.9299; found 510.9299.
Benzyl 1-(4-(benzyloxy)phenyl)-2,2-dibromo-2-nitroethylcarbamate (3l).
Colorless oil (78% yield). 1H NMR (300 MHz, CDCl3): δ 7.27–7.58 (m, 12H), 6.94–7.08 (m, 2H), 6.53 (d, J = 10.3 Hz, 1H), 6.44 (d, J = 10.2 Hz, 1H), 5.03–5.21 (m, 4H). 13C NMR (75 MHz, CDCl3): δ 156.79, 155.06, 136.15, 135.79, 131.01, 130.62, 128.80, 128.59, 128.37, 128.24, 127.35, 121.05, 112.91, 94.01, 70.69, 67.68, 62.11. IR (KBr): ν = 3412, 3316, 3033, 2955, 2882, 1716, 1575, 1497, 1227, 1043 cm−1. HRMS (ESI/[M+Na]+) Calcd. For C23H20N2O5Br2Na: 586.9605; found 586.9612.
Benzyl 2,2-dibromo-2-nitro-1-(4-(trifluoromethyl)phenyl)ethylcarbamate (3m).
Colorless oil (83% yield). 1H NMR (300 MHz, CDCl3): δ 7.66 (d, J = 8.52 Hz, 2H), 7.59 (d, J = 8.52 Hz, 2H), 7.29–7.43 (m, 5H), 6.12 (d, J = 10.4 Hz, 1H), 5.85 (d, J = 10.2 Hz, 1H), 5.05–5.19 (m, 2H). 13C NMR (75 MHz, CDCl3): δ 154.69, 137.36, 135.32, 131.85 (q, 2JCF = 32.91 Hz), 129.64, 128.68, 128.4, 125.59 (q, 3JCF = 3.27 Hz), 123.65 (q, 1JCF = 273.29 Hz), 92.46, 68.18, 64.64. IR (KBr): ν = 3415, 3311, 3035, 2959, 1712, 1578, 1326, 1131, 1071, 1018 cm−1. HRMS (ESI/[M+Na]+) Calcd For C17H13N2O4Br2F3Na: 548.9075; found 548.9067.
Benzyl 2,2-dibromo-1-(furan-2-yl)-2-nitroethylcarbamate (3n).
Brown oil (68% yield). 1H NMR (300 MHz, CDCl3): δ 7.42 (dd, J = 0.75, 1.78 Hz, 1H), 7.31–7.41 (m, 5H), 6.44 (d, J = 3.32 Hz, 1H), 6.38 (dd, J = 1.9, 3.35 Hz, 1H), 6.24 (d, J = 10.44 Hz, 1H), 5.84 (d, J = 10.23 Hz, 1H), 5.11–5.26 (m, 2H). 13C NMR (75 MHz, CDCl3): δ 155.07, 146.03, 143.74, 135.49, 128.68, 128.58, 128.39, 111.54, 110.95, 91.18, 68.11, 60.45. IR (KBr): ν = 3254, 3034, 2956, 1694, 1572, 1538, 1323, 1257, 1016 cm−1. HRMS (ESI/[M+Na]+) Calcd For C14H12N2O5Br2Na: 470.8971; found 470.8986.
Benzyl 2,2-dibromo-2-nitro-1-(thiophen-2-yl)ethylcarbamate (3o).
Yellow solid (72% yield). mp 98–100 °C. 1H NMR (300 MHz, CDCl3): δ 7.29–7.42 (m, 6H), 7.18 (d, J = 3.72 Hz, 1H), 7.01 (dd, J = 3.62, 5.13 Hz, 1H), 6.38 (d, J = 10.44 Hz, 1H), 5.7 (d, J = 10.39 Hz, 1H), 5.07–5.24 (m, 2H). 13C NMR (75 MHz, CDCl3): δ 154.72, 135.42, 129.58, 128.66, 128.57, 128.38, 127.37, 126.9, 126.83, 92.62, 68.11, 62.18. IR (KBr): ν = 3269, 3033, 2955, 1693, 1577, 1522, 1323, 1249 cm−1. HRMS (ESI/[M+Na]+) Calcd For C14H12N2O4Br2SNa: 486.8753; found 486.8757.
Benzyl 1,1-dibromo-1-nitrononan-2-ylcarbamate (3p).
Colorless oil (82% yield). 1H NMR (300 MHz, CDCl3): δ 7.31–7.42 (m, 5H), 5.08–5.25 (m, 2H), 4.98 (d, J = 10.47 Hz, 1H), 4.65–4.88 (m, 1H), 1.76–1.97 (m, 1H), 1.07–1.55 (m, 11H), 0.88 (t, J = 6.97 Hz, 3H). 13C NMR (75 MHz, CDCl3): δ 155.57, 135.83, 128.62, 128.44, 128.21, 93.84, 67.66, 62.43, 32.27, 31.65, 29.00, 28.92, 25.81, 22.60, 14.09. IR (KBr): ν = 3404, 3309, 2955, 2928, 2857, 1709, 1575, 1326, 1247, 1052 cm−1. HRMS (ESI/[M+Na]+) Calcd For C17H24N2O4Br2Na: 502.9978; found 502.9976.
Benzyl 2,2-dibromo-1-(3-bromo-4-methoxyphenyl)-2-nitroethylcarbamate (3q).
Colorless oil (87% yield). 1H NMR (300 MHz, CDCl3): δ 7.65 (d, J = 2.36 Hz, 1H), 7.28–7.42 (m, 6H), 6.86 (d, J = 8.57 Hz, 1H), 5.99 (d, J = 10.42 Hz, 1H), 5.84 (d, J = 10.26 Hz, 1H), 5.04–5.18 (m, 2H), 3.9 (s, 3H). 13C NMR (75 MHz, CDCl3): δ 156.88, 154.67, 135.4, 133.62, 129.65, 128.67, 128.57, 128.39, 126.69, 111.79, 111.48, 93.51, 68.07, 64.12, 56.34. IR (KBr): ν = 3409, 3309, 2209, 2958, 2841, 1712, 1576, 1498, 1294, 1056 cm−1. HRMS (ESI/[M+Na]+) Calcd For C17H15N2O5Br3Na: 588.8406; found 588.8404.
Benzyl 2,2-dibromo-1-(4-cyanophenyl)-2-nitroethylcarbamate (3r).
Colorless oil (81% yield). 1H NMR (300 MHz, CDCl3): δ 7.68 (d, J = 8.35 Hz, 2H), 7.59 (d, J = 8.35 Hz, 2H), 7.28–7.41 (m, 5H), 6.11 (d, J = 10.51 Hz, 1H), 5.92 (d, J = 10.41 Hz, 1H), 5.06–5.18 (m, 2H). 13C NMR (75 MHz, CDCl3): δ 154.59, 138.48, 135.19, 132.30, 129.95, 128.69, 128.41, 117.89, 113.83, 91.88, 68.27, 64.62. IR (KBr): ν = 3312, 2957, 2232, 1731, 1715, 1576, 1506, 1232, 1050 cm−1. HRMS (ESI/M+Na]+) Calcd. For C17H13N3O4Br2Na: 505.9150; found 505.9146.
To a flask containing 3a (2 mmol), a solution of HBr in AcOH (5mL, 33% w/w) was added. The mixture was stirred at room temperature for 2 h. When evolution of bubbles stopped, excess HBr and HOAc were filtered, giving white powder 4 with 85% yield.
2,2-dibromo-2-nitro-1-phenylethanaminium bromide (4).
White solid (85% yield). 1H NMR (300 MHz, D2O): δ 7.48–7.53 (m, 5H), 5.33 (s, 1H). 13C NMR (75 MHz, D2O): δ 131.66, 130.6, 129.7, 128.42, 127.67, 59.26. IR (KBr): ν = 3264, 3013, 2908, 1959, 1630, 1573, 1499, 1399, 997cm−1.
Conclusions
In summary, we have developed a new aminobromination reaction with benzyl carbamate/NBS as nitrogen/bromine source. This facile and efficient system tolerated a broad range of substrates (aromatic, aliphatic and heterocyclic), giving good to excellent yields, even on the large scale preparation. Furthermore, free bromoamine product could be easily obtained by removing the N-protecting group under convenient and mild conditions. Further study on aminobromination of this nitrogen source is under progress.
Acknowledgements
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (No. 21102071) and the Fundamental Research Funds for the Central Universities (No. 1107020522 and No. 1082020502). The Jiangsu 333 program (for Pan) and Changzhou Jin-Feng-Huang program (for Han) are also acknowledged.
References
- J. Qui and R. B. Silverman, J. Med. Chem., 2000, 43, 706 CrossRef.
-
(a) D. Chen, C. Timmons, L. Guo, X. Xu and G. Li, Synthesis, 2004, 2749 Search PubMed;
(b) D. Chen, L. Guo, J. Liu, S. Kirtane, J. F. Cannon and G. Li, Org. Lett., 2005, 7, 921 CrossRef CAS;
(c) H. B. Mei, L. J. Yan, J. L. Han, G. Li and Y. Pan, Chem. Biol. Drug Des., 2010, 76, 392 CrossRef CAS;
(d) J. L. Han, Y. F. Li, S. J. Zhi, Y. Pan, C. Timmons and G. Li, Tetrahedron Lett., 2006, 47, 7225 CrossRef CAS.
-
J. E. Kemp, In Comprehensive Organic Synthesis, Vol. 3; B. M. Trost and I. Fleming, ed.; Pergamon Press: Oxford, 1991, 469–513 Search PubMed.
-
(a) Y. Y. Yeung, X. Gao and E. J. Corey, J. Am. Chem. Soc., 2006, 128, 9644 CrossRef CAS;
(b) D. A. Griffith and S. J. Danishefsky, J. Am. Chem. Soc., 1991, 113, 5863 CrossRef CAS;
(c) J. Lessard, H. Driguez and J. P. Vermes, Tetrahedron Lett., 1970, 11, 4887 CrossRef.
-
(a) F. A. Daniher and P. E. Butler, J. Org. Chem., 1968, 33, 4336 CrossRef CAS;
(b) F. A. Daniher and P. E. Butler, J. Org. Chem., 1968, 33, 2637 CrossRef CAS;
(c) F. A. Daniher, M. T. Melchior and P. E. Butler, Chem.Commun., 1968, 931 RSC;
(d) B. S. Orlek and G. Stemp, Tetrahedron Lett., 1991, 32, 4045 CrossRef CAS.
- For recent review on aminohalogenation, see: G. Li, S. R. S. Saibabu Kotti and C. Timmons, Eur. J. Org. Chem., 2007, 2745 CrossRef CAS.
-
(a) G. Li, H. X. Wei, S. H. Kim and M. Neighbors, Org. Lett., 1999, 1, 395 CrossRef CAS;
(b) H. X. Wei, S. H. Kim and G. Li, Tetrahedron, 2001, 57, 3869 CrossRef CAS;
(c) G. Li, H. X. Wei and S. H. Kim, Tetrahedron, 2001, 57, 8407 CrossRef CAS.
- J. L. Han, S. J. Zhi, L. Y. Wang, Y. Pan and G. Li, Eur. J. Org. Chem., 2007, 1332 CrossRef CAS.
-
(a) H. Sun, G. Q. Zhang, S. J. Zhi, J. L. Han, G. Li and Y. Pan, Org. Biomol. Chem., 2010, 8, 4236 RSC;
(b) D. Chen, C. Timmons, S. Chao and G. Li, Eur. J. Org. Chem., 2004, 3097 CrossRef CAS;
(c) Z. G. Chen, J. F. Wei, R. T. Li, X. Y. Shi and P. F. Zhao, J. Org. Chem., 2009, 74, 1371 CrossRef CAS.
-
(a) Q. Li, M. Shi, C. Timmons and G. Li, Org. Lett., 2006, 8, 625 CrossRef CAS;
(b) M. H. Qi, L. X. Shao and M. Shi, Chin. J. Chem., 2011, 29, 2739 CrossRef CAS.
- S. J. Zhi, J. L. Han, C. Lin, G. H. An, Y. Pan and G. Li, Synthesis, 2008, 1570 CAS.
-
(a) For selected examples on the aminohalogenation with sulfonamides as nitrogen source, see: L. Revesz, E. Blum and R. Wicki, Tetrahedron Lett., 2005, 46, 5577 CrossRef CAS;
(b) S. Minakata, Y. Yoneda, Y. Oderaotoshi and M. Komatsu, Org. Lett., 2006, 8, 967 CrossRef CAS;
(c) G. W. Wang and X. L. Wu, Adv. Synth. Catal., 2007, 349, 1977 CrossRef CAS;
(d) V. V. Thakur, S. K. Talluri and A. Sudalai, Org. Lett., 2003, 5, 861 CrossRef CAS;
(e) X. L. Wu, J. J. Xia and G. W. Wang, Org. Biomol. Chem., 2008, 6, 548 RSC;
(f) J. F. Wei, L. H. Zhang, Z. G. Chen, X. Y. Shi and J. J. Cao, Org. Biomol. Chem., 2009, 7, 3280 RSC;
(g) Z. Wang, Y. M. Zhang, H. Fu, Y. Y. Jiang and Y. F. Zhao, Synlett, 2008, 2667 CAS.
-
(a) T. A. Foglia and D. Swern, J. Org. Chem., 1966, 31, 3625 CrossRef CAS;
(b) T. A. Foglia and D. Swern, J. Org. Chem., 1968, 33, 766 CrossRef CAS;
(c) B. Crookes, T. P. Seden and R. W. Turner, Chem. Commun., 1968, 324 Search PubMed.
- S. J. Zhi, H. Sun, C. Lin, G. Q. Zhang, G. Li and Y. Pan, Sci. China: Chem., 2010, 53, 140 CrossRef CAS.
- Z. G. Chen, Y. Wang, J. F. Wei, P. F. Zhao and X. Y. Shi, J. Org. Chem., 2010, 75, 2085 CrossRef CAS.
- S. J. Zhi, H. B. Mei, G. Q. Zhang, H. Sun, J. L. Han, G. Li and Y. Pan, Sci. China: Chem., 2010, 53, 1946 CrossRef CAS.
-
(a) H. B. Mei, J. L. Han, G. Li and Y. Pan, RSC Adv., 2011, 1, 429 RSC;
(b) H. B. Mei, Y. W. Xiong, Y. Qian, J. L. Han, G. Li and Y. Pan, RSC Adv., 2012, 2, 151 RSC.
- Z. G. Chen, P. F. Zhao and Y. Wang, Eur. J. Org. Chem., 2011, 5887 CrossRef CAS.
- X. Y. Ji, H. B. Mei, Y. Qian, J. L. Han, G. Li and Y. Pan, Synthesis, 2011, 3680 CAS.
-
(a) D. Enders and J. Wiedemann, Synthesis, 1996, 1443 CrossRef CAS;
(b) D. Lucet, L. Toupet, T. L. Gall and G. Mioskowski, J. Org. Chem., 1997, 62, 2682 CrossRef CAS.
- A. Schmauder, L. D. Sibley and M. E. Maier, Chem.–Eur. J., 2010, 16, 4328 CrossRef CAS.
- S. L. Deng, B. Isabelle, N. Mohammed and C. Christian, Heteroat. Chem., 2008, 19, 55 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.