
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carbene-catalyzed enantioselective oxidative coupling of enals and di(hetero)arylmethanes†
Qiao
Chen‡
 b,
Tingshun
Zhu‡§
b,
Pankaj Kumar
Majhi
b,
Chengli
Mou
a,
Huifang
Chai
a,
Jingjie
Zhang
a,
Shitian
Zhuo
b and
Yonggui Robin
Chi
*b
b,
Tingshun
Zhu‡§
b,
Pankaj Kumar
Majhi
b,
Chengli
Mou
a,
Huifang
Chai
a,
Jingjie
Zhang
a,
Shitian
Zhuo
b and
Yonggui Robin
Chi
*b
aGuiyang College of Traditional Chinese Medicine, Guizhou, P. R. China
bDivision of Chemistry & Biological Chemistry, School of Physical & Mathematical Sciences, Nanyang Technological University, Singapore 637371, Singapore. E-mail: robinchi@ntu.edu.sg
First published on 18th September 2018
Abstract
Di(hetero)arylmethane is a unique structural motif for natural and synthetic functional molecules. To date, it remains challenging to functionalize the diaryl methyl sp3 carbon–hydrogen bond directly in an enantioselective manner. This is likely due to the relatively inert nature of the carbon–hydrogen bond and the difficult enantiofacial discrimination of two sterically similar aryl substituents. Here we disclose an N-heterocyclic carbene-catalyzed direct oxidative coupling of enals and di(hetero)arylmethanes. Our method allows for highly enantioselective transformation of diaryl methanes and quick access to benzimidazole fused lactams.
Introduction
Diarylmethyl units are widely present in functional molecules and natural products. Examples of such molecules include the antidepressant drug Sertraline,1 Bz–IB conjugates with anti-inflammatory activities2 and the natural product voacamine3 with cannabinoid CB1 receptor antagonistic activity (Fig. 1a). Notably, the diaryl methyl sp3 carbon in these bioactive molecules is often present as a chiral center.4 To date, the most well-studied approach for the incorporation of diaryl methane unit relies on reactions between diarylmethyl carbocations and nucleophilic substrates5 (Fig. 1b). Diarylmethyl carbocation intermediates are typically generated in situ from their precursors such as diarylmethanols.6 In contrast, as a potentially more straightforward approach, direct modification of diarylmethanes is much less studied (Fig. 1c). This is in part due to the weak acidity of the methyl C–H and the difficult enantiofacial discrimination of two sterically similar aryl substituents. Deprotonation of such a benzylic C–H bond typically requires the use of a strong base such as nBuLi or Na.7 As an alternative choice, oxidative transformations of diarylmethanes via benzylic organometallic species8 or benzylic radicals9 can be realized under milder conditions. However, in all these approaches, direct enantioselective modification of diarylmethane remains challenging. Limited examples in this direction include oxidative or photoredox transformations via o-quinone methides (o-QM)10 or o-quinodimethanes(o-QDM)11 as the key intermediates, and recent Rh-mediated asymmetric C–H arylation of diarylmethane.12 | ||
| Fig. 1 Diarylmethyl compounds and their enantioselective synthesis. | ||
Herein we disclose an N-heterocyclic carbene (abbreviated as NHC or carbene) organic catalyst-mediated formal oxidative coupling of enal β-carbon and diarylmethanes (Fig. 1d). Our initial designing consideration is to generate a diarylmethyl radical from a diarylmethane substrate as a key intermediate under oxidative conditions (Fig. 1c). It is known that the tendency for deprotonation and radical formation of benzylic carbons can be well-tuned by altering the electronic properties of the aryl substituents13 (Fig. 1c). We therefore eventually introduce heterocyclic benzimidazole as one of the aryl units of the diarylmethane substrate in order to achieve suitable reactivity. The selection of benzimidazole units also provides two additional benefits: benzimidazole is a structural analogue of purine and a potential pharmacophore;14 the NH group in benzimidazole can facilitate the turnover of the NHC catalyst via the formation of a lactam product at the end of the catalytic cycle.15 Mechanistic studies suggest that some of our carbene-catalyzed reactions likely go through single-electron-transfer (SET) processes and radical intermediates in the key steps, although an electron pair pathway cannot be completely ruled out.
Results and discussion
Key results in searching for suitable conditions by using cinnamaldehyde (1a) and diarylmethane 2 as model substrates are summarized in Table 1. We firstly used achiral triazolium salt A16 as the NHC pre-catalyst, DABCO as the base, and 3,3′,5,5′-tetra-tert-butyl-4,4′-diphenoquinone (DQ) as the oxidant.17 With 2-benzyl benzimidazole 2a as the substrate, the proposed lactam product was not observed in an appreciable amount under various conditions. We then introduced an electron-withdrawing nitro group into the phenyl ring of diarylmethane (using 2b as the substrate), and obtained the corresponding lactam product in 22% yield (entry 2). The use of amino indanol-derived triazolium carbene pre-catalyst B18 led to the lactam product in 36% yield with excellent ee (95% ee, entry 3). Several chiral NHC catalysts evaluated here did not provide better results in terms of both yields and ee values (entries 4–6). We then decided to use pre-catalyst B for further condition optimization (entries 7–12). The switch of the base from DABCO to DMAP significantly improved the yield from 36% (entry 3) to 60% (entry 7) with a slight increase of ee (from 95% to 98% ee) as well. Further optimization by using 1.5 equivalents of 1a and DQ and performing the reaction at 40 °C gave a better result of 84% yield and 98% ee (entry 8). Several oxidants (phenazine,19 hexachloroethane20 and 4-nitro pyridine N-oxide21) previously used in oxidation and single-electron-transfer (SET) radical reactions could also mediate the reaction with low to moderate yields and excellent ee values (entries 9–11). Finally, the reaction outcome with B as the catalyst could be further improved by lowering the reaction concentration (entry 12).|
|
|||||
|---|---|---|---|---|---|
| Entry | 2 | NHC, base | Oxidant | Yieldb/% | eec/% |
| a Reaction conditions: 1a (0.06 mmol, 1.2 equiv.), 2 (0.05 mmol, 1.0 equiv.), NHC (20 mol%), base (0.5 equiv.) and oxidant (1.2 equiv.) in 0.5 mL THF at rt. b Determined by 1H NMR, with 1,3,5-trimethoxybenzene as an internal standard. c Determined by chiral-phase HPLC analysis. d 1.5 equiv. of 1a and oxidant were used; reaction temperature was 40 °C. e In 1 mL THF. DABCO = 1,4-diazabicyclo[2.2.2]octane; DMAP = 4-dimethylaminopyridine. | |||||
| 1 | 2a | A, DABCO | DQ | Trace | — |
| 2 | 2b | A, DABCO | DQ | 22 | — |
| 3 | 2b | B, DABCO | DQ | 36 | 95 |
| 4 | 2b | C, DABCO | DQ | 27 | −73 |
| 5 | 2b | D, DABCO | DQ | 18 | −77 |
| 6 | 2b | E, DABCO | DQ | 25 | −95 |
| 7 | 2b | B, DMAP | DQ | 60 | 98 |
| 8d | 2b | B, DMAP | DQ | 84 | 98 |
| 9d | 2b | B, DMAP | Phenazine | 46 | 96 |
| 10d | 2b | B, DMAP | CCl3CCl3 | 7 | nd |
| 11d | 2b | B, DMAP | 4-Nitropyridine N-oxide | 40 | 98 |
| 12d,e | 2b | B, DMAP | DQ | 92 | 98 |

With an acceptable condition in hand (Table 1, entry 12), we evaluated the scope of enals by using 2b as a model diarylmethane substrate (Table 2). Placing halogens, carboxylic esters, nitro groups, methyl, and methoxyl units as substituents at the para-position of the enal β-phenyl ring was well tolerated (3a–3h). The absolute configuration of 3c was unambiguously confirmed by single-crystal X-ray diffraction analysis (for details, see the ESI†). The use of enals with different substitution patterns on the phenyl ring all gave products with excellent yield, dr, and ee values (3i–3k). Heterocyclic, naphthalene, and alkene substituents were all tolerated in β-substitution of enals (3l–3o). Enal with a β-carboxylate substituent gave product 3p in moderate yield with excellent dr and good ee. The use of enals with a β-alkyl substituent led to products with low yields (around 10% yields as estimated via NMR).

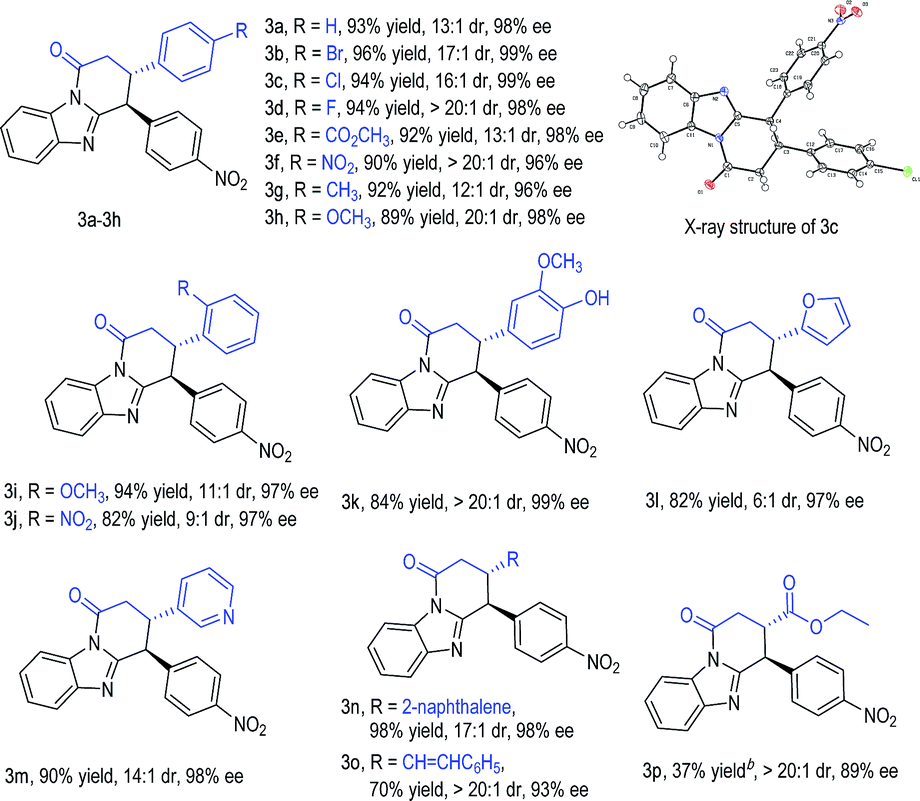
We next studied the scope of diarylmethanes by using cinnamaldehyde 1a as the model enal substrate (Table 3). The nitro substituent in 2b can be replaced with other electron-withdrawing units such as CN, CO2CH3, SO2CH3, CF3, and Br (4b–4f). Placing nitro groups at the ortho- or meta-position of the benzyl group of substrate 2 led to some decrease in reaction yields without affecting enantioselectivities (4g–4h). We also investigated the effect of substituents on the benzimidazole framework, and found that various substituents and substation patterns were well tolerated (4i–4o). When unsymmetrical benzimidazoles were used, the corresponding lactam products were obtained as a mixture of two regio-isomers (4k–4o, see the ESI† for details). This regio-isomer issue can be circumvented via one additional step in a one-pot operation: the lactam ring opened in acid alcohol in quantitative yield with ee and dr retained (as exemplified in 2k to 5, Table 3, one-pot procedure). Substrate bearing simple imidazolidine can also be used, giving product 4p in 69% yield, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr, and 98% ee.
:1 dr, and 98% ee.


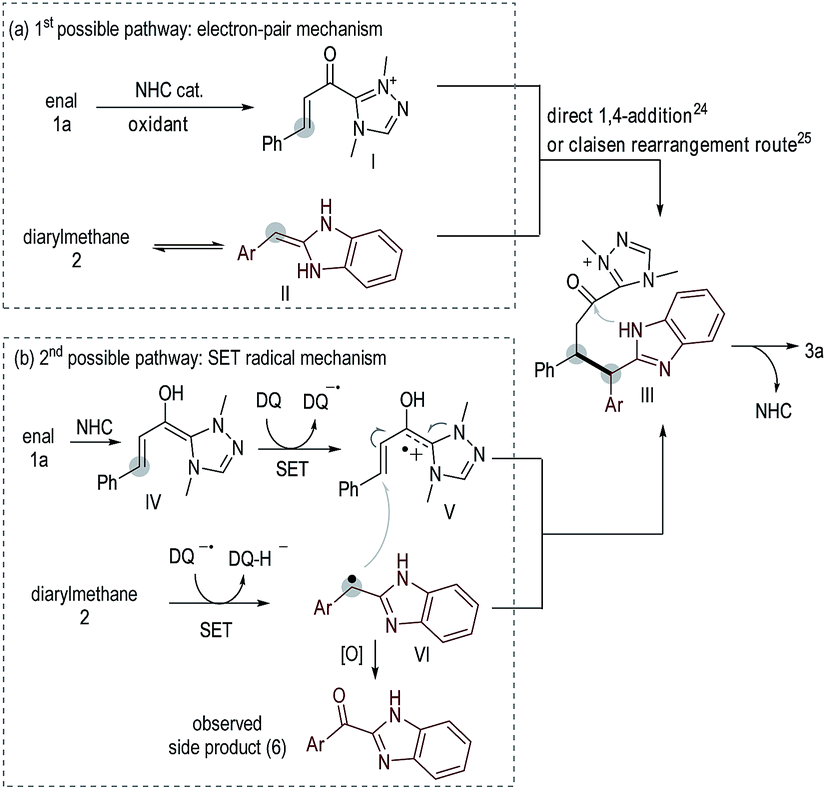
To understand the reaction pathways, we performed several control experiments. We performed H/D exchange experiments for diarylmethane substrates 2b and 2f (Scheme 1a). With 2b as the substrate, deuterated adduct D-2b was observed in 50% yield (NMR yield, see the ESI† for details). However, when 2f (an effective substrate in our oxidative coupling reaction, Table 3, product 4f) was used, no H/D exchange was observed. This result suggested that the formation of an enamine intermediate was not necessary in our catalytic coupling reaction. When 2b was mixed with the radical scavenger BHT (butylated hydroxytoluene) in the presence of a DQ oxidant, adduct 7 could be obtained in 47% yield (Scheme 2b), suggesting the existence of benzylic radical intermediates. It is also worth noting that ketones 6 derived from 2via oxidation of the benzylic carbon were observed as the main side products in nearly all examples. 2q-dimer generated from homo-coupling of benzylic carbon was also observed when 2q was used as a substrate (Scheme 1c). This evidence suggested the presence of benzylic radical intermediates (see the ESI† for details of other mechanism study).
 | ||
| Scheme 1 Control test. | ||
 | ||
| Scheme 2 Possible reaction pathways. | ||
Two possible reaction pathways are illustrated in Scheme 2. In the 1st possible pathway (Scheme 2a), oxidation of 1a under NHC catalysis leads to α,β-unsaturated azolium ester intermediate I.22 Isomerization of 2 leads to an enamine intermediate II. Nucleophilic addition of II to Ivia an electron-pair pathway23 (direct 1,4-addition24 or Claisen rearrangement25 route) leads to III. Cyclization of III leads to product 3a with regeneration of the NHC catalyst. In the 2nd possible pathway (Scheme 2b), NHC-bound radical cation intermediate V is formed via a SET oxidative process21,26 starting from enal 1a. Under oxidative conditions, diarylmethane 2 is converted to a benzylic radical intermediate VI.9 Coupling of radical intermediates V and VIvia a SET process27 leads to intermediate III, which is subsequently converted to product 3a. Ketones 6 derived from 2via oxidation of the benzylic carbon, as well as the homo-coupling generated 2-dimer (Scheme 1c), were observed as the main side products in nearly all examples, suggesting the presence of radical intermediate VI.9
The oxidative coupling products from our catalytic reactions can undergo further transformations under simple conditions (Scheme 3). For example, adduct 3a was reduced by DIBAL-H to N,O-acetal 8 in 84% yield. Nitro group reduction with simultaneously ring opening of lactam can also transform 3a to ester 9 in 86% yield. Reduction of adduct 4e with LiAlH4 efficiently gave alcohol 10 (89% yield), which was further cyclized under MsCl to afford benzimidazole fused piperidine 11 in 98% yield. In all cases, the ee and dr of the molecules were completely retained during the transformations.
 | ||
| Scheme 3 Product Transformation. | ||
Conclusions
In summary, we have developed an NHC-catalyzed highly enantioselective oxidative coupling of di(hetero)arylmethanes and enals to give benzimidazole fused lactams. Ongoing studies include the development of effective methods for enantioselective oxidative coupling of more challenging inactivated carbon–hydrogen bonds, application of this method for assembly or modification of pesticides and active components of Chinese medicines, and bioactivity evaluations of relevant molecules.Conflicts of interest
There are no conflicts of interest to declareAcknowledgements
We acknowledge financial support by the Singapore National Research Foundation (NRF-NRFI2016-06), Ministry of Education of Singapore (MOE2013-T2-2-003; MOE2016-T2-1-032; RG108/16), A*STAR Individual Research Grant (A1783c0008), Nanyang Technological University, Singapore; Guizhou Province First-Class Disciplines Project (YiLiu Xueke Jianshe Xiangmu)-GNYL(2017)008, and Guiyang College of Traditional Chinese Medicine, China.Notes and references
- D. Murdoch and D. McTavish, Drug, 1992, 44, 604 CrossRef PubMed.
- Y. Bansal, M. Kaur and O. Silakari, Eur. J. Med. Chem., 2015, 89, 671 CrossRef PubMed.
- M. Kitajima, M. Iwai, R. Kikura-Hanajiri, Y. Goda, M. Iida, H. Yabushita and H. Takayama, Bioorg. Med. Chem. Lett., 2011, 21, 1962 CrossRef PubMed.
- D. Ameen and T. J. Snape, MedChemComm, 2013, 4, 893 RSC.
- (a) Y. Li, F.-Q. Shi, Q.-L. He and S.-L. You, Org. Lett., 2009, 11, 3182 CrossRef PubMed; (b) F.-L. Sun, X.-J. Zheng, Q. Gu, Q.-L. He and S.-L. You, Eur. J. Org. Chem., 2010, 2010, 47 CrossRef; (c) M.-H. Zhuo, Y.-J. Jiang, Y.-S. Fan, Y. Gao, S. Liu and S. Zhang, Org. Lett., 2014, 16, 1096 CrossRef PubMed; (d) Y. Huang and T. Hayashi, J. Am. Chem. Soc., 2015, 137, 7556 CrossRef PubMed; (e) L. Caruana, M. Mondatori, V. Corti, S. Morales, A. Mazzanti, M. Fochi and L. Bernardi, Chem.–Eur. J., 2015, 21, 6037 CrossRef PubMed; (f) M. Nambo, Z. T. Ariki, D. Canseco-Gonzalez, D. D. Beattie and C. M. Crudden, Org. Lett., 2016, 18, 2339 CrossRef PubMed.
- For selected examples, see: (a) G. Bergonzini, S. Vera and P. Melchiorre, Angew. Chem., Int. Ed., 2010, 49, 9685 CrossRef PubMed; (b) C. Xing, H. Sun, J. Zhang, G. Li and Y. R. Chi, Chem.–Eur. J., 2011, 17, 12272 CrossRef PubMed; (c) Z. Wang, Y. F. Wong and J. Sun, Angew. Chem., Int. Ed., 2015, 54, 13711 CrossRef PubMed; (d) H.-H. Liao, A. Chatupheeraphat, C.-C. Hsiao, I. Atodiresei and M. Rueping, Angew. Chem., Int. Ed., 2015, 54, 15540 CrossRef PubMed; (e) M. Chen and J. Sun, Angew. Chem., Int. Ed., 2017, 56, 4583 CrossRef PubMed.
- (a) W. G. Kofron and J. Mathew, J. Org. Chem., 1976, 41, 114 CrossRef; (b) M. S. Hill and P. B. Hitchcock, Organometallics, 2002, 21, 220 CrossRef.
- (a) H. Sterckx, J. D. Houwer, C. Mensch, I. Caretti, K. A. Tehrani, W. A. Herrebout, S. V. Doorslaer and B. U. W. Maes, Chem. Sci., 2016, 7, 346 RSC; (b) A. Vasilopoulos, S. L. Zultanski and S. S. Stahl, J. Am. Chem. Soc., 2017, 139, 7705 CrossRef PubMed.
- (a) Y.-Z. Li, B.-J. Li, X.-Y. Lu, S. Lin and Z.-J. Shi, Angew. Chem., Int. Ed., 2009, 48, 3817 CrossRef PubMed; (b) S. Guo, Y. Li, Y. Wang, X. Guo, X. Meng and B. Chen, Adv. Synth. Catal., 2015, 357, 950 CrossRef; (c) L. Ren, L. Wang, Y. Lv, G. Li and S. Gao, Org. Lett., 2015, 17, 2078 CrossRef PubMed; (d) D. P. Hruszkewycz, K. C. Miles, O. R. Thiel and S. S. Stahl, Chem. Sci., 2017, 8, 1282 RSC; (e) E. Larionov, M. M. Mastandrea and M. A. Pericàs, ACS Catal., 2017, 7, 7008 CrossRef; (f) D. Uraguchi, M. Torii and T. Oii, ACS Catal., 2017, 7, 2765 CrossRef.
- (a) K. Gebauer, F. Reuß, M. Spanka and C. Schneider, Org. Lett., 2017, 19, 4588 CrossRef PubMed; (b) B. Wu, X. Gao, Z. Yan, M.-W. Chen and Y.-G. Zhou, Org. Lett., 2015, 17, 6134 CrossRef PubMed.
- (a) L. Dell'Amico, A. Vega-Peñaloza, S. Cuadros and P. Melchiorre, Angew. Chem., Int. Ed., 2016, 55, 3313 CrossRef PubMed; (b) L. Dell'Amico, V. M. Fernández-Alvarez, F. Maseras and P. Melchiorre, Angew. Chem., Int. Ed., 2017, 56, 3304 CrossRef PubMed; (c) X. Yuan, S. Dong, Z. Liu, G. Wu, C. Zou and J. Ye, Org. Lett., 2017, 19, 2322 CrossRef PubMed.
- J. H. Kim, S. Freßies, M. Boultadakis-Arapinis, C. Daniliuc and F. Glorius, ACS Catal., 2016, 6, 7652 CrossRef.
- F. G. Bordwell, J.-P. Cheng, M. J. Bausch and J. E. Bares, J. Phys. Org. Chem., 1988, 1, 209 CrossRef.
- W. Akhtar, M. F. Khan, G. Verma, M. Shaquiquzzaman, M. A. Rizvi, S. H. Mehdi, M. Akhter and M. M. Alam, Eur. J. Med. Chem., 2017, 126, 705 CrossRef PubMed.
- For selected examples, see: (a) D. E. A. Raup, B. Cardinal-David, D. Holte and K. A. Scheidt, Nat. Chem., 2010, 2, 766 CrossRef PubMed; (b) A. G. Kravina, J. Mahatthananchai and J. W. Bode, Angew. Chem., Int. Ed., 2012, 51, 9433 CrossRef PubMed; (c) P. Wheeler, H. U. Vora and T. Rovis, Chem. Sci., 2013, 4, 1674 RSC; (d) C. Guo, B. Sahoo, C. G. Daniliuc and F. Glorius, J. Am. Chem. Soc., 2014, 136, 17402 CrossRef PubMed; (e) L. Wang, S. Li, M. Blümel, A. R. Philipps, A. Wang, R. Puttreddy, K. Rissanen and D. Enders, Angew. Chem., Int. Ed., 2016, 55, 1110 Search PubMed.
- P.-C. Chiang, M. Rommel and J. W. Bode, J. Am. Chem. Soc., 2009, 131, 8714 CrossRef PubMed.
- (a) S. D. Sarkar and A. Studer, Angew. Chem., Int. Ed., 2010, 49, 9266 CrossRef PubMed; (b) S. D. Sarkar, S. Grimme and A. Studer, J. Am. Chem. Soc., 2010, 132, 1190 CrossRef PubMed.
- (a) M. He, J. R. Struble and J. W. Bode, J. Am. Chem. Soc., 2006, 128, 8418 CrossRef PubMed; (b) J. R. Struble and J. W. Bode, Org. Synth., 2010, 87, 362 CrossRef.
- (a) C. Noonan, L. Baragwanath and S. J. Connon, Tetrahedron Lett., 2008, 49, 4003 CrossRef; (b) X. Zhao, K. E. Ruhl and T. Rovis, Angew. Chem., Int. Ed., 2012, 51, 12330 CrossRef PubMed.
- X. Wu, B. Liu, Y. Zhang, M. Jeret, H. Wang, P. Zheng, S. Yang, B.-A. Song and Y. R. Chi, Angew. Chem., Int. Ed., 2016, 55, 12280 CrossRef PubMed.
- (a) N. A. White and T. Rovis, J. Am. Chem. Soc., 2014, 136, 14674 CrossRef PubMed; (b) N. A. White and T. Rovis, J. Am. Chem. Soc., 2015, 137, 10112 CrossRef PubMed.
- For recent review, see: C. Zhang, J. F. Hooper and D. W. Lupton, ACS Catal., 2017, 7, 2583 CrossRef.
- (a) M. D. Greenhalgh, S. Qu, A. M. Z. Slawin and A. D. Smith, Chem. Sci., 2018, 9, 4909 RSC; (b) S. Vera, Y. Liu, M. Marigo and E. C. Escudero-Adán, Synlett, 2011, 4, 489 Search PubMed.
- R. C. Samantha, B. Maji, S. D. Sarkar, K. Bergander, R. Fröhlich, C. Mück-Lichtenfeld and H. Mayr, Angew. Chem., Int. Ed., 2012, 51, 5234 CrossRef PubMed.
- J. Mahatthananchai, J. Kaeobamrung and J. W. Bode, ACS Catal., 2012, 2, 464 CrossRef.
- Y. Zhang, Y. Du, Z. Huang, J. Xu, X. Wu, Y. Wang, M. Wang, S. Yang, R. D. Webster and Y. R. Chi, J. Am. Chem. Soc., 2015, 137, 2416 CrossRef PubMed.
- H. Fischer, Chem. Rev., 2001, 101, 3581 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1843274 and 1843275. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc03480j |
| ‡ Qiao Chen and Tingshun Zhu contributed equally. |
| § Present address: School of Chemistry, Sun Yat-Sen University, Guangzhou, 510275, P. R. China. |
| This journal is © The Royal Society of Chemistry 2018 |