Magnetic ZnO surface-imprinted polymers prepared by ARGET ATRP and the application for antibiotics selective recognition†
Longcheng
Xu
,
Jianming
Pan
,
Jiangdong
Dai
,
Zhijing
Cao
,
Hui
Hang
,
Xiuxiu
Li
and
Yongsheng
Yan
*
School of Chemistry and Chemical Engineering, Jiangsu University, Zhenjiang, 212013, People's Republic of China. E-mail: xulongcheng123@126.com; Fax: +86 511 88791800; Tel: +86 511 8890683
First published on 4th April 2012
Abstract
This paper reports a molecularly imprinted polymers (MIPs) based fluorescence sensor which is synthesized by grafting MIP layers on the surface of ZnO nanorods embedded γ-Fe2O3 nanoparticles via activators regenerated by electron transfer atom transfer radical polymerization (ARGET ATRP). Methacrylic acid (MAA, functional monomer), 3-(trimethoxysilyl)propyl mathacrylate (MPS) modified γ-Fe2O3 (γ-Fe2O3/MPS, assistant magnetic monomer) and ethylene glycol dimethacrylate (EGDMA, cross-linking monomer) were co-polymerized in anisole at 313 K in the presence of sulfamethazine as a template molecule. Sulfamethazine was then solvent-extracted to obtain ZnO-grafted molecularly imprinted polymers (ZnO-MIPs). ZnO-MIPs were characterized by FE-SEM, TEM, FT-IR, TGA/DSC, VSM, fluorescence spectroscopy and Raman spectroscopy. It was observed that sulfamethazine can quench the luminescence of ZnO-MIPs in a concentration-dependent manner that can be described by a Stern–Volmer-type equation. ZnO-MIPs were used to determine sulfamethazine in a spiked pork sample with good recognition ability. This study therefore demonstrates the potential application in the recognition and separation of antibiotics based on molecularly imprinted polymers.
Introduction
In recent years, molecular imprinting polymers (MIPs) have attracted great interest because of their potential applications in protein separation,1 extraction of small organic molecules,2 highly selective sensing,3 catalysts4 and enzyme inhibitors.5 Traditionally, bulk polymerization has been known as a useful strategy to obtain MIPs. However, the extraction of template suffers from obstacles due to the high cross-linking nature of MIPs. Recently, many efforts have been made on surface imprinting,6,7 in which the imprinted templates are situated at the surface or in the proximity of the material's surface. The surface imprinting technique provides an alternative method to reduce mass transfer limitations and reduce permanent entrapment of templates.8MIPs can be produced by emulsion polymerization,9 distillation precipitation polymerization10 or free radical polymerization.11 Free radical polymerization is the most investigated technology and was performed in preparing the polymeric materials prior to use as artificial receptors. Surface-initiated atom transfer radical polymerization (SI-ATRP) is one of the controlled radical polymerization (CRP) methods providing polymers with controlled molecular weights and low polydispersities for a wide range of vinyl monomers and solvents.12 CRP methods13 have previously been used to fabricate MIPs and have led to a greater understanding of the imprinting mechanism and optimization of binding parameters. Particularly, Husson et al.14 have used ATRP to control the thickness of polymer by surface plasmon resonance. One crucial disadvantage of ATRP, however, is that a relatively large amount of transition-metal catalyst is needed. Consequently, the materials become coloured and it is necessary to remove the residual catalyst. Another disadvantage is that the ATRP reaction must be conducted in a completely inert atmosphere. A new technology based on “activators regenerated by electron transfer atom transfer radical polymerization” (ARGET ATRP) was developed by Matyjazewski et al.15 In ARGET ATRP, an inactive Cu(II) species is rapidly reduced to an active Cu(I) species in the presence of a reducing agent and only a few ppm of Cu(II) is needed to mediate the polymerization.15 Ascorbic acid is a strong reducing agent and can convert Cu(II) to Cu(I) species very quickly, as such this would fail to control polymerization if high concentrations of radicals were generated. However, ascorbic acid has been successfully applied to ARGET ATRP under heterogeneous conditions when anisole is used as solvent.16 The solubility of ascorbic acid in anisole is limited so avoiding too high a concentration of radicals, while the large excess of ascorbic acid could allow the reaction to be conducted in the presence of limited amounts of air.15
As we know, high selectivity and sensitivity play a key role in molecular recognition. To date, functional MIPs have aroused great interest, including temperature response,17 pH response,18 photoresponse,19 magnetic response,20 and fluorescence characteristics.21 When magnetic nanoparticles are encapsulated into MIPs, the resulting magnetic molecularly imprinted polymers will not only have selectivity for the template polymers, but can be separated by an external magnetic field. The unique electronic and optical properties of metal and semiconductor nanoparticles and nanorods, add new interests to the area of detection. ZnO is a wide-band-gap semiconductor material (Eg = 3.35 eV) with a large excitation binding energy (60 meV) at room temperature.22 Because ZnO is abundant, environmentally friendly and exhibits high chemical stability, it has potential use in efficient UV detection, so ZnO-polymer composite systems are considered important for UV optoelectronic applications.23 However, the selectivity of composites has, as yet, not been satisfactory. It is hoped that for ZnO combined with MIPs, the obtained materials can achieve highly selective optoelectronic detection of analytes. Surface molecularly imprinted polymers embedded QDs have been used to detect 4-nitrophenol24 and pyrethroid25 based on room-temperature fluorescence quenching mechanism of electron transfer. Composites combining ZnO with MIPs are hoped to sensitively and selectively detect analytes.
Sulfonamide antibiotics are widely used in treating infections in human therapy,26 and livestock production.27 However, due to concerns about sulfonamide antibiotics inducing high level of resistance,28 recent interests have been on developing selective materials for their recognition and separation.29,30 Consequently, materials targeting selective recognition and separation of sulfonamide antibiotics from complex matrices prior to their detection are required.
Here, we report the formation of molecular recognition sites on the surface of SiO2-modified ZnO nanorods by ARGET ATRP. Sulfamethazine (SMZ) was the chosen target molecule while methacrylic acid (MAA) was selected as the functional monomer, 3-(trimethoxysilyl)propyl mathacrylate (MPS) modified γ-Fe2O3 as assistant monomer on the support surface provides basic functional groups, which can interact with the sulfamethazine, and can significantly improve the imprinting effect of the resulting ”stick–ball” ZnO-MIPs. Bath binding tests were carried out to evaluate the binding characteristics of the ZnO-MIPs. The obtained ZnO-MIPs were characterized by FE-SEM, TEM, FT-IR, TGA/DSC, VSM, fluorescence spectroscopy and Raman spectroscopy. The resultant imprinted polymer coupled to ZnO fluorescent property can be optimized to the detection of sulfonamide-based chemical warfare agents. Under optimal conditions, the relative fluorescence intensity decreased linearly with the increasing concentration of SMZ in the range of 0.002–0.1 mM with a detection limit of 19.0 μg L−1. Finally, the ZnO-MIPs were applied for selective recognition of SMZ in an SMZ-spiked pork sample. More specifically, the main focus of ARGET ATRP herein is, however, not to obtain a system with perfect control, but rather to be able to form a layer of a high affinity imprinted polymer on the surface of ZnO with a binding site population that is as homogeneous as possible.
Experimental
Materials
Ethylene glycol dimethacrylate (EGDMA, 98%), 2-bromoisobutyryl bromide (BiB), N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA, 99%), sulfamethazine (SMZ) and γ-Fe2O3 were purchased from Aladdin reagent Co., Ltd. (Shanghai, China). Methacrylic acid (MAA), CuCl2, triethylamine (TEA), ascorbic acid (AsAc), anisole, dichloromethane (CH2Cl2), zinc nitrate hexahydrate (Zn(NO3)2·6H2O), anhydrous sodium sulfate (Na2SO4), 2-propyl alcohol, hydrazine monohydrate (N2H4·H2O) (85% v/v), ammonia solution (25.0–28.0%), tetraethyl orthosilicate (TEOS) and 3-(methacryloxyl)propyl trimethoxysilane (MPS) were all obtained from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China).PMDETA and BiB were dried by CaH2 overnight and distilled under reduced pressure, respectively. Doubly distilled water was used for preparing all aqueous solutions and cleaning processes.
Characterization
The morphology of ZnO-MIPs A was carried out on a transmission electron microscope (TEM, JEOL, JEM-200CX) and a field-emission scanning electron microscope (FE-SEM, S-4800). Fluorescence spectroscopy of ZnO-MIPs and ZnO-NIPs B was measured on a PTI QuantaMaster model C-60/2000 spectrofluorometer (Photon Technology International, Lawrenceville, NJ, USA). Infrared spectra (4000–500 cm−1) were collected on a Nicolet NEXUS-470 FT-IR apparatus (USA) using KBr disks. Magnetic measurements was carried out using a vibrating sample magnetometer (VSM, 7300, Lakeshore) under a magnetic field. Raman spectra were recorded in the range of 200–800 cm−1 at ambient temperature using a WITEC Spectra Pro 2300I spectrometer equipped with an Ar-ion laser, which provided a laser beam of 514 nm wavelength. Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) were carried out using a DSC/DTA-TG (STA 449C Jupiter, Netzsch, Germany). Chromatographic analysis was carried out on an Agilent 1200 LC system (Agilent, Germany) equipped with UV detection. The UV-vis adsorption spectra were obtained with a UV-vis spectrophotometer. (UV-2450, Shimadzu, Japan).Preparation of ZnO nanorods
In a typical procedure,31 1.188 g of Zn(NO3)2·6H2O and 0.568 g of Na2SO4 were loaded into a Teflon-lined stainless steel autoclave of 100 mL capacity and dissolved in 60 mL deionized water. Then 20 mL of hydrazine monohydrate (85% v/v) solution was added dropwise during vigorous stirring. 10 min later, the autoclave was sealed and maintained at 453 K for 24 h. The resulting precipitate was collected by filtration and washed with absolute ethanol several times. The final product was dried in a vacuum oven at 333 K overnight.Preparation of ARGET ATRP initiator
0.6 g of ZnO nanorods were dispersed in 75 mL 2-propyl alcohol and 6.0 mL deionized water by sonication for 10 min, followed by the dropwise addition of 2.25 mL ammonia solution and 1.5 mL TEOS. The resulting ZnO/SiO2 was placed in a three-necked round-bottom flask containing 30 CH2Cl2. TEA (1.0 mL) was then added and after the mixture was bubbled three times with high-purity nitrogen in an ice-bath, 2-bromopropionyl bromide (1.0 mL) was added dropwise to start the reaction. The mixture was stirred at 273 K for 24 h under N2 protection. The initiator was thoroughly washed with CH2Cl2, ethanol and then dried under a stream of N2.Preparation of γ-Fe2O3/MPS
30 mgc of γ-Fe2O3 nanoparticles was dispersed in 100 mL ethanol and 10 mL deionized water and the mixture was sonicated for 10 min. Then 0.3 mL ammonia solution and 0.2 mL MPS were added slowly. The reaction was kept at room temperature for 12 h. The resulting material was dried in a vacuum oven at 333 K overnight.Preparation of ZnO-grafted molecularly imprinted polymers (ZnO-MIPs A) and ZnO non-imprinted polymers (ZnO-NIPs B)
The molecular imprinting polymerization reaction was carried out in organic solution using anisole as the solvent, MAA as the functional monomer, γ-Fe2O3/MPS as the assistant monomer, EGDMA as the cross-linking monomer, CuCl2 as the catalyst, PMDETA as the ligand and AsAc as the reducing agent. Typically, the SMZ (0.2783 g, 1.0 mmol), MAA (0.3410 mL, 4.0 mmol), EGDMA (3.7 mL, 20 mmol) were dissolved in 30 mL anisole. Then γ-Fe2O3/MPS (10 mg), PMDETA (52 μL, 0.25 mmol) were added, CuCl2 (3.4 mg, 25 μmol) and AsAc (44 mg, 0.25 mmol) were quickly transferred into the flask under the protection of N2. The reaction proceeded at 313 K for 24 h with vigorous vibration. After the polymerization, the mixture was redispersed in ethanol and the product was collected by magnet. Then the obtained ZnO-MIPs A were washed with the mixture solution of methanol/acetic acid (95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5, v/v) using Soxhlet extraction to remove the template molecules. Finally, the obtained ZnO-MIPs A were dried at 333 K under vacuum overnight. Moreover, the nonimprinted polymers based on ZnO nanorods (Zn-NIPs B) were also prepared by a similar way but without the addition of SMZ.
:5, v/v) using Soxhlet extraction to remove the template molecules. Finally, the obtained ZnO-MIPs A were dried at 333 K under vacuum overnight. Moreover, the nonimprinted polymers based on ZnO nanorods (Zn-NIPs B) were also prepared by a similar way but without the addition of SMZ.
Binding behaviour of ZnO-MIPs A and ZnO-NIPs B
A binding kinetic study was carried out to test the rate of the adsorption process for ZnO-MIPs A and ZnO-NIPs B. The experiments were performed using an initial SMZ concentration of 0.05 mmol L−1 and the temperature was kept at 298 K in a water-bath. To obtain the adsorption capacity, ZnO-MIPs A (5 mg) were dispersed in a single-component solution (10 mL) with various concentrations of SMZ from 0.0005 to 0.1 mmol L−1, and the temperature was also kept at 298 K in a water bath. The concentration of SMZ was detected by UV-vis spectrophotometer at 238 nm. The equilibrium binding amounts were calculated according to eqn (1), and the binding isotherms were plotted.
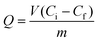
| (1) |
Determination of sulfamethazine by fluorescence measurement
The fluorescence spectra analysis of the samples were carried out by dispersing 20 mg of ZnO-MIPs A in 1.0 mL ethanol, then 0.1 mL SMZ solutions with concentrations ranging from 0.002 to 0.1 mmol L−1 were added. The samples were mixed end-over-end at room temperature for 12 h, and afterwards the sorbent was dried at room temperature and measured with a QuantaMaster C-60/2000 spectrofluorometer under a 364 nm excitation light source with slit width of 2 nm.Selectivity experiments
Sulfadiazine (SD), sulfamethizol (SMI) and tetracycline (TC), whose structures were shown in Fig. S5 (ESI†) were chosen as structural analogues to evaluate the selectivity of ZnO-MIPs. ZnO-MIPs A and ZnO-NIPs B (5 mg) were dispersed in the mixture of SMZ and other three analogues, respectively. The binding amounts of SMZ and structural analogues were compared. Competitive binding of ZnO-MIPs A and ZnO-NIPs B for SMZ in the presence of 0.1 mmol L−1 of SD, SMI and TC were also investigated. The experiments were all carried out in a tube at 298 K for 12 h.Sample preparation and SPE procedure
In order to evaluate the potential applications of ZnO-MIPs A in real sample analysis a fresh pork sample was crushed into homogenate paste, and then 10 g of the homogenate was dispersed in 15% (w/v) trichloroacetic acid aqueous solution. After stirring for 6.0 h at 298 K, the mixture was filtered and the solution was collected. 0.2 mL of the extraction solution and an appropriate amount of SMZ (concentration of 2 × 10−4 mmol L−1) was added to the aqueous solution to obtain 100 mL spiked pork sample.The SPE process was as follows. 50 mg of ZnO-MIPs A (or ZnO-NIPs B) were added into a tube and then 40 mL of SMZ solution was added and kept for 6.0 h at 298 K. Then the reacted ZnO-MIPs A (or ZnO-NIPs B) were collected by a magnet and washed with 10 mL methanol–acetic acid solution (v/v = 9:1). The extracts were dried at 298 K and the residues were redissolved in 400 μL methanol and then filtered through a 0.22 μm membrane filter for further chromatographic analysis.
A C18 column (150 mm × 4.6 mm ID, 3.5 μm) was used as the analytical column. HPLC conditions employed for the SMZ separation were as follows: mobile phase, methanol–water (30:70, v/v), (pH adjusted to 3.0 with acetic acid (30:70, v/v) for the simultaneous separation of other antibiotics); flow rate, 1.0 mL min−1; room temperature; UV detection, at 280 nm; injection volume, 20 μL.
Results and discussion
Synthesis and characterization of imprinted polymer, non-imprinted polymer and precursors
The schematic procedure of preparing ZnO-MIPs A is shown in Scheme 1. The ZnO nanorods were first prepared by a hydrothermal method. Then ARGET ATRP initiator was derived from silica-modified ZnO nanorods. The surface-imprinted polymers prepared via ARGET ATRP were obtained by immersing initiator-functionalized ZnO nanorods in a one-pot mixture of SMZ, anisole, MAA, γ-Fe2O3/MPS, EGDMA, CuCl2, PMDETA and AsAc, and then the solution were purged with nitrogen for 20 min. Anisole, a weak polar solvent, was helpful for the formation of hydrogen bonds between SMZ and functional monomer which would be disrupted by the use of a more strongly polar solvent. The reaction was kept at 313 K for 24 h with vigorous stirring. The obtained imprinted polymers were washed with methanol–acetic acid (95:5, v/v) using Soxhlet extraction to remove the template molecules. The aim of the study was not to obtain truly monodisperse grafts but rather to accomplish a sufficiently good control over the polymerization so that an even surface layer of polymers was obtained.
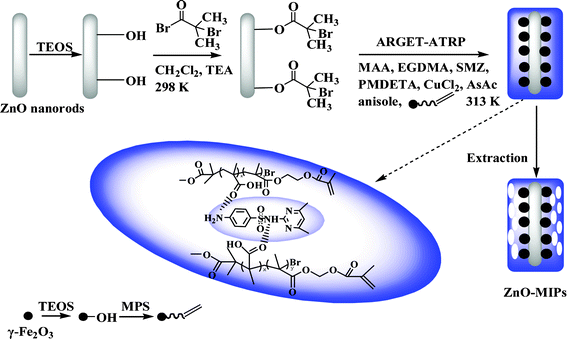 | ||
| Scheme 1 Schematic illustration of the fabrication of magnetic ZnO surface-imprinted polymers via ARGET ATRP. | ||
The morphologies of ZnO-MIPs A were observed by SEM and TEM, which are shown in Fig. 1 while a typical SEM image of wurtzite ZnO is shown in Fig. S1 (ESI†). The images of stick–ball magnetic fluorescence molecules imprinted polymers based on ZnO nanorods are shown in Fig. 1a and b. MPS modified γ-Fe2O3 nanoparticles were attached to the surface of ZnO in the process of polymerization reaction. The thickness of the imprinted polymer layer was about 200 nm as observed from Fig. 1c and d. Therefore, it could be concluded that the polymerization was well controlled by ARGET ATRP and it was possible to obtain MIP layers with nanoscale thicknesses.
 | ||
| Fig. 1 FE-SEM image of ZnO-MIPs A at low magnification (a), FE-SEM image of ZnO-MIPs at a higher magnification (b), TEM images of ZnO-MIPs (c, d). | ||
The FT-IR spectra of ZnO-MIPs A, ZnO-NIPs B and precursors were measured and shown in Fig. 2 and Fig. S2.† The 500–1700 cm−1 spectral region of ZnO-Br (Fig. S2, ESI†) exhibited a striking difference in comparison to ZnO nanorods, in that it had four peaks at 1620, 1395, 1098 and 572 cm−1 which corresponded to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C–CH3, Si–O–Si and C–Br stretching modes, respectively, while the peak at 2983 cm−1 was characteristic of a C–H stretch. These supported the successful attachment of ARGET ATRP initiator to ZnO surface. In Fig. 2, the characteristic peak at 1727 cm−1 was assigned to the ester carbonyl group of poly-methacrylic acid. The medium peaks at 1635 cm−1, which corresponded to the CC stretching mode, indicated that not all of the bonded EGDMA molecules were cross-linked.32 The small peak at 1452 cm−1 ascribed to CH3 of MAA, indicating a few functional monomers were successfully grafted. The initial EGDMA/MAA molar ratio was 5.0, so that higher molar mounts of EGDMA compared to MAA were expected among the polymers. The bands at 1153 and 1098 cm−1 were characteristic of C–O and Si–O–Si stretching vibrations, respectively, and the peak at 2958 cm−1 was characteristic of the C–H stretch. The broad band at 3422 cm−1 indicated the –OH group at the surface of polymers. The Raman spectra of ZnO-MIPs A and ZnO-NIPs B are shown in Fig. S3 )ESI†) and the characteristic peaks located at 430, 498 and 606 cm−1 suggested that γ-Fe2O3 nanoparticles were present in polymers.33
O, C–CH3, Si–O–Si and C–Br stretching modes, respectively, while the peak at 2983 cm−1 was characteristic of a C–H stretch. These supported the successful attachment of ARGET ATRP initiator to ZnO surface. In Fig. 2, the characteristic peak at 1727 cm−1 was assigned to the ester carbonyl group of poly-methacrylic acid. The medium peaks at 1635 cm−1, which corresponded to the CC stretching mode, indicated that not all of the bonded EGDMA molecules were cross-linked.32 The small peak at 1452 cm−1 ascribed to CH3 of MAA, indicating a few functional monomers were successfully grafted. The initial EGDMA/MAA molar ratio was 5.0, so that higher molar mounts of EGDMA compared to MAA were expected among the polymers. The bands at 1153 and 1098 cm−1 were characteristic of C–O and Si–O–Si stretching vibrations, respectively, and the peak at 2958 cm−1 was characteristic of the C–H stretch. The broad band at 3422 cm−1 indicated the –OH group at the surface of polymers. The Raman spectra of ZnO-MIPs A and ZnO-NIPs B are shown in Fig. S3 )ESI†) and the characteristic peaks located at 430, 498 and 606 cm−1 suggested that γ-Fe2O3 nanoparticles were present in polymers.33
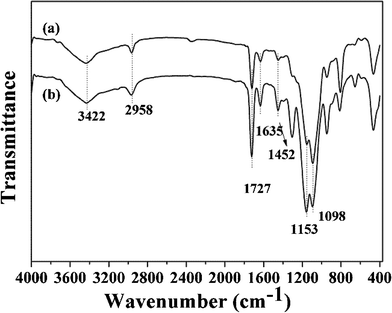 | ||
| Fig. 2 FT-IR spectra of ZnO-NIPs B (a) and ZnO-MIPs A (b). | ||
Fig. 3a and b show the magnetic hysteresis loops of γ-Fe2O3, ZnO-MIPs A and ZnO-NIPs B, respectively. The saturation magnetization values of γ-Fe2O3, ZnO-MIPs A and ZnO-NIPs B were 37.6, 1.60 and 1.47 emu g−1, respectively. The magnetic separability of ZnO-MIPs A was tested in water by placing a magnet near the glass bottle. The ZnO-MIPs A could be fully attracted toward the magnet in about 60 s (Fig. 3c). This property of ZnO-MIPs provided us an efficient method to separate polymers without filtering and inspired us to prepare the functionalized polymerization monomer.
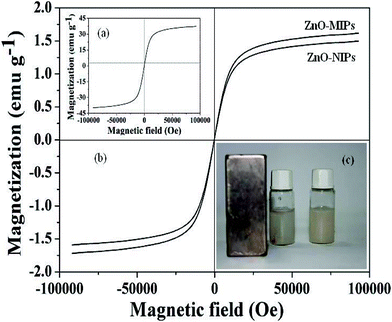 | ||
| Fig. 3 Magnetization curves at room temperature of γ-Fe2O3 (a), ZnO-MIPs A and ZnO-NIPs B (b). ZnO-MIPs A suspended in water (right) and in the presence (left) of an externally placed magnet (c). | ||
TGA and DSC curves were employed to further estimate the grafting yield of imprinted polymer coating (Fig. 4). It can be observed that ZnO-MIPs A and ZnO-NIPs B were relatively stable within the initial temperature range (<100 °C). When the temperature was increased to 800 °C, weight losses could be observed for ZnO-MIPs A (17.18%) (Fig. 4a) and ZnO-NIPs B (14.91%) (Fig. 4b), indicating that the grafting yields of ZnO-MIPs A and ZnO-NIPs B coating to ZnO nanorods were about 17.18 and 14.91 wt%, respectively. The lost weight would correspond to the polymer and the residual mass could be attributed to the stability of γ-Fe2O3, ZnO nanorods and carbon. Moreover, the two exothermic peaks around 666 °C and 685 °C in DSC curve of ZnO-MIPs A (Fig. 4c) and the two exothermic peaks around 651 °C and 677 °C in DSC curve of ZnO-NIPs B (Fig. 4d) were observed, while there were no corresponding changes in the TGA curves, indicating the possibility of the phase transition from γ-Fe2O3 to α-Fe2O3,34 from wurtzite phase to other phase transition, respectively.35
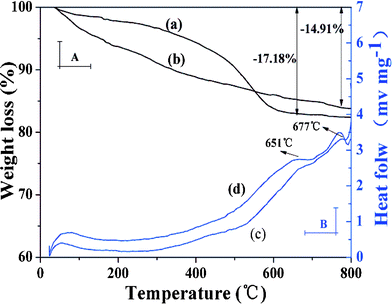 | ||
| Fig. 4 TGA and DSC curves at a heating rate of 10 °C min−1 from room temperature to 800 °C under N2 atmosphere. TGA curve of ZnO-MIPs A (a), TGA curve of ZnO-NIPs B (b), DSC curve of ZnO-MIPs A (c) and DSC curve of ZnO-NIPs B (d). | ||
A simple schematic illustration of the fluorescence quenching process is shown in Fig. 5a. When there is no template SMZ around the ZnO-MIPs A, a green emission is generated. After adding the template SMZ, there will be a strong interaction between the template molecule and the carboxyl groups with electron transfer from the ZnO-MIPs A to SMZ, and so a quenching mechanism.36 As shown in Fig. 5b, when ZnO-MIPs A were irradiated with high energy photons (<368 nm), greater than the band gap energy, an electron is promoted from the valence band to the conduction band. The electrons were excited from the valence band to the conduction band and subsequently relax via green emission. The fluorescence quenching of the ZnO-MIPs A was mainly achieved because the addition of SMZ led to a strong interaction between the carboxyl groups and the template SMZ. The UV-vis maximal adsorption peak of SMZ was around 238 and 263 nm (Fig. S4, ESI†) and since all the energy bands of the SMZ were higher than the green emission of the ZnO-MIPs A around 470 nm, the electrons at the conductive band of the ZnO-MIPs A can directly transfer to the lowest unoccupied molecular orbital (LUMO) of the UV band of the SMZ molecules (dashed arrows), and quenching is observed. According to the mechanism above, the large quenching constant means that there are suitable binding sites on the ZnO-MIPs. The above experimental results indicate that the surface-modification process has a significant impact on the luminescence properties of ZnO and should be further studied.
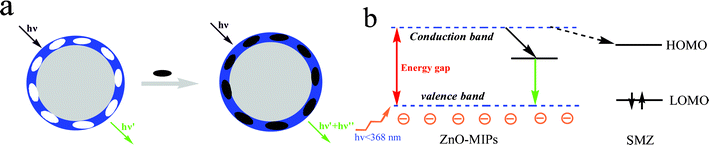 | ||
| Fig. 5 The process of ZnO-MIPs A fluorescence quenching (a) and a possible quenching mechanism of ZnO-MIPs A (b). | ||
Binding characteristics of ZnO-MIPs A and ZnO-NIPs B for SMZ
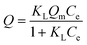 | (2) |
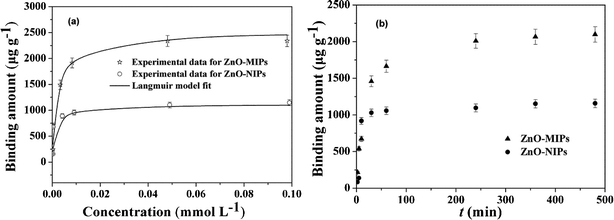 | ||
| Fig. 6 Adsorption isotherms of SMZ on the ZnO-MIPs A and ZnO-NIPs B (a). Adsorption conditions: V = 10 mL, m = 5.0 mg, Ci = 0.0005–0.10 mmol L−1, time 12 h, temperature 25 °C. Adsorption kinetics of SMZ on the ZnO-MIPs A and ZnO-NIPs B (b). Adsorption conditions: V = 10 mL, m = 5.0 mg, C = 0.05 mmol L−1, temperature 25 °C. | ||
It was apparent that the saturation curves fitted well to the Langmuir monolayer adsorption model,40 and the maximum binding capacity of ZnO-MIPs A and ZnO-NIPs B for SMZ were 2350 and 1085 μg g−1, respectively. These values were larger than imprinted nanoparticles studied by Chen's group through RAFT.38 The ZnO-MIPs A exhibited much higher binding amounts than ZnO-NIPs B, suggesting that recognition sites were generated on the surface of ZnO-MIPs A. Thus, we could prepare more favourable imprinted polymer by ARGET ATRP and there may exist a vast potential for preparing imprinted polymers including nanoparticles, films and layers by ARGET ATRP.
Determination of sulfamethazine by fluorescence spectra
The aim of this process was to determinate the sensitivity of SMZ by ZnO-MIPs A. As shown in Fig. 7, the fluorescence intensity of ZnO-MIPs A and ZnO-NIPs B increased in comparison with that of ZnO nanorods and showed slight blue shifts of the maximum emission wavelength (2 nm) of fluorescence spectrum of ZnO-MIPs A (or ZnO-NIPs B) in comparison with that of ZnO nanorods. Owing to the protection layer of polymer on the surface of ZnO, the fluorescence intensity increased after coating ZnO with EGDMA and MAA, and similar phenomena were observed by Zhang et al.23 Thus, the slight blue shift of ZnO-MIPs A could be attributed to the electron donor characteristics of the polymer being changed by the ZnO environment.41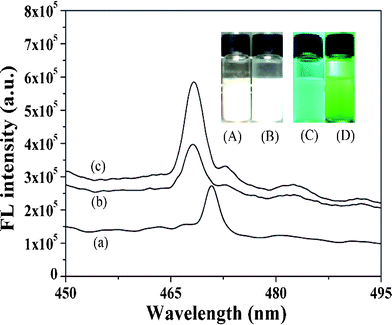 | ||
| Fig. 7 Photoluminescence spectra of the ZnO nanorods (a), ZnO-NIPs B (b) and ZnO-MIPs A (c). Inset: fluorescence photographs of ZnO in water under room light (A) and under UV light (C) irradiation (λ = 365 nm), ZnO-MIPs A in water under room light (B) and under UV light (D) irradiation (λ = 365 nm). | ||
Fig. 7A–D present fluorescence photographs of ZnO and ZnO-MIPs A in water under room light and under UV light irradiation (λ = 365 nm). Yan's group prepared a MIP-based room-temperature phosphorescence optosensor and the Mn-doped ZnS quantum dot room-temperature phosphorescence quenching followed the Stern–Volmer equation.42 Similarly a quenching fluorescence of ZnO-MIPs A in the presence of SMZ can also be described by a Stern–Volmer type equation:43
| I0/I = 1 + KSV[Q] | (3) |
The binding constant (K) and binding sites (n) are calculated by the double-logarithm equation for static quenching:44
| log[(I0 − I)/I] = logK + nlog[Q] | (4) |
The fluorescence spectra of ZnO-MIPs A with increasing concentrations of SMZ are shown in Fig. 8. It is expected that the imprinted binding sites play a key role in the luminescence response to SMZ. The well-defined structure of the imprinted cavities could be developed to rebind template molecules, which results in the selective fluorescence response to SMZ. The Ksv value was found to be 15524 M−1 while logK and n were 2.968 and 0.701, respectively. The detection limit (DL), calculated following the 3σ IUPAC criteria, was 19.0 μg L−1, which was inferior to enzyme-linked immunosorbent assay for detecting SMZ with DL < 0.03 μg L−1,45 but achieved the level of HPLC for determination of SMZ. For example, Dong's group used HPLC to detect SMZ in milk using surface-imprinted silica with DL of 25.0 μg L−1.46 However, the luminescence method has a potential application in analysis concerning on-site applicability and rapid detection analysis, alongside simple sample treatment and little solvent consumption in comparison to chromatographic analysis.
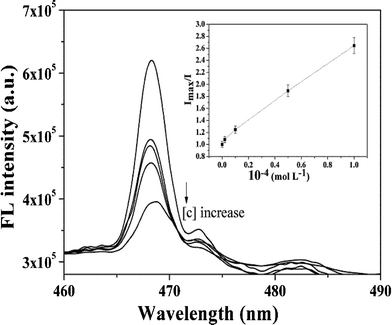 | ||
| Fig. 8 Fluorescence quenching spectra of ZnO-MIPs A at various concentrations of SMZ. Inset: Stern–Volmer-type plot of the data showing a linear fit throughout the SMZ concentration range, R2 = 0.993. | ||
Selectivity comparison of ZnO-MIPs A to SMZ and other antibiotics
Sulfamethazine (SMZ), sulfamethizol (SMI), sulfadiazine (SD) and tetracycline (TC) (structures in Fig. S5, ESI†) were used to test the selectivity of the imprinted polymers. Fig. 9 showed the adsorption capacities of ZnO-MIPs A and ZnO-NIPs B for different antibiotics with concentration of 0.1 mmol L−1 in aqueous solution. The larger antibiotics displayed less adsorption capacity, in other words, adsorption capacity of ZnO-MIPs A for single antibiotics was increased and followed the order TC < SMI < SD < SMZ. However, while SD has a similar structure to that of SMZ, the adsorption capacity of SD onto ZnO-MIPs A was lower than that of SMZ. Thus, ZnO-MIPs A only had significant adsorption specificity toward the template SMZ. Hydrogen bonding is a plausible binding mechanism rather than non-covalent interactions such as hydrophobic and ion interactions.11 The antibiotics adsorption to ZnO-NIPs B is likely driven by non-specific interactions.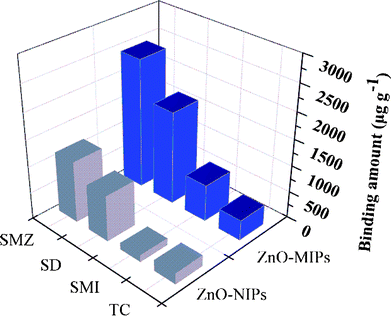 | ||
| Fig. 9 Adsorption capacity of ZnO-MIPs A and ZnO-NIPs B towards sulfamethazine, sulfadiazine, sulfamethizol and tetracycline. | ||
Selectivity capability of ZnO-MIPs A to SMZ in mixed solutions
In order to further investigate the importance of the template structure on target binding of the ZnO-MIPs A, we also carried out competitive antibiotics binding experiments in which an excess of various antibiotics (SMI, SD and TC) (0.1 mmol L−1) were added to a mixture of ZnO-MIPs A (5.0 mg) and SMZ (0.1 mmol L−1) in water. When binding equilibrium was reached the concentrations of each antibiotic in mixed solutions were determined by HPLC. The adsorption capability of each antibiotic is shown in Fig. 10 from which it is seen that the adsorption capacities for SMZ on ZnO-MIPs A slightly decreased compared to the single adsorption. The results illustrated that the presence of other antibiotics had only little influence on the adsorption of SMZ onto ZnO-MIPs A. This was a good proof of the successful formation on imprinted cavities and the importance of the shape memory effect on ZnO-MIPs A. On the contrary, the other four antibiotics were adsorbed by ZnO-NIPs B and the adsorption capacity of SMZ onto ZnO-NIPs B was lower than those in single solution, suggesting that no selective imprinted sites existed on the surface of ZnO-NIPs B. The same phenomenon was reported by Fu's group.47 The results illustrated that ZnO-MIPs A exhibited high selectivity to template SMZ in the presence of the other three antibiotics.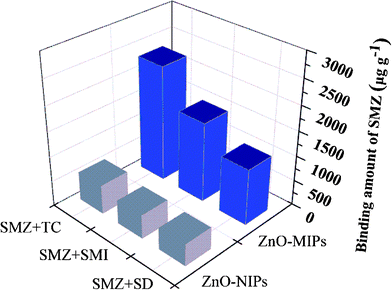 | ||
| Fig. 10 Competitive binding of template sulfamethazine with non-template tetracycline, sulfamethizol and sulfadiazine on ZnO-MIPs A and ZnO-NIPs B. | ||
Analysis of SMZ in pork sample
To demonstrate the applicability of HPLC for detection of SMZ in meat samples, the spiked pork sample was cleaned with ZnO-MIPs A. As no SMZ in the pork sample was detected by HPLC, a study was carried out on the sample spiked with 0.1 μmol L−1 SMZ to evaluate the developed method. SMZ solutions were tested by ZnO-MIPs A and ZnO-NIPs B while traditional C18 column separation was used for comparison. As can be clearly seen from Fig. 11a, no SMZ was detected in the pork sample because of very low concentration. However, the peak of SMZ increased after extraction by ZnO-MIPs A (Fig. 11(c)). ZnO-NIPs B showed only slightly selectively enriched ability (Fig. 11(b)), which may result from the absence of specific binding sites on ZnO-NIPs B. The chromatogram confirmed that SMZ in pork was enriched by the ZnO-MIPs A and can be recovered by washing. For analysis of SMZ spiked pork sample, the SMZ recovery was higher than 85%, and was unaffected by the nature of meat in which analytes were dissolved. Consequently, ZnO-MIPs A could be used as a selective recognition material and have a potential use for direct analysis of SMZ from meat.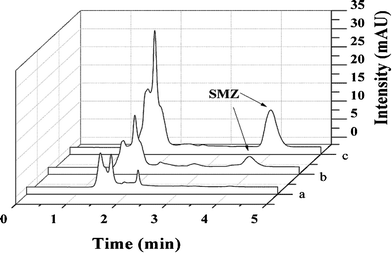 | ||
| Fig. 11 HPLC-UV chromatograms of pork sample (pork solution containing 0.1 μmol L−1 SMZ) (a), extraction with ZnO-NIPs B (b) and extraction with ZnO-MIPs A (c). Experimental conditions: 20 mL solution; 50 mg adsorbent; wash solution, 1 mL methanol; elution solution, 10 mL methanol–acetic acid (9:1, v/v); re-dissolution solution 800 μL methanol. | ||
Conclusions
ZnO-grafted sulfamethazine-imprinted polymers (ZnO-MIPs A) were prepared by ARGET (activators regenerated by electron transfer) ATRP (atom transfer radical polymerization). When sulfamethazine was extracted by methanol–acetic acid (95:5, v/v), imprinted binding sites were left on the surface of ZnO-MIPs, which were capable of selectively rebinding sulfamethazine. The combination of the magnetism, excellent fluorescence and high selectivity not only makes the ZnO-MIPs applicable to selective detection of the target molecule, but also improves the potential application of separation of molecularly imprinted polymers. This study showed ZnO nanorods and ARGET ATRP devised so far could be efficiently extended to designing novel and multifunctional recognition devices based on molecularly imprinted polymer grafts.
Acknowledgements
This work was financially supported by National Natural Science Foundation of China (No. 21077046, No. 21107037, No. 21176107, No. 21174057), Ph.D. Programs Foundation of Ministry of Education of China (No. 20093227110015) and Natural Science Foundation of Jiangsu Province (BK2011461, SBK2011459, BK2011514).References
- Q. Q. Gai, F. Qu, Z. J. Liu, R. J. Dai and Y. K. Zhang, J. Chromatogr., A, 2010, 1217, 5035 CrossRef CAS
.
- K. G. Yang, M. M. Berg, C. S. Zhao and L. Ye, Macromolecules, 2009, 42, 8739 CrossRef CAS
- G. Y. Jin, W. Li, S. N. Yu, Y. Y. Peng and J. L. Kong, Analyst, 2008, 133, 1367 RSC
- S. Ogasawara and S. J. Kato, J. Am. Chem. Soc., 2010, 132, 4608 CrossRef CAS
- A. Cutivet, C. Schembri, J. Kovensky and K. Haupt, J. Am. Chem. Soc., 2009, 131, 14699 CrossRef CAS
- Y. Mao, Y. Bao, S. Y. Gan, F. H. Li and L. Niu, Biosens. Bioelectron., 2011, 28, 291 CrossRef CAS
- X. B. Luo, S. L. Luo, Y. C. Zhan, H. Y. Shu, Y. N. Huang and X. M. Tu, J. Hazard. Mater., 2011, 192, 949 CrossRef CAS
- B. J. Gao, J. Wang, F. Q. An and Q. Liu, Polymer, 2008, 49, 1230 CrossRef CAS
- N. Perez-Moral and A. G. Mayes, Langmuir, 2004, 20, 3775 CrossRef CAS
- K. G. Yang, M. M. Berg, C. S. Zhao and L. Ye, Macromolecules, 2009, 42, 8739 CrossRef CAS
- J. M. Pan, H. Yao, L. C. Xu, H. X. Ou, P. W. Huo, X. X. Li and Y. S. Yan, J. Phys. Chem. C, 2011, 115, 5440 CAS
- M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721 CrossRef CAS
- A. D. Vaughan, S. P. Sizemore and M. E. Byrne, Polymer, 2007, 48, 74 CrossRef CAS
- X. Wei, X. Li and S. M. Husson, Biomacromolecules, 2005, 6, 1113 CrossRef CAS
- W. Jakubowski, K. Min and K. Matyjaszewski, Macromolecules, 2006, 39, 39 CrossRef CAS
- K. Min, H. Gao and K. Matyjaszewski, Macromolecules, 2007, 40, 1789 CrossRef CAS
- X. Y. Liu, Y. Guan, X. B. Ding, Y. X. Peng, X. P. Long, X. C. Wang and K. Chang, Macromol. Biosci., 2004, 4, 680 CrossRef CAS
- G. Demirel, G. Özçetin, E. Turan and T. Çaykara, Macromol. Biosci., 2005, 5, 1032 CrossRef CAS
- L. J. Fang, S. J. Chen, Y. Zhang and H. Q. Zhang, J. Mater. Chem., 2011, 21, 2320 RSC
- L. G. Chen, J. Liu, Q. L. Zeng, H. Wang, A. M. Yu, H. Q. Zhang and L. Ding, J. Chromatogr., A, 2009, 1216, 3710 CrossRef CAS
- J. X. Liu, H. Chen, Z. Lin and J. M. Lin, Anal. Chem., 2010, 82, 7380 CrossRef CAS
- J. B. Liang, J. W. Liu, Q. Xie, S. Bai, W. C. Yu and Y. T. Qian, J. Phys. Chem. B, 2005, 109, 9463 CrossRef CAS
- L. Zhang, F. Li, Y. W. Chen and X. F. Wang, J. Lumin., 2011, 131, 1701 CrossRef CAS
- J. X. Liu, H. Chen, Z. Lin and J. M. Lin, Anal. Chem., 2010, 82, 7380 CrossRef CAS
- H. B. Li, Y. L. Li and J. Cheng, Chem. Mater., 2010, 22, 2451 CrossRef CAS
- B. A. Hemstreet, Pharmacotherapy, 2006, 26, 551 CrossRef CAS
- C. Accinelli, W. C. Koskinen, J. M. Becker and M. J. Sadowsky, J. Agric. Food Chem., 2007, 55, 2677 CrossRef CAS
- J. Acar and B. Rostel, Rev. Sci. Tech., 2001, 20, 797 CAS
- Z. Zhang, J. F. Liu, B. Shao and G. B. Jiang, Environ. Sci. Technol., 2010, 44, 1030 CrossRef CAS
- Q. Gao, D. Luo, J. Ding and Y. Q. Feng, J. Chromatogr., A, 2010, 1217, 5602 CrossRef CAS
- R. Yi, G. Z. Qiu and X. H. Liu, J. Solid State Chem., 2009, 182, 2791 CrossRef CAS
- S. Gam-Derouich, M. N. Nguyen, A. Madani, N. Maouche, P. Lang, C. Perruchot and M. M. Chehimi, Surf. Interface Anal., 2010, 42, 1050 CrossRef CAS
- X. Liang, B. J. Xi, S. L. Xiong, Y. C. Zhu, F. Xue and Y. T. Qian, Mater. Res. Bull., 2009, 44, 2233 CrossRef CAS
- S. Basak, K. S. Rane and P. Biswas, Chem. Mater., 2008, 20, 4906 CrossRef CAS
- Y. Wang, Y. Zhang, W. J. Chang, G. L. Lu, J. Z. Jiang, Y. C. Li, J. Liu and T. D. Hu, J. Phys. Chem. Solids, 2005, 66, 1775 CrossRef CAS
- Y. P. Zhang, S. Y. Shi, X. Q. Chen, W. Zhang, K. L. Huang and M. J. Peng, J. Agric. Food. Chem., 2011, 59, 8499 CrossRef CAS
- G. Q. Fu, H. Y. He, Z. H. Chai, H. C. Chen, J. Kong, Y. Wang and Y. Z. Jiang, Anal. Chem., 2011, 83, 1431 CrossRef CAS
- S. F. Xu, J. H. Li and L. X. Chen, J. Mater. Chem., 2011, 10, 1039 Search PubMed
- M. Mazzotti, J. Chromatogr., A, 2006, 1126, 311 CrossRef CAS
- J. X. Liu, K. G. Yang, Q. L. Deng, Q. R. Li, L. H. Zhang, Z. Liang and Y. K. Zhang, Chem. Commun., 2011, 47, 3969 RSC
-
J. I. Pankove, Optical Processes in Semiconductors, Prentice-Hall, Englewood Cliffs, NJ, 1971 Search PubMed
- H. F. Wang, Y. He, T. R. Ji and X. P. Yan, Anal. Chem., 2009, 81, 1615 CrossRef CAS
-
J. R. Lakowicz and B. R. Masters, Principles of Fluorescence Spectroscopy, 3rd edn, Springer, New York, 2006 Search PubMed
- U. S. Mote, S. L. Bhattar, S. R. Patil and G. B. Kolekar, Luminescence, 2010, 25, 1 CAS
- W. L. Shelver, N. W. Shappell, M. Franek and F. R. Rubio, J. Agric. Food Chem., 2008, 56, 6609 CrossRef CAS
- S. F. Su, M. Zhang, B. L. Li, H. Y. Zhang and X. C. Dong, Talanta, 2008, 76, 1141 CrossRef CAS
- G. Q. Fu, H. Y. He, Z. H. Chai, H. C. Chen, J. Kong, Y. Wang and Y. Z. Jiang, Anal. Chem., 2011, 83, 1431 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1: FE-SEM image of ZnO nanorods; Fig. S2: FT-IR spectra of ZnO nanorods (a), ZnO/SiO2 (b), ZnO-Br (c); Fig. S3: Raman spectra of ZnO-MIPs A (a) and ZnO-NIPs B (b); Fig. S4: UV-vis adsorption spectra of SMZ, ZnO-MIPs, ZnO-SiO2 and ZnO; Fig. S5: Chemical structures of SMZ, SMI, SD and TC. See DOI: 10.1039/c2ra20282d |
| This journal is © The Royal Society of Chemistry 2012 |