
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sulfur-doped silicon oxycarbide by facile pyrolysis process as an outstanding stable performance lithium-ion battery anode†
Jungjin Park‡
a,
Won Young An‡a,
Keunho Leea,
Seungman Parka,
Minjun Bae a,
Seon Jae Hwanga,
Hwichan Honga,
Yonghwan Kima,
Taehyun Yooa,
Dohyeong Kima,
Jong Min Kim*c and
Yuanzhe Piao*ab
a,
Seon Jae Hwanga,
Hwichan Honga,
Yonghwan Kima,
Taehyun Yooa,
Dohyeong Kima,
Jong Min Kim*c and
Yuanzhe Piao*ab
aGraduate School of Convergence Science and Technology, Seoul National University, 145 Gwanggyo-ro, Yeongtong-gu, Suwon-Si, Gyeonggi-do 16229, Republic of Korea. E-mail: parkat9@snu.ac.kr
bAdvanced Institutes of Convergence Technology, 145 Gwanggyo-ro, Yeongtong-gu, Suwon-si, Gyeonggi-do 16229, Republic of Korea
cSamsung Electro-Mechanics, 150, Maeyeong-ro, Yeongtong-gu, Suwon-si, Gyeonggi-do 16674, Republic of Korea. E-mail: vitamin66@snu.ac.kr
First published on 20th September 2024
Abstract
Silicon oxycarbide (SiOC) is drawing significant attention as a potential anode material for lithium-ion batteries due to its remarkable cycle life and the distinctive Si–O–C hybrid bonding within its structure. However, a notable drawback of SiOC-based electrodes is their poor electrical conductivity. In this study, we synthesized sulfur-doped silicon oxycarbide (S-SiOC) via facile one-pot pyrolysis from a mixture of commercial silicone oil with 1-dodecanethiol. Upon testing the S-SiOC electrode materials, we observed significant attributes, including an outstanding specific capacity (650 mA h g−1 at 1 A g−1), exceptional capacity retention (89.2% after 2000 cycles at 1 A g−1), and substantial potential for high mass loading of active materials (up to 2.2 mg cm−2). Sulfur doping led to enhanced diffusivity of lithium ions, as investigated through cyclic voltammetry (CV) and galvanostatic intermittent titration technique (GITT) tests. Consequently, this sulfur-doped silicon oxycarbide, exhibiting excellent electrochemical performance, holds promising potential as an anode material for lithium-ion batteries.
1. Introduction
Lithium-ion batteries (LIBs), renowned for their high energy density, power density, and exceptional cycling performance, are one of the most potent energy storage systems (ESS). These batteries have been extensively explored for diverse applications, including portable devices and electric vehicles (EVs).1–4 Graphite has seen widespread adoption and commercialization as an anode material in the battery domain due to factors such as its low operating potential vs. Li+/Li, limited volumetric change during cycling, and cost-effectiveness.4,5 Nevertheless, with the rising demand for increasingly advanced electronics, the limitations of graphite have become evident, primarily due to its low theoretical capacity of 372 mA h g−1. As a result, a plethora of studies have been undertaken to explore alternative candidates. The silicon-based anode has emerged as one of the most enticing materials, boasting a theoretical capacity surpassing 3579 mA h g−1. Nonetheless, the substantial volume change observed during cycling, reaching up to 400%, remains a significant challenge as it can lead to particle fractures and decreased electrical contact. To address this issue, several innovative strategies have been implemented in the development of Si/C composite materials.6–8 However, despite these efforts, the problem of capacity fading in Si/C composite materials still persists, primarily attributed to structural breakdown during cycling.In this context, silicon oxycarbide (SiOC) ceramics have recently garnered attention as potential anode materials due to their potential to replace carbonaceous and silicon-based anodes. SiOC exhibits thermal and structural stability, good cycle performance, a simple synthesis process, and an impressive reversible capacity of approximately 600 mA h g−1, attributed to its distinctive structure.9 SiOC ceramics can be represented by SiOxC4−x + yCfree (0 ≤ x ≤ 4), wherein SiOxC4−x refers to the Si–O–C glass phase and Cfree corresponds to the segregated free carbon phase. However, despite these promising features, SiOC-based electrodes still face some significant challenges.10 One of the primary issues with SiOC electrodes is their low initial coulombic efficiency, which means that a significant amount of the initial lithium introduced during the first cycle is not efficiently utilized and results in inefficiencies in energy storage. Additionally, SiOC electrodes often exhibit voltage hysteresis, where the voltage during charge and discharge does not align precisely, resulting in energy losses and reduced overall efficiency. Another significant challenge is the inferior electrical conductivity of SiOC electrodes. This limitation is primarily due to their inherently electrically insulating characteristics and limited ionic transport capabilities.9,11,12 SiOC materials typically have lower conductivities, particularly for LIB applications, because of the open structure of free carbon and the lack of a sufficient volume fraction of conductive carbon necessary to form a continuous conducting network.13 This low conductivity can lead to poor rate capability and hinder their overall performance in LIBs.
In response to these challenges, diverse strategies have been employed to enhance SiOC-based electrodes. To augment electrical conductivity, various types of carbonaceous materials, including carbon nanotubes,11,14 carbon nanofibers,15 and graphene,16–18 have been integrated into composite structures with SiOC. Moreover, the creation of hybrid anodes by combining SiOC with metals has been explored as a viable route to enhance electrochemical performance.19,20 Kaspar et al. proposed a stable SiOC/Sn nanocomposite achieved through chemical modifications of distinct polysiloxane precursors with tin(II) acetate, followed by an annealing pyrolysis process at 1000 °C. The resulting SiOC/Sn nanocomposite anode demonstrated a capacity of 562 mA h g−1 at a current density of 74 mA g−1, exhibiting minimal capacity fading over 20 cycles, and retaining a capacity of 133 mA h g−1 even under high current rates of 744 mA g−1.20 Doping also presents a noteworthy approach to achieve enhanced electrical conductivity and improved electrochemical activity. The introduction of heteroatoms (such as nitrogen, sulfur, and phosphorus) can establish active sites and modify electronic structures.21,22 In the realm of energy storage systems, various carbonaceous materials used as anode components, such as graphene, carbon nanotubes, and porous carbon, have utilized heteroatom doping to enhance their performance.23–29 Among these, sulfur has gained attention as a promising dopant for electrochemical applications, owing to its smaller atomic radius and electronegativity.30,31 Additionally, sulfur introduces supplementary electron transfer pathways via C–S–C bonding.32 Despite the various advantages of sulfur doping in boosting electrochemical performance, the utilization of H2S gas as a sulfur source in doping procedures raises safety concerns due to its inherent toxicity and flammability.33,34
In this study, we present, for the first time, sulfur-doped silicon oxycarbide (S-SiOC) as a promising anode material for lithium-ion batteries. We employed a novel sulfur doping approach for silicon oxycarbide by utilizing a facile pyrolysis process involving commercial silicone oil and 1-dodecanethiol under an inert atmosphere. Importantly, this sulfur doping was achieved without the use of H2S gas. Through this process, sulfur atoms were successfully incorporated into the silicon oxycarbide lattice, forming covalent bonds with carbon atoms, and generating a partially heterocyclic structure. This strategic sulfur doping not only facilitated efficient electron transfer by creating additional pathways through C–S–C bonding but also resulted in increased (002) interlayer spacing and the provision of extra active sites, thereby enhancing Li+ accommodation. As a result of these modifications, the prepared S-SiOC material exhibited remarkable electrochemical performance, substantial cycle stability, and exceptional rate capability. Impressively, even after 1000 cycles, the electrode maintained a capacity of 650 mA h g−1 without any observable capacity fading. Notably, the electrode displayed an impressive capacity retention rate of 89.2% after 2000 cycles at 1 A g−1. Moreover, during 250 cycles at 1 A g−1 (at a mass loading of 2.2 mg cm−2), the electrode demonstrated an areal capacity exceeding 0.88 mA h cm−2.
2. Experimental section
2.1. Materials
Silicone oil (AP1000, polyphenyl-methylsiloxane) was purchased from Aldrich Chemical Co., and 1-dodecanethiol (>98.5%) was purchased from Samchun Co. All materials were handled without any purification.2.2. Preparation of SiOC and S-SiOC
Both bare SiOC and S-SiOC were synthesized using a facile one-pot pyrolysis involving liquid precursors. To prepare bare SiOC, 4 g of silicone oil were directly added to an alumina boat without any pretreatment. The oil was subjected to pyrolysis in a quartz tube furnace under Ar atmosphere, maintaining a temperature of 800 °C for 5 hours with a heating rate of 5 °C min−1. Similarly, for the synthesis of S-SiOC, with no pretreatment, 4 g of silicone oil with 0.8 g of 1-dodecanethiol were blended using a mortar and pestle. Subsequently, the mixture was transfered into an alumina boat and positioned at the inside of a tube furnace. The pyrolysis process was conducted under the same conditions used for SiOC, and the sample was retrieved after cooling down to room temperature.2.3. Characterization
The weight loss of the materials within the temperature range of 25 to 1100 °C was measured using a thermogravimetric analysis (TGA) analyzer (Mettler Toledo). The heating rate was set at 5 °C min−1, and the measurements were conducted in an air atmosphere. To determine the Brunauer–Emmett–Teller (BET) surface areas and microstructural properties, a BELSORP-mini II instrument (MicrotracBEL Corp) was employed. Morphological analysis of the samples was carried out using a field-emission scanning electron microscopy (FE-SEM) with an energy dispersive X-ray spectrometer (EDS), specifically the Hitachi S-4800 model. For further characterization of morphology and structure, field-emission transmission electron microscopy (FE-TEM) was conducted using the JEOL JEM-2100F instrument, equipped with high-resolution transmission electron microscopy (HRTEM) and selected area electron diffraction (SAED) capabilities. X-ray diffraction (XRD) patterns were acquired utilizing a diffractometer, the Bruker D8-Advance 2020 model, with a wavelength (λ) of 1.5406 Å. Raman spectra were obtained employing a Raman spectrometer, specifically the LabRAM HR Evolution instrument, with a 532 nm laser source. Lastly, X-ray photoelectron spectroscopy (XPS) analysis was performed utilizing an X-ray photoelectron spectrometer from ThermoFisher Scientific, the NEXSA model.2.4. Electrochemical measurement
The S-SiOC anode was prepared by creating a homogeneous mixture of active materials, Super-P as a conductive additive, and sodium carboxymethylcellulose (with a molecular weight of approximately 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, sourced from Sigma Aldrich) as a binder. The weight ratio of these components was 70:15:15. This mixture was thoroughly blended in deionized (DI) water using a Mini-Mill (PULVERISETTE 23 FRITSCH) for a duration of 30 minutes. Subsequently, the resulting mixture was spread onto a copper (Cu) foil using a doctor blade. The wet electrode was then dried at 60 °C in a vacuum oven for 4 hours. Following drying, the electrode was shaped into a disk with a diameter of 11 mm. The mass loading of the active material was maintained within the range of 0.7 to 2.2 mg cm−2. The assembly of CR2016 coin cells was carried out within an argon-filled glovebox (O2 and H2O < 1 ppm). These cells consisted of the S-SiOC material as the anode, a lithium (Li) metal disk serving as the counter electrode, and Celgard 2400 acting as the separator. The electrolyte was employed a 1.3 M solution of LiPF6 in a 3:7 (v/v) mixture of ethylene carbonate and diethyl carbonate, with the addition of 10% fluoroethylene carbonate obtained from PANAX. Electrochemical measurements were conducted using a WBCS3000S cycler from WonAtech. These measurements encompassed a voltage window spanning from 0.01 to 3 V (versus Li+/Li) and were conducted at room temperature. Cyclic voltammetry (CV) measurements were performed using the same WBCS3000S cycler over a voltage range of 0.01 to 3 V, employing scan rates ranging from 0.1 to 4.0 mV s−1. The galvanostatic intermittent titration technique (GITT) was employed to evaluate lithium-ion diffusivity during the second discharge–charge cycle, with the measurements also conducted using the WBCS3000S cycler. Finally, electrochemical impedance spectroscopy (EIS) analysis was carried out using a ZIVE SP1 instrument. The frequency range covered by this analysis spanned from 100 mHz to 100 kHz.
000, sourced from Sigma Aldrich) as a binder. The weight ratio of these components was 70:15:15. This mixture was thoroughly blended in deionized (DI) water using a Mini-Mill (PULVERISETTE 23 FRITSCH) for a duration of 30 minutes. Subsequently, the resulting mixture was spread onto a copper (Cu) foil using a doctor blade. The wet electrode was then dried at 60 °C in a vacuum oven for 4 hours. Following drying, the electrode was shaped into a disk with a diameter of 11 mm. The mass loading of the active material was maintained within the range of 0.7 to 2.2 mg cm−2. The assembly of CR2016 coin cells was carried out within an argon-filled glovebox (O2 and H2O < 1 ppm). These cells consisted of the S-SiOC material as the anode, a lithium (Li) metal disk serving as the counter electrode, and Celgard 2400 acting as the separator. The electrolyte was employed a 1.3 M solution of LiPF6 in a 3:7 (v/v) mixture of ethylene carbonate and diethyl carbonate, with the addition of 10% fluoroethylene carbonate obtained from PANAX. Electrochemical measurements were conducted using a WBCS3000S cycler from WonAtech. These measurements encompassed a voltage window spanning from 0.01 to 3 V (versus Li+/Li) and were conducted at room temperature. Cyclic voltammetry (CV) measurements were performed using the same WBCS3000S cycler over a voltage range of 0.01 to 3 V, employing scan rates ranging from 0.1 to 4.0 mV s−1. The galvanostatic intermittent titration technique (GITT) was employed to evaluate lithium-ion diffusivity during the second discharge–charge cycle, with the measurements also conducted using the WBCS3000S cycler. Finally, electrochemical impedance spectroscopy (EIS) analysis was carried out using a ZIVE SP1 instrument. The frequency range covered by this analysis spanned from 100 mHz to 100 kHz.
3. Results and discussion
The schematic representation of the S-SiOC synthesis process is presented in Fig. 1a. Commercial silicone oil (polyphenyl-methylsiloxane) was utilized as the precursor, while 1-dodecanethiol served as the source of sulfur. Given the transparent nature of both silicone oil and 1-dodecanethiol, they were conveniently mixed without the need for supplementary solvents or surfactants.35,36 Subsequently, the mixture underwent pyrolysis at 800 °C under an Ar atmosphere. This process yielded black powders, indicative of the formation of carbon networks.37 The specific surface area of both SiOC and S-SiOC samples was assessed through N2 adsorption/desorption measurements. As illustrated in Fig. S1a,† the SiOC and S-SiOC materials were characterized by type II and type IV isotherms, respectively. BET analysis of SiOC and S-SiOC revealed nonporous attributes, featuring low specific surface areas of 0.257 m2 g−1 and 2.033 m2 g−1, respectively. The pore size distribution of SiOC and S-SiOC was examined using the BJH plot, as displayed in Fig. S1b.† The total pore volumes are 0.00141 cm3 g−1 and 0.00371 cm3 g−1, respectively. | ||
| Fig. 1 (a) Schematic illustration of preparing S-SiOC. (b) SEM image of S-SiOC. (c) TEM and (d) HRTEM images of S-SiOC (SAED pattern inset). (f) Carbon, (g) oxygen, (h) silicon, (i) sulfur elemental mappings of S-SiOC corresponding (e). | ||
As shown in Fig. 1b and S2a,† morphologies and particle dimensions are illustrated in the SEM images. Both samples display particles primarily in a spherical form, featuring micron-sized distributions. In particular, the introduction of sulfur through doping manifests no prominent differences in particle morphology between two samples, as evident in the SEM images. The particle sizes of SiOC and S-SiOC can be determined via analysis of SEM images. The particle sizes of SiOC and S-SiOC range from 1.4 to 5.4 μm and 2.9 to 9.6 μm, respectively, demonstrating a diverse distribution of particles in terms of size. For a more comprehensive exploration of morphology and structure, TEM images of SiOC and S-SiOC are presented in Fig. 1c and S2b.† In both cases, bulk particles exhibiting spherical configurations were consistent with observations from SEM images. HRTEM images and SAED analyses of SiOC and S-SiOC are displayed in Fig. 1d and S2c,† respectively. These images showed that both samples possess a fully amorphous structure devoid of crystallinity. This outcome shows that the pyrolysis temperature did not suffice to induce crystalline formation. To further verify elemental composition, EDS elemental mapping was employed, and results are shown in Fig. 1f–i and S2e–h.† This analysis was confirmed the uniform distribution of Si, O, and C elements, while S was not detected within SiOC. In contrast, S-SiOC exhibited homogeneous distributions of Si, O, C, and S elements. EDS spectra of both samples are presented in Fig. S3.† In pure SiOC, distinct peaks were observed for Si, O, and C, with no signal indicative of S. Conversely, in sulfur-doped SiOC, all the expected peaks were observed, including a visible S spectrum (∼1.83% in atomic ratio). Notably, EDS elemental mapping images provided compelling evidence that sulfur atoms have been effectively doped into the silicon oxycarbide structure.
Fig. 2a illustrates TGA curves of SiOC and S-SiOC across the temperature range of 25 to 1100 °C under an air atmosphere. For both samples, the initial weight loss occurring below 200 °C was attributed to water evaporation. Meanwhile, the primary weight reduction observed above 500 °C was attributed to the oxidation of free carbon in the presence of air.37 Notably, the reduced weight percentages for SiOC and S-SiOC are 33.8% and 32.7%, respectively, indicating the establishment of a carbon network with minimal variation in carbon content between the two samples. Meanwhile, the content of the free carbon phase in SiOC plays a vital role in influencing electrochemical activity. This finding suggests that the difference in electrochemical activity between the two samples is not influenced by the free carbon phase.
 | ||
| Fig. 2 (a) TGA curves of SiOC and S-SiOC at a heating rate of 5 °C min−1 under the air atmosphere. (b) Raman spectra of SiOC and S-SiOC. High resolution XPS spectra of (c) C 1s, (d) O 1s, (e) Si 2p, (f) S 2p in S-SiOC. | ||
Fig. S4† presents the X-ray diffraction (XRD) patterns of the as-prepared SiOC and S-SiOC samples. In SiOC, the broad diffraction peak at 2θ = 22.8° corresponds to the (002) reflection and signifies the presence of locally graphitized carbon structures within the amorphous carbon phase. Upon sulfur doping, the intensity of the (002) peak in S-SiOC decreased and shifted to a lower value of 21.4°. The calculated d002 values (representing the interlayer spacing) for SiOC and S-SiOC are 0.390 nm and 0.415 nm, respectively. This expanded interlayer spacing is beneficial for lithium-ion storage and diffusion. Sulfur's larger ionic radius in comparison to carbon contributes to the extension of the carbon interlayer spacing.38–41 Notably, the XRD patterns for S-SiOC do not exhibit peaks indicative of pristine sulfur, confirming that sulfur does not exist in an elemental state.29 The broadness and intensity of the XRD patterns signify that SiOC maintains its amorphous structure, consistent with previous reports.36,42–44 Similar patterns persist in S-SiOC, indicating that doped sulfur atoms do not alter the amorphous structure of SiOC. Furthermore, the absence of SiC crystallization aligns with the SAED patterns, as the pyrolysis temperature is insufficient for SiC carbothermal reduction.37,45 The absence of SiC is advantageous for achieving enhanced reversible capacity due to its inertness towards lithium ions.46 To provide more qualitative information, the domain sizes of the samples were analyzed utilizing the Scherrer equation.47–49 The domain sizes of SiOC and S-SiOC, as determined from the XRD patterns, are measured to be 2.12 nm and 1.83 nm, respectively.
The Raman spectra of both SiOC and S-SiOC are shown in Fig. 2b. Two major peaks at 1330 cm−1 and 1592 cm−1 were observed, which are generally observed in carbonaceous materials.25,44,45,50,51 The former peak is related to the disordered carbon (D band) and the latter is associated with the graphitized carbon (G band), respectively. The intensity ratio of the D and G band (ID/IG), which generally refers to the ordering degree of carbon, increased from 1.01 to 1.09 after sulfur doping. This result reflects the increase of defects and degree of disorder in S-SiOC material due to doped-sulfur atoms, which can supply more diffusion channels and active sites for Li+ insertion.42 It is consistent well with the XRD result.
The surface chemical composition of S-SiOC was assessed via X-ray photoelectron spectroscopy (XPS). Fig. 2c–f presents high-resolution spectra of C 1s, O 1s, Si 2p, and S 2p in S-SiOC along with the corresponding fitted results. In Fig. 2c, the C 1s peak was deconvoluted into six distinct peaks: C–Si (283.4 eV), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (284.2 eV), C–C (284.8 eV), C–S (285.9 eV), C–O (286.9 eV), and CO (288.7 eV). The O 1s peak yielded two clear peaks: Si–O (532.38 eV) and SiO2 (533.08 eV) in Fig. 2d. The C 1s spectrum revealed an elevated peak intensity for C–C/CC in S-SiOC, along with a distinctly observable C–S bonding peak, as compared to SiOC shown in Fig. S5a.† This observation suggests that doped sulfur atoms influenced the distribution of C–Si and C–C/CC bonds, thereby modifying the structure of both the Si–O–C glass phase and the free carbon phase. Similarly, the O 1s spectrum in Fig. 2d shows comparable Si–O/SiO2 bonding peaks post-sulfur doping, concerning bare SiOC shown in Fig. S5b.† The Si 2p peak in Fig. 2e can be resolved into four peaks: SiOC3 (101.6 eV), SiO2C2 (102.4 eV), SiO3C (102.9 eV), and SiO4 (103.7 eV), consistent with previous findings.37,52,53 In particular, Fig. S5c† shows that the deconvoluted peaks display minimal changes following sulfur doping in comparison to bare SiOC. This suggests that structural adjustments between the two phases occurred without the formation of covalent bonds between silicon and sulfur atoms. However, a distinct contrast emerges in the S 2p spectrum, highlighting the pronounced differences between the two samples. The high-resolution S 2p spectrum for S-SiOC is characterized by four peaks at 163.4 eV, 164.6 eV, 168.6 eV, and 170.1 eV in Fig. 2f. The initial two peaks, separated by 1.2 eV, are attributed to S 2p3/2 and S 2p1/2, corresponding to C–S–C bonding with a heterocyclic structure.23,28,29,41 The latter two peaks correspond to oxidized sulfur groups.54 This observation indicates the successful establishment of covalent bonds between sulfur and carbon atoms.23,51 Conversely, in Fig. S5d,† the S 2p spectrum for SiOC does not display any obvious peak.
C (284.2 eV), C–C (284.8 eV), C–S (285.9 eV), C–O (286.9 eV), and CO (288.7 eV). The O 1s peak yielded two clear peaks: Si–O (532.38 eV) and SiO2 (533.08 eV) in Fig. 2d. The C 1s spectrum revealed an elevated peak intensity for C–C/CC in S-SiOC, along with a distinctly observable C–S bonding peak, as compared to SiOC shown in Fig. S5a.† This observation suggests that doped sulfur atoms influenced the distribution of C–Si and C–C/CC bonds, thereby modifying the structure of both the Si–O–C glass phase and the free carbon phase. Similarly, the O 1s spectrum in Fig. 2d shows comparable Si–O/SiO2 bonding peaks post-sulfur doping, concerning bare SiOC shown in Fig. S5b.† The Si 2p peak in Fig. 2e can be resolved into four peaks: SiOC3 (101.6 eV), SiO2C2 (102.4 eV), SiO3C (102.9 eV), and SiO4 (103.7 eV), consistent with previous findings.37,52,53 In particular, Fig. S5c† shows that the deconvoluted peaks display minimal changes following sulfur doping in comparison to bare SiOC. This suggests that structural adjustments between the two phases occurred without the formation of covalent bonds between silicon and sulfur atoms. However, a distinct contrast emerges in the S 2p spectrum, highlighting the pronounced differences between the two samples. The high-resolution S 2p spectrum for S-SiOC is characterized by four peaks at 163.4 eV, 164.6 eV, 168.6 eV, and 170.1 eV in Fig. 2f. The initial two peaks, separated by 1.2 eV, are attributed to S 2p3/2 and S 2p1/2, corresponding to C–S–C bonding with a heterocyclic structure.23,28,29,41 The latter two peaks correspond to oxidized sulfur groups.54 This observation indicates the successful establishment of covalent bonds between sulfur and carbon atoms.23,51 Conversely, in Fig. S5d,† the S 2p spectrum for SiOC does not display any obvious peak.
XPS analysis was performed on the interfaces of SiOC and S-SiOC anodes following the initial cycle.55,56 Firstly, Fig. S6a† shows the F 1s peak of SiOC and S-SiOC. In the F 1s spectrum of SiOC, two peaks appear at 685.9 and 687.6 eV, corresponding to LiF and CF3, respectively. For S-SiOC, the F 1s peaks are located at 684.7 eV and 687.2 eV for LiF and CF3, respectively. Here, LiF indicates the formation of the anode's SEI, while CF3 refers to byproducts resulting from the decomposition of the electrolyte.57 Comparing the LiF to CF3 ratio between SiOC and S-SiOC, it is evident that S-SiOC exhibits a higher LiF ratio of 87.7% compared to SiOC's 77.4%. Therefore, it can be concluded that sulfur doping positively influences SEI formation.58 Fig. S6b† displays the O 1s peaks for SiOC and S-SiOC. The O 1s spectrum for SiOC reveals peaks at 528.5 and 531.9 eV, which correspond to Li2O and LiCOx, respectively. Similar peaks at 528.5 (Li2O) and 531.9 eV (LiCOx) are also observed in S-SiOC. Given that Li2O is one of the components of the SEI layer, the relatively larger Li2O peak in S-SiOC suggests that it has been influenced by sulfur doping.58 Fig. S6c† displays the C 1s peak of SiOC and S-SiOC. While it resembles the C 1s spectrum of original SiOC, we observed the emergence of C–H and R–O–C peaks at 283.2 eV and 287 eV, respectively, after cycling.58–60 These peaks represent the bonds of carbon and organic substances formed alongside the SEI layer post-cycling. In Li-ion batteries, the combination of carbon and organic materials adversely affects the SEI layer, unlike inorganic bonds.61 The increased peaks of carbon-organic bonds in SiOC compared to S-SiOC suggests that the SEI stability of SiOC may be some what lower than that of S-SiOC.
CV was conducted at a scan rate of 0.1 mV s−1 to provide insights into the electrochemical reactions. In Fig. 3b, The CV curves for the S-SiOC electrode exhibited a broad anodic peak and a distinct cathodic peak, both occurring at around 0.01 V (vs. Li+/Li). These features was attributed to the insertion or extraction of Li+ within the amorphous SiOC structure.44,62,63 Likewise, as shown in Fig. 3a, the SiOC electrode displays comparable CV curves, characterized by a broad anodic peak and a sharp cathodic peak. The faint cathodic peak observed at 0.7–0.8 V indicates the formation of a solid electrolyte interface (SEI) layer.64 Moreover, overlapping curves of both samples from the second to fifth cycles indicate a highly reversible lithiation/delithiation reaction. Especially, response current was elevated in the S-SiOC electrode, a trend attributable to enhanced electrical conductivity resulting from the doping process.
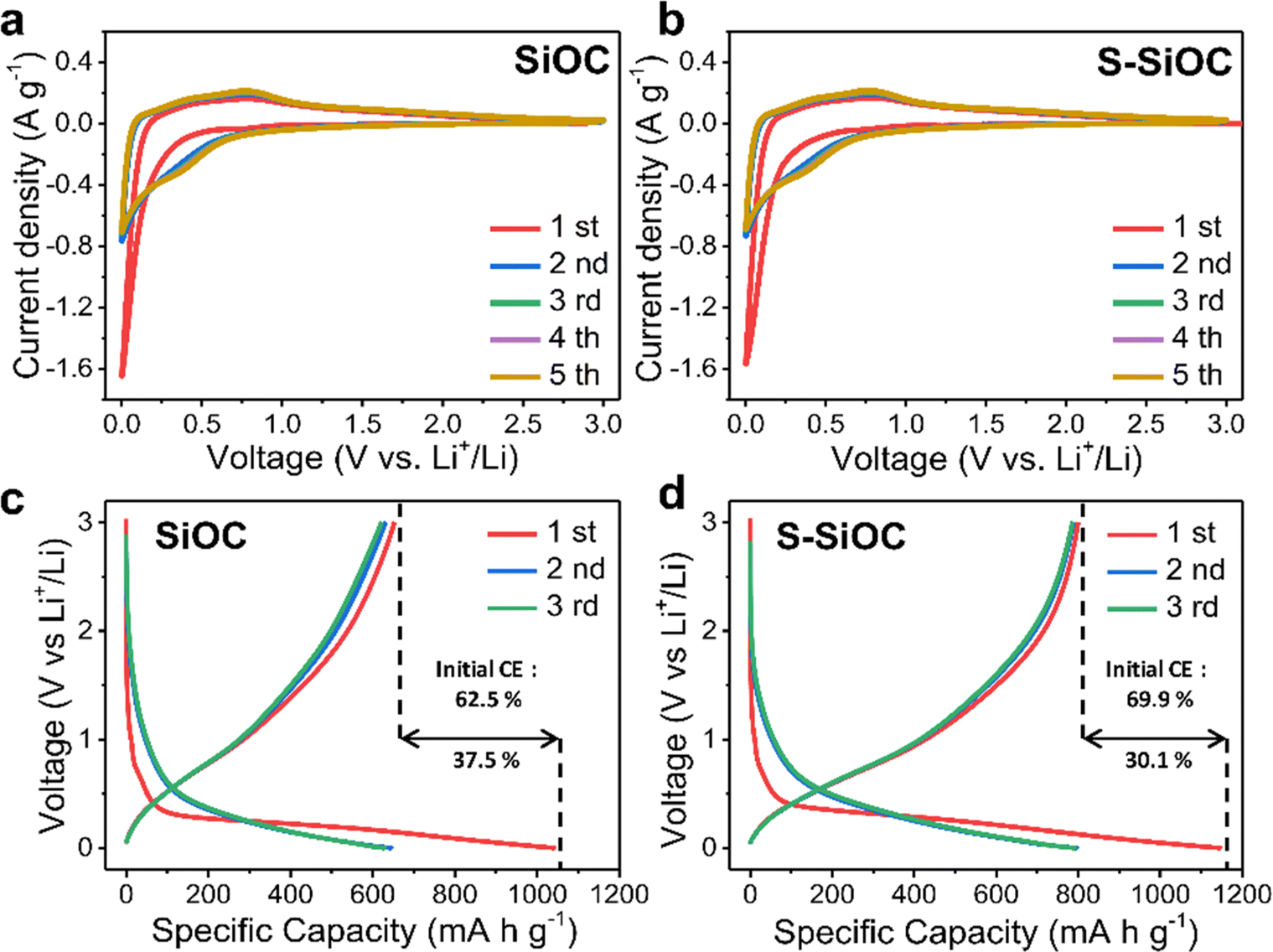 | ||
| Fig. 3 Cyclic voltammetry for the initial 5 cycles of (a) SiOC and (b) S-SiOC, at a scan rate of 0.1 mV s−1. Galvanostatic profiles of (c) SiOC and (d) S-SiOC for the initial 3 cycles. The current density is 0.1 A g−1. | ||
Galvanostatic charging and discharging curves for both SiOC and S-SiOC are demonstrated in Fig. 3c and d during the initial three cycles at a current rate of 0.1 A g−1 within the voltage range of 0.01 to 3 V. In the initial cycle, the discharge curve displays a plateau at 0.7–0.8 V, aligning with the CV curves. The initial cycle discharge capacities for SiOC and S-SiOC at 0.1 A g−1 was recorded as 1042 mA h g−1 and 1146 mA h g−1, respectively. SiOC demonstrates a reversible specific capacity of 652 mA h g−1, accompanied by an initial coulombic efficiency (ICE) of 62.5%. On the other hand, S-SiOC exhibited an elevated reversible capacity of 800 mA h g−1 and an improved ICE of 69.9%. The observed low ICE could stem from irreversible side reactions associated with the binding of lithium to oxygen-containing functional groups (e.g., CO, C–OH) or active carbon sites, which subsequently leads to the formation of solid electrolyte interface (SEI) layers on the anode surface.44,46 These functional groups, contributing to the non-reversible storage of lithium ions, could be eliminated through sulfur doping reactions, either by SOx oxidation or S substitution, resulting in an enhancement of coulombic efficiency.65 Furthermore, considering the similarity in specific surface area between the two samples based on BET results, the distinction in ICE could likely be attributed to the sulfur doping reaction. The second cycle discharge capacities for SiOC and S-SiOC was measured at 645 mA h g−1 and 797 mA h g−1, respectively. Correspondingly, ensuing reversible specific capacities and coulombic efficiencies for these cycles was measured as 630 mA h g−1 with 97.7% and 788 mA h g−1 with 98.9%. Importantly, as cycles progress, the coulombic efficiencies demonstrate an inclination to increase.
To assess the impact of doping on electrochemical properties, CV curves were conducted at different scan rates for both SiOC and S-SiOC, and are shown in Fig. 4a and b. The scan rate ranged from 0.5 mV s−1 to 4 mV s−1. Especially, as the scan rate escalates, there is a tendency for the peak current to increase in both samples. This phenomenon is likely linked to the activation of electrochemical reactions.44 For a deeper understanding of the role of sulfur doping in electrochemical performance, a linear correlation between the anodic peak current and the square root of the scan rate was established and displayed in Fig. 4c. Specifically, Fig. 4a and b are depicted Peak A of SiOC, and Peak B of S-SiOC, respectively. The diffusion coefficient of lithium-ion, DLi+, was subsequently calculated from the CV curves with varying scan rates using the Randles–Sevcik equation (eqn (1)).
| Ip = 2.69 × 105n1.5ADLi0.5v0.5CLi | (1) |
 | ||
| Fig. 4 Cyclic voltammetry curves of (a) SiOC and (b) S-SiOC with different scan rates from 0.5 mV s−1 to 4 mV s−1. (c) Linear fitting of the current at each anodic peak to the corresponding scan rate. (d) GITT data of SiOC and S-SiOC during the second discharge–charge cycle, current pulse of 0.1 A g−1. (e) Diffusivity plot of SiOC and S-SiOC during charge and discharge, and which evaluated by GITT. (f) Nyquist plots of SiOC and S-SiOC electrodes after 100 cycles at 1 A g−1 and equivalent circuit model (inset). (Rs: electrolyte resistance, CPESEI: SEI layer capacitance, RSEI: SEI layer resistance, CPEct: double layer capacitance, Rct: charge transfer resistance, and Zw: Warburg diffusion element.) | ||
For a more in-depth exploration of lithium-ion diffusivity, GITT was executed on both SiOC and S-SiOC throughout the second discharge–charge cycle. The GITT curve for both samples within the voltage range of 0.01 to 3.0 V is depicted in Fig. 4d. This technique involved applying a current pulse of 0.1 A g−1 for a pulse time of 10 minutes, followed by a relaxation time of 60 minutes. To determine the diffusivity of lithium-ion (DLi+), the calculation employed eqn (2), initially formulated by Weppner and Huggins. This method aids in assessing the rate at which lithium ions diffuse within the material.
 | (2) |
 | (3) |
Subsequently, electrochemical impedance spectroscopy (EIS) was conducted to gain insights into the enhanced electrochemical kinetics of both SiOC and S-SiOC electrodes after 100 cycles. The Nyquist plots of the electrodes, accompanied by their equivalent circuit, are displayed in Fig. 4f, while the EIS fitting outcomes are provided in Table S1.† These plots split into distinct components, including electrolyte resistance (Rs), resistance associated with the solid–electrolyte interface (RSEI), and charge transfer resistance (Rct). Furthermore, the straight line observed in the low-frequency region, known as the Warburg line, corresponds to lithium-ion diffusion occurring between the electrolyte and the electrode interface.73 After 100 cycles, it's noteworthy that the Rs and RSEI values for the S-SiOC electrode are closely aligned with those of the SiOC electrode, measuring 4.57 Ω and 12.2 Ω, respectively, and 4.69 Ω and 12.8 Ω, respectively. However, the Rct value of the S-SiOC electrode, standing at 43.0 Ω, is approximately half the magnitude of the SiOC electrode's Rct value of 87.2 Ω. These outcomes strongly indicate that sulfur doping contributes to an enhancement in electrical conductivity and an increase in ionic diffusion within the electrode. This phenomenon can be attributed to the sulfur-induced expansion of the carbon interlayer spacing, coupled with its potential to lower the energy barrier for lithium-ion diffusion. Additionally, sulfur can provide supplementary pathways for electron transfer, facilitated by the presence of C–S–C bonding, as highlighted in the XRD results.32,38,72
The rate performance of both SiOC and S-SiOC electrodes was evaluated across different current densities, as depicted in Fig. 5a–c. Specifically, SiOC electrodes delivered reversible specific capacities of 529, 416, 322, 238, and 164 mA h g−1 at current densities of 0.2, 0.5, 1, 2, and 4 A g−1, respectively. Through the heightened electrical conductivity afforded by sulfur doping, the S-SiOC electrodes exhibited significantly elevated capacities of 626, 505, 406, 312, and 231 mA h g−1 at the same respective current densities. Furthermore, as the current density increased, it became evident from Fig. 5b and c that the degree of overpotential was more pronounced in bare SiOC compared to S-SiOC. Interestingly, when the current density was lowered from 4 A g−1 to 0.2 A g−1, a notable increase in reversible specific capacities was observed. Specifically, larger reversible specific capacities of 570 mA h g−1 and 680 mA h g−1 were recorded for SiOC and S-SiOC electrodes, respectively, upon the current density's reduction. These outcomes could likely be attributed to the activation of the electrodes during cycling, which contributes to the increased capacities.
 | ||
| Fig. 5 (a) Rate performance of SiOC and S-SiOC at different current densities. Corresponding voltage profiles of (b) SiOC and (c) S-SiOC at various current densities. (d) Cycling performance and coulombic efficiency of SiOC and S-SiOC at 1 A g−1 for 1000 cycles (0.7 mg cm−2 of loading mass) and (e and f) corresponding voltage profiles at current density of 1 A g−1. (g) Discharge areal capacity plot of SiOC and S-SiOC at 1 A g−1 for 250 cycles (2.2 mg cm−2 of loading mass) and (h and i) corresponding voltage profiles at current density of 1 A g−1. | ||
The cycling performances of the electrodes were assessed at a current density of 1 A g−1, as illustrated in Fig. 5d–f. Both the SiOC and S-SiOC electrodes display activation processes and impressive cycling stability, attributable to the inherent structural stability within the SiOC-based electrode.7 This activation process could potentially be attributed to the micro-sized nature of the produced SiOC particles, as well as their dense surfaces.63 Remarkably high coulombic efficiencies of 99.6% were consistently observed for both types of electrodes throughout the 1000 consecutive cycles. However, S-SiOC electrode exhibited a remarkable increase in reversible capacity throughout cycling, indicating ongoing activation. As results of Fig. 5e and f, this dynamic activation process contributed to the S-SiOC electrode achieving a higher specific capacity of 650 mA h g−1 with no capacity fading, in contrast to the SiOC electrode's specific capacity of 480 mA h g−1 and capacity retention of 83.9% after 1000 cycles. Furthermore, as shown in Fig. S8,† S-SiOC electrode showed exceptional capacity retention, with an impressive 89.2% retention after 2000 cycles at 1 A g−1. To evaluate the high loading potential of the S-SiOC electrode, we fabricated electrodes with a mass loading of 2.2 mg cm−2. As depicted in Fig. 5g, the S-SiOC electrode attained a discharge areal capacity surpassing 0.88 mA h cm−2 during 250 cycles at 1 A g−1. In contrast, SiOC electrode demonstrated relatively lower areal capacity under the same conditions. Moreover, Fig. 5h and i indicates that the cycle stability of S-SiOC electrode was notably superior, with maintaining a capacity retention of 80.6% after 250 cycles, while bare SiOC showed 69.5% remaining capacity. This exhibits the enhanced electrical performance of the S-SiOC electrode, which can be largely attributed to the effect of sulfur doping. While many SiOC-based electrodes contend with challenges related to electrically insulating characteristics and poor ionic transport, the S-SiOC electrodes exhibit improved electrical conductivity, thereby leading to exceptional cycle and rate performance. Our synthesis method and electrochemical performance data on S-SiOC as the anode are compared to the data found in the literature, as shown in Table S2.† To provide qualitative information on the durability of the S-SiOC electrode, surface analysis was conducted following cycling. The SEM images of the S-SiOC electrode before and after cycling are shown in Fig. S9a and b.† Both images display the morphologies of the S-SiOC electrode fabricated using a CMC binder and carbon. After cycling test, it was observed that there were no substantial agglomerations or cracks compared to the pre-cycling state, and the morphology displayed similarities. The TEM analysis findings of the S-SiOC electrode post cycling indicated that the spherical morphology of the S-SiOC electrode was sustained, as depicted in Fig. S9c.† Consequently, the SEM and TEM analyses conducted post cycling indicated that the morphology of the S-SiOC remained predominantly unchanged, thereby highlighting the stability of S-SiOC.74,75
4. Conclusion
In summary, successful synthesis of sulfur-doped silicon oxycarbide (S-SiOC) through a facile pyrolysis process of commercial silicone oil marks a significant advancement in the field. This study has demonstrated the exceptional capabilities of S-SiOC as an anode material for lithium-ion batteries (LIBs). The electrode exhibits noteworthy attributes including a substantial specific capacity, remarkable capacity retention, and the ability to accommodate a high mass loading of active materials. These outstanding electrochemical performances are directly linked to the enhanced conductivity of S-SiOC, a consequence of the sulfur doping strategy employed. The experimental findings offer compelling evidence that the introduction of sulfur atoms enhances the material's capacity to store Li+ ions, resulting in improved Li+ diffusion rates and overall rate capability. Additionally, the formation of covalent bonds between carbon and sulfur atoms contributes to the heightened electrical conductivity of the material. As a result, the S-SiOC electrode emerges as a highly promising candidate for anode applications in lithium-ion batteries. The demonstrated electrochemical performance of S-SiOC, coupled with its facile synthesis process, positions it as a promising potential for advancing the performance of energy storage systems and driving the evolution of battery technologies.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2021R1A2C1008380 & no. 2022R1A6A1A03063039), the Higher Education DisciplineInnovation Project (111 Project, D18012), and Nano Material Technology Development Program (NRF-2015M3A7B6027970). Also, this research was partly supported by the Korea Institute of Energy Technology Evaluation and Planning (KETEP) grant funded by the Korean government (MOTIE) (20215710100170).References
- J. M. Tarascon and M. Armand, Issues and challenges facing rechargeable lithium batteries, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- P. G. Bruce, B. Scrosati and J. M. Tarascon, Nanomaterials for rechargeable lithium batteries, Angew Chem. Int. Ed. Engl., 2008, 47, 2930–2946 CrossRef CAS PubMed.
- J. B. Goodenough and Y. Kim, Challenges for rechargeable Li Batteries, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- N. Nitta, F. Wu, J. T. Lee and G. Yushin, Li-ion battery materials: present and future, Mater. Today, 2015, 18, 252–264 CrossRef CAS.
- H. Fukui, Y. Harimoto, M. Akasaka and K. Eguchi, Lithium species in electrochemically lithiated and delithiated silicon oxycarbides, ACS Appl. Mater. Interfaces, 2014, 6, 12827–12836 CrossRef CAS PubMed.
- C. K. Chan, H. Peng, G. Liu, K. McIlwrath, X. F. Zhang, R. A. Huggins and Y. Cui, High-performance lithium battery anodes using silicon nanowires, Nat. Nanotechnol., 2008, 3, 31–35 CrossRef CAS.
- Y. Yao, M. T. McDowell, I. Ryu, H. Wu, N. Liu, L. Hu, W. D. Nix and Y. Cui, Interconnected silicon hollow nanospheres for lithium-ion battery anodes with long cycle life, Nano Lett., 2011, 11, 2949–2954 CrossRef CAS.
- H. Kim and J. Cho, Superior lithium electroactive mesoporous Si@carbon core−shell nanowires for lithium battery anode material, Nano Lett., 2008, 8, 3688–3691 CrossRef CAS PubMed.
- H. Sun and K. Zhao, Atomistic origins of high capacity and high structural stability of polymer-derived SiOC anode materials, ACS Appl. Mater. Interfaces, 2017, 9, 35001–35009 CrossRef CAS PubMed.
- R. Sujith, J. Gangadhar, M. Greenough, R. K. Bordia and D. K. Panda, A review of silicon oxycarbide ceramics as next generation anode materials for lithium-ion batteries and other electrochemical applications, J. Mater. Chem. A, 2023, 11, 20324–20348 RSC.
- R. Bhandavat and G. Singh, Stable and efficient Li-ion battery anodes prepared from polymer-derived silicon oxycarbide–carbon nanotube shell/core composites, J. Phys. Chem. C, 2013, 117, 11899–11905 CrossRef CAS.
- G. Shao, D. A. H. Hanaor, J. Wang, D. Kober, S. Li, X. Wang, X. Shen, M. F. Bekheet and A. Gurlo, Polymer-derived SiOC integrated with a graphene aerogel as a highly stable Li-ion battery anode, ACS Appl. Mater. Interfaces, 2020, 12, 46045–46056 CrossRef CAS PubMed.
- A. Maheshwari, E. P. Gopikrishnan, J. Gangadhar and R. Sujith, Highly conducting graphene dispersed silicon oxycarbide glasses, Mater. Chem. Phys., 2020, 239, 121963 CrossRef CAS.
- R. Bhandavat, M. Cologna and G. Singh, Polymer-derived SiOC–CNT paper as lithium-ion battery anodes, Nanomater. Energy, 2012, 1, 57–61 CrossRef CAS.
- Y. Li, Y. Hu, Y. Lu, S. Zhang, G. Xu, K. Fu, S. Li, C. Chen, L. Zhou, X. Xia and X. Zhang, One-dimensional SiOC/C composite nanofibers as binder-free anodes for lithium-ion batteries, J. Power Sources, 2014, 254, 33–38 CrossRef CAS.
- L. David, R. Bhandavat, U. Barrera and G. Singh, Silicon oxycarbide glass-graphene composite paper electrode for long-cycle lithium-ion batteries, Nat. Commun., 2016, 7, 10998 CrossRef CAS PubMed.
- Y. Ren, B. Yang, X. Huang, F. Chu, J. Qiu and J. Ding, Intercalated SiOC/graphene composites as anode material for li-ion batteries, Solid State Ionics, 2015, 278, 198–202 CrossRef CAS.
- Z. Sang, X. Yan, L. Wen, D. Su, Z. Zhao, Y. Liu, H. Ji, J. Liang and S. X. Dou, A graphene-modified flexible SiOC ceramic cloth for high-performance lithium storage, Energy Storage Mater., 2020, 25, 876–884 CrossRef.
- R. J. Dubey, P. V. W. Sasikumar, F. Krumeich, G. Blugan, J. Kuebler, K. V. Kravchyk, T. Graule and M. V. Kovalenko, Silicon oxycarbide-tin nanocomposite as a high-power-density anode for Li-ion batteries, Adv. Sci., 2019, 6, 1901220 CrossRef CAS PubMed.
- J. Kaspar, C. Terzioglu, E. Ionescu, M. Graczyk-Zajac, S. Hapis, H. J. Kleebe and R. Riedel, Stable SiOC/Sn nanocomposite anodes for lithium-ion batteries with outstanding cycling stability, Adv. Funct. Mater., 2014, 24, 4097–4104 CrossRef CAS.
- J. Wang, R. Ma, Z. Zhou, G. Liu and Q. Liu, Magnesiothermic synthesis of sulfur-doped graphene as an efficient metal-free electrocatalyst for oxygen reduction, Sci. Rep., 2015, 5, 9304 CrossRef PubMed.
- C. Hu, D. Liu, Y. Xiao and L. Dai, Functionalization of graphene materials by heteroatom-doping for energy conversion and storage, Prog. Nat. Sci., 2018, 28, 121–132 CrossRef CAS.
- B. Quan, A. Jin, S. H. Yu, S. M. Kang, J. Jeong, H. D. Abruna, L. Jin, Y. Piao and Y. E. Sung, Solvothermal-derived S-doped Graphene as an anode material for sodium-ion batteries, Adv. Sci., 2018, 5, 1700880 CrossRef PubMed.
- Y. Yang, S. Jin, Z. Zhang, Z. Du, H. Liu, J. Yang, H. Xu and H. Ji, Nitrogen-doped hollow carbon nanospheres for high-performance Li-ion batteries, ACS Appl. Mater. Interfaces, 2017, 9, 14180–14186 CrossRef CAS PubMed.
- Y. Tan, C. Xu, G. Chen, Z. Liu, M. Ma, Q. Xie, N. Zheng and S. Yao, Synthesis of ultrathin nitrogen-doped graphitic carbon nanocages as advanced electrode materials for supercapacitor, ACS Appl. Mater. Interfaces, 2013, 5, 2241–2248 CrossRef CAS.
- X. Miao, D. Sun, X. Zhou and Z. Lei, Designed formation of nitrogen and sulfur dual-doped hierarchically porous carbon for long-life lithium and sodium ion batteries, Chem. Eng. J., 2019, 364, 208–216 CrossRef CAS.
- J. Lin, Y. Xu, J. Wang, B. Zhang, D. Li, C. Wang, Y. Jin and J. Zhu, Nitrogen-doped hierarchically porous carbonaceous nanotubes for lithium ion batteries, Chem. Eng. J., 2018, 352, 964–971 CrossRef CAS.
- X. Zhou, Z. Zhang, P. Yan, Y. Jiang, H. Wang and Y. Tang, Sulfur-doped reduced graphene oxide/Sb2S3 composite for superior lithium and sodium storage, Mater. Chem. Phys., 2020, 244, 122661 CrossRef CAS.
- X. Wang, G. Li, F. M. Hassan, J. Li, X. Fan, R. Batmaz, X. Xiao and Z. Chen, Sulfur covalently bonded graphene with large capacity and high rate for high-performance sodium-ion batteries anodes, Nano Energy, 2015, 15, 746–754 CrossRef CAS.
- J. P. Paraknowitsch and A. Thomas, Doping carbons beyond nitrogen: an overview of advanced heteroatom doped carbons with boron, sulphur and phosphorus for energy applications, Energy Environ. Sci., 2013, 6, 2839–2855 RSC.
- Q. Hu, Y. Meng, H. Zhang, G. Zhao, J. Hu, F. Zhu and Y. Zhang, Encapsulated Ni3S2 nanoparticles with N, S dual-doped carbon nanotubes: a robust structure for lithium storage, J. Electroanal. Chem., 2020, 873, 114383 CrossRef CAS.
- H. Tao, M. Zhou, K. Wang, S. Cheng and K. Jiang, Nickel sulfide nanospheres anchored on reduced graphene oxide in situ doped with sulfur as a high performance anode for sodium-ion batteries, J. Mater. Chem. A, 2017, 5, 9322–9328 RSC.
- C. H. Wong, Z. Sofer, K. Klimova and M. Pumera, Microwave exfoliation of graphite oxides in H2S plasma for the synthesis of sulfur-doped graphenes as oxygen reduction catalysts, ACS Appl. Mater. Interfaces, 2016, 8, 31849–31855 CrossRef CAS PubMed.
- H. L. Poh, P. Simek, Z. Sofer and M. Pumera, Sulfur-doped graphene via thermal exfoliation of graphite oxide in H2S, SO2, or CS2 gas, ACS Nano, 2013, 7, 5262–5272 CrossRef CAS PubMed.
- Y. Lee, K. Y. Lee and W. Choi, One-pot synthesis of antimony-embedded silicon oxycarbide materials for high-performance sodium-ion batteries, Adv. Funct. Mater., 2017, 27, 1702607 CrossRef.
- D. Kim, H. Kim, H. Lim, K. J. Kim, H. G. Jung, D. Byun, C. Kim and W. Choi, A facile control in free-carbon domain with divinylbenzene for the high-rate-performing Sb/SiOC composite anode material in sodium-ion batteries, Int. J. Energy Res., 2020, 44, 11473–11486 CrossRef CAS.
- C. Chandra and J. Kim, Silicon oxycarbide produced from silicone oil for high-performance anode material in sodium ion batteries, Chem. Eng. J., 2018, 338, 126–136 CrossRef CAS.
- Q. Jin, W. Li, K. Wang, P. Feng, H. Li, T. Gu, M. Zhou, W. Wang, S. Cheng and K. Jiang, Experimental design and theoretical calculation for sulfur-doped carbon nanofibers as a high performance sodium-ion battery anode, J. Mater. Chem. A, 2019, 7, 10239–10245 RSC.
- Z. Sang, Z. Zhao, D. Su, P. Miao, F. Zhang, H. Ji and X. Yan, SiOC nanolayer wrapped 3D interconnected graphene sponge as a high-performance anode for lithium ion batteries, J. Mater. Chem. A, 2018, 6, 9064–9073 RSC.
- W. Li, M. Zhou, H. Li, K. Wang, S. Cheng and K. Jiang, A high performance sulfur-doped disordered carbon anode for sodium ion batteries, Energy Environ. Sci., 2015, 8, 2916–2921 RSC.
- L. Qie, W. Chen, X. Xiong, C. Hu, F. Zou, P. Hu and Y. Huang, Sulfur-doped carbon with enlarged interlayer distance as a high-performance anode material for sodium-ion batteries, Adv. Sci., 2015, 2, 1500195 Search PubMed.
- M. Wang, Y. Xia, X. Wang, Y. Xiao, R. Liu, Q. Wu, B. Qiu, E. Metwalli, S. Xia, Y. Yao, G. Chen, Y. Liu, Z. Liu, J. Q. Meng, Z. Yang, L. D. Sun, C. H. Yan, P. Muller-Buschbaum, J. Pan and Y. J. Cheng, Silicon oxycarbide/carbon nanohybrids with tiny silicon oxycarbide particles embedded in free carbon matrix based on photoactive dental methacrylates, ACS Appl. Mater. Interfaces, 2016, 8, 13982–13992 CrossRef CAS PubMed.
- H. Fukui, H. Ohsuka, T. Hino and K. Kanamura, A Si-O-C composite anode: high capability and proposed mechanism of lithium storage associated with microstructural characteristics, ACS Appl. Mater. Interfaces, 2010, 2, 998–1008 CrossRef CAS PubMed.
- H. Shi, A. Yuan and J. Xu, Tailored synthesis of monodispersed nano/submicron porous silicon oxycarbide (SiOC) spheres with improved Li-storage performance as an anode material for Li-ion batteries, J. Power Sources, 2017, 364, 288–298 CrossRef CAS.
- J. Kaspar, M. Graczyk-Zajac and R. Riedel, Lithium insertion into carbon-rich SiOC ceramics: influence of pyrolysis temperature on electrochemical properties, J. Power Sources, 2013, 244, 450–455 CrossRef CAS.
- X. Liu, M.-C. Zheng and K. Xie, Mechanism of lithium storage in Si–O–C composite anodes, J. Power Sources, 2011, 196, 10667–10672 CrossRef CAS.
- W. Li, D. Peng, W. Huang, X. Zhang, Z. Hou, W. Zhang, B. Lin and Z. Xing, Adjusting coherence length of expanded graphite by self-activation and its electrochemical implication in potassium ion battery, Carbon, 2023, 204, 315–324 CrossRef CAS.
- L. Cheng, C. Ma, W. Lu, X. Wang, H. Yue, D. Zhang and Z. Xing, A graphitized hierarchical porous carbon as an advanced cathode host for alkali metal-selenium batteries, Chem. Eng. J., 2022, 433, 133527 CrossRef CAS.
- U. Holzwarth and N. Gibson, The Scherrer equation versus the 'Debye-Scherrer equation', Nat. Nanotechnol., 2011, 6, 534 CrossRef CAS PubMed.
- Z. Li, Z. Xu, X. Tan, H. Wang, C. M. B. Holt, T. Stephenson, B. C. Olsen and D. Mitlin, Mesoporous nitrogen-rich carbons derived from protein for ultra-high capacity battery anodes and supercapacitors, Energy Environ. Sci., 2013, 6, 871–878 RSC.
- J. Yang, X. Zhou, D. Wu, X. Zhao and Z. Zhou, S-doped N-rich carbon nanosheets with expanded interlayer distance as anode materials for sodium-ion batteries, Adv. Mater., 2017, 29, 1604108 CrossRef PubMed.
- M. Halim, C. Hudaya, A.-Y. Kim and J. K. Lee, Phenyl-rich silicone oil as a precursor for SiOC anode materials for long-cycle and high-rate lithium ion batteries, J. Mater. Chem. A, 2016, 4, 2651–2656 RSC.
- S. Yu, R. Tu and T. Goto, Preparation of SiOC nanocomposite films by laser chemical vapor deposition, J. Eur. Ceram. Soc., 2016, 36, 403–409 CrossRef CAS.
- W. Gu, M. Sevilla, A. Magasinski, A. B. Fuertes and G. Yushin, Sulfur-containing activated carbons with greatly reduced content of bottle neck pores for double-layer capacitors: a case study for pseudocapacitance detection, Energy Environ. Sci., 2013, 6, 2465–2476 RSC.
- F. Wang, B. Wang, Z. Yu, Q. Lv, F. Jun, C. Bao and D. Wang, A simple and green self-conversion method to construct silicon hollow spheres for high-performance Li-ion battery anodes, Electrochim. Acta, 2023, 443, 141950 CrossRef CAS.
- C. Bao, J. Wang, B. Wang, J. Sun, L. He, Z. Pan, Y. Jiang, D. Wang, X. Liu, S. X. Dou and J. Wang, 3D sodiophilic Ti3C2 MXene@g-C3N4 Hetero-interphase raises the stability of sodium metal anodes, ACS Nano, 2022, 16, 17197–17209 CrossRef CAS PubMed.
- J. Wang, Y. Gao, Z. He, X. Feng, D. Lin, D. Kong, X. Zhang, H. Hu, Z. Guo and D. Chen, Regulation structural units and morphology of SiOC anode for enhanced high-rate storage and long-life lithium ion batteries, J. Power Sources, 2024, 621, 235238 CrossRef CAS.
- M. Bae, S.-J. Park, M. Kim, E. Kwon, S. Yu, J. Choi, Y. Chang, Y. Kim, Y. J. Choi, H. Hong, L. Lin, W. Zhang, S. Park, J. Y. Maeng, J. Park, S.-Y. Lee, S.-H. Yu and Y. Piao, Synergistic regulation of intrinsic lithiophilicity and mass transport kinetics of non-lithium-alloying nucleation sites for stable operation of low N/P ratio lithium metal batteries, Adv. Energy Mater., 2024, 14, 2304101 CrossRef CAS.
- M. Nie, D. P. Abraham, Y. Chen, A. Bose and B. L. Lucht, Silicon solid electrolyte interphase (SEI) of lithium ion battery characterized by microcopy and spectroscopy, J. Phys. Chem. C, 2013, 117, 13403–13412 CrossRef CAS.
- T. Jaumann, J. Balach, M. Klose, S. Oswald, U. Langklotz, A. Michaelis, J. Eckert and L. Giebeler, SEI-component formation on sub 5 nm sized silicon nanoparticles in Li-ion batteries: the role of electrode preparation, FEC addition and binders, Phys. Chem. Chem. Phys., 2015, 17, 24956–24967 RSC.
- S. J. An, J. Li, C. Daniel, D. Mohanty, S. Nagpure and D. L. Wood III, The state of understanding of the lithium-ion-battery graphite solid electrolyte interphase (SEI) and its relationship to formation cycling, Carbon, 2016, 105, 52–76 CrossRef CAS.
- M. Wilamowska-Zawlocka, P. Puczkarski, Z. Grabowska, J. Kaspar, M. Graczyk-Zajac, R. Riedel and G. D. Sorarù, Silicon oxycarbide ceramics as anodes for lithium ion batteries: influence of carbon content on lithium storage capacity, RSC Adv., 2016, 6, 104597–104607 RSC.
- B. Dong, Y. Han, T. Wang, Z. Lei, Y. Chen, F. Wang, H. Abadikhah, S. A. Khan, L. Hao, X. Xu, R. Cao, L. Yin and S. Agathopoulos, Hard SiOC microbeads as a high-performance lithium-ion battery anode, ACS Appl. Energy Mater., 2020, 3, 10183–10191 CrossRef CAS.
- M. Ma, H. Wang, M. Niu, L. Su, X. Fan, J. Deng, Y. Zhang and X. Du, High rate capabilities of HF-etched SiOC anode materials derived from polymer for lithium-ion batteries, RSC Adv., 2016, 6, 43316–43321 RSC.
- G. Ning, X. Ma, X. Zhu, Y. Cao, Y. Sun, C. Qi, Z. Fan, Y. Li, X. Zhang, X. Lan and J. Gao, Enhancing the Li storage capacity and initial coulombic efficiency for porous carbons by sulfur doping, ACS Appl. Mater. Interfaces, 2014, 6, 15950–15958 CrossRef CAS PubMed.
- H. Kim, J. Lee, H. Ahn, O. Kim and M. J. Park, Synthesis of three-dimensionally interconnected sulfur-rich polymers for cathode materials of high-rate lithium-sulfur batteries, Nat. Commun., 2015, 6, 7278 CrossRef CAS PubMed.
- H. Li, Z. Cheng, A. Natan, A. M. Hafez, D. Cao, Y. Yang and H. Zhu, Dual-function, tunable, nitrogen-doped carbon for high-performance Li metal-sulfur full cell, Small, 2019, 15, 1804609 CrossRef PubMed.
- C.-W. Chang-Jian, B.-C. Ho, C.-K. Chung, J.-A. Chou, C.-L. Chung, J.-H. Huang, J.-H. Huang and Y.-S. Hsiao, Doping and surface modification enhance the applicability of Li4Ti5O12 microspheres as high-rate anode materials for lithium ion batteries, Ceram. Int., 2018, 44, 23063–23072 CrossRef CAS.
- J.-Y. Lin, C.-C. Hsu, H.-P. Ho and S.-h. Wu, Sol–gel synthesis of aluminum doped lithium titanate anode material for lithium ion batteries, Electrochim. Acta, 2013, 87, 126–132 CrossRef CAS.
- J. Kaspar, M. Graczyk-Zajac and R. Riedel, Determination of the chemical diffusion coefficient of Li-ions in carbon-rich silicon oxycarbide anodes by electro-analytical methods, Electrochim. Acta, 2014, 115, 665–670 CrossRef CAS.
- C. Chandra, H. S. Cahyadi, S. Alvin, W. Devina, J.-H. Park, W. Chang, K. Y. Chung, S. K. Kwak and J. Kim, Revealing the sodium storage mechanism in high-temperature-synthesized silicon oxycarbides, Chem. Mater., 2019, 32, 410–423 CrossRef.
- X. Yuan, S. Chen, J. Li, J. Xie, G. Yan, B. Liu, X. Li, R. Li, L. Pan and W. Mai, Understanding the improved performance of sulfur-doped interconnected carbon microspheres for Na-ion storage, Carbon Energy, 2021, 3, 615–626 CrossRef CAS.
- J. Jang, H. Kim, H. Lim, K. J. Kim, H.-G. Jung, S.-O. Kim and W. Choi, Surfactant-based selective assembly approach for Si-embedded silicon oxycarbide composite materials in lithium-ion batteries, Chem. Eng. J., 2020, 401, 126091 CrossRef CAS.
- F. Wang, B. Wang, Z. Yu, C. Zhu, P. Liu, J. Li, B. Wang, Y. Zhou, D. Wang, H. K. Liu and S. Dou, Construction of air-stable pre-lithiated SiOx anodes for next-generation high-energy-density lithium-ion batteries, Cell Rep. Phys. Sci., 2022, 3, 100872 CrossRef CAS.
- F. Wang, B. Wang, T. Ruan, T. Gao, R. Song, F. Jin, Y. Zhou, D. Wang, H. Liu and S. Dou, Construction of structure-tunable Si@Void@C anode materials for lithium-ion batteries through controlling the growth kinetics of resin, ACS Nano, 2019, 13, 12219–12229 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04608k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |