
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Base-induced reversible H2 addition to a single Sn(II) centre†
Roland C.
Turnell-Ritson
a,
Joshua S.
Sapsford
a,
Robert T.
Cooper
a,
Stella S.
Lee
a,
Tamás
Földes
b,
Patricia A.
Hunt
 a,
Imre
Pápai
*b and
Andrew E.
Ashley
*a
a,
Imre
Pápai
*b and
Andrew E.
Ashley
*a
aDepartment of Chemistry, Imperial College London, London, SW7 2AZ, UK. E-mail: a.ashley@imperial.ac.uk
bResearch Center for Natural Sciences, Hungarian Academy of Sciences, Magyar tudósok körútja 2, H-1117 Budapest, Hungary. E-mail: papai.imre@ttk.mta.hu
First published on 18th September 2018
Abstract
A range of amines catalyse the oxidative addition (OA) of H2 to [(Me3Si)2CH]2Sn (1), forming [(Me3Si)2CH]2SnH2 (2). Experimental and computational studies point to ‘frustrated Lewis pair’ mechanisms in which 1 acts as a Lewis acid and involve unusual late transition states; this is supported by the observation of a kinetic isotope effect 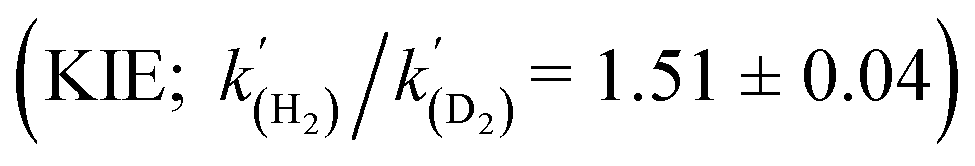 for Et3N. When DBU is used the energetics of H2 activation are altered, allowing an equilibrium between 1, 2 and adduct [1·DBU] to be established, thus demonstrating reversible oxidative addition/reductive elimination (RE) of H2 at a single main group centre.
for Et3N. When DBU is used the energetics of H2 activation are altered, allowing an equilibrium between 1, 2 and adduct [1·DBU] to be established, thus demonstrating reversible oxidative addition/reductive elimination (RE) of H2 at a single main group centre.
Introduction
In the past decade there has been significant interest in transition metal (TM) free systems which activate H2.1 Two main strategies have emerged to facilitate this reactivity: the use of low-valent main group (MG) compounds,2 and so-called ‘frustrated Lewis pairs’ (FLPs).3 In both cases, reactivity arises from simultaneously having access to a high-lying HOMO and low-lying LUMO (Fig. 1). Various low-valent MG compounds containing multiple E–E bonds (E = Al, Si, Ga, Ge, Sn),4,5 or single-site low-valent centres such as carbenes and heavier tetrylene analogues, have been shown to react with H2.6 The scope of Lewis bases (LBs) and, to a lesser extent, Lewis acids (LAs), which can be used in H2-activating FLPs has expanded to include a number of elements from across the periodic table. This is principally due to the readily tuneable steric and electronic profiles of the individual LA and LB sites.7–9 Many FLP systems display reversible H2 cleavage, which has facilitated their rapid expansion into the field of catalytic hydrogenation.10 The same is not true for low-valent MG compounds; examples of reversible H2 activation are very rare and limited to antiaromatic boracycles,11 a phosphorus-based singlet biradicaloid,12 and only one low-valent group 14 compound: a dinuclear Sn(I) distannyne.13 The design of single-site MG systems which are ergoneutral for H2 activation requires fine-tuning of thermodynamic (e.g. weak E–H bond strengths promoting an accessible formal En+2/En couple) and kinetic factors, both of which are constrained to a mononuclear species, and is hence especially challenging.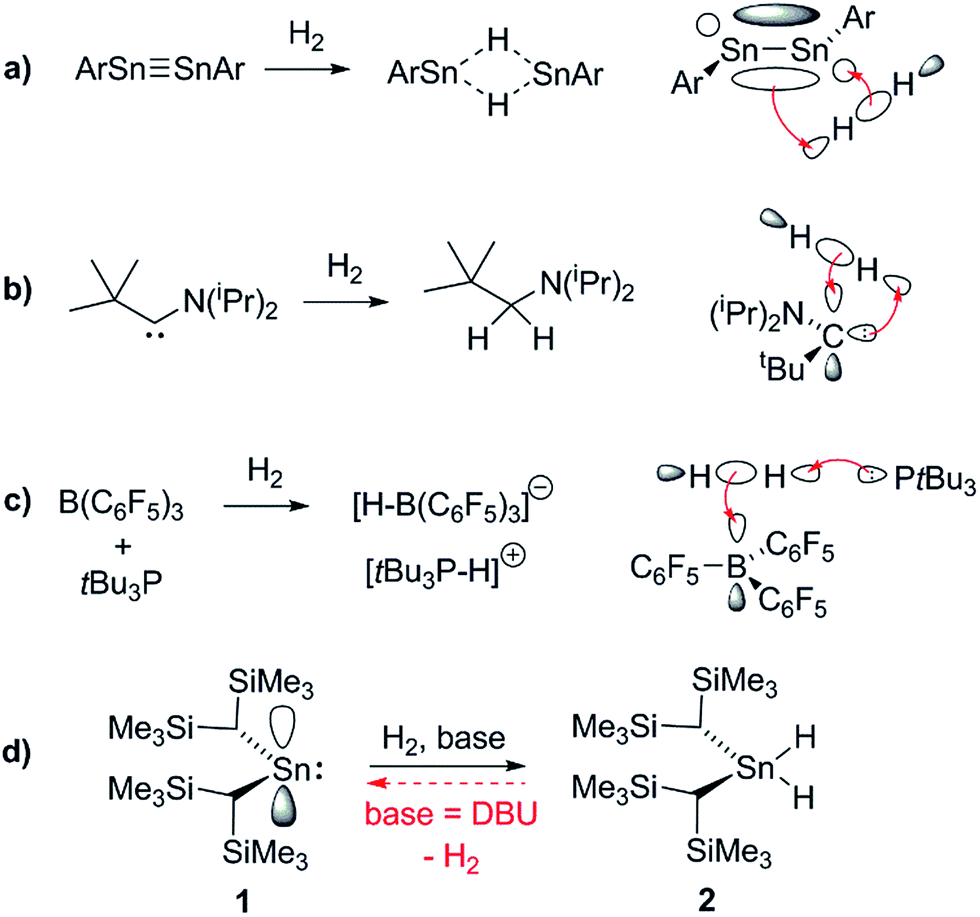 | ||
| Fig. 1 Representative orbital interactions between H2 and main group compounds: (a) unsaturated E–E compounds e.g. distannynes (Ar = C6H2-2,6-(C6H3-2,6-iPr2)2-4-X; X = H, SiMe3, F); for X = H, the reaction is reversible at 80 °C;5a,13 (b) single site low-valent centres e.g. carbenes;6a (c) sterically hindered LAs and LBs (FLPs); (d) this work. | ||
The ability of L2Sn(II) compounds to undergo OA has been inversely correlated with the size of the singlet–triplet (HOMO–LUMO) gap, which may be diminished through the use of extremely strong σ-donor ligands. Aldridge et al. have employed a bis(boryl)tin(II) system to achieve the only example of direct OA of H2 to a mononuclear Sn(II) centre, irreversibly forming the Sn(IV) dihydride; boryl ligands are even stronger σ-donors than hydride or alkyl ligands, permitting a successful reaction outcome.6d
Conversely, the irreversible base-induced RE of H2 from organostannanes is well-known.14 Wesemann and others have studied RE from ArSnH3 and [(Me3Si)2CH]SnH3 compounds to yield various mononuclear Sn and Sn–Sn bound species (Ar = terphenyl).15 Nevertheless, there has yet to be a report of reversible OA and RE occurring on a single Sn(II) scaffold. Lappert's stannylene [(Me3Si)2CH]2Sn (1), which can act as both Lewis acid (LA) and base (LB), is a paradigmatic system for investigating OA to low-valent MG centres, yet to date its reactivity with H2 has been unexplored.16 Herein we report the use of FLP methodology to promote formal OA of H2 to this simple dialkylstannylene. Furthermore we document the first example of reversible H2 addition to a single-site MG complex, which accesses an FLP via reversible dissociation of a classical 1·LB adduct; formation of the latter renders OA of H2 to 1 energetically less favourable, enabling RE to occur from the Sn(IV) dihydride and reform 1, which is in equilibrium with 1·LB.17
Results and discussion
1 is in a rapid solution-phase equilibrium with its dimer [1]2, which has been crystallographically characterised and contains a formal Sn![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Sn double bond.18 When a d8-toluene solution of 1/[1]2 was placed under an atmosphere of H2 (4 bar) in a sealed NMR tube, no change was observed in the 1H NMR spectrum, even after prolonged periods (>48 h), confirming that neither 1 nor [1]2 can react with H2 alone. Separately, addition of Et3N (20 mol%) to a solution of 1 resulted in no perturbation of their 1H NMR resonances, suggesting no interaction between the components; i.e. the formation of an FLP.19 Placing this new mixture under H2 (4 bar, RT) resulted in the solution turning from deep red to colourless over the course of 24 h, with the 1H NMR spectrum revealing complete consumption of 1 and a new Sn–H triplet resonance at δ = 5.10 ppm [3J(1H–1H) = 2.2 Hz] with attendant satellites [1J(117Sn–1H) = 1704 Hz; 1J(119Sn–1H) = 1784 Hz], in addition to signals for the Si(CH3)3 and methine protons [δ/ppm = 0.17 (s) and −0.42 (t, 3J(1H–1H) = 2.2 Hz), respectively] (see Fig. 2). 119Sn NMR spectroscopy showed only a triplet of triplets at −196 ppm [1J(119Sn–1H) = 1784 Hz, 2J(119Sn–1H) = 87 Hz] which collapsed to a singlet upon 1H decoupling. Collectively these data correspond to the previously unreported dihydride [(Me3Si)2CH]2SnH2 (2), which was confirmed by comparison with an authentic sample prepared by the reaction of LiAlH4 and [(Me3Si)2CH]2SnCl2 (see ESI† for details).
Sn double bond.18 When a d8-toluene solution of 1/[1]2 was placed under an atmosphere of H2 (4 bar) in a sealed NMR tube, no change was observed in the 1H NMR spectrum, even after prolonged periods (>48 h), confirming that neither 1 nor [1]2 can react with H2 alone. Separately, addition of Et3N (20 mol%) to a solution of 1 resulted in no perturbation of their 1H NMR resonances, suggesting no interaction between the components; i.e. the formation of an FLP.19 Placing this new mixture under H2 (4 bar, RT) resulted in the solution turning from deep red to colourless over the course of 24 h, with the 1H NMR spectrum revealing complete consumption of 1 and a new Sn–H triplet resonance at δ = 5.10 ppm [3J(1H–1H) = 2.2 Hz] with attendant satellites [1J(117Sn–1H) = 1704 Hz; 1J(119Sn–1H) = 1784 Hz], in addition to signals for the Si(CH3)3 and methine protons [δ/ppm = 0.17 (s) and −0.42 (t, 3J(1H–1H) = 2.2 Hz), respectively] (see Fig. 2). 119Sn NMR spectroscopy showed only a triplet of triplets at −196 ppm [1J(119Sn–1H) = 1784 Hz, 2J(119Sn–1H) = 87 Hz] which collapsed to a singlet upon 1H decoupling. Collectively these data correspond to the previously unreported dihydride [(Me3Si)2CH]2SnH2 (2), which was confirmed by comparison with an authentic sample prepared by the reaction of LiAlH4 and [(Me3Si)2CH]2SnCl2 (see ESI† for details).
 | ||
Fig. 2
1H/2H NMR spectra from the reaction of 1 and 20 mol% Et3N: (a) CH(SiMe3)2 region under H2 (left); H2/D2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (middle); HD (right). (b) SnH region under H2 (left); H2/D2 (1:1) (middle; 2H NMR); HD (right). ● denotes trace formation of 2 from H2 in commercial HD gas. :1) (middle); HD (right). (b) SnH region under H2 (left); H2/D2 (1:1) (middle; 2H NMR); HD (right). ● denotes trace formation of 2 from H2 in commercial HD gas. | ||
Isotopic investigation
When D2 was used in place of H2, the methine peak present in the 1H NMR spectrum of the product mixture resolved as a singlet, while the Sn–H signal was absent and replaced by a Sn–D signal at δ = 5.11 ppm [1J(117Sn–2H) = 262 Hz, 1J(119Sn–2H) = 274 Hz] in the 2H NMR spectrum. These results demonstrate the formation of dideuteride 2-D2,20 and that the Sn-bound protons in 2 must originate from the hydrogen atmosphere.In order to probe the mechanism further, a d8-toluene solution of 1/[1]2 and Et3N was reacted with a 1:1 mixture of H2/D2. The resultant 1H NMR spectrum was very similar in appearance to that of 2, with two exceptions: the relative integration of the Sn–H peak did not match that of the methine signal (1.2:2; consistent with the faster rate of reaction with H2vs. D2 – vide infra), and the C–H resonance was composed of overlapping peaks commensurate with a mixture of 2 and 2-D2. No spectroscopic evidence was seen for the formation of 2-HD, which was independently and selectively obtained by analogous reaction of 1/[1]2 under an HD atmosphere. These observations provide strong evidence that delivery of both atoms from H2/D2/HD to a single Sn centre occurs either simultaneously, or in a near-concerted fashion.
Kinetic analysis
By analogy with established FLP systems, and the microscopic reverse of the polar mechanism by which dehydrogenation of ArSnH3 species is proposed to occur,15a we envisaged a reaction mechanism in which 1 and Et3N form a weakly associated ‘encounter complex’ which subsequently reacts with H2 (Scheme 1).21 Assuming that encounter complex formation is a rapid pre-equilibrium prior to rate-limiting H2 activation gives the expected rate law: rate = k′[1][Et3N][H2], where k′ = (k1k2)/k−1. Calorimetric studies on H2 activation by the FLP Mes3P/B(C6F5)3 (Mes = 2,4,6-C6Me3H2) found the rate to be very accurately modelled as a single, termolecular step, which formally gives the same rate law.22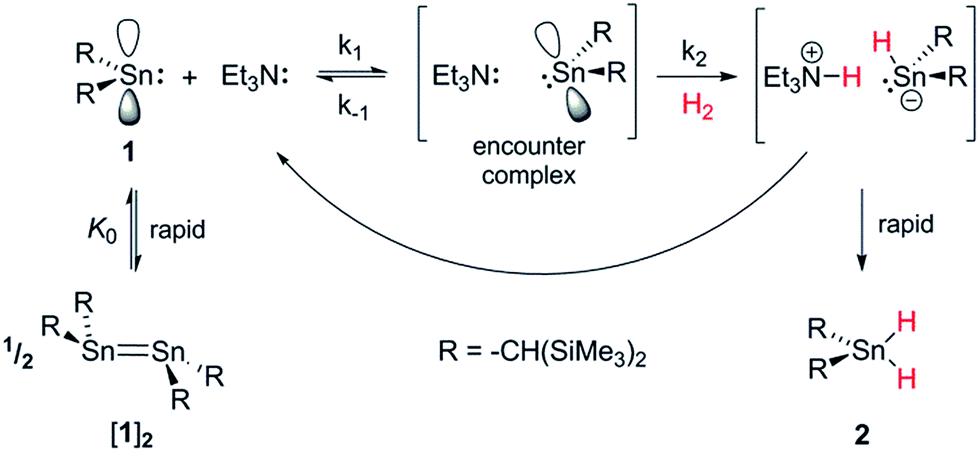 | ||
| Scheme 1 Proposed reaction mechanism for H2 heterolysis by 1, catalysed by Et3N. | ||
To confirm the order of catalytic Et3N, the method of time (t) scale normalisation was used;23 normalisation to the scale of t·[Et3N]x resulted in the superposition of all reactant traces only when x = 1, confirming the rate to be first order with respect to the amine (Fig. 3a). Determination of reaction order with respect to 1 requires its concentration to be known accurately at any given time in a reaction mixture. However, since the observed 1H NMR resonances are a weighted average of the signals from 1 and [1]2 (ΔG293K = 3.1 kcal mol−1), with both species present at significant concentrations under reaction conditions, simple observation of the concentration of 1 is not directly possible by 1H NMR spectroscopy.18a The concentration of 1 can, however, be calculated from the total concentration of “R2Sn” species in solution, [Tot], present as either monomer or dimer, which are related to the concentrations of 1 and [1]2 by:
| [Tot] = [1] + 2[(1)2] | (1) |
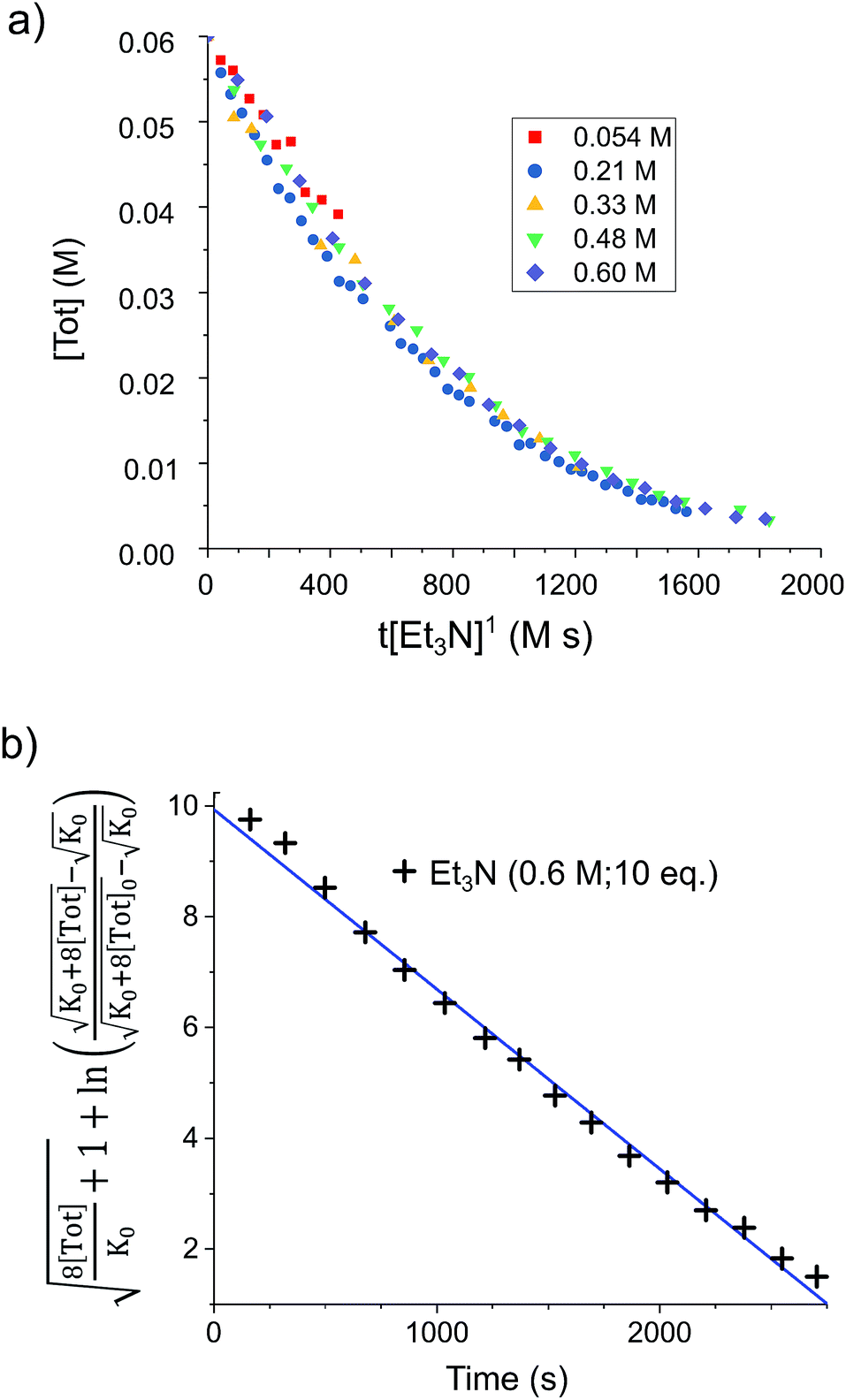 | ||
| Fig. 3 (a) Solutions of 1 (0.03 mmol) in d8-toluene (0.5 mL) under H2 (4 bar) containing various base concentrations were prepared. When the stannylene concentration, [Tot], is plotted against the normalised timescale t·[Et3N]1, all traces overlap, confirming the order in base to be one. (b) Linearised rate data for a similar solution of 1 (0.03 mmol) in d8-toluene (0.5 mL) under H2 (4 bar) containing Et3N (0.6 M; 10 eq.). | ||
The dimerisation equilibrium of 1 can be expressed as:
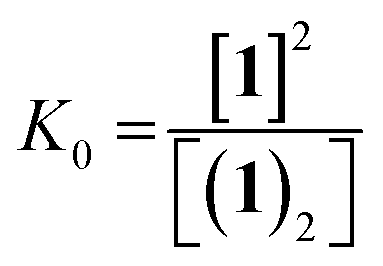 | (2) |
Combining eqn (1) and (2) and solving for [1] yields:
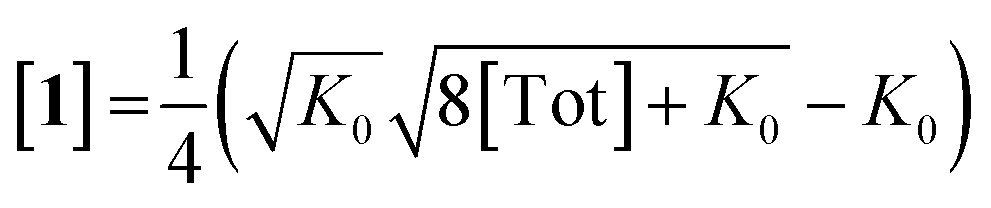 | (3) |
Inserting eqn (3) into the expected rate law (vide supra) gives:
 | (4) |
 | (5) |
Rearrangement and integration by substitution of eqn (4) (see ESI†) gives:
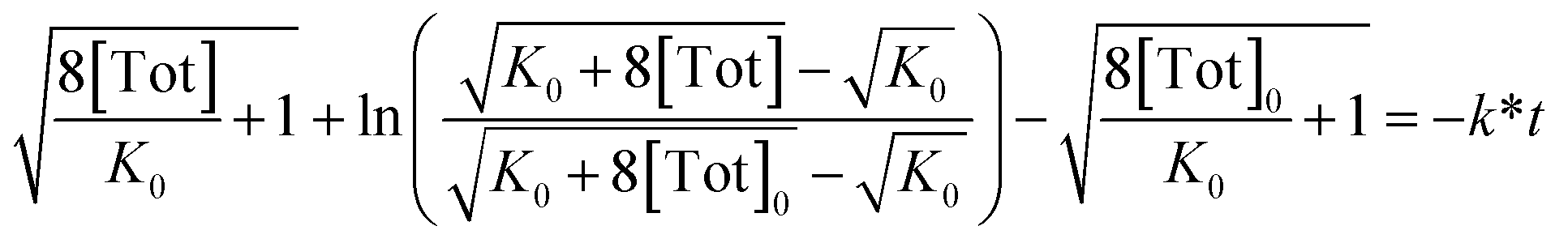 | (6) |
Therefore, plotting the variable portion of the LHS of this expression against t gives a straight line of gradient −k∗, confirming the proposed first-order dependence on 1 (Fig. 3b).
Using the known value of [H2] in toluene at 4 bar (293 K)24 provides a value of 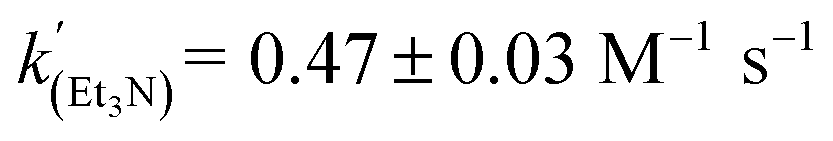 .25 As well as Et3N, 2-tert-butyl-1,1,3,3-tetramethylguanidine (Barton's base, TBTMG) and 1,2,2,6,6-pentamethylpiperidine (PMP), were also found to form FLPs with 1/[1]2, with corresponding rates of H2 cleavage:
.25 As well as Et3N, 2-tert-butyl-1,1,3,3-tetramethylguanidine (Barton's base, TBTMG) and 1,2,2,6,6-pentamethylpiperidine (PMP), were also found to form FLPs with 1/[1]2, with corresponding rates of H2 cleavage: 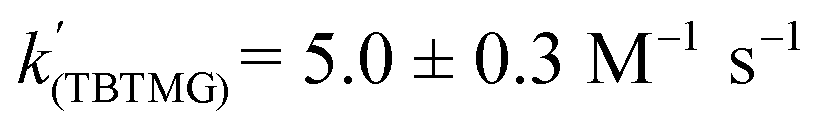 ,
,  .26 Despite the similar basicity to Et3N, the bulkier Hünig's base (iPr2EtN; pKa(MeCN): 18.0)27 was ineffective for H2 heterolysis, as was the weaker base 2,4,6-collidine (pKa(MeCN): 14.98).28 Clearly H2 activation requires that the LB be sufficiently basic and not too sterically encumbered, in line with observations of other FLP systems.29
.26 Despite the similar basicity to Et3N, the bulkier Hünig's base (iPr2EtN; pKa(MeCN): 18.0)27 was ineffective for H2 heterolysis, as was the weaker base 2,4,6-collidine (pKa(MeCN): 14.98).28 Clearly H2 activation requires that the LB be sufficiently basic and not too sterically encumbered, in line with observations of other FLP systems.29
A kinetic analysis of the isotopic systems permitted quantification of the KIE: 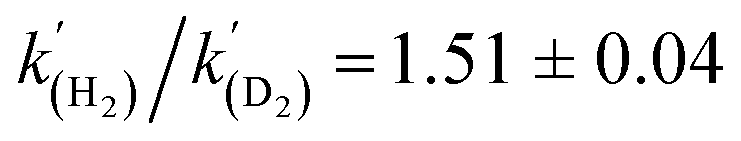 when Et3N was used as the base. In addition, the acceleration in rate from a more polar solvent could also be quantified:
when Et3N was used as the base. In addition, the acceleration in rate from a more polar solvent could also be quantified: 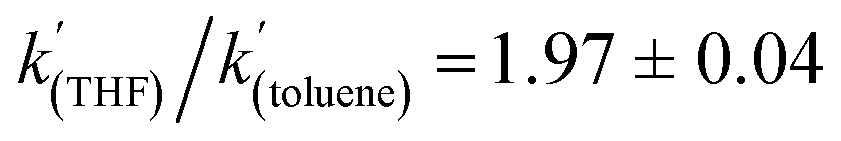 (when Et3N was used).
(when Et3N was used).
Coordinating bases
When the less sterically bulky 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) is used, an interaction with 1 can be clearly seen in the 13C{1H} NMR spectrum: upon gradual addition of DBU to 1/[1]2, the methine resonance undergoes a substantial upfield shift, reaching a limiting value of δ = 18.5 ppm (10-fold excess of DBU). Using the established 13C NMR chemical shift values for 1 and [1]2 (60.0 ppm and 28.7 ppm, respectively),18a this is consistent with a fast equilibrium between 1·DBU, 1 and [1]2 (Scheme 2; see ESI† for full details). A value of ΔG = −3.7 ± 0.2 kcal mol−1 for the formation of 1·DBU from [1]2 was obtained from a van't Hoff analysis of variable temperature UV-Vis spectra. | ||
| Scheme 2 Equilibrium between product 2 + DBU and the dehydrogenated mixture 1·DBU ↔ 1 ↔ [1]2. | ||
While the reaction of 1/DBU mixtures (containing 0.1–10 equivalents of DBU) with H2 proceed rapidly, they do not reach completion, indicative of a reversible process (see Fig. S7 in ESI†).
The reversibility can be explicitly demonstrated by the (CH3)3Si region of the 1H NMR spectrum, whereby addition of DBU to a solution of 2 led to the appearance of a signal corresponding to the dehydrogenated mixture 1·DBU ↔ 1 ↔ [1]2; this increased in intensity at the expense of the (CH3)3Si peak of 2 (Fig. 4a–c). No H2 is observed in the 1H NMR spectrum as the solution was degassed multiple times in order to accelerate the reaction – however, the very small amount of H2 generated (approx. 0.3 bar) would likely hamper detection. Furthermore, the methine resonance of the 1·DBU ↔ 1 ↔ [1]2 mixture is subject to a significant upfield shift compared to [1]/[1]2 (dependent upon the DBU concentration), and so is obscured beneath the relatively intense (CH3)3Si region. Upon charging this reaction with H2, restoration of 2 was rapidly observed (Fig. 4d). For the equilibrium involving H2 (Scheme 2), an equilibrium constant, Keq = 164 ± 5, in favour of 2 can be calculated from the relative intensities of the (CH3)3Si resonances, providing ΔG = −3.0 kcal mol−1 (1 bar H2).
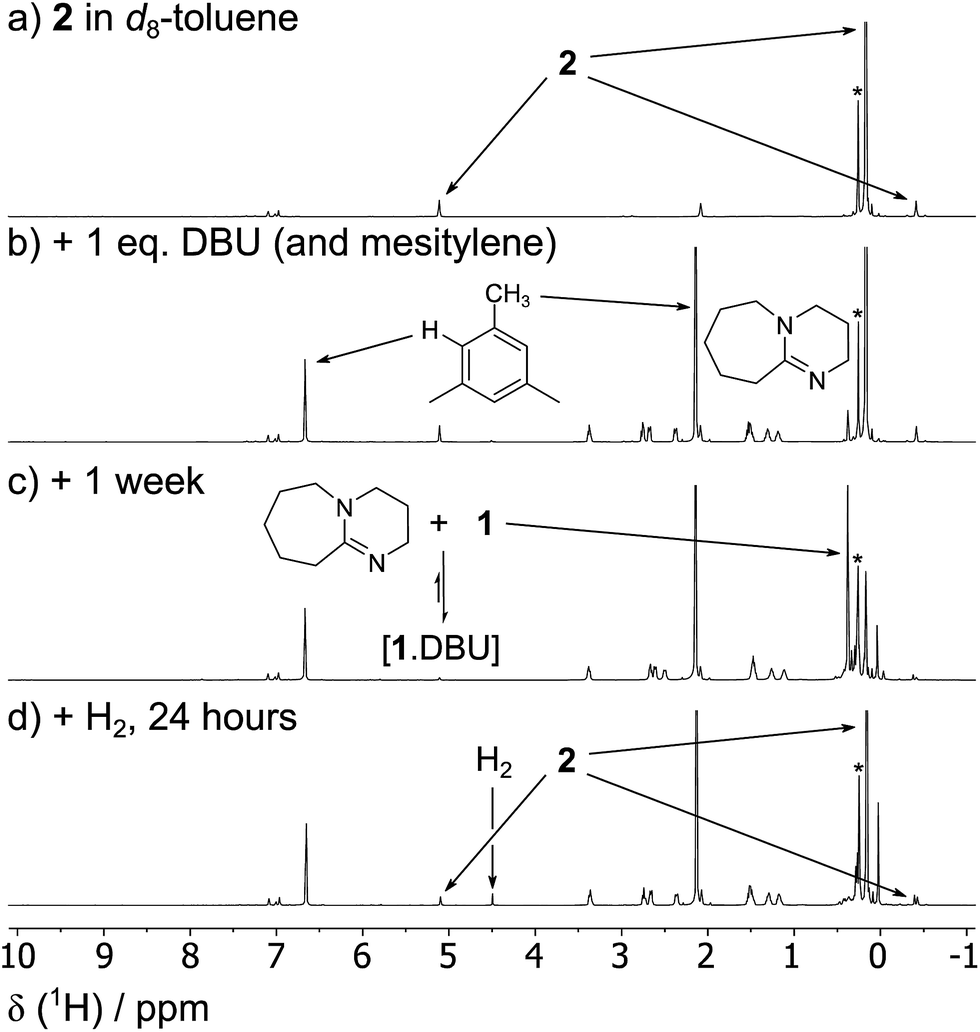 | ||
| Fig. 4 The reversibility of the reaction between 1/[1]2, DBU and H2 can be shown explicitly by a series of 1H NMR spectra depicting: (a) a solution of 2 (0.03 mmol) in d8-toluene (0.5 mL) (b) the same solution with added DBU (0.03 mmol, 1 equivalent) and mesitylene (2%) as an internal standard; (c) after degassing three times over the course of one week, showing the formation of 1·DBU ↔ 1 ↔ [1]2; (d) reformation of 2 after the addition of H2 (4 bar). *Small amount of silicone grease from the independent synthesis of 2. | ||
Using the similarly unhindered but less basic 4-(dimethyl amino)pyridine (DMAP) also gave an adduct 1·DMAP, but no reaction with H2 at room temperature. However, heating a solution of 1 with excess DMAP (4 bar H2, 2 h, 100 °C) yielded 2 in 31% conversion.
Computational investigation
To gain further insight into the mechanism of H2 activation, DFT calculations were performed for various 1/LB pairs;31 the computed reaction profiles for both the Et3N- and DBU-mediated reactions are depicted in Fig. 5. When LB = Et3N, the reaction was found to proceed via initial H2 heterolysis leading to a tight ion pair intermediate [1H]−[Et3NH]+ (int1). Facile rearrangement to int2 and subsequent delivery of the H+ to the lone pair on the [1H]− moiety furnishes 2 (Fig. 6a); a very similar mechanism was found when LB = DBU. In support of this polar mechanism, the rate using Et3N as the LB was found to be faster in THF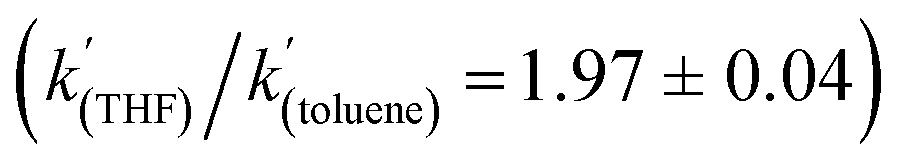 . The low barriers to rearrangement of the intermediates also offer an explanation as to why H/D exchange is not observed upon reaction with an H2/D2 mixture or HD: collapse of the ion pairs is likely much faster than solvent cage escape.
. The low barriers to rearrangement of the intermediates also offer an explanation as to why H/D exchange is not observed upon reaction with an H2/D2 mixture or HD: collapse of the ion pairs is likely much faster than solvent cage escape.
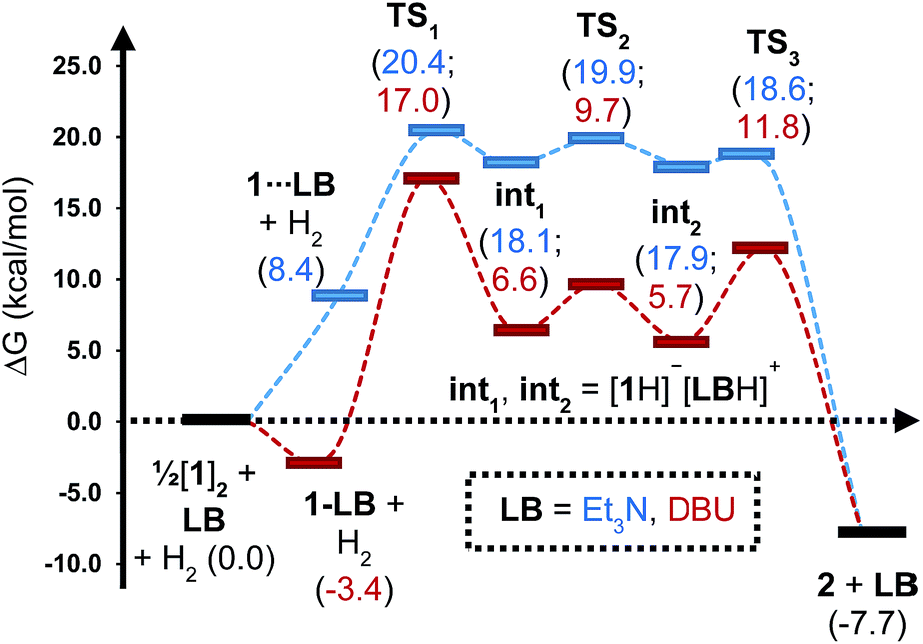 | ||
| Fig. 5 Computed free energy profile for Et3N- and DBU-assisted H2 activation with 1. Relative free energies (in kcal mol−1) are with respect to 0.5·[1]2 + LB + H2. | ||
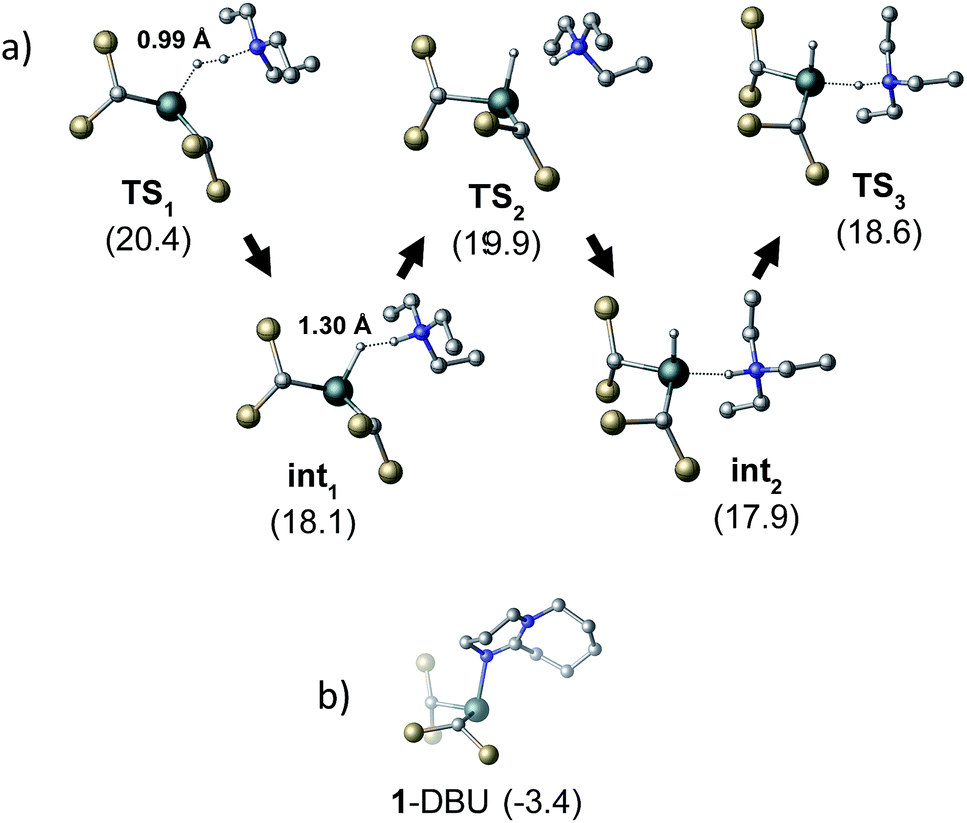 | ||
| Fig. 6 (a) Structural representations of the computed transition states for the heterolysis of H2 by 1 and Et3N. H–H distances are given for TS1 and int1. (b) The computed adduct formed between 1 and DBU. All energies (in kcal mol−1) are relative to 0.5·[1]2 + LB + H2. Si–CH3 and C–H groups omitted for clarity. | ||
Although the located transition states (TSs) are energetically close-lying, the overall reaction barrier appears to be determined by the H2 splitting step, which is in line with kinetic measurements. Free energy data computed for the H2 splitting step for reactions with different bases are compiled in Table 1 alongside other properties. For Et3N, TBTMG and PMP, no favourable adduct formation was found with 1, and the ΔG‡ values follow the order TBTMG < Et3N < PMP, which is consistent with experimental reaction rates. For the coordinating bases DBU (Fig. 6b) and DMAP, adducts favourable relative to free [1]2 and base were computationally determined. This reduces the absolute value of ΔGreaction such that an equilibrium is experimentally observed in the case of DBU. For DMAP, the activation barrier is found to be much higher, paralleling results seen by experiment where elevated temperatures are required to obtain product 2.
| Property | Et3N | TBTMG | PMP | DBU | DMAP |
|---|---|---|---|---|---|
| a Free energy data relative to 0.5·[1]2 + base + H2 (kcal mol−1); ΔG‡ is activation free energy. b Proton affinity is defined as the free energy of base + H+ → baseH+. c Measured in MeCN.30 d H–H distance in TS1 (0.76 Å in free H2). | |||||
| 1·LBa | — | — | — | −3.4 | −3.6 |
| TS1a | 20.4 | 18.3 | 21.4 | 17.0 | 20.1 |
| int1 | 18.1 | 5.4 | 16.1 | 6.6 | 16.8 |
| ΔG‡a | 20.4 | 18.3 | 21.4 | 20.4 | 23.7 |
| ΔGreactiona | −7.7 | −7.7 | −7.7 | −4.3 | −4.1 |
| PAa,b | −270.1 | −286.0 | −272.7 | −283.5 | −272.2 |
| pKac | 18.8 | 23.6 | 18.7 | 24.3 | 18.0 |
| d(HH)d/Å | 0.99 | 0.87 | 0.96 | 0.88 | 0.99 |
The energies of all intermediates int1 are computed to be well above the reference state, which follows from the weak Lewis acidity of 1. The stabilities of int1 species correlate very well with the general trend in PA and pKa, but this is not strictly true for the TSs, where steric factors are more important. Unstable int1 intermediates imply late TSs for the H2 activation step, which is shown by significantly elongated H–H distances in the TS structures. The experimentally observed KIE (1.51 ± 0.04) supports this finding, which is commensurate with rate-limiting H2/D2 activation involving considerable H–H/D–D bond breaking.32
Conclusions
In conclusion, we have demonstrated the ability of FLP-mediated reactivity to enable the formal oxidative addition of H2 to an otherwise inert MG centre, and in doing so have also observed the first example of reversible H2 addition to a single-site MG complex. We have utilised experimental and computational means to comprehensively explore the mechanism of this transformation and found that H2 activation in this system differs from those based on more typical FLPs, due to the high-energy nature of the immediate H2 splitting products, resulting in rare examples of late TSs. The development of methods to harness this FLP-promoted OA/RE H2 reactivity for hydrogenation catalysis is currently underway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We wish to thank the EPSRC (J. S. S. and R. T. C; EP/N026004) and an Imperial College President's PhD Scholarship (R. C. T.-R.) for PhD funding and the Royal Society for a University Research Fellowship (AEA; UF/160395). This work was also supported by NKFIH (K-115660).Notes and references
- (a) A. L. Kenward and W. E. Piers, Angew. Chem., Int. Ed., 2008, 47, 38 CrossRef PubMed; (b) P. P. Power, Nature, 2010, 463, 171 CrossRef PubMed; (c) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46 CrossRef PubMed; (d) D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2011, 2, 389 RSC.
- (a) P. P. Power, Acc. Chem. Res., 2011, 44, 627 CrossRef PubMed; (b) T. Chu and G. I. Nikonov, Chem. Rev., 2018, 118, 3608 CrossRef PubMed.
- (a) F.-G. Fontaine and D. W. Stephan, Philos. Trans. R. Soc., A, 2017, 375, 2101 CrossRef PubMed; (b) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400 CrossRef PubMed; (c) D. W. Stephan, Science, 2016, 354, 1248 CrossRef PubMed.
- First example of a MG compound to activate H2 under facile conditions: G. H. Spikes, J. C. Fettinger and P. P. Power, J. Am. Chem. Soc., 2005, 127, 12232 CrossRef PubMed.
- (a) Y. Peng, M. Brynda, B. D. Ellis, J. C. Fettinger, E. Rivard and P. P. Power, Chem. Commun., 2008, 45, 6042 RSC; (b) Z. Zhu, X. Wang, Y. Peng, H. Lei, J. C. Fettinger, E. Rivard and P. P. Power, Angew. Chem., Int. Ed., 2009, 48, 2031 CrossRef PubMed; (c) K. Nagata, T. Murosaki, T. Agou, T. Sasamori, T. Matsuo and N. Tokitoh, Angew. Chem., Int. Ed., 2016, 55, 12877 CrossRef PubMed; (d) A. Rit, J. Campos, H. Niu and S. Aldridge, Nat. Chem., 2016, 8, 1022 CrossRef PubMed; (e) T. Kosai and T. Iwamoto, J. Am. Chem. Soc., 2017, 139, 18146 CrossRef PubMed; (f) D. Wendel, T. Szilvási, C. Jandl, S. Inoue and B. Rieger, J. Am. Chem. Soc., 2017, 139, 9156 CrossRef PubMed.
- (a) G. D. Frey, V. Lavallo, B. Donnadieu, W. W. Schoeller and G. Bertrand, Science, 2007, 316, 439 CrossRef PubMed; (b) Y. Peng, J. D. Guo, B. D. Ellis, Z. Zhu, J. C. Fettinger, S. Nagase and P. P. Power, J. Am. Chem. Soc., 2009, 131, 16272 CrossRef PubMed; (c) A. V. Protchenko, K. H. Birjkumar, D. Dange, A. D. Schwarz, D. Vidovic, C. Jones, N. Kaltsoyannis, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2012, 134, 6500 CrossRef PubMed; (d) A. V. Protchenko, J. I. Bates, L. M. A. Saleh, M. P. Blake, A. D. Schwarz, E. L. Kolychev, A. L. Thompson, C. Jones, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2016, 138, 4555 CrossRef PubMed; (e) D. Wendel, A. Porzelt, F. A. D. Herz, D. Sarkar, C. Jandl, S. Inoue and B. Rieger, J. Am. Chem. Soc., 2017, 139, 8134 CrossRef PubMed.
- First example of FLP-mediated H2 heterolysis: G. C. Welch, R. R. San Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124 CrossRef PubMed.
- For a review detailing the scope of MG LAs in FLP chemistry see: S. A. Weicker and D. W. Stephan, Bull. Chem. Soc. Jpn., 2015, 88, 1003 CrossRef.
- For FLP H2 activation mediated by a Sn LA see: D. J. Scott, N. A. Phillips, J. S. Sapsford, A. C. Deacy, M. J. Fuchter and A. E. Ashley, Angew. Chem., Int. Ed., 2016, 55, 14738 CrossRef PubMed.
- (a) D. W. Stephan, S. Greenberg, T. W. Graham, P. Chase, J. J. Hastie, S. J. Geier, J. M. Farrell, C. C. Brown, Z. M. Heiden, G. C. Welch and M. Ullrich, Inorg. Chem., 2011, 50, 12338 CrossRef PubMed; (b) D. J. Scott, M. J. Fuchter and A. E. Ashley, Chem. Soc. Rev., 2017, 46, 5689 RSC.
- (a) A. Y. Houghton, V. A. Karttunen, W. E. Piers and H. M. Tuononen, Chem. Commun., 2014, 50, 1295 RSC; (b) E. von Grotthuss, M. Diefenbach, M. Bolte, H. W. Lerner, M. C. Holthausen and M. Wagner, Angew. Chem., Int. Ed., 2016, 55, 14067 CrossRef PubMed.
- A. Hinz, A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2016, 55, 12214 CrossRef PubMed.
- S. Wang, T. J. Sherbow, L. A. Berben and P. P. Power, J. Am. Chem. Soc., 2018, 140, 590 CrossRef PubMed.
- (a) H. G. Kuivila, A. K. Sawyer and A. G. Armour, J. Org. Chem., 1961, 26, 1426 CrossRef; (b) W. P. Neumann and K. König, Justus Liebigs Ann. Chem., 1964, 677, 1 CrossRef; (c) W. P. Neumann and K. König, Justus Liebigs Ann. Chem., 1964, 677, 12 CrossRef.
- (a) C. P. Sindlinger, A. Stasch, H. F. Bettinger and L. Wesemann, Chem. Sci., 2015, 6, 4737 RSC; (b) J.-J. Maudrich, C. P. Sindlinger, F. S. W. Aicher, K. Eichele, H. Schubert and L. Wesemann, Chem.–Eur. J., 2017, 23, 2192 CrossRef PubMed; (c) C. P. Sindlinger and L. Wesemann, Chem. Sci., 2014, 5, 2739 RSC; (d) C. P. Sindlinger, W. Grahneis, F. S. W. Aicher and L. Wesemann, Chem.–Eur. J., 2016, 22, 7554 CrossRef PubMed; (e) J. Klösener, M. Wiesemann, M. Niemann, B. Neumann, H.-G. Stammler and B. Hoge, Chem.–Eur. J., 2018, 24, 4412 CrossRef PubMed.
- (a) P. J. Davidson, D. H. Harris and M. F. Lappert, J. Chem. Soc., Dalton Trans., 1976, 21, 2268 RSC; (b) The OA of reactive heteropolar and homopolar bonds (e.g. alkyl halides, Br2) has been documented: M. J. S. Gynane, M. F. Lappert, S. J. Miles, A. J. Carty and N. J. Taylor, J. Chem. Soc., Dalton Trans., 1977, 2009 RSC; (c) J. D. Cotton, P. J. Davidson and M. F. Lappert, J. Chem. Soc., Dalton Trans., 1976, 2275 RSC.
- A cyclic dialkylsilylene, Z. Dong, Z. Li, X. Liu, C. Yan, N. Wei, M. Kira and T. Müller, Chem.–Asian J., 2017, 12, 1204 CrossRef PubMed , has been shown to act as either a LA or LB to activate H2 in partnership with other LBs/LAs, forming . However, this reaction was irreversible; see . In part, the different reversible behaviour of R2Sn in this work is likely a consequence of the weaker Sn–H vs. Si–H bond strength, which renders the thermodynamics of RE more favourable (see ref. 6d).
- (a) K. W. Zilm, G. A. Lawless, R. M. Merrill, J. M. Millar and G. G. Webb, J. Am. Chem. Soc., 1987, 109, 7236 CrossRef; (b) R. Sedlak, O. A. Stasyuk, C. Fonseca Guerra, J. Řezáč, A. Růžička and P. Hobza, J. Chem. Theory Comput., 2016, 12, 1696 CrossRef PubMed.
- The 119Sn NMR resonance for 1 can only be observed in dilute samples and at high temperature (T > 345 K), while that for [1]2 can only be resolved at T < 225 K. At intermediate temperatures no signal is observable, which prevents an assessment of interaction between the two tin species and Et3N using 119Sn NMR under relevant reaction conditions. See ref. 18a.
- A 1:2:3:2:1 multiplet was also observed in the 119Sn NMR spectrum from 1J(119Sn–2H) coupling. See Fig. S3 in ESI.†.
- (a) L. Rocchigiani, G. Ciancaleoni, C. Zuccaccia and A. MacChioni, J. Am. Chem. Soc., 2014, 136, 112 CrossRef PubMed; (b) T. A. Rokob, A. Hamza, A. Stirling, T. Soós and I. Pápai, Angew. Chem., Int. Ed., 2008, 47, 2435 CrossRef PubMed.
- A. Y. Houghton and T. Autrey, J. Phys. Chem. A, 2017, 121, 8785 CrossRef PubMed.
- J. Burés, Angew. Chem., Int. Ed., 2016, 55, 2028 CrossRef PubMed.
- E. Brunner, J. Chem. Eng. Data, 1985, 30, 269 CrossRef.
- Fitting the data to a model based on [1]2 gave non-linear plots (see ESI† for further details).
- Assuming the same rate law applies. Supported by computational calculations.
- T. M. Alligrant and J. C. Alvarez, J. Phys. Chem. C, 2011, 115, 10797 CrossRef.
- I. Kaljurand, A. Kütt, L. Sooväli, T. Rodima, V. Mäemets, I. Leito and I. A. Koppel, J. Org. Chem., 2005, 70, 1019 CrossRef PubMed.
- T. A. Rokob, A. Hamza and I. Pápai, J. Am. Chem. Soc., 2009, 131, 10701 CrossRef PubMed.
- Et3N and DMAP: Z. Glasovac, M. Eckert-Maksić and Z. B. Maksić, New J. Chem., 2009, 33, 588 RSC ; PMP: M. Dworniczak and K. T. Leffeck, Can. J. Chem., 1990, 68, 1657 CrossRef ; DBU and TBTMG: C. F. Lemaire, J. J. Aerts, L. C. Libert, F. Mercier, D. Goblet, A. R. Plenevaux and A. J. Luxen, Angew. Chem., Int. Ed., 2010, 49, 3161 CrossRef PubMed.
- The energy values reported correspond to solution phase Gibbs free energies that are based on ωB97X-D/Def2TZVPP electronic energies, and all additional terms obtained at the ωB97X-D/Def2SVP level. See ESI† for full computational details.
- In FLPs with early TSs, such as Mes3P/B(C6F5)3, a KIE of only 1.1 has been obtained (see ref. 22).
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details. See DOI: 10.1039/c8sc03110j |
| This journal is © The Royal Society of Chemistry 2018 |