
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStibine-protected Au13 nanoclusters: syntheses, properties and facile conversion to GSH-protected Au25 nanocluster†
Ying-Zhou
Li
 ,
Rakesh
Ganguly
,
Kar Yiu
Hong
,
Yongxin
Li
,
Malcolm Eugene
Tessensohn
,
Richard
Webster
and
Weng Kee
Leong
*
,
Rakesh
Ganguly
,
Kar Yiu
Hong
,
Yongxin
Li
,
Malcolm Eugene
Tessensohn
,
Richard
Webster
and
Weng Kee
Leong
*
Division of Chemistry & Biological Chemistry, Nanyang Technological University, 21 Nanyang Link, Singapore 637371. E-mail: chmlwk@ntu.edu.sg
First published on 17th September 2018
Abstract
Monostibine-protected ionic Au13 nanoclusters, namely, [Au13(L)8(Cl)4][Cl] (L= SbPh3, 2a·Cl; Sb(p-tolyl)3, 2b·Cl) were prepared by the direct reduction of Au(L)Cl with NaBH4 in dichloromethane. Anion exchange with 2a·Cl afforded [Au13(SbPh3)8(Cl)4][X] (X = PF6, 2a·PF6; BPh4, 2a·BPh4). All these have been characterized by multinuclear NMR, ESI-MS and UV-Vis spectroscopy. Crystallographic analysis of 2a·BPh4 reveals that the cation possesses C2v symmetry and the tridecagold core adopts a closed icosahedron configuration. The weaker coordinating ability of the stibine ligands leads to the ready reaction of 2b·Cl with PPh3 or glutathione (GSH) to form the smaller phosphine-protected cluster [Au11(PPh3)8Cl2][Cl] or larger thiolate-protected cluster Au25(SG)18, respectively. In the latter reaction, the addition of a small amount (0.5 to 3.5 equivalents) of a suitable oxidant such as K3(Fe(CN)6 accelerates the conversion rate significantly.
Introduction
The past decade has witnessed an explosion in the research output on thiolate-,1 and alkynyl-protected2 gold nanoclusters. The earliest work on what is now generally referred to as Au nanoclusters can, however, be traced back to the pioneering work of Malatesta on phosphine-protected species.3 A large number of these, with a variety of metal core configurations, have already been structurally characterized. They include some cationic4 and neutral species5 with 8–39 gold atoms, as well as some even larger ones for which their sizes have only been estimated, such as, [Au55(PPh3)12Cl6],6 and [Au101(PPh3)21(Cl)5].7 Among these, the icosahedral Au13 clusters are of special interest as their cluster core represents an important sub-unit in some of the larger clusters, such as, the homometallic Au25, Au37, Au38, Au39 and Au60 clusters,1b–d,4i,8 and the heterometallic Au13Cun (n = 2, 4, 8, 12) clusters.9Although the Au13 cluster core is expected to be thermodynamically stable due to its closed-shell geometry,10 the synthesis of monodispersed Au13 clusters remains a challenge. Most of the studies have been on those protected by diphosphines as they exhibit higher thermodynamic stability arising from the chelating effect of the diphosphine.4d The first diphosphine-protected cluster, with the probable formulation [Au13(dppm)6][NO3]4, was prepared by NaBH4 reduction of the Au(I) complex [Au2(dppm)][NO3]2 (dppm = 1,1-bis(diphenylphosphino)methane) in ethanol.11 Two other structurally related examples, viz., [Au13(dppm)6][BPh4]3 and [Au13(dppm)6][Cl]5, which contain open icosahedral metal cores, have also been reported recently; the former was obtained from a dppm-protected Au18 cluster by thiol-induced size focusing, while the latter resulted from the direct reduction of [Au2(dppm)(Cl)2] with NaBH4 in a solution of methanol and dichloromethane (DCM).12,13 Interestingly, these three cationic Au13 clusters have the same composition but carry different charges. A facile two-step approach to diphosphine-protected Au13 clusters has also been reported; the addition of aqueous HCl to a polydispersed mixture of Aun clusters (n = 9–15), obtained from reduction of the corresponding diphosphine-Au(I) complex, was found to induce nuclearity convergence to Au13 clusters. This method yielded the diphosphine-protected Au13 clusters [Au13(L2)5(Cl)2][PF6]3 and [Au13(L)4(Cl)4][Cl] (L = L3, L4, L5), where Lm denotes an alkyl-bridged diphosphine (Ph2P(CH2)mPPh2).14 The method failed, however, for monophosphines, and for diphosphines with a longer (L6) or shorter (L1) alkyl bridge.
In comparison, Au13 clusters stabilized by monophosphines are much less well-characterised. Reduction of [Au(L)X] (L = monophosphine, X = halides, SCN or NO3) with NaBH4 gave mainly Au11 or other smaller clusters,4a–d,5a,b,15 although minor amounts of Au13 clusters may also be formed in particular cases (vide infra). To our knowledge, only three monophosphine-protected icosahedral Au13 clusters have been reported. The first was [Au13(PPhMe2)10Cl2][PF6]3, obtained from reaction of the initially formed [Au11(PPhMe2)10][PF6]3 with [Et4N]Cl in alcohol.4f This was followed 15 years later by [Au13(PPh2Me)8Cl4][C2B9H12], which was obtained from the reaction of [Au11(PPh2Me)10][C2B9H12]3 with [Au(PPh2Me)Cl].16 The third example, [Au13(TOP)8Cl4][Cl] (where TOP = trioctylphosphine), was obtained as a minor product from the NaBH4 reduction of [Au(TOP)Cl] in aqueous THF.14b Of these three examples, only the first (with a P![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cl ratio of 10:2) has been fully characterized, while the other two (with P:Cl ratios of 8:4) have only been characterized spectroscopically and the exact ligand arrangements have not been unambiguously determined.
:Cl ratio of 10:2) has been fully characterized, while the other two (with P:Cl ratios of 8:4) have only been characterized spectroscopically and the exact ligand arrangements have not been unambiguously determined.
In comparison, no stibine-protected Au nanoclusters are known. We present here the first study on monostibine-protected Au13 nanoclusters with Sb:Cl ratios of 8:4, viz., [Au13(L)8Cl4][X] (where L = Ph3Sb, (p-tolyl)3Sb; X = Cl, PF6, BPh4), and their conversion to the larger glutathione-stabilised nanocluster Au25(SG)18.
Results and discussion
Syntheses and characterization
The Au(I) stibine complexes [Au(L)Cl] (L = SbPh3, 1a; Sb(p-tolyl)3, 1b) could be prepared by the slow addition of an equimolar amount of the stibine into a DCM solution of Me2SAuCl at room temperature (ca. 21 °C) and in the dark. The products were obtained as white solids after removal of the solvent but quickly decomposed on exposure to light, and especially in solution. Crystallographic analyses show that while 1a is a monomer, the p-tolyl analogue 1b is a dimer with a strong Au…Au interaction (Fig. S1–S3†). This difference in structure is probably the result of polymorphism, as both stacking modes have been observed previously in the phosphine analogue [(p-tolyl)3PAuCl].17 An ORTEP plot depicting one of the two crystallographically distinct dimeric units observed for 1b is shown in Fig. 1. The Au…Au distance (2.9441(14) Å and 2.9784(16) Å) is typical for the Au…Au interaction but is much shorter than that found in its phosphine analogue (3.375 Å). This aurophilic interaction is believed to bear significant influence on the reduction process for the preparation of Au nanoclusters.18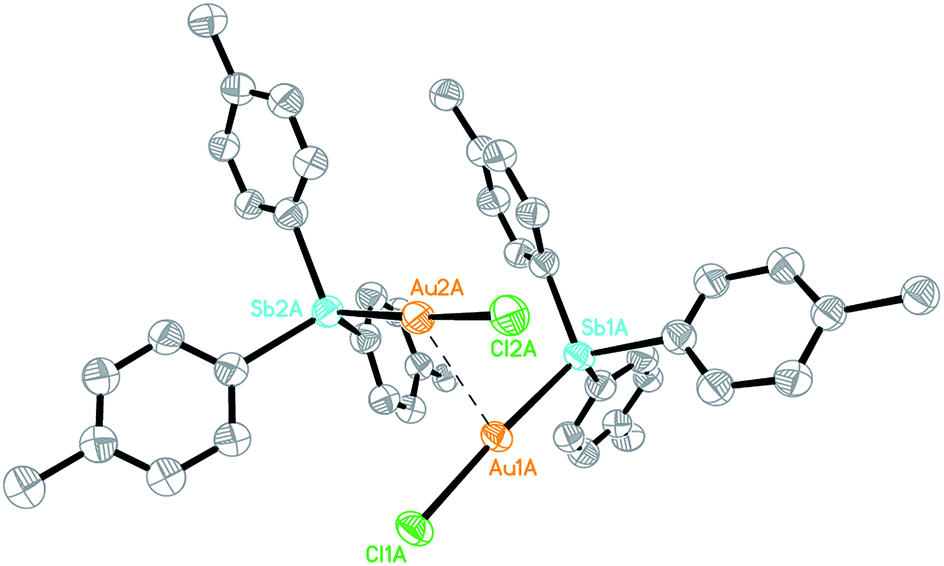 | ||
| Fig. 1 ORTEP plot depicting one of the two crystallographically distinct dimers of 1b. Thermal ellipsoids are drawn at the 50% probability level. Organic hydrogen atoms are omitted for clarity. Selected bond lengths (Å): Sb(1A)–Au(1A) = 2.460 (2), Sb(2A)–Au(2A) = 2.507(2), Au(1A)–Cl(1A) = 2.327(7), Au(2A)–Cl(2A) = 2.259(7), Au(2A)–Au(2A) = 2.9441(14). | ||
The reduction of 1a and 1b with 0.25 equivalents of NaBH4 in the presence of the free stibine gave the corresponding ionic nanoclusters [Au13(L)8(Cl)4][Cl] (L = SbPh3, 2a·Cl; Sb(p-tolyl)3, 2b·Cl) (Scheme 1). Unlike the phosphine-protected Au11 nanoclusters [Au11(PPh3)8Cl2][Cl], these stibine-protected nanoclusters could not be purified by column chromatography but they could be obtained in good purity by precipitation with hexane followed by washing with suitable solvents. Although the isolated yields were relatively low (15% and 30% for 2a·Cl and 2b·Cl, respectively), the MS and UV-Vis spectra of the crude products did not show any detectable amounts of nanoclusters of other sizes such as Au11 (Fig. S30 and S37†). Indeed, fractional crystallization of the crude from DCM/hexane (8/4, v/v) in the dark gave mainly 2·Cl and a colorless crystalline side product identified as the mononuclear Au(I) complexes [Au(SbPh3)4][SbPh2Cl2], 3a,19 and [(p-tolyl)2Cl2Sb]Au[Sb(p-tolyl)3]3, 3b, respectively. Both 3a and 3b were characterized spectroscopically and crystallographically (Fig. S4, S5, S26–S28, S34 and S35†).
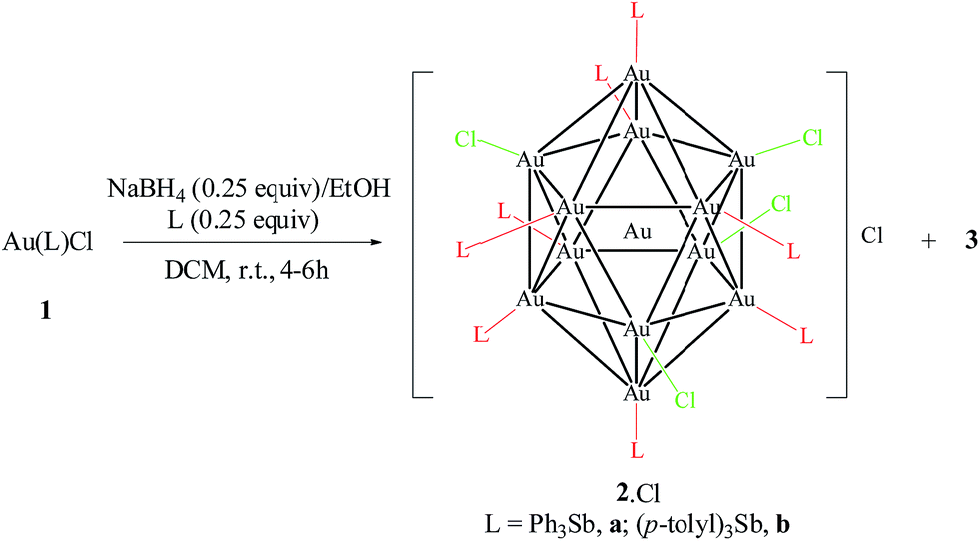 | ||
| Scheme 1 Reduction of 1 with NaBH4. | ||
The reduction of 1 exhibited markedly different behavior from that of the phosphine analogue [Au(PPh3)Cl]. The latter was reported to give the ionic nanocluster [Au11(PPh3)8Cl2][Cl] with 0.25 equivalent of NaBH4 in DCM but the relatively less stable, neutral nanocluster [Au11(PPh3)7Cl3] with 5 equivalents of NaBH4 in THF.15 For 1, reduction with more than 0.25 equivalent of NaBH4 led to a decrease in yield without any new nanocluster isolated or observed (Fig. S38†). The use of 5 equivalents of NaBH4 (with either DCM or THF as the solvent) led to the immediate formation of an insoluble black precipitate and a colorless supernatant, indicating full decomposition into bulk Au particles. This indicates that while NaBH4 can reduce 1 to form 2·Cl, it also leads to the destruction of 2·Cl; presumably, the weakly coordinating stibine allows for more ready decomposition and/or aggregation of the gold cores. Addition of a small amount (0.25 equiv.) of the stibine prior to reduction was found to aid the formation of 2·Cl, but larger amounts (1.0 to 3.0 equiv.) led to lower yields. This is probably related to favoring the formation and/or enhancing the stability of intermediate species, and is consistent with a recent report that the reduction of [Au(PPh3)Cl] and [Au(PPh3)2Cl] gave different products.20 We have similarly observed that reduction of the tetra-stibine coordinated 3a under the same reaction conditions led to a more complicated mixture, as revealed by the UV-Vis spectrum of the crude mixture, and much less 2a·Cl was obtained (Fig. S39†). It is also known that steric bulk of the ligand can have a significant influence on the course of the reaction;21 we tested this with SbMes3 (Mes = mesityl), which has a larger percent buried volume (40% for SbMes3vs. 28% for SbPh3 and Sb(p-tolyl)3).22 The reduction of [Au(SbMes3)Cl] (1c) under similar reaction conditions, however, afforded a black precipitate in a colorless supernatant; the 1H NMR spectrum of the latter showed only SbMes3 (Fig. S11†). Presumably, the larger steric bulk of SbMes3 prevented nucleation of the intermediate to form nanoclusters.23
The chloride anion in 2a·Cl could be exchanged with [PF6]− or [BPh4]− to give 2a·PF6 or 2a·BPh4, respectively, as verified by their multinuclear NMR spectra (Fig. S12–S21†). Irrespective of the anion, the 1H NMR spectrum is invariant and exhibits three doublets, in an integration ratio of 1:2:1, in the 7.15–7.50 ppm region (Fig. 2). These resonances are assignable to the ortho-H's of the SbPh3 ligand, and suggest the presence of three chemically non-equivalent stibine ligand environments. The spectrum of 2b·Cl similarly exhibits three singlets in a 1:2:1 integration ratio, in the 2.04–2.11 ppm region, which are ascribable to the methyl protons in three chemically non-equivalent Sb(p-tolyl)3 ligand environments (Fig. S20†). This is in contrast to the phosphine-protected nanoclusters [Au11(PPh3)7Cl3] and [Au11(PPh3)8Cl2]Cl, which were reported to show only one set of resonances in their 1H and 31P NMR spectra even down to −80 °C. Presumably, therefore, the metal cores of 2 are structurally more rigid than the undecagold core in those phosphine-protected nanoclusters.5a,15
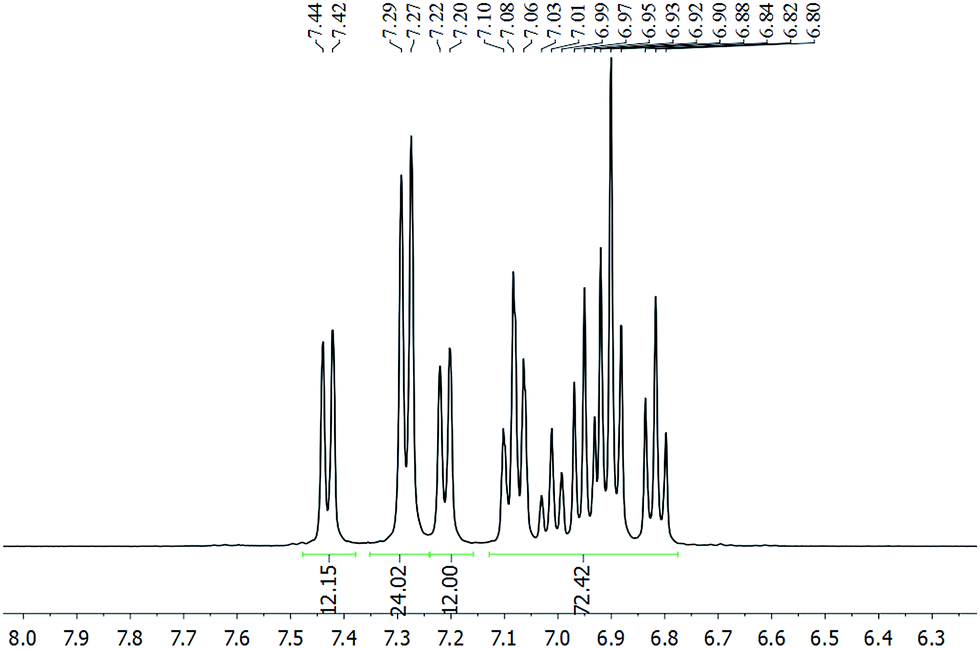 | ||
| Fig. 2 1H NMR spectrum of 2a·PF6 in CD2Cl2 (aromatic region). | ||
Crystallographic analysis of a dark red, diffraction-quality crystal of 2a·BPh4 confirmed its chemical formulation, although the crystal exhibited disorder; seven of the peripheral Au atoms were modelled with ∼90:10 disorder. The thirteen gold atoms of the cluster cation are in an icosahedral arrangement, with eight SbPh3 and four chlorides forming the ligand sphere (Fig. 3). The presence of one BPh4 counterion confirmed that the nanocluster is monocationic and consistent with a valence electron count of 8 in accordance with the superatom rule.24 The electronic structure of 2a, calculated at the MPW1PW91/LANL2DZ level of theory, clearly shows frontier orbitals corresponding to this superatom complex (Fig. S6†). In fact, the three “HOMO” orbitals may be regarded as comprising a single HOMO (−0.258 eV) and a pair of near-degenerate HOMO − 1 (−0.265 eV), with p orbital characteristics. This is consistent with a breakdown of the icosahedrdal symmetry of the gold core by the ligand sphere; the nanocluster exhibits non-crystallographic mm2 point symmetry. The four Cl ligands lie in two mutually perpendicular planes, dividing the eight SbPh3 ligands into three groups in a 1:2:1 ratio, in agreement with the 1H NMR spectrum. The peripheral Au–Au bond lengths (2.8487(10)–2.9248(11) Å) are clearly longer than those from the central Au atom (2.7151(10)–2.7629(10) Å), and comparable to those in [Au13(PPhMe2)10Cl2][PF6]3, the only other crystallographically characterized monophosphine-protected icosahedral Au13 cluster (see also Table S3†).4f
 | ||
| Fig. 3 Views of the crystal structure of the cationic fragment of 2a·BPh4 (left: full structure with hydrogen atoms omitted; right: structure of the core showing the mm2 point symmetry). Au: yellow, Sb: blue, Cl: green, C: grey. | ||
Consistent with the formulation, the ESI-MS(+) spectra of 2a·Cl and 2a·PF6 both show a main peak at m/z ∼2746 with an isotopic interval of 0.5 amu, indicating a +2 charge (Fig. 4 and S31†). This is thus attributable to the fragment ion [M−Cl]2+, i.e., [Au13(SbPh3)8Cl3]2+; the experimental isotopic pattern matches well with the calculated pattern. Similarly, the mass spectrum for 2b·Cl shows a major ion fragment at m/z ∼2914 assignable to the [M−Cl]2+ ion (Fig. S32†).
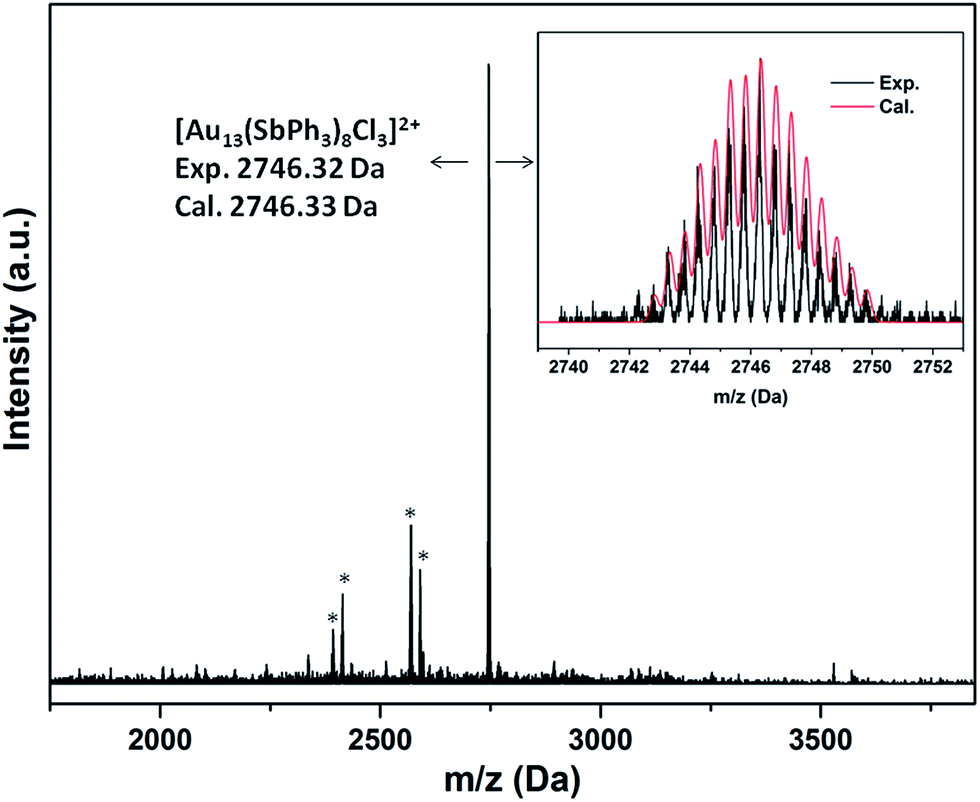 | ||
| Fig. 4 ESI-MS(+) spectrum of 2a·PF6. The asterisks indicate fragments corresponding to loss of SbPh3 ligands. Inset: calculated and experimental patterns of the main ion fragment. | ||
While a solid sample of 2a·Cl kept at 4 °C without exclusion of air remained intact after ten months, as manifested by the 1H NMR spectrum (Fig. S22†), a solution in DCM or acetone showed the formation of a small amount of an unidentified product after several days under ambient conditions. Decomposition in acetone was almost complete after four months; the mass spectrum of the resulting brown mixture suggests the presence of Ph3SbClX (X = Cl or OH), indicating dissociation and oxidation of the ligands (Fig. S33†). The decomposition process also appears to be solvent dependent although the nature of this is unclear (Fig. S23 and S24†). The p-tolyl analogue 2b·Cl displayed similar stability in solution (Fig. S25†). These stibine-protected Au13 clusters are stable to natural light but somewhat sensitive to UV light (254 nm) in solution. The lower thermal stability of 2·Cl compared to phosphine-protected Au11 clusters such as [Au11(PPh3)8Cl2][Cl] may be ascribable to the lower coordinating ability of the stibine ligands. Consistent with this is the observation that treatment of 2b·Cl with excess PPh3 led readily to the formation of [Au11(PPh3)8Cl2][Cl] (Fig. S29 and S40†). TGA and DTG measurements on 2a·PF6 and [Au11(PPh3)8Cl2][Cl] also clearly demonstrate that the former possesses lower thermal stability; decomposition corresponding to loss of all ligands occurred at ∼155 °C and ∼172 °C, respectively (Fig. S7†).
Optical and electrochemical properties
The electronic spectra of 2a·X (X = Cl, PF6, BPh4) and 2b·Cl, are essentially identical (Fig. 5), pointing to a common (Au13) metal core. There are two main absorption peaks at 352 and 454 nm, together with several weaker absorptions around 490, 530, 580 and 620 nm. The two main peaks are red-shifted compared to the 340 and 430 nm reported for the phosphine-protected clusters [Au13(L)4Cl4][Cl] (L = diphosphine) and [Au13(L)8Cl4][Cl] (L = monophosphine), all of which contain a closed icosahedral Au13 core.14b,16 The UV-Vis spectra also appear to be solvent-independent; the main absorption peak of 2b·Cl in acetonitrile is slightly blue-shifted compared to that in DCM or ethanol (Fig. S41†). A time-dependant DFT (TDDFT) calculation performed on the cationic fragment [Au13(SbPh3)8Cl4]+, 2a, at the MPW1PW91/LANL2DZ level of theory shows two strong peaks in the calculated spectrum at ca. 338 nm and 436 nm, which may correspond to the observed peaks at 352 nm and 454 nm (Fig. 5). Both peaks are due to several transitions; the transitions for the former are primarily LMCT in nature, and those for the latter are mainly metal-to-metal transitions (Table S2 and Fig. S8†). The red shift with respect to the phosphine-protected clusters can thus be understood in terms of the difference in the Au–L interactions and is believed to be related to the weaker σ-basicity and stronger π-acidity of the stibines compared to phosphines.25 Such a “heavier ligand displacement” effect has previously been observed in thiolate-protected clusters.26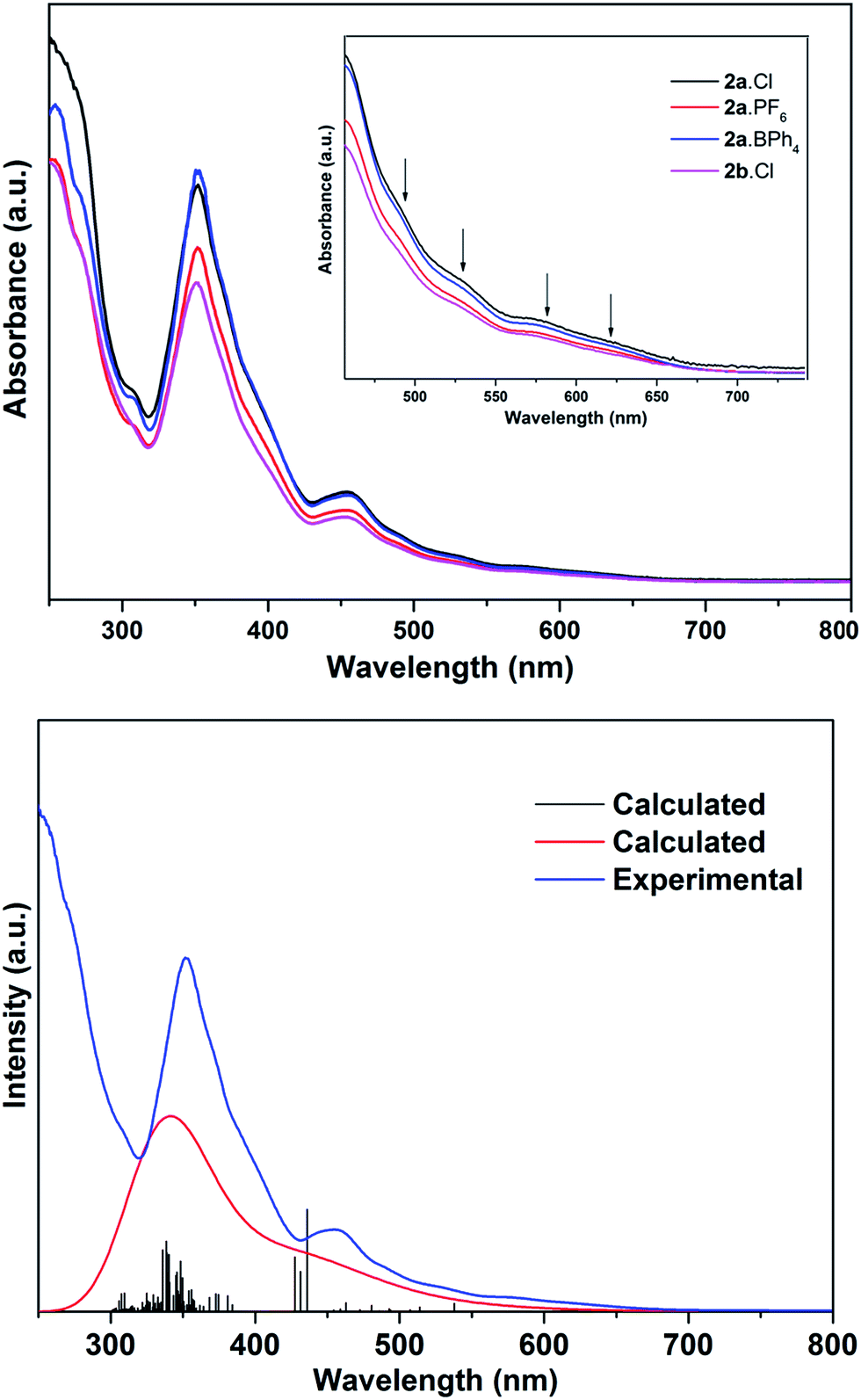 | ||
| Fig. 5 Top: electronic spectra of 2a·X (X = Cl, PF6, BPh4) and 2b·Cl in dichloromethane. Inset: the enlarged spectra for the lower-energy adsorption peaks. Bottom: comparison of the calculated (TDDFT) and experimental UV-Vis electronic spectra of 2a. | ||
Thiolate-protected gold nanoclusters display weak photoluminescence in the visible to near-infrared (NIR) region with a low quantum yield (<0.1%),27 although some with specific core–shell structures,28 or specific ligands such as peptides,29 have been reported to show stronger emissions, demonstrating the importance of the protecting ligands in tuning the luminescence.30 An example is that of [Au25(SC2H4Ph)18][TOA] and [Au25(SePh)18][TOA], with the latter displaying a 25 nm red-shifted luminescence compared to the former.26b Similarly, the diphosphine-protected clusters [Au13(dppe)5(X)2]3+ (X= Cl; CCPh) have been reported to show an emission maximum at ca. 800 nm, while that in [Au13(dppp)4(Cl)4][Cl] and [Au13(TOP)8Cl4][Cl] are blue-shifted to ca. 775 nm.14b In comparison, the stibine-protected Au13 clusters 2a·X (X = Cl, PF6, BPh4) and 2b·Cl show an NIR emission at ca. 740 nm, along with a shoulder at ca. 825 nm, i.e., a further blue shift of ca. 35 nm (Fig. 6). Thus a more electron-donating ligand leads to lower energy transitions and emissions. In addition, it is noted that 2a·Cl and 2b·Cl show weaker luminescence intensities than 2a·PF6 and 2a·BPh4, indicating that the counterion Cl− may have a quenching effect.
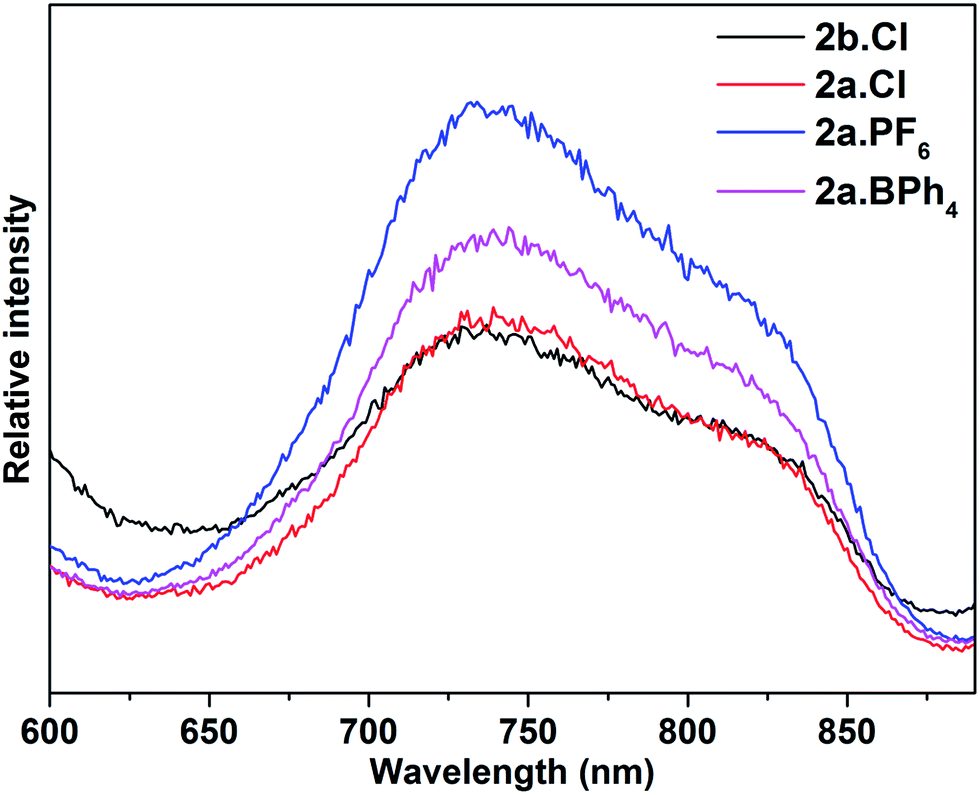 | ||
| Fig. 6 Photoluminescence spectra of 2a·X (X = Cl, PF6, BPh4) and 2b·Cl (λex = 470 nm) in DCM solution at room temperature (concentration: ca. 1.0 mg ml−1). | ||
The cyclic voltammogram (CV) of 2a·PF6 in DCM with nBu4NPF6 as the supporting electrolyte shows that the cluster undergoes irreversible oxidation and reduction (Fig. 7). While electron transfers occurring between −2.5 and +1.0 V are reproducible, i.e., the redox peaks in both the positive and negative potential directions overlap, those occurring beyond +1.0 V are not, presumably arising from breakdown of the cluster compound (Fig. S36†). The redox behavior of 2a·PF6 between −2.5 and +1.0 V is therefore reliable for analysis of its electrochemical properties. In this region, three oxidation peaks were found at +0.34, +0.58 and +0.78 V in the square-wave voltammogram (SWV), probably corresponding to oxidation to the +2, +3 and +4 states, respectively; seven reduction peaks (−1.51, −1.61, −1.68, −1.77, −1.97, −2.11, −2.22 V) were observed in the same region. The electrochemical HOMO–LUMO energy gap was determined to be about 1.85 eV (without subtracting the charge energy) from the first oxidation and reduction potentials (O1 and R1), which is in excellent agreement with that derived from the UV-Vis spectral data (620 nm absorption peak) by extrapolation to zero absorbance. In comparison, the other two Au13 clusters [Au13(PPh3)4(SC12H25)2Cl2] and [Au13(dppm)6][Cl]5, with closed and open icosahedral metal core geometries, have lower HOMO–LUMO gaps of 1.76 and 1.66 eV, respectively.13,31
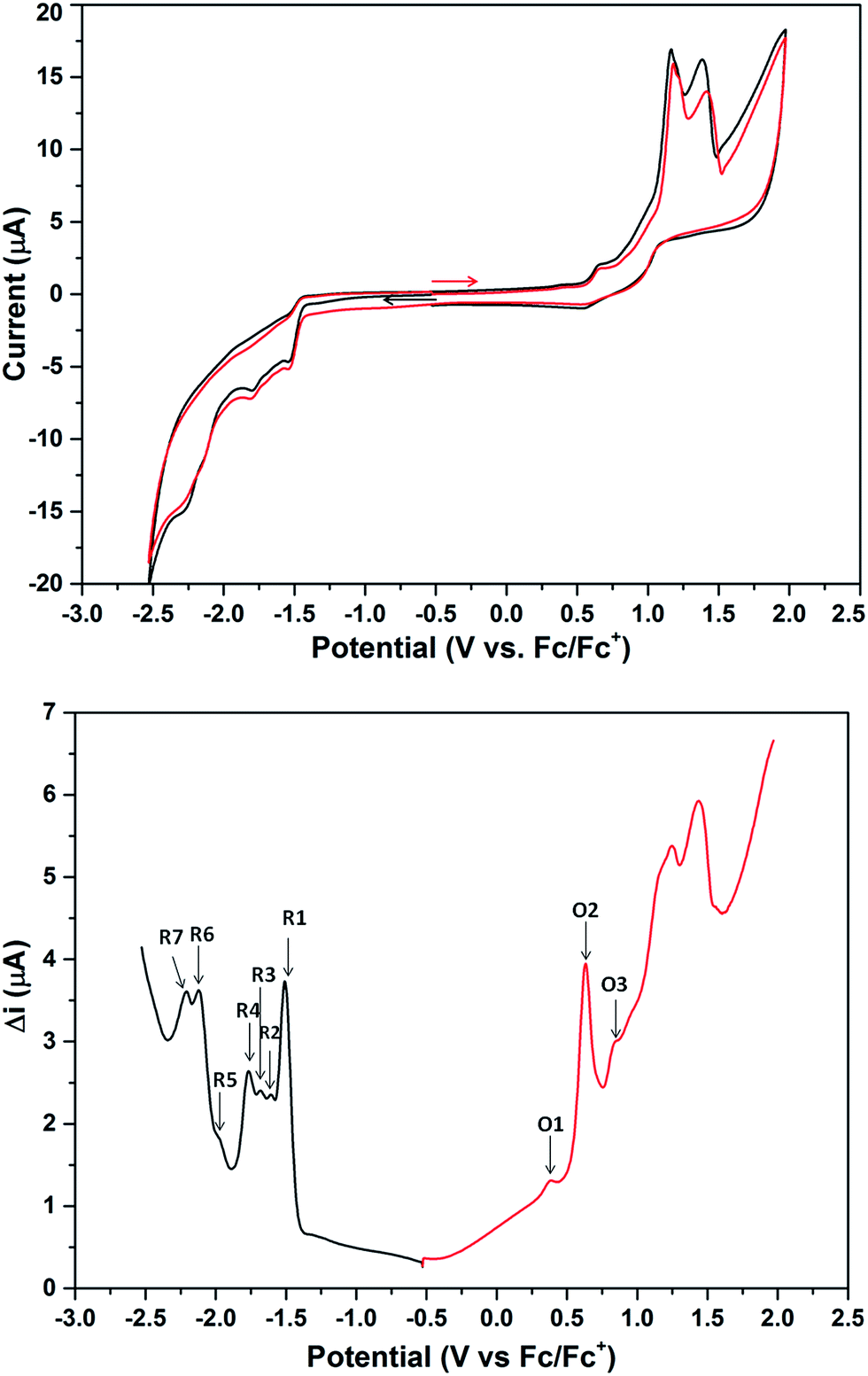 | ||
| Fig. 7 CV (top) and DSV (bottom) spectra of 2a·PF6 in DCM (1.0 mM) with 0.10 M nBu4NPF6 as the supporting electrolyte. Working electrode: 1 mm diameter planar glassy carbon; auxiliary electrode: Pt wire; pseudo-reference electrode: Ag wire (in 0.50 M nBu4NPF6 in CH3CN); scan rate: 0.10 V s−1; ferrocene (Fc) was added as an internal reference at the end of the experiment. For CV, the scans were initiated in the positive (red) and negative (black) potential directions, respectively. For SWV, the red and blue traces were scanned in the positive and negative potential directions, respectively. Pulse period (τ) = 25 Hz; potential step = 5.0 mV; pulse amplitude = 20 mV. | ||
Conversion to Au25 cluster
Water-soluble, peptide-protected Au25 clusters possessing relatively strong fluorescence, such as [Au25(SG)18], 4, have wide applications in sensing, imaging and biomedicine.32 Although they can be prepared by the modified Brust method, viz., reduction of the putative [Au(I)-SR]n oligomer with suitable reducing agents,33 it has been found recently that treatment of PPh3-protected Au11 nanoclusters or larger nanoparticles,15,34 or thiolate-protected colloidal clusters [Aun(SG)m],35 with excessive glutathione (GSH) is a more efficient synthetic route to 4. Although the exact mechanism for the ligand exchange is unclear,36 it is strongly associated with the stability of the reactant clusters, as indicated by the observation that while [Au11(PPh3)7Cl3] undergoes ligand exchange with GSH to give 4, the more structurally stable ionic clusters [Au11(PPh3)8Cl2][Cl] and [Au11(dppf)4Cl2][Cl] do not react with GSH under the same conditions.15,4d Given the weaker coordinating ability of stibines compared to phosphines, the ligand exchange reaction between 2 and thiolates may be expected to occur more readily. Indeed, ligand exchange in 2b·Cl with GSH does occur, and the formation of 4 can be monitored by its characteristic peak at ∼670 nm (Scheme 2). In comparison, unlike the case with PPh3, the reaction of [Au25(SC2H4Ph)18][TOA] with Sb(p-tolyl)3 (20 and 60 equiv.) did not appear to give any of a biicosahedral rod [Au25{Sb(tolyl)3}10(SR)5Cl2]2+.37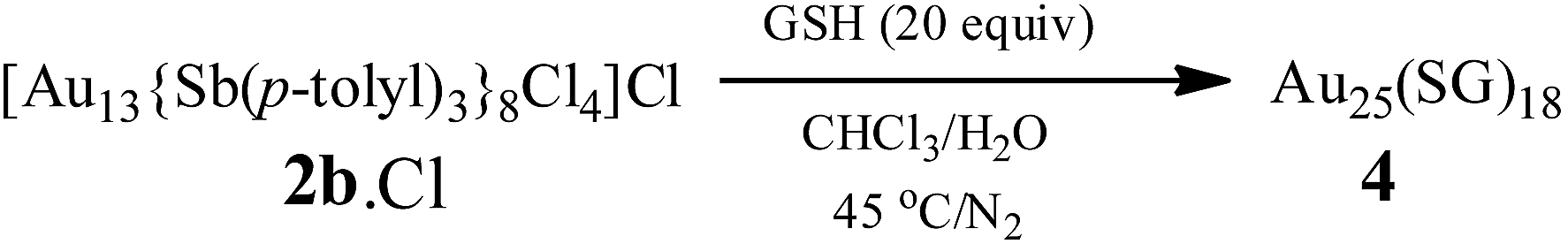 | ||
| Scheme 2 Ligand exchange reaction of 2b·Cl with GSH. | ||
The formation of 4 was detectable after 4 h (Fig. S42†). In comparison, [Au11(PPh3)7Cl3] was reported to show the formation of 4 after more than 6 h at 50 °C, while the cationic [Au11(PPh3)8Cl2][Cl] showed even lower reactivity although air was found to aid the reaction.34a The important role of air was also apparent for 2b·Cl; under aerobic conditions, the formation of 4 was apparent from about 90 min (Fig. S43†), with a higher reaction rate than under anaerobic conditions (Fig. S44 and S45†). Since the fraction of Au(I) to Au atoms changed from 0.38 to 0.72 in the conversion of 2b to 4, we hypothesize that the oxygen in air acts as an oxidant to facilitate the conversion; such an oxidation-induced transformation of smaller clusters to larger clusters has received much attention recently.38 To test the idea that oxidation is involved, the conversion was carried out with a variety of oxidants (0.50 equiv.) with differing oxidation potential and solubility. The addition of an oxidant clearly affected the rate of conversion, although we could not find any clear correlation between the conversion rate and the oxidation potential or solubility of the oxidant (Fig. 8 and S46–S53†). Besides air, the oxidants K3[Fe(CN)6] and I2 showed significant acceleration of the conversion while others showed only moderate to poor effects. The addition of more K3[Fe(CN)6] from 0.5 equiv. to 3.5 equiv. (the theoretical amount required to change the fraction of Au(I) to Au(0) atoms in 2b·Cl from 0.38 to 0.72) increased the conversion rate further but further addition to 7.0 equiv. led to a decrease in the conversion rate (Fig. 9 and S54–S57†).
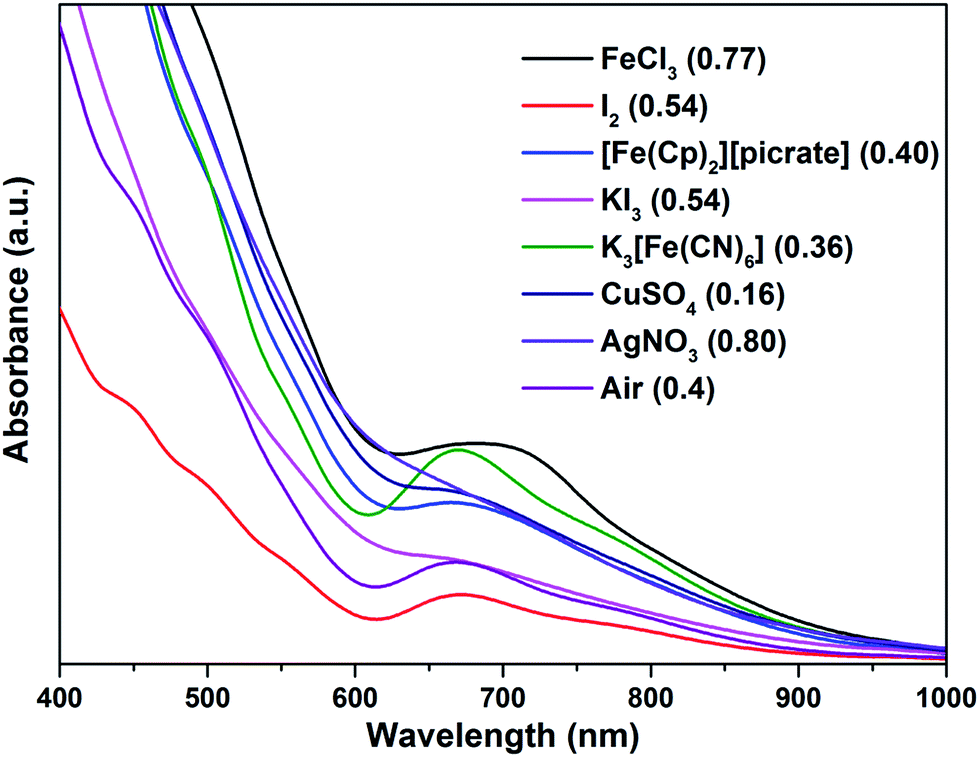 | ||
| Fig. 8 Electronic spectra of the aqueous solution for the conversion of 2b·Cl to 4 with different oxidants, at 240 min reaction time.Redox potential (V) vs. SHE for oxidants given in parenthesis.39 | ||
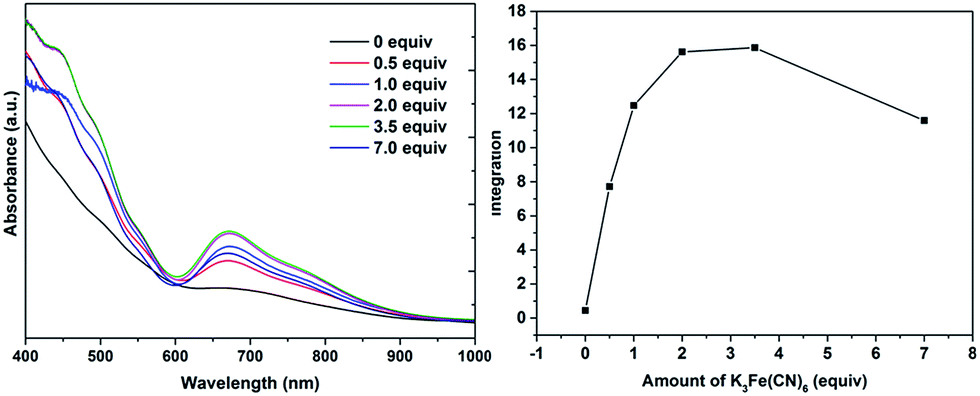 | ||
| Fig. 9 Electronic spectra of the aqueous solution for the conversion of 2b·Cl to 4 with varying amount of K3[Fe(CN)6], at 240 min reaction time (left), and the characteristic peak integration vs. amount of K3[Fe(CN)6] (right). | ||
Although antigalvanic reduction is a possibility,40 the mechanism for the conversion is not entirely clear. In the conversion mediated by K3[Fe(CN)6], a small amount of Prussian blue was formed, identified by comparison of its IR spectral characteristics with that of an authentic sample (Fig. S58†). This is presumably formed from the reduced [Fe(CN)6]4−and hydrolysed forms of the more labile [Fe(CN)6]3−. Although the stibine and GSH were found to be able to reduce K3[Fe(CN)6] to K4[Fe(CN)6] under the same reaction conditions, no Prussian blue formation was observed (Fig. S58†), indirectly pointing to involvement of the Au core rather than the ligand sphere of 2b·Cl. An inner sphere electron transfer, presumably involving bridging CN-ligands, may therefore be operative. Consistent with this was that treatment of 2b·Cl with K3[Fe(CN)6] (3.5 equiv) indeed led to decomposition of 2b·Cl and the formation of Prussian blue (Fig. S58†).
Conclusion
We have reported here, for the first time, Au nanoclusters with stibines as protecting ligands. In contrast to phosphines, the use of stibines as the protecting ligand favors the formation of icosahedral Au13 clusters rather than Au11 or a polydispersed mixture of smaller clusters and they made for interesting comparison with, and a better understanding of, the structures of the phosphine analogues. The weaker coordinating ability of the stibine ligands also made them promising precursors to other Au nanoclusters, for example, to the thiolate protected Au25 cluster. More importantly, we have found that the addition of a suitable oxidant can significantly increase the conversion rate by aiding the size-focusing process.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a research grant (SERC grant no. 1521200076) from the Agency for Science, Technology and Research (A*STAR), Singapore.Notes and references
-
(a) P. D. Jadzinsky, G. Calero, C. J. Ackerson, D. A. Bushnell and R. D. Kornberg, Science, 2007, 318, 430–433 CrossRef PubMed
; (b) M. Zhu, C. M. Aikens, F. J. Hollander, G. C. Schatz and R. Jin, J. Am. Chem. Soc., 2008, 130, 5883–5885 CrossRef PubMed
-
(a) X.-K. Wan, Q. Tang, S.-F. Yuan, D.-e. Jiang and Q.-M. Wang, J. Am. Chem. Soc., 2015, 137, 652–655 CrossRef PubMed
- L. Naldini, F. Cariati, G. Simonetta and L. Malatesta, Chem. Commun., 1966, 647–648 RSC
-
(a) J. W. A. Van der Velden, P. T. Beurskens, J. J. Bour, W. P. Bosman, J. H. Noordik, M. Kolenbrander and J. A. K. M. Buskes, Inorg. Chem., 1984, 23, 146–151 CrossRef
-
(a) B. S. Gutrath, U. Englert, Y. Wang and U. Simon, Eur. J. Inorg. Chem., 2013, 2013, 2002–2006 CrossRef
- G. Schmid, R. Pfeil, R. Boese, F. Bandermann, S. Meyer, G. H. M. Calis and J. W. A. van der Velden, Chem. Ber., 1981, 114, 3634–3642 CrossRef
- W. W. Weare, S. M. Reed, M. G. Warner and J. E. Hutchison, J. Am. Chem. Soc., 2000, 122, 12890–12891 CrossRef
-
(a) Y. Shichibu, Y. Negishi, T. Watanabe, N. K. Chaki, H. Kawaguchi and T. Tsukuda, J. Phys. Chem. C, 2007, 111, 7845–7847 CrossRef
-
(a) H. Yang, Y. Wang, J. Lei, L. Shi, X. Wu, V. Makinen, S. Lin, Z. Tang, J. He, H. Hakkinen, L. Zheng and N. Zheng, J. Am. Chem. Soc., 2013, 135, 9568–9571 CrossRef PubMed
- B. Fresch, E. Hanozin, F. Dufour and F. Remacle, Eur. Phys. J. D, 2012, 66, 326–335 CrossRef
- J. W. A. van der Velden, F. A. Vollenbroek, J. J. Bour, P. T. Beurskens, J. M. M. Smits and W. P. Bosnian, Recl. Trav. Chim. Pays-Bas, 1981, 100, 148–152 CrossRef
- S. Jin, W. Du, S. Wang, X. Kang, M. Chen, D. Hu, S. Chen, X. Zou, G. Sun and M. Zhu, Inorg. Chem., 2017, 56, 11151–11159 CrossRef PubMed
- S.-S. Zhang, L. Feng, R. D. Senanayake, C. M. Aikens, X.-P. Wang, Q.-Q. Zhao, C.-H. Tung and D. Sun, Chem. Sci., 2018, 9, 1251–1258 RSC
-
(a) Y. Shichibu and K. Konishi, Small, 2010, 6, 1216–1220 CrossRef PubMed
- L. C. McKenzie, T. O. Zaikova and J. E. Hutchison, J. Am. Chem. Soc., 2014, 136, 13426–13435 CrossRef PubMed
- R. C. B. Copley and D. M. P. Mingos, J. Chem. Soc., Dalton Trans., 1996, 491–500 RSC
-
(a) P. D. Cookson and E. R. T. Tiekink, Acta Crystallogr., Sect. C: Struct. Chem., 1994, 50, 1896–1898 CrossRef
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370–412 RSC
- Crystallographically, the anion in 3a was modelled as disordered between a [Ph2SbCl2]−and a chloride; with appropriate restraints, the final occupancies were ∼93% and 7%, respectively. See Fig. S4† for the Ortep diagram.
- T. Huang, L. Huang, Y. Jiang, F. Hu, Z. Sun, G. Pan and S. Wei, Dalton Trans., 2017, 46, 12239–12244 RSC
- E. B. Guidez, A. Hadley and C. M. Aikens, J. Phys. Chem. C, 2011, 115, 6305–6316 CrossRef
- L. Falivene, R. Credendino, A. Poater, A. Petta, L. Serra, R. Oliva, V. Scarano and L. Cavallo, Organometallics, 2016, 35, 2286–2293 CrossRef
-
(a) B. M. Barngrover and C. M. Aikens, J. Phys. Chem. Lett., 2011, 2, 990–994 CrossRef
-
(a) M. Walter, J. Akola, O. Lopez-Acevedo, P. D. Jadzinsky, G. Calero, C. J. Ackerson, R. L. Whetten, H. Grönbeck and H. Häkkinen, Proc. Natl. Acad. Sci., 2008, 105, 9157–9162 CrossRef PubMed
- S. L. Benjamin and G. Reid, Coord. Chem. Rev., 2015, 297, 168–180 CrossRef
-
(a) X. Meng, Q. Xu, S. Wang and M. Zhu, Nanoscale, 2012, 4, 4161–4165 RSC
-
(a) S. Link, A. Beeby, S. FitzGerald, M. A. El-Sayed, T. G. Schaaff and R. L. Whetten, J. Phys. Chem. B, 2002, 106, 3410 CrossRef
- Z. Gan, Y. Lin, L. Luo, G. Han, W. Liu, Z. Liu, C. Yao, L. Weng, L. Liao, J. Chen, X. Liu, Y. Luo, C. Wang, S. Wei and Z. Wu, Angew. Chem., Int. Ed., 2016, 55, 11567–11571 CrossRef PubMed
-
(a) Y. Yu, S. Y. New, J. Xie, X. Su and Y. N. Tan, Chem. Commun., 2014, 50, 13805 RSC
- Z. Wu and R. Jin, Nano Lett., 2010, 10, 2568 CrossRef PubMed
- L. D. Menard, S.-P. Gao, H. Xu, R. D. Twesten, A. S. Harper, Y. Song, G. Wang, A. D. Douglas, J. C. Yang, A. I. Frenkel, R. G. Nuzzo and R. W. Murray, J. Phys. Chem. B, 2006, 110, 12874–12883 CrossRef PubMed
-
(a) L.-Y. Chen, C.-W. Wang, Z. Yuan and H.-T. Chang, Anal. Chem., 2015, 87, 216–229 CrossRef PubMed
- Z. Wu, J. Suhan and R. Jin, J. Mater. Chem., 2009, 19, 622–626 RSC
-
(a) Y. Shichibu, Y. Negishi, T. Tsukuda and T. Teranishi, J. Am. Chem. Soc., 2005, 127, 13464–13465 CrossRef PubMed
- S. Yukatsu, N. Yuichi, T. Hironori, K. Masayuki, T. Toshiharu and T. Tatsuya, Small, 2007, 3, 835–839 CrossRef PubMed
- G. H. Woehrle, M. G. Warner and J. E. Hutchison, J. Phys. Chem. B, 2002, 106, 9979–9981 CrossRef
- M.-B. Li, S.-K. Tian, Z. Wu and R. Jin, Chem. Mater., 2016, 28, 1022–1025 CrossRef
-
(a) M.-B. Li, S.-K. Tian, Z. Wu and R. Jin, Chem. Commun., 2015, 51, 4433–4436 RSC
-
W. M. Haynes, CRC handbook of chemistry and physics: a ready-reference book of chemical and physical data, CRC Press, Boca Raton, Florida, 97th edn, 2016–2017, pp. 5–85 Search PubMed
- Z. Wu, Angew. Chem., Int. Ed., 2012, 51, 2934–2938 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, crystallographic and computational details; table of crystal and refinement data for 1a, 1b, 2a·BPh4, 3a and 3b; ORTEP and stacking diagrams of 1a, 1b, 3a and 3b; NMR, ESI-MS and UV-Vis spectra of products and some monitoring reactions; TD-DFT excitation transition data of 2a; CV spectra of 2a·PF6; percent buried volumes of stibine ligands; optimized coordinates of 1a–c and 2a. CCDC (CCDC 1852139–1852143). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc03132k |
| This journal is © The Royal Society of Chemistry 2018 |