
An electrografted monolayer of polyaniline as a tuneable platform for a glucose biosensor†
Elžbieta
Ragauskaitė
 a,
Samuelis
Marčiukaitis
a,
Ingrida
Radveikienė
a and
Gintautas
Bagdžiūnas
*ab
a,
Samuelis
Marčiukaitis
a,
Ingrida
Radveikienė
a and
Gintautas
Bagdžiūnas
*ab
aGroup of Supramolecular Analysis, Institute of Biochemistry, Life Sciences Centre, Vilnius University, Saulėtekio av. 7, LT-10257, Vilnius, Lithuania. E-mail: gintautas.bagdziunas@gmc.vu.lt; Tel: +370 5 2234393
bDepartment of Functional Materials and Electronics, Center for Physical Sciences and Technology, Saulėtekio av. 3, LT-10257, Vilnius, Lithuania
First published on 1st February 2024
Abstract
Polyaniline (PANI), a nanostructured conducting polymer, has shown significant potential in optical and bioelectrochemical devices. However, its performance and stability on various substrates are hindered by weak adhesion to the surface. In this study, a strongly adherent polyaniline conducting polymer layer with a thickness of five nanometers was electrografted onto an initiating monolayer on gold and tin-doped indium oxide substrates. These electrografted monolayers consist of vertically oriented fully oxidized-protonated (pernigraniline salt) and deprotonated (pernigraniline base) forms of polyaniline. The monolayer exhibits pH-dependent colour changes and it is suitable for enzyme compatibility. In light of these findings, we have developed and characterized an electrochemical glucose biosensor based on the monolayer of polyaniline on a gold electrode. The biosensor utilizes glucose oxidase as the biorecognition element for the selective detection of glucose concentrations in real blood plasma samples.
1. Introduction
Nanostructured conducting polymers have been extensively studied and widely applied in state-of-the-art technologies due to the processing advantages of polymeric conductors and the nano-size effect of nanomaterials.1 Vertically aligned and ordered conducting polymer monolayers, known as polymer brushes, have received special attention due to their fast charge transfer and the associated mass transport phenomenon characteristic of nanolayers.2 In recent years, the fabrication of polymer brushes using surface-initiated polymerization techniques has led to significant advancements in surface science and applications.3 On the other hand, the deposition of conducting polymers as thin layers onto various materials, such as glass and metals, has garnered considerable attention in the field of nanotechnology.4 However, a major issue with these layers is the typically weak and short-term adhesion between the polymers and the substrate. To address this problem, covalent bonding to the surface of the substrate and surface graft polymerization through chemical or electrochemical deposition, achieved by oxidizing the monomer, have been employed. In these approaches, polyaniline (PANI) has garnered great interest as a conducting polymer for a broad range of applications, including gas, pH, and bioanalyte sensors, printable electronics, electrochromics and photovoltaics, actuators, and supercapacitors.5Grafting molecules onto carbon-based surfaces through the electroreduction of aromatic diazonium salts is a widely used electrografting method, resulting in the formation of an aromatic organic layer that is covalently bonded to the surface.6 This diazonium approach is commonly employed for electrografting PANI onto carbon substrates by means of electrochemical polymerization of aniline.7,8 A facile method for chemically grafting conjugated tetraaniline as the smallest conjugated repeat unit of polyaniline decorated with a reactive azide group onto important materials including graphite, carbon nanotubes, reduced graphite oxide and polymers was reported by Lin et al. The authors found that these tetraaniline modified membranes are hydrophilic and exhibit extraordinarily low bovine serum albumin (BSA) and Escherichia coli adhesions.9 Additionally, this method is suitable for electrografting on metal oxide electrodes such as tin-doped indium oxide (ITO) for enzyme immobilization and bioelectrocatalysis.10 Through covalent bonding to metal oxide substrates, new and innovative methods have been proposed and developed. Zhang et al. demonstrated the modification of an ITO substrate with 4-aminobenzylphosphonic acid, followed by the covalent bonding of a stable PANI thin layer with a thickness of 30 nm through electropolymerization.11 Gold (Au) substrates are also suitable for PANI electrografting. Vacca et al. coated a gold substrate with PANI using the electrografting of aryl diazonium salts.12 Furthermore, Pillalamarri et al. synthesized PANI polymers on gold nanoparticles using surfactants with a side peroxide group.13
High blood glucose levels, exceeding 6 mM, indicate diabetes mellitus as a metabolic disorder resulting from defects in insulin secretion, insulin action, or both. Consequently, the quick and convenient determination of glucose levels in human blood holds great importance in bioanalysis. In 1962, Clark and Lyons published the first enzymatic glucose sensor utilizing glucose oxidase (GOx) as a biorecognition element.14 This seminal work paved the way for intensive scientific research on the development and application of biosensors. Meanwhile, PANI has found widespread use as an interlayer between biomolecules and electrode substrates in glucose biosensors.15 This is owing to its favourable conductivity, cost-effectiveness, and straightforward fabrication. Numerous fabrication methods for PANI-based bioelectrodes in glucose biosensor applications have been extensively studied and discussed in the scientific literature.16 However, their commercialization remains challenging due to issues such as ageing and stability effects, low electrochemical stability of PANI, and the absence of well-optimized deposition techniques.
In this study, a stable electrografted monolayer is designed, prepared, and characterized as conducting polymer brushes on ITO and Au substrates. This PANI monolayer demonstrates efficient oxygen reduction and pH-response, while being biocompatible with enzymes. Consequently, a stable biosensor for glucose detection is tested and developed using this approach.
2. Results and discussion
2.1 Electrografting of PANI monolayers
To prepare the grafted polyaniline (PANI) monolayer on indium-tin-oxide (ITO) and gold (Au) surfaces, a combination of chemical and electrochemical methods was employed. Scheme 1 depicts a schematic visualization of the process for the deposition of the initiating monolayer and polymerization of electrografted polyaniline monolayers on the Au and ITO surfaces. It illustrates the sequential steps involved in the preparation of the monolayers. First, the ITO and Au surfaces were chemically and electrochemically cleaned. Then, 3-(phenylamino)propyltrimethoxysilane (PhNHPrSi) and 4-aminothiophenol (SPhN) containing N-substituted phenyl groups were deposited on the activated ITO and Au surfaces, respectively, serving as the polymerization initiating monolayers. Finally, aniline monomers were electrochemically polymerized via cyclic voltammetry (CV) on the initiating monolayers to obtain the electrografted polyaniline monolayers on Au and ITO surfaces.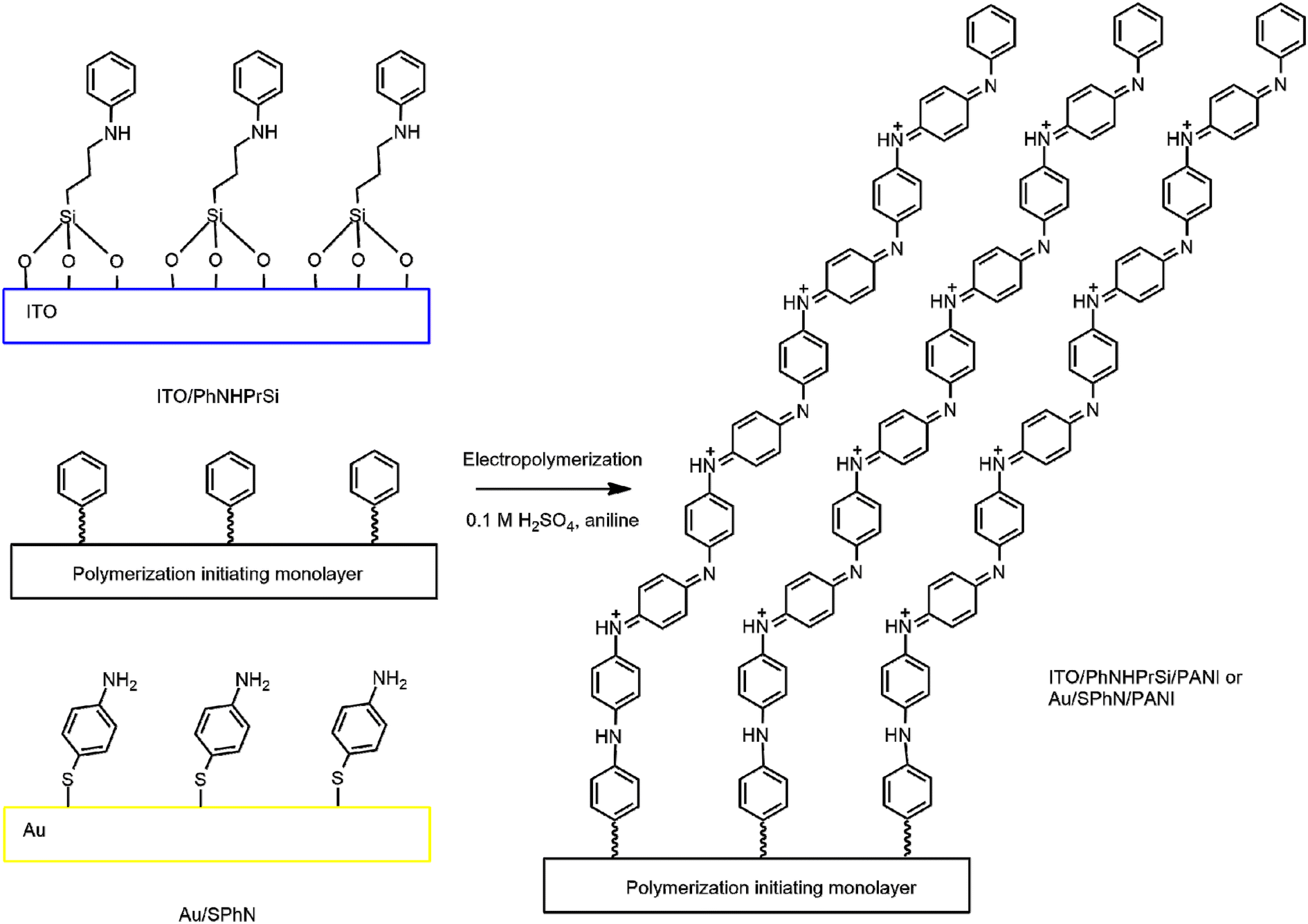 | ||
| Scheme 1 Schematic visualization of the polymerization initiating and electrografted polyaniline monolayers on Au and ITO. | ||
As the monomer, aniline was then subjected to electrochemical polymerization through cyclic voltammetry (CV) in 0.1 M H2SO4 electrolyte solution on the aforementioned monolayers. Fig. S1† shows the electrochemical synthesis of the electrografted PANI monolayer on the ITO and gold substrates with correspondingly grafting PhNHPrSi and SPhN molecules using CV. However, no significant difference in the electrochemical response was observed. This process resulted in the formation of electrografted and non-grafted regular PANI. The resulting ITO/PhNHPrSi/PANI and Au/SPhN/PANI electrodes were immersed in water and subjected to ultrasonic cleaning to remove any non-grafted PANI polymers. The progress of polymer removal from the ITO/PhNHPrSi/PANI surface was monitored using UV-vis absorption spectroscopy in pH 4 buffer at 5-minute intervals (Fig. S2a†). It was observed that the absorption of the layer remained unchanged after 35 minutes, indicating the removal of additional polymers. The resulting ITO/PhNHPrSi/PANI monolayer exhibited a light green colour and remained firmly attached to the ITO glass. In contrast, Fig. S2b and c† illustrate that the unattached regular electropolymerized PANI layer on ITO has weak adhesion to the surface and is not stable after an ultrasonic 30 min washing. Mechanical removal of this layer was only possible when removing the entire ITO layer from the glass. On the other hand, the monolayer on the Au surface could only be removed through abrasive cleaning followed by electrochemical oxidation and reduction in 0.5 M H2SO4 solution.
2.2 Characterization of the surfaces
The surfaces were characterized to understand their properties and morphology. ITO and Au electrodes were chosen for further research and characterization due to the optical transparency of ITO and the large surface area of the electrode. After the deposition of ITO/PhNHPrSi with hydrophobic groups, the average water contact angles increased from 38 ± 5° for the bare activated ITO to 78 ± 4°. However, after the deposition of the PANI monolayer, the surface became more hydrophilic than that of the bare activated ITO, with a water contact angle of 23 ± 4°. Fig. 1a illustrates that the ITO/PhNHPrSi/PANI surface is more hydrophilic than the original ITO surface.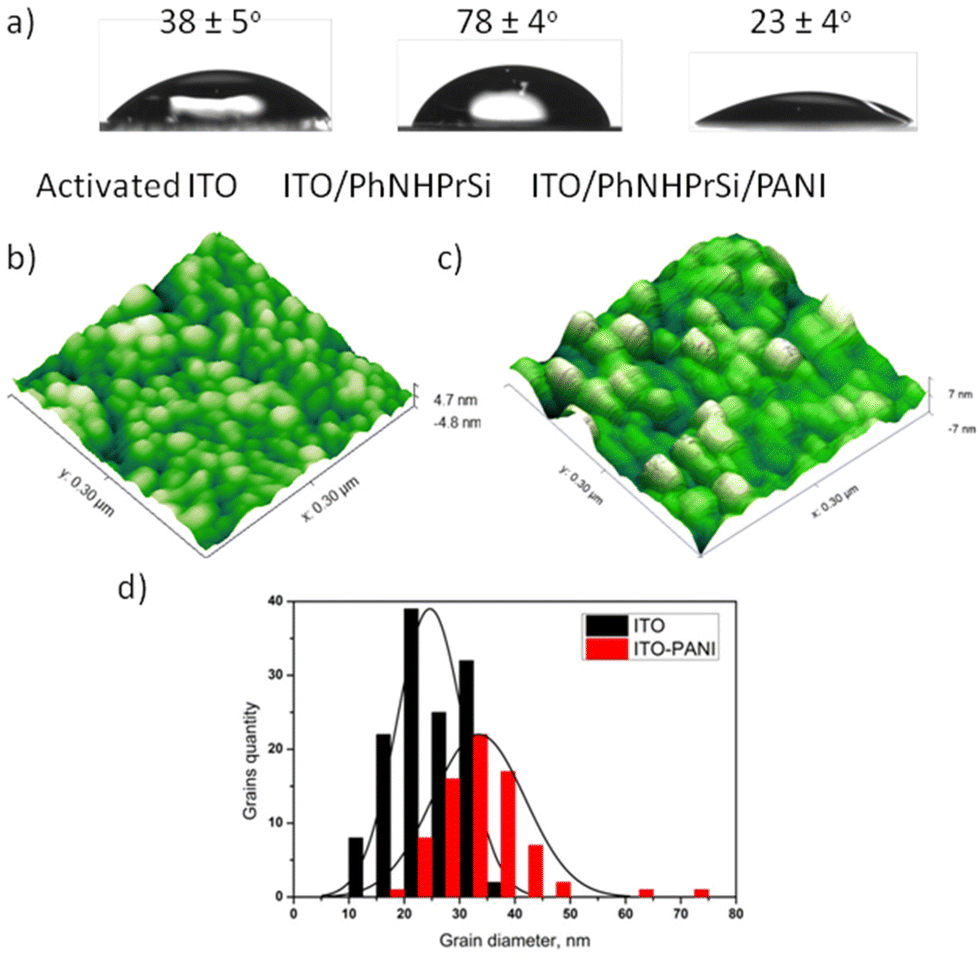 | ||
| Fig. 1 Properties of the ITO/PhNHPrSi/PANI surface: (a) the averaged water contact angles of the bare ITO, ITO/PhNHPrSi, and ITO/PhNHPrSi/PANI surfaces; (b) AFM image of the bare ITO and (c) the ITO/PhNHPrSi/PANI surfaces; (d) the statistical distributions of grain diameters (the histograms are mean distributions of the grains diameters) on the surfaces. | ||
To estimate the average lengths of the grafted PANI polymer, the surface morphologies of the bare ITO and ITO/PhNHPrSi/PANI surfaces were investigated using atomic force microscopy (AFM) in a 300 nm × 300 nm area, as shown in Fig. 1b and c. Both surfaces exhibited characteristic grains, with average diameters of 23.8 ± 0.5 nm and 33.5 ± 0.7 nm for the bare ITO and ITO/PhNHPrSi/PANI surfaces, respectively. The increase in the grain diameter is proportional to the length of the PANI molecules.17 Therefore, by analyzing the statistical distributions of grain diameters, the average length of the grafted PANI polymer was calculated to be 4.8 ± 0.1 nm. Based on density functional theory (DFT) computation, this length corresponds to eight aniline monomers attached to the anchor PhNHPrSi molecule (Fig. S3b†).
To confirm the molecular structures of the monolayer, Fourier-transform infrared spectroscopy (FT-IR) was performed on the starting ITO/PhNHPrSi silane monolayer, as well as the ITO/PhNHPrSi/PANI samples before and after ultrasonic washing (Fig. 2). After electropolymerization, the electrografted PANI and unattached regular electropolymerized PANI layers formed on this electrode. Subsequent to ultrasonic washing, only the monolayer of PANI adhered to the electrode persisted. Consequently, the FT-IR spectrum (Fig. 2b) of the sample exhibited a greater number of bands before the washing process. The spectra of the samples before (Fig. 2b) and after (Fig. 2c) ultrasonic washing exhibit characteristic peaks indicating a fully oxidized pernigraniline base form.18 Specifically, the peak at 1586 cm−1 with a shoulder at 1604 cm−1 corresponds to the C–C stretching vibration of the phenyl ring in PANI in the sample after polymerization without washing (Fig. 2b). After washing, the peak at 1586 cm−1 remains (Fig. 2c) indicating the presence of both non-grafted and grafted PANI polymers in the layer. Additionally, characteristic peaks at 1514 cm−1, 1349 cm−1, and 1172 cm−1, corresponding to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibration of the quinoneimine ring, stretching of C–N bonds, and scissoring of C–H bonds, respectively, are observed in the spectrum of the ITO/PhNHPrSi/PANI self-assembly (SAM) monolayer (Fig. 2c). The broad peak at 851 cm−1 is assigned to ITO and silane vibrations. The spectra were also computationally simulated using density functional theory (DFT) at the B3LYP/6-31G(d,p) level to confirm the structural analysis. The experimental FT-IR spectrum of the monolayer (Fig. 2c) closely matches the theoretical spectrum of the proposed fully oxidized and protonated PANI (pernigraniline salt) form (Fig. 2g). The monolayer may also contain the pernigraniline base form (Fig. 2e) and not fully protonated pernigraniline base and salt forms (Fig. 2f). However, peaks corresponding to the leucoemeraldine form (Fig. 2d) are not observed in the experimental spectrum. Summing up, the FT-IR spectroscopy, in conjunction with theoretical calculations, confirms the presence of the fully oxidized and protonated PANI (pernigraniline salt) form in the ITO/PhNHPrSi/PANI monolayer. The monolayer may also contain other protonated PANI forms, but the not reduced PANI form was detected in the experimental spectrum. It is worth noting that the emeraldine form of PANI is not necessary to explain all the color changes in our case.
C stretching vibration of the quinoneimine ring, stretching of C–N bonds, and scissoring of C–H bonds, respectively, are observed in the spectrum of the ITO/PhNHPrSi/PANI self-assembly (SAM) monolayer (Fig. 2c). The broad peak at 851 cm−1 is assigned to ITO and silane vibrations. The spectra were also computationally simulated using density functional theory (DFT) at the B3LYP/6-31G(d,p) level to confirm the structural analysis. The experimental FT-IR spectrum of the monolayer (Fig. 2c) closely matches the theoretical spectrum of the proposed fully oxidized and protonated PANI (pernigraniline salt) form (Fig. 2g). The monolayer may also contain the pernigraniline base form (Fig. 2e) and not fully protonated pernigraniline base and salt forms (Fig. 2f). However, peaks corresponding to the leucoemeraldine form (Fig. 2d) are not observed in the experimental spectrum. Summing up, the FT-IR spectroscopy, in conjunction with theoretical calculations, confirms the presence of the fully oxidized and protonated PANI (pernigraniline salt) form in the ITO/PhNHPrSi/PANI monolayer. The monolayer may also contain other protonated PANI forms, but the not reduced PANI form was detected in the experimental spectrum. It is worth noting that the emeraldine form of PANI is not necessary to explain all the color changes in our case.
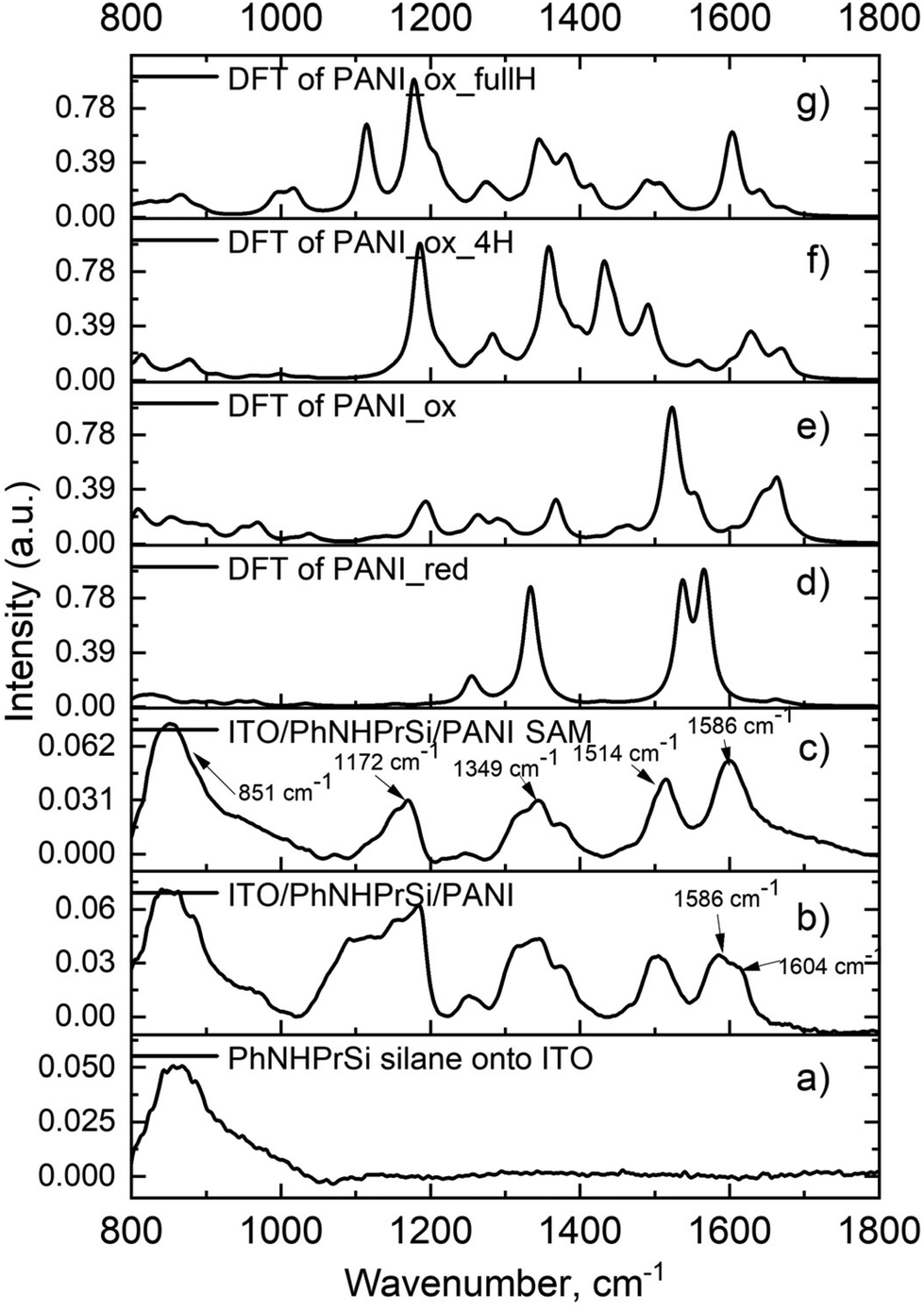 | ||
| Fig. 2 Experimental FT-IR spectra recorded in the ATR mode: (a) PhNHPrSi silane on ITO; (b) ITO/PhNHPrSi/PANI layer after electropolymerization; (c) ITO/PhNHPrSi/PANI self-assembly monolayer (SAM) after washing, and theoretical spectra of various PANI polymer forms: (d) reduced PANI_red (leucoemeraldine); (e) deprotonated PANI_ox (pernigraniline base); (f) not fully protonated PANI_ox_4H (pernigraniline half base and salt); (g) fully oxidized and protonated PANI_ox_fullH (pernigraniline salt) forms. | ||
2.3 Optical and electrochemical properties of the PANI monolyers
The optical properties of the ITO/PhNHPrSi/PANI monolayer were investigated by varying the buffer pH from 3.4 to 10 (Fig. 3a). Observable colour changes were noted on the slide, with the monolayer appearing light green at acidic pH 3.4, blue at neutral pH 7, and purple at alkaline pH 10. These colour variations are indicative of different protonation states of the PANI polymer. UV-vis spectra (Fig. 3a) reveal a broad band at around 800 nm and a shoulder at around 420 nm for the ITO/PhNHPrSi/PANI monolayer under acidic conditions (pH 3.4, 4, and 5). As the pH increases, the band maximum undergoes a hypsochromic shift to 690 nm at pH 6 and 640 nm at pH 7. Under more alkaline conditions, the band maximum shifts and stabilizes at around 600 nm. The pH dependence of the band maximum at 600 nm and 800 nm is illustrated in Fig. 3b, representing the protonation and deprotonation processes of the PANI polymer. The point of intersection of these curves allows estimation of the negative logarithm of the acid dissociation constant (pKa) for the electrografted PANI, which was determined to be 6.1. This value is lower than the pKa value of 6.9 observed for the electrochemically deposited PANI layer (∼1 μm thick) on ITO in a previous study.19 Furthermore, slight variations in the absorption band maxima at 455 nm, 630 nm, and 855 nm were measured as the pH increased for the PANI monolayer.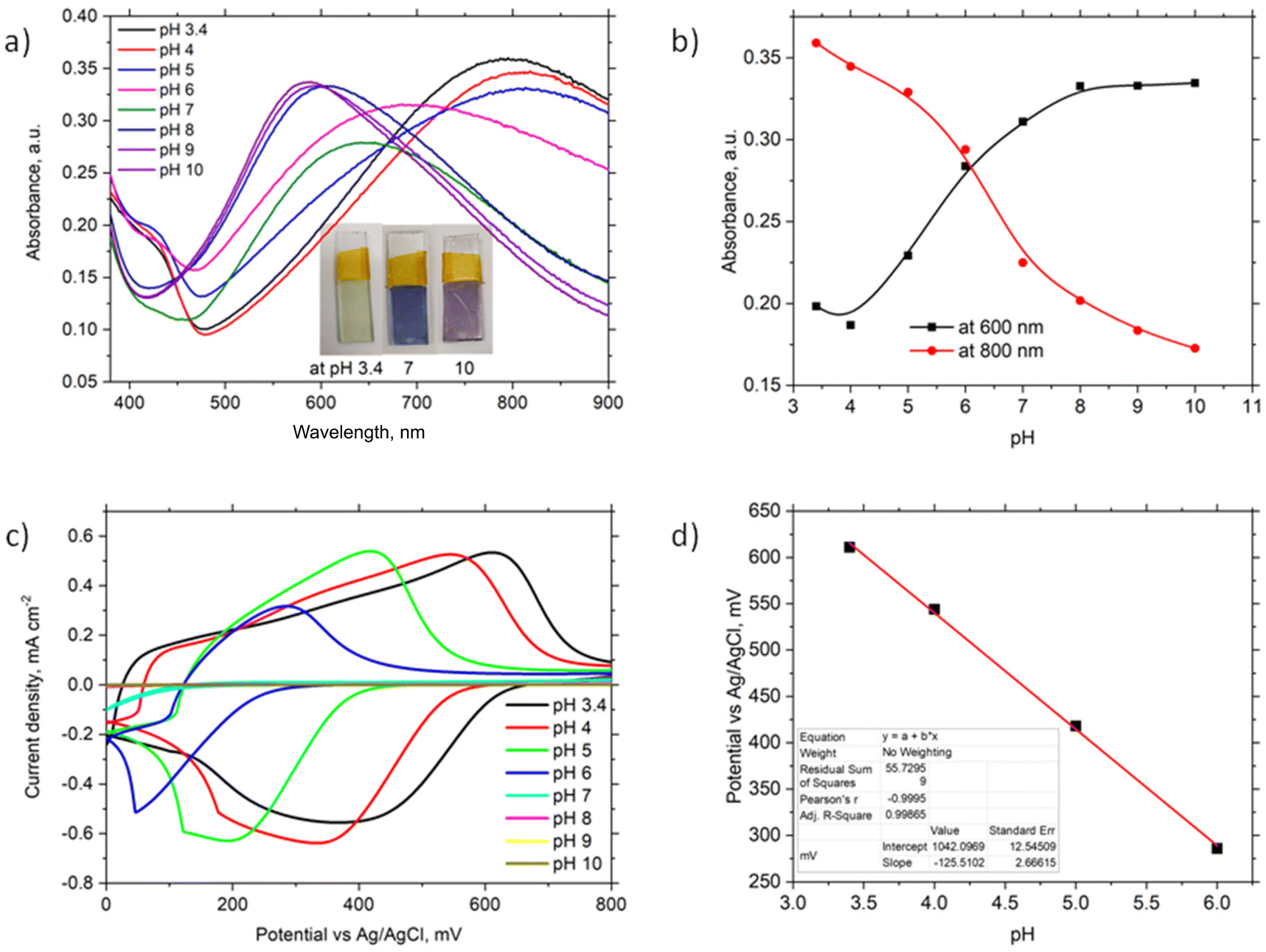 | ||
| Fig. 3 Optical and electrochemical properties of ITO/PhNHPrSi/PANI: (a) UV-vis spectra of the monolayer by changing the buffer pH from 3.4 to 10 (inset: visualization of the samples after exposure to the buffer of pH 3.4, 7, and 10); (b) the pH and band maximum trends for protonation and deprotonation of the samples at 600 nm and 800 nm; (c) CVs of the electrodes by changing the buffer pH from 3.4 to 10; (d) the potential against pH plot. | ||
Theoretical UV-vis spectra were computed for the optimized structures20 of the reduced PANI-red as the leucoemeraldine form, oxidized deprotonated PANI-ox as the pernigraniline base form, oxidized-tertraprotonated PANI-ox-4H as the pernigraniline half base and salt, and oxidized fully protonated PANI-ox-8H as the pernigraniline salt with a silane moiety (Fig. S3b†). The time dependent density functional theory TD-CAM-B3LYP/6-31+G(d,p) method with a polarizable continuum solvation model for water was used for the computation of UV-vis spectra of such PANI forms (Fig. S3a†). The obtained theoretical spectra show good agreement with the experimental results (Fig. 3a). The experimental spectra exhibit bands at around 430 nm and 800 nm, which correspond to the oxidized-tertraprotonated PANI-ox-4H form. This form predominates in solutions with pH values ranging from 3.4 to 5. On the other hand, the oxidized deprotonated PANI-ox form is predominant in solutions with pH values of 8 to 10. A mixture of these forms is observed at an intermediate pH of 6 to 7. It is important to note that the absorption bands of the reduced PANI-red and the oxidized fully protonated PANI-ox-8H forms would be expected to appear at around 330 nm and 1300 nm, respectively. However, due to the limited absorption window of ITO, these bands were not observed in the experimental spectra.
To investigate the electrochemical properties of the ITO/PhNHPrSi/PANI monolayer as the working electrode, cyclic voltammetry (CV) was conducted by varying the pH of the electrolyte from 3.4 to 10 and applying working potentials ranging from 0 to 800 mV versus Ag/AgCl. During the measurements, a decrease in the redox potentials of the positive current density peaks was observed as the pH increased from 3.4 to 6 (Fig. 3c). However, no redox signal was observed at pH values higher than 7 because the deprotonated form of PANI, known as the pernigraniline base, is non-conductive. Furthermore, a single wide redox signal indicated a synchronous transfer of protons and electrons. The redox potential values were plotted against pH (Fig. 3d). The ITO/PhNHPrSi/PANI electrode exhibited a linear Nernstian response of 125.5 ± 2.5 mV per pH with an excellent correlation coefficient of R2 = 0.999 within the pH range of 3.4 to 6. Assuming a temperature of 298 K and combining the ideal gas and Faraday constants into a single value, the potential against pH plot should increase according to the equation: 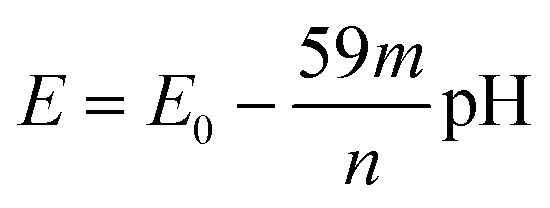 , where m and n represent the number of protons and electrons involved in the redox reaction, respectively. In our case, the redox reaction involved two electrons (n = 2) and four protons (m = 4). Therefore, the linear Nernstian response of the ITO/PhNHPrSi/PANI electrode is twice as high as the typical response for most pH sensors based on regular PANI.21 This indicates that the electrografted polymer is fully deprotonated and then protonated. We attempted to use these glass electrodes as optical sensors for pH determination in juice samples. Unfortunately, the juices had pH values between 2 and 3, and their optical absorption interfered with the analysis, making it challenging to accurately determine the pH using this method.
, where m and n represent the number of protons and electrons involved in the redox reaction, respectively. In our case, the redox reaction involved two electrons (n = 2) and four protons (m = 4). Therefore, the linear Nernstian response of the ITO/PhNHPrSi/PANI electrode is twice as high as the typical response for most pH sensors based on regular PANI.21 This indicates that the electrografted polymer is fully deprotonated and then protonated. We attempted to use these glass electrodes as optical sensors for pH determination in juice samples. Unfortunately, the juices had pH values between 2 and 3, and their optical absorption interfered with the analysis, making it challenging to accurately determine the pH using this method.
It is worth noting that the ITO-based electrodes are not stable at a negative potential due to the reduction of indium and tin oxides. Therefore, during electrochemistry at a negative potential, the used electrode loses conductivity and the attached monolayer. These ITO electrodes are suitable only for optical measurements. For a wider application of the PANI monolayer, the structures were deposited on the Au surface. So, similar optical properties were observed for the electrografted monolayer on Au. Following the electrochemical synthesis of Au/SPhN/PANI in an acidic solution, the monolayer appeared transparent, revealing the gold surface (Fig. S2d†). However, after ultrasonication of the electrode, the monolayer became nontransparent, resulting in a blue surface appearance (Fig. S2d†). Additionally, the Au-based electrodes are stable to −0.4 V vs. Ag/AgCl potential. Therefore, the Au/SPhN/PANI electrode was used for further work on biosensors.
2.4 A glucose biosensor based on the PANI monolayer
The Au/SPhN/PANI electrode was used to fabricate an electrochemical biosensor based on glucose oxidase (GOx). GOx is an affordable and stable enzyme that finds widespread use in the analysis of glucose concentrations in various biological samples.14 In our previous studies, we have extensively employed this enzyme to assess the biocompatibility and efficacy of polymeric interlayers of electrodes in biosensor applications.22 To evaluate the performance of our system, a drop of GOx solution (1 mg mL−1) was deposited on the PANI monolayer and allowed to dry. This step facilitated the immobilization of the enzyme on the electrode surface. To prevent the adsorbed enzyme from being washed away, a dialysis membrane was added as a protective barrier (Fig. 4). This configuration ensured the retention of the enzyme on the electrode surface and enabled the detection of glucose through enzymatic reactions.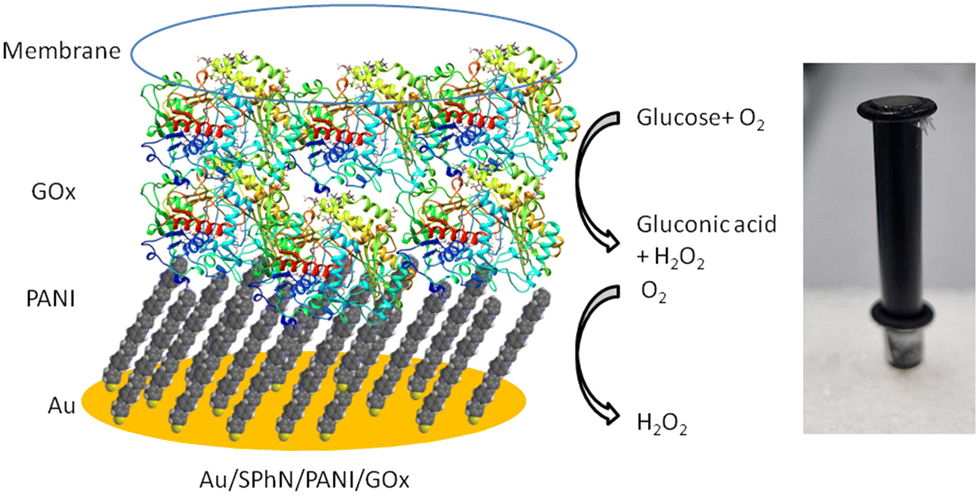 | ||
| Fig. 4 Schematic presentation and photography of the Au/SPhN/PANI/GOx bioelectrode with the reactions occurring in the corresponding layers. | ||
To investigate the electrochemical properties of the Au/SPhN/PANI/GOx bioelectrode, CV measurements were conducted in a pH 7 buffer solution, both without and with the addition of 10 mM glucose (Glu). Fig. 5a illustrates changes in currents at negative and positive potentials, but no bioelectrocatalytic currents indicative of direct charge carrier transfer between the electrode and the enzyme were observed.23 Also, chronoamperometry measurements were performed at different potentials (−0.4 V, 0.25 V, −0.25 V vs. Ag/AgCl) to assess the response to Glu concentration. Notably, there was no change in current upon adding Glu at 0.25 V and −0.25 V. Still a noticeable current response was observed at −0.4 V. Also, no current response of the Au/SPhN/PANI electrode after the addition of 1 mM Glu at −0.4 V vs. Ag/AgCl and pH 7 was indicated (Fig. S4†).
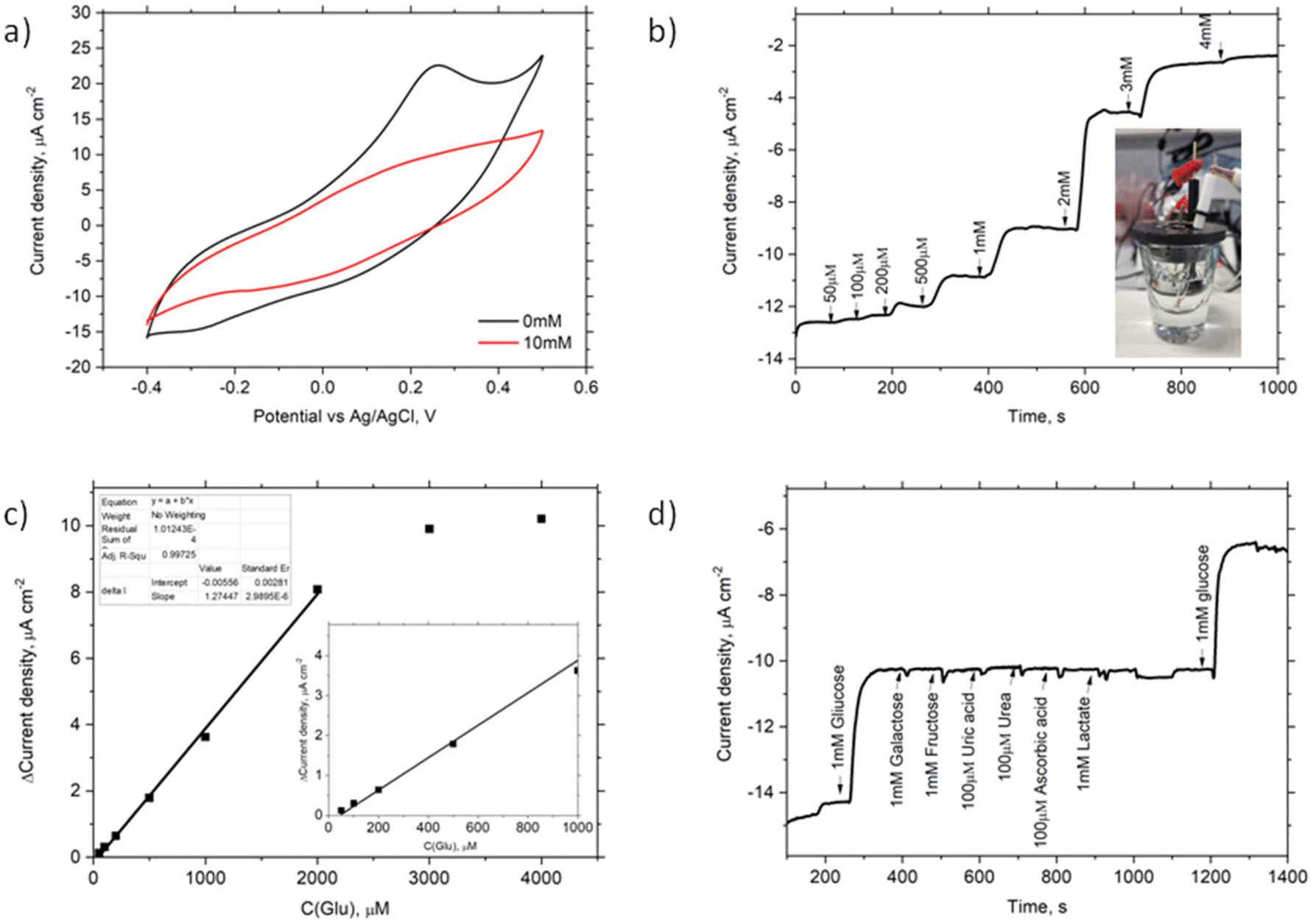 | ||
| Fig. 5 Electrochemical characterization of the Au/SPhN/PANI/GOx bioelectrode: (a) CV of this bioelectrode without and with 10 mM Glu in PPB pH 7 from −0.4 V to 0.5 V vs. Ag/AgCl at a scan rate of 5 mV s−1; (b) the chronoamperometric titration from 50 μM to 4 mM of Glu in PPB pH = 7 (inset: photography of the electrochemical cell used in the experiments); (c) changes in currents at −0.4 V vs. Ag/AgCl upon the addition of glucose (inset: data of linear fitting and the change in currents at low Glu concentrations); (d) chronoamperometry after adding interfering reagents at −0.4 V vs. Ag/AgCl. | ||
To determine the optimal buffer pH, current responses at this potential were measured using four different buffer solutions ranging from pH 5 to pH 8. Notably, the current response at pH 5 was discernible; however, the most significant response to the addition of 1 mM glucose was observed at pH 7 (see Fig. S5a†). Furthermore, it is widely recognized that the enzyme exhibits greater stability and reactivity at a neutral pH, which is why this pH was selected for subsequent experiments. Fig. 5b displays the current density response obtained by chronoamperometry as the Glu concentration was varied from 50 μM to 4 mM. A linear relationship between current and Glu concentration was observed in the range of 50 μM to 2 mM, with a squared correlation coefficient (R2) of 0.999. Based on this linear range, the sensitivity and limit of detection (LOD) of the bioelectrode were calculated to be 3.7 μA mM−1 cm−2 and 17 μM, respectively, with a signal-to-noise ratio (S/N) of 3.0.
The repeatability of the bioelectrode was evaluated by performing five measurements at a fixed concentration of 1 mM Glu. As shown in Fig. S5b,† the maximum difference in response currents was only 3%. The statistical repeatability, i.e. relative standard deviation (RSD%), for 1 mM Glu samples (N = 6) remained below 1.5%. Additionally, after storing the bioelectrode in a PPB solution at 4 °C for seven days, the response currents only decreased by 11%. These results indicate that the electrode surface decorated with the electrografted polymer is biocompatible with biomolecules and suitable for use in biosensor technology. To assess the selectivity of the Au/SPhN/PANI/GOx bioelectrode, interfering reagents that may be present in blood samples, such as galactose, fructose, uric acid, urea, ascorbic acid, and lactate, were tested. Fig. 5d shows that these reagents did not interfere or inhibit the glucose bioanalysis. Finally, the prepared Au/SPhN/PANI/GOx bioelectrode was employed to detect Glu concentrations in a pH 7 buffer solution and bovine blood serum. In the real Cormay Serum HN sample, the declared range of Glu concentration was 4.13–5.05 mM, and the measured Glu concentration was found to be 4.6 ± 0.2 mM (N = 3).
The classification of enzymatic electrochemical biosensors into three generations based on their mechanisms of action is well-established in the field.14 In the first-generation biosensors, the electrochemical signal is directly related to the concentration of a product or reagent of the enzymatic reaction, such as hydrogen peroxide or oxygen. In the second-generation biosensors, an electrochemically active mediator is used to transfer the charge between the enzyme and the electrode. In the third-generation biosensors, the charge is transferred directly from the enzyme to the electrode.24 Additional experiments were performed to determine the mechanism of action of the Au/SPhN/PANI/GOx-based biosensor. Hydrogen peroxide was added to the solution, and the dissolved oxygen was removed by purging the electrolyte with argon gas, respectively. The observed low drop in currents after the addition of hydrogen peroxide and the smaller increment in currents after the addition of glucose in the absence of oxygen confirm that the biosensor operates based on the first-generation mechanism, where the response is dependent on the concentration of oxygen (Fig. S6a and b†). First, glucose oxidizes with oxygen to gluconic acid and hydrogen peroxide by catalyzing GOx. Second, the decrease of oxygen concentration is electrochemically registered during the reduction of the oxidized PANI layer by oxygen (Fig. 4). This finding is consistent with previous studies that have shown PANI to be an effective electrocatalyst for the reduction of oxygen to hydrogen peroxide. Furthermore, comparison of the performance of the Au/SPhN/PANI/GOx-based biosensor with that of other PANI-based biosensors has demonstrated its good and reliable biosensor performance. Overall, these findings support the conclusion that the Au/SPhN/PANI/GOx biosensor operates based on the first-generation mechanism and exhibits comparable performance to other published PANI-based biosensors.16
According to the review,16 the characteristics obtained for the Au/SPhN/PANI/GOx-based biosensor, including sensitivity, linear range, and limit of detection, are comparable to the results reported in the literature for similar glucose biosensors based on PANI. These characteristics have been provided and compared with the literature results of glucose biosensors based on the PANI polymer and GOx without metallic nanostructures in Table 1. Horng et al. demonstrated an amperometric enzyme biosensor based on GOx-incorporated polyaniline nanowires (PANI-NWs) on a carbon cloth (CC) electrode at 0.5 V vs. Ag/AgCl with a linear range up to 8 mM by detecting H2O2 (Table 1, entry 2).25 However, this high positive potential cannot avoid interference from ascorbic acid, a common interfering substance in the detection of glucose. Kuwahara et al. provided an amperometric glucose bioelectrode based on a PANI/PAA (poly(acrylic acid)) composite film with immobilized GOx and catalase (Cat). The performance of this biosensor is based on a competitive oxygen consumption reaction with PANI. The authors utilized immobilized Cat to reproduce O2 from H2O2 by GOx-catalytic glucose oxidation. The addition of Cat significantly enhances the performance of this biosensor (Table 1, entry 3).26 Zhang and colleagues chemically prepared PANI microtubes. The immobilization of GOx on the surface of a glassy carbon (GC) electrode with these PANI microtubes was utilized to prepare the glucose biosensor. The authors showed that increasing the nanostructured surface area affects the sensitivity of this biosensor, but a narrower linearity up to 0.8 mM was observed (Table 1, entry 4).27 The fact that our biosensor shows good sensitivity, a wide linear range, and a low limit of detection demonstrates its potential for accurate and reliable glucose concentration measurements.
3. Conclusions
Through comprehensive characterization and performance analysis, we have gained insights into the electrochemical and optical properties of the electrografted PANI monolayer on ITO and Au substrates. Based on our AFM measurements, the average length of the grafted PANI polymers was calculated to be 4.8 ± 0.1 nm. The optical properties of grafted PANI exhibited pH-dependent colour changes, while the electrochemical properties demonstrated a high linear Nernstian response of 125.5 ± 2.5 mV per pH, indicating the suitability for pH sensing applications. The fully oxidized and protonated PANI (pernigraniline salt) form of this grafted monolayer was proved by vibrational spectroscopy and theoretical computation. Furthermore, we have successfully developed an electrochemical glucose biosensor based on a monolayer of polyaniline (PANI) on a gold electrode. The biosensor utilizes glucose oxidase (GOx) as the biorecognition element for the selective detection of glucose concentrations at −0.4 V vs. Ag/AgCl. The GOx-based biosensor showed excellent sensitivity and a wide linear range, from 0.05 mM to 2 mM, of glucose detection, with a sensitivity of 3.7 μA mM−1 cm−2 and a limit of detection of 16 μM. The biosensor also exhibited good repeatability and stability, and can be used to detect glucose in real blood plasma samples. Based on the mechanistic studies, the biosensor operates through the first-generation mechanism, as the response is dependent on the presence of oxygen. The PANI monolayer on the electrode surface effectively catalyzes the reduction of oxygen to hydrogen peroxide, enabling the measurement of glucose concentrations by detecting oxygen consumption. Overall, this research has demonstrated the successful development of a highly sensitive and selective electrochemical glucose biosensor based on the electrografted monolayer of polyaniline. We believe that this PANI-based platform will be suitable for biosensors based on other oxidase enzymes as well.4. Methods
The used reagents, instruments, all the experimental procedures of electrode preparation and electrochemical grafting of PANI, and theoretical computational methods are provided in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the European Regional Development Fund under the “Promotion of Centers of Excellence and Innovation and Technology Transfer Centers” grant No. 01.2.2-CPVA-K-703-03-0010. Ragauskaitė is thankful for the support of the project through the Student Research Project funding agreement No. P-ST-23-78. Thanks are expressed to M. Jankunec for the AFM, and I. Plikusienė and V. M. Mačiulis for the ellipsometry experiments.References
- Y. Xue, S. Chen, J. Yu, B. R. Bunes, Z. Xue, J. Xu, B. Lu and L. Zang, J. Mater. Chem. C, 2020, 8, 10136–10159 RSC.
- F. S. Kim, G. Ren and S. A. Jenekhe, Chem. Mater., 2011, 23, 682–732 CrossRef CAS.
- R. Wang, Q. Wei, W. Sheng, B. Yu, F. Zhou and B. Li, Angew. Chem., Int. Ed., 2023, 62, e202219312 CrossRef CAS PubMed.
- S. Wang, Z. Wang, J. Li, L. Li and W. Hu, Mater. Chem. Front., 2020, 4, 692–714 RSC.
- C. O. Baker, X. Huang, W. Nelson and R. B. Kaner, Chem. Soc. Rev., 2017, 46, 1510–1525 RSC.
- J. Pinson and F. Podvorica, Chem. Soc. Rev., 2005, 34, 429–439 RSC.
- W. Ullah, G. Herzog, N. Vilà and A. Walcarius, Electrochem. Commun., 2021, 122, 106896 CrossRef CAS.
- L. M. Santos, J. Ghilane, C. Fave, P.-C. Lacaze, H. Randriamahazaka, L. M. Abrantes and J.-C. Lacroix, J. Phys. Chem. C, 2008, 112, 16103–16109 CrossRef CAS.
- C.-W. Lin, S. Aguilar, E. Rao, W. H. Mak, X. Huang, N. He, D. Chen, D. Jun, P. A. Curson, B. T. McVerry, E. M. V. Hoek, S.-C. Huang and R. B. Kaner, Chem. Sci., 2019, 10, 4445–4457 RSC.
- T. G. A. A. Harris, N. Heidary, S. Frielingsdorf, S. Rauwerdink, A. Tahraoui, O. Lenz, I. Zebger and A. Fischer, ChemElectroChem, 2021, 8, 1329–1336 CrossRef CAS.
- W. Zhang, W. Ju, X. Wu, Y. Wang, Q. Wang, H. Zhou, S. Wang and C. Hu, Appl. Surf. Sci., 2016, 367, 542–551 CrossRef CAS.
- A. Vacca, M. Mascia, S. Rizzardini, S. Palmas and L. Mais, Electrochim. Acta, 2014, 126, 81–89 CrossRef CAS.
- S. K. Pillalamarri, F. D. Blum and M. F. Bertino, Chem. Commun., 2005, 4584–4585 RSC.
- J. Wang, Chem. Rev., 2008, 108, 814–825 CrossRef CAS PubMed.
- C. Dhand, M. Das, M. Datta and B. D. Malhotra, Biosens. Bioelectron., 2011, 26, 2811–2821 CrossRef CAS PubMed.
- V. Osuna, A. Vega-Rios, E. A. Zaragoza-Contreras, I. A. Estrada-Moreno and R. B. Dominguez, Biosensors, 2022, 12, 137 CrossRef CAS PubMed.
- I. Radveikienė, D. Palinauskas, E. Ragauskaitė and G. Bagdžiūnas, Appl. Surf. Sci., 2022, 600, 154170 CrossRef.
- S. Quillard, G. Louarn, S. Lefrant and A. G. Macdiarmid, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 12496–12508 CrossRef PubMed.
- M. Gicevicius, R. Celiesiute, J. Kucinski, A. Ramanaviciene, G. Bagdziunas and A. Ramanavicius, J. Electrochem. Soc., 2018, 165, H903 CrossRef CAS.
- G. Bagdžiūnas, Mol. Syst. Des. Eng., 2020, 5, 1504–1512 RSC.
- F. Mazzara, B. Patella, C. D'Agostino, M. G. Bruno, S. Carbone, F. Lopresti, G. Aiello, C. Torino, A. Vilasi, A. O'Riordan and R. Inguanta, Chemosensors, 2021, 9, 169 CrossRef CAS.
- G. Bagdžiūnas and D. Palinauskas, Biosensors, 2020, 10, 104 CrossRef PubMed.
- G. Bagdžiūnas, Š. Žukauskas and A. Ramanavičius, Biosens. Bioelectron., 2018, 102, 449–455 CrossRef PubMed.
- G. Bagdžiūnas and A. Ramanavičius, Phys. Chem. Chem. Phys., 2019, 21, 2968–2976 RSC.
- Y.-Y. Horng, Y.-K. Hsu, A. Ganguly, C.-C. Chen, L.-C. Chen and K.-H. Chen, Electrochem. Commun., 2009, 11, 850–853 CrossRef CAS.
- T. Kuwahara, K. Ogawa, D. Sumita, M. Kondo and M. Shimomura, J. Electroanal. Chem., 2018, 811, 62–67 CrossRef CAS.
- L. Zhang, C. Zhou, J. Luo, Y. Long, C. Wang, T. Yu and D. Xiao, J. Mater. Chem. B, 2015, 3, 1116–1124 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr03680d |
| This journal is © The Royal Society of Chemistry 2024 |