
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient strategies for the preparation of non-noble metal catalysts for electrocatalytic glycerol oxidation towards high-value-added chemicals
Mingyang Songa,
Yitong Baia,
Jiefei Li *a and
Xingyue Qi*b
*a and
Xingyue Qi*b
aSchool of Petrochemical Engineering, Shenyang University of Technology, Liaoyang, 111003, China. E-mail: lijf@sut.edu.cn
bSchool of Public Health, Inner Mongolia Medical University, Hohhot, 010110, China. E-mail: xingyueqichem@163.com
First published on 17th June 2025
Abstract
Glycerol is an important by-product of the biodiesel production process, is cheap and abundant, and can be converted into a variety of high-value-added fine chemicals. The glycerol electrooxidation reaction (GEOR) has significant application potential owing to its lack of oxidant requirements and the generation of clean hydrogen energy at the cathode. Currently, noble metal materials serve as effective catalysts for the GEOR, but their high cost and scarcity limit their commercial applications. Consequently, there is an urgent necessity to develop efficient non-noble metal catalysts for the GEOR to replace noble metal catalysts. This paper briefly introduces the reaction mechanism of the GEOR and reviews recent research progress on the synthesis of transition metal-based GEOR catalysts, focusing on Ni-, Co-, Cu-, and Mn-based catalysts. A particular emphasis is placed on the strategies to improve the GEOR performance of different non-noble metal catalysts, which include alloying, doping engineering, defect engineering, heterostructure construction, crystal phase regulation, morphology control and composite synthesis. Finally, we provide a comprehensive discussion on the challenges and future prospects of non-noble metal catalysts in the GEOR.
With the rapid depletion of fossil fuels and increasingly severe environmental problems, renewable resources as a green and clean energy source have gained significant attention.1–3 Among them, biodiesel has significant advantages such as renewability and biodegradability, making it an important substitute for fossil fuels. During biodiesel production, 10% of the output consists of glycerol, which is considered an important biomass platform molecule with unlimited potential for producing high-value chemicals.4,5
Glycerol is a highly functionalized molecule with three hydroxyl (–OH) functional groups, which can undergo reactions such as oxidation,6–8 hydrogenolysis,9 pyrolysis,10 etherification,11 and esterification.12 Among them, selective oxidation of glycerol can produce organic acids such as glycolic acid (GA), tartronic acid (TAR), dihydroxyacetone (DHA), glyceraldehyde (GLAD), glyceric acid (GLA), lactic acid (LA), and formic acid (FA), which have very high added value and are widely used in the cosmetic, pharmaceutical, fine chemical, and food industries.13–16
Glycerol oxidation can be primarily achieved through four catalytic approaches: enzymatic, thermal, photocatalytic, and electrocatalytic methods. However, enzymatic oxidation suffers from prolonged reaction cycles, low product selectivity, and unsatisfactory yields, complicating product separation. Additionally, enzyme deactivation remains a critical limitation. Thermal catalytic oxidation typically relies on noble metal catalysts and requires harsh reaction conditions (elevated temperatures and pressures), resulting in higher energy consumption compared with electrocatalysis. Photocatalytic oxidation exhibits slow surface reaction kinetics, necessitating extended reaction durations, which consequently lead to low catalytic efficiency. Among the many ways to achieve glycerol oxidation, the glycerol electrooxidation reaction (GEOR) has great application prospects because it does not require oxidants such as O2 and H2O2, its product selectivity can be flexibly controlled by regulating the potential at the anode, and clean H2 energy can be produced at the cathode.
However, current GEOR catalysts are mainly noble metals (Pd, Pt, and Au) and their alloys (e.g., PtAu, PtSb, PtxBi, and PdxBi),17–20 but the high cost and scarcity of these catalysts hinder their practical application. As reported by Kimura's team,21 although noble metal catalysts can be reused for up to 10 cycles, their cost still accounts for 95% of the production cost of conversion products such as dihydroxyacetone, tartronic acid, and mesoxalic acid. Besides, the GEOR involves multiple complex reaction pathways, producing various C1, C2, and C3 intermediate oxidation products, resulting in low selectivity for the target products. Therefore, it is necessary to develop novel, low-cost catalysts with high activity, selectivity, and stability for GEOR. In this case, research has been focused on constructing highly active non-noble metal electrocatalysts with abundant reserves, low cost, and stable performance. Recently, numerous innovative research works ranging from synthesis strategies (doping engineering, alloying strategies, defect engineering, heterostructure construction, phase regulation, morphology control engineering and composite strategies) to performance testing have emerged on transition metal-based catalysts (e.g., Ni-, Co-, Cu-, and Mn-based catalysts),22–29 which demonstrates the tremendous application prospects of non-noble metal catalysts (Fig. 1).
 | ||
| Fig. 1 Design principles and synthesis strategies for GEOR electrocatalysts. | ||
Based on this, herein, we initially discuss the mechanism of GEOR, and then comprehensively review the design and optimization of catalyst synthesis strategies through case studies, detailing the roles of doping engineering, alloying strategies, defect engineering, heterostructure construction, phase regulation, morphology control engineering and composite strategies in improving the GEOR performance. Finally, the research progress to date on non-noble metal catalysts for GEOR is summarized, and the challenges and future prospects in this field are discussed. This review will provide theoretical guidance for the rational design of efficient electrocatalysts, thus promoting the further development of high-value-added products from GEOR.
1 Glycerol oxidation reaction mechanism
The complete oxidation of glycerol to carbonate requires 14 electrons, as shown in eqn (1).| CH2OH–CHOH–CH2OH + 20OH− = 3CO32− + 14H2O + 14e− | (1) |
Due to the differences in the adsorption of glycerol and its intermediates on the catalyst, as well as C–C bond cleavage, glycerol oxidation yields different products. Under alkaline conditions, the possible reaction pathways are shown in Fig. 2. Firstly, glycerol is oxidized to glyceraldehyde (GLAD) or dihydroxyacetone (DHA), with GLAD and DHA in equilibrium under basic conditions, and they can interconvert.30 Subsequently, these compounds undergo base-catalyzed dehydration to form 2-hydroxypropenal/acetol intermediates, which may either undergo Cannizzaro rearrangement to produce lactic acid (LA) or be further oxidized to generate glyceric acid (GLA), tartronic acid (TA), and C–C bond cleavage products such as glycolic acid (GA), formic acid (FA), and oxalic acid (OA).31,32 Catalysts are one of the main factors influencing the glycerol oxidation reaction; therefore, optimizing catalysts based on the target products to achieve high selectivity is crucial in the electrocatalytic oxidation of glycerol.
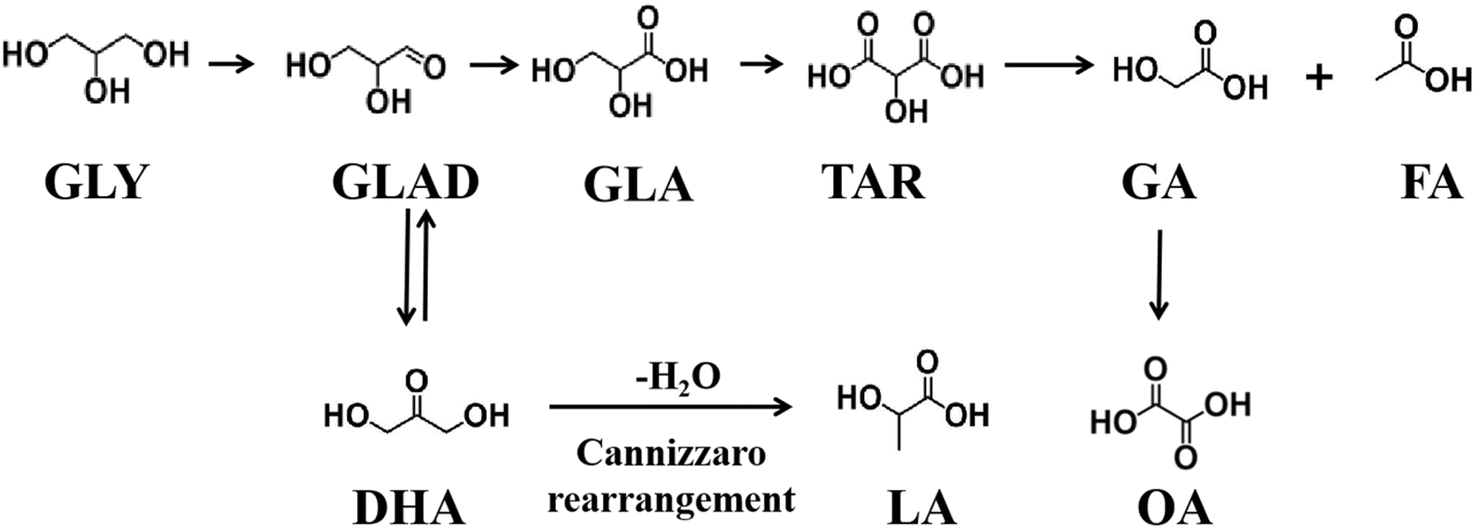 | ||
| Fig. 2 Possible reaction pathways for glycerol oxidation. | ||
The adsorption behavior of glycerol and its oxidation intermediates on the catalyst surface plays a vital role in determining both the catalytic activity and product selectivity during the glycerol electrooxidation reaction (GEOR). For instance, the construction of heterojunction structures prolongs the residence time of glycerol molecules on their surface, which facilitates subsequent steps in the glycerol electrooxidation reaction (GEOR). An upward shift in the d-band center of the catalyst further indicates that the formation of a heterojunction enhances the glycerol adsorption capacity, thereby promoting GEOR catalysis.54 Through alloying strategies, catalysts can be tuned to generate key reactive species such as *OH rather than NiOOH or CoOOH during glycerol oxidation. The *OH species selectively promote C–C bond cleavage in glycerol prior to O–H and C–H bond dehydrogenation. This reduces the formation of C3 intermediates and simplifies the reaction pathway, ultimately increasing the overall yield of formate.37 A similar effect was observed using the doping strategy, where the substitution of Co ions in Co3O4 with single-atom Bi enhanced the formation of *OH species, thereby lowering the energy barrier of GEOR. The *OH species accelerate both hydroxyl oxidation and C–C bond cleavage, improving both the GEOR activity and formate selectivity.44
The enhanced selectivity toward other oxidation products is often governed by the adsorption strength of specific intermediates. For example, a vertical chemisorption configuration facilitates the dissociation of key intermediates, thus promoting the formation of glycolate.38 Manganese oxide catalysts with different crystal phases exhibit high selectivity toward dihydroxyacetone (DHA), which is attributed to their stronger affinity for secondary –OH groups compared to primary ones, favoring DHA formation over glyceraldehyde (GLYD). At high anodic potentials, phase transitions suppress C–C bond cleavage, thereby increasing the selectivity toward C3 products.63 Similarly, in the compositing strategy, MnO2–CuO/CF composite electrodes exhibit high selectivity for DHA, given that DHA has a lower adsorption energy on MnO2 (100), facilitating its desorption from the catalyst surface.68
Thus, different synthesis strategies can modify the surface properties and crystal phase structure of the catalyst, thereby influencing the adsorption of glycerol and its oxidation intermediates, which enhances the catalytic performance and selectivity toward specific products.
2 Design strategies for non-precious metal catalysts in GEOR
2.1 Alloy strategy
Pure nickel catalysts exhibit limited activity for GEOR, but their performance can be improved through alloying effects. Transition metal alloys exhibit various ligand and electronic effects, integrating the catalytic properties of multiple metal elements. Additional adsorption sites can be established on the surface of nickel catalysts, enabling the regulation of the adsorption of different intermediates on the catalyst.Oliveira and colleagues33,34 previously prepared nickel-based catalysts (CoNi/C, FeNi/C, FeCoNi/C) and compared their performance with Ni/C catalysts, finding that Fe and Co in binary and ternary nickel-based catalysts promoted the formation of oxides and hydroxides, thereby enhancing the electrocatalytic glycerol activity. HPLC analysis revealed that the main reaction product was formate, with tartrate, glycolate, oxalate, and glycerate also detected. CoNi/C exhibited the highest glycerol conversion (17.9%). The effect on the Ni hydroxyl group (β-NiOOH) and its impact on glycerol conversion was investigated. The Fe and Co elements altered the morphology of the oxy-hydroxide region. In particular, in the case of Fe-containing materials, the NiFe alloy can catalyze the oxygen evolution reaction (OER), which tends to dominate and obscure the oxy-hydroxide region. Among the studied Ni-based materials, Ni/C and FeCoNi/C showed the best conversion of glycerol to formate and glycolate, and CoNi/C exhibited the highest glycerol conversion. Habibi et al.35 alloyed Ni with Cu and Co on a carbon–ceramic electrode substrate, which showed lower glycerol oxidation overpotentials and higher anodic peak currents and significantly improved catalytic activity for glycerol oxidation (Fig. 3a and b). Due to the presence of Cu and Co, the surface concentration of β-NiOOH in Ni–Cu/CCE and Ni–Co/CCE was higher, enabling these electrodes to exhibit greater GEOR electrocatalytic activity in NaOH solution. Additionally, Ni-based alloys exhibited smaller Tafel slopes compared to Ni/CCE. The chronoamperometry (CP) and cyclic voltammetry (CV) results further indicated that the prepared Ni-based alloy catalysts had good long-term stability and storage performance. Ghaith et al.36 modified 3D carbon felt (CF) with nickel–copper (Ni–Cu@CF) bimetallic nanostructures using sequential and co-electrodeposition methods (Fig. 3c). Researchers found that inserting Cu into Ni resulted in strong interactions between the two metals, providing Ni–Cu@CF with more Ni active surface sites compared to Ni@CF, thereby exhibiting higher GEOR activity. Compared with Ni@CF, the glycerol oxidation activity of NiCu@CF prepared via sequential electrodeposition increased by 1.6 times, and the onset potential was reduced by approximately 63 mV. Similarly, the Ni85%Cu15%@CF catalyst prepared via co-electrodeposition exhibited 1.4 times higher glycerol oxidation activity than the Ni@CF electrode. Moreover, both NiCu@CF and Ni85%Cu15%@CF demonstrated good stability.
 | ||
| Fig. 3 Effect of the scan rate on the electrooxidation of 0.1 M glycerol in 1.0 M NaOH at (a) Ni–Cu/CCE and (b) Ni–Co/CCE.35 (c) XRD patterns of Ni@CF, Cu@CF, NiCu@CF, CuNi@CF, and Ni85%Cu15%@CF electrodes.36 (d) Schematic of the MEA flow electrolyzer.37 (e) Free energy diagram of the GOR on the four catalyst models, in which the inserted configurations are the corresponding intermediate structures adsorbed on the Ni0.1Co0.9@NiSACoSA-NCNTs catalyst; the Ni, Co, N and C atoms are shown in green, pink, blue and grey, respectively; for distinction, the C atoms from glycerol are given in black.37 (f) Glycolic acid on Au(111) and AuCu(111) crystalline surfaces.38 Reproduced with permission from ref. 35–38. Copyright 2016, Royal Society of Chemistry, Copyright 2023, Royal Society of Chemistry, Copyright 2025, Wiley-VCH, Copyright 2024, American Chemical Society. | ||
Single-atom catalysts (SACs) exhibit remarkable advantages in the glycerol electrooxidation reaction (GEOR) due to their unique electronic structures and maximized atomic utilization efficiency. Zhang et al.37 constructed a dual-atomic-site catalyst comprised of atomically dispersed nickel (NiSA) and cobalt (CoSA) anchored on nitrogen-doped carbon nanotubes (NCNTs), with an embedded Ni0.1Co0.9 alloy core (denoted as Ni0.1Co0.9@NiSACoSA-NCNTs). This catalyst exhibited an outstanding glycerol conversion efficiency of 98.81% at an applied potential of 1.45 V vs. RHE, while maintaining a high formate selectivity of 93.27%. Moreover, it demonstrated excellent long-term stability at a constant current density of 100 mA cm−2, sustaining continuous operation for over 108 h. The integrated electrocatalytic system required only a low cell voltage of 1.52 V to achieve the same current density, highlighting its energy efficiency. Crucially, the authors proposed a reaction mechanism distinct from conventional transition metal-based systems. Unlike the widely reported activation pathways involving the in situ formation of high-valent species such as Ni3+–OOH or Co3+–OOH under anodic conditions, the key reactive species in this system was identified to be surface-adsorbed hydroxyl (*OH). The *OH species selectively promoted the cleavage of the C–C bond in the glycerol molecule, preceding the dehydrogenation of O–H and C–H bonds. This unique reaction pathway minimized the formation of undesired C3 intermediates and reduced the number of elementary steps, thereby enhancing the overall formate yield. The synergistic interaction between the NiSA and CoSA active sites within NiSACoSA-NCNTs was found to modulate the electronic structure of CoSA, effectively lowering the energy barriers associated with the rate-determining steps (RDS), specifically the C–C bond cleavage and the dehydrogenation of C1 intermediates. Additionally, the coupling effect between the NiCo alloy core and the NiSACoSA-NCNT shell induced a moderately electron-deficient state in the CoSA sites. This not only enhanced the adsorption affinity for glycerol but also further reduced the energetic barrier for C–C bond cleavage, thereby markedly improving the formate production efficiency (Fig. 3d and e).
The product distribution of GEOR on non-precious metals is typically restricted to formate. Introducing noble metal heteroatoms modifies the electronic structure of the active sites, selectively stabilizing the C2/C3 intermediates and suppressing C–C bond cleavage. Shen et al.38 reported the synthesis of a hollow spherical bimetallic Au1Cu1 catalyst that exhibited high activity and selectivity for the electrooxidation of glycerol, 1,2-propanediol, and ethylene glycol. Under the optimized conditions, the catalyst achieved a glycerol conversion rate of 90% and glycolic acid selectivity of 45%. Density functional theory (DFT) calculations revealed that the adsorption energy of glycerol on the AuCu(111) surface (−0.29 eV) was significantly lower than that on the Au(111) surface (0.23 eV), indicating the stronger adsorption affinity of AuCu(111) toward glycerol molecules (Fig. 3f). On the Au(111) surface, glycerol exhibited weak adsorption, characterized by a molecular–surface distance of approximately 3 Å, consistent with the physisorption state. In contrast, the formation of the AuCu(111) alloy led to a more robust interaction, wherein glycerol adsorbed in an upright configuration and formed a Cu–O bond with the bond length of 2.09 Å. This chemisorbed configuration facilitated the dissociation of key intermediates, thereby promoting the formation of glycolic acid.
A self-supported high-entropy alloy (HEA) electrode for GEOR by growing CoNiCuMnMo nanoparticles (NPs) on carbon cloth (CC) was developed by Fan et al.39 X-ray diffraction (XRD) results indicated that the catalyst possessed a face-centered cubic (FCC) crystal structure. Transmission electron microscopy (TEM) further revealed that the particle size of HEA-CoNiCuMnMo NPs was approximately 10 nm (Fig. 4a) and they interconnected through graphitized carbon layers derived from MOFs. Based on the strong synergistic effects among the metals, this electrode exhibited an excellent electrocatalytic performance in alkaline electrolyte, requiring only 1.25 V to achieve a current density of 10 mA cm−2 (Fig. 4b). In a broad potential range (1.27–1.47 V vs. RHE), the faradaic efficiency (FE) for formic acid exceeded 90% (Fig. 4c). The authors further investigated the ideal electrocatalytic activity of HEA-CoNiCuMnMo NPs for GEOR on carbon cloth using machine learning (ML) combined with Monte Carlo (MC) simulations. The advantages of the synergistic effects of each metal in the HEA were verified. ML-based MC simulations revealed that the catalytic active centers were Mo sites coordinated by Mn, Mo, and Ni. Additionally, an alkali/acid mixed electrolyzer was constructed, and the long-term stability of the system was further tested using chronoamperometry. At a fixed current density of 50 mA cm−2, the electrolyzer could maintain stable electrolysis for 300 h. Additionally, Yao et al.40 used a hydrothermal method for the first time to in situ grow a self-supported high-entropy selenide electrode (CoNiCuMnMo)Se/CF on copper foam (CF). This electrode demonstrated an excellent glycerol oxidation reaction electrocatalytic performance in alkaline electrolyte with 0.1 M glycerol and 1 M KOH, achieving a current density of 10 mA cm−2 at only 1.20 V vs. RHE. It also maintained high selectivity for the formic acid product over a wide potential range (1.27–1.57 V vs. RHE). The authors further used in situ infrared spectroscopy to propose the reaction pathway of glycerol in alkaline medium (Fig. 4d). Glycerol was first oxidized to glyceraldehyde, which was then further oxidized to glycolate, and eventually to various carboxyl-containing products (Fig. 4e). CP analysis showed that the potential of (CoNiCuMnMo)Se/CF did not significantly increase during 25 h of electrolysis, indicating its good electrochemical stability (Fig. 4f). The alloying of Cu and Ni significantly reduced the grain size of the metal nanoparticles and improved their dispersion. Additionally, in high-pH electrolyte, the high concentration of hydroxide ions retained on the electrode surface facilitated the formation of copper hydroxide and nickel hydroxide at lower potentials, providing more active sites for the electrooxidation of glycerol. This electrocatalyst exhibited excellent catalytic activity and significant selectivity for formic acid (97.4%). Thus, various alloying strategies have created numerous approaches to enhance the activity of catalysts, with continuous breakthroughs in improving the product selectivity through the rich synergistic effects among components.
 | ||
| Fig. 4 (a) Bright-field TEM image showing the dispersion of HEA NPs at a low magnification.39 (b) Comparison of glycerol anodic oxidation activities among various catalysts in 1 M KOH with 0.1 M glycerol addition (IR correction). Scan rate = 2 mV s−1.39 (c) FEs for formate production at different potentials.39 (d) In situ IR spectra as a function of potential of (CoNiCuMnMo)Se/CF in 1 M KOH with 0.1 M glycerol.40 (e) Possible reaction pathway of the glycerol oxidation reaction in an alkaline medium.40 (f) Stability test of (CoNiCuMnMo)Se/CF for anodic glycerol oxidation at a current density of 10 mA cm−2.40 Reproduced with permission from ref. 39 and 40. Copyright 2022, American Chemical Society, Copyright 2023, Springer Nature. | ||
2.2 Doping engineering
Researchers have found that modifying the surface of non-noble metal catalysts with p-block metals such as bismuth (Bi), antimony (Sb), and tin (Sn) can regulate the adsorption of GEOR intermediates and the product selectivity. Houache et al.41 incorporated small amounts of Bi into Ni nanoparticles to form Ni–Bi bimetallic catalysts. Thus, glycerol electrooxidation on the Ni–Bi catalysts exhibited a lower onset potential and higher current density (Fig. 5a). After aging, the catalyst structure evolved from an Ni–Bi core–shell configuration to Bi-modified Ni sheets, with activity significantly higher than the undoped Ni catalysts. The incorporation of Bi inhibited C–C bond cleavage in glycerol, suppressing the formation of carbonates and formates, and thereby promoting the formation of C2 and C3 products. Most of the GEOR products of Fe, Co, Cu catalysts are FA. Although FA is an important industrial chemical, its added value is still less than that of C2 and C3 products, and Mn-based catalysts contribute to the preparation of high-added-value C3 products in glycerol oxidation (Fig. 5b). Goetz et al.42 doped manganese-based materials with Sb to successfully prepare the MnSb2O6 catalyst. Compared to MnOx catalysts, the structure of MnSb2O6 (where no Mn sites are nearby) and the oxidation state characteristics (Mn2+ remaining stable in the MnSb2O6 lattice) inhibited C–C bond cleavage, achieving a selectivity of C3 product glyceric acid up to 82% (Fig. 5c). Recently, Wang et al.43 activated nickel molybdate (NiMoO4) using cyclic voltammetry, developing a molybdenum-doped nickel oxyhydroxide (Mo–NiOOH) catalyst. Mo–NiOOH exhibited a higher FE for formate (84.7% vs. 59.6%) compared to NiOOH, which was attributed to the incorporation of Mo, which created more active sites. This accelerated the GEOR kinetics, facilitated the oxidation of Ni2+ to Ni3+, enhanced C–C bond cleavage, and effectively improved the activity and selectivity for formic acid. Additionally, at a high current density of 400 mA cm−2, the oxidation potential of Mo–NiOOH (1.51 V vs. RHE) was lower than that of NiOOH (1.84 V vs. RHE). | ||
| Fig. 5 (a) Linear sweep voltammetry at 1 mV s−1.41 (b) Product distribution and glycerol conversion (dashed line) at different applied potentials and temperatures over Ni90Bi10 and in 1 M KOH + 0.1 M glycerol.41 (c) Direct comparison of product distribution on MnOx (1.6 VRHE) and MnSb2O6 (2.0 VRHE) at pH 14, showing the relative selectivity (left axis) and FE (right axis), with percentages given in gray.42 Reproduced with permission from ref. 41 and 42. Copyright 2020, American Chemical Society, Copyright 2023, American Chemical Society. | ||
Doping strategies are also widely applied to spinel oxides with tetrahedral (Td) and octahedral (Oh) sites, where substituting Td or Oh atoms with heteroatoms or single atoms is an attractive design strategy for regulating the GEOR activity and selectivity. Wang et al.44 incorporated single-atom Bi into Co3O4 to form the Bi-Co3O4 catalyst. Glycerol (mainly in the form of glycerol alkoxide) adsorbed at the bridge sites between the CoOh3+ and BiOh sites. Bi3+ substitution for octahedral Co (CoOh3+) promoted the generation of OH* on adjacent tetrahedral Co sites (CoTd2+), strengthening the Co–O bond between CoTd2+ and OH*, thus lowering the reaction barrier for GEOR (Fig. 6a). OH* accelerated the oxidation of hydroxyl groups and the cleavage of glycerol C–C bonds, enhancing the FE and selectivity for formates. The FE for formic acid of Bi–Co3O4 (97.05%) was significantly higher than that of Co3O4 (<80%). Furthermore, a chronoamperometric test was conducted at 1.35 V for 200 h, demonstrating the excellent long-term stability of the Bi–Co3O4 catalyst (Fig. 6b). Luo et al.45 replaced octahedral Co (CoOh3+) in Co3O4 with Ni, resulting in an NiCo2O4/NF bimetallic oxide nanowire array. The nanowire array exhibited excellent catalytic activity under high current densities (E300 = 1.42 V, E600 = 1.62 V), with a total FE of 97.5% at 1.42 V (Fig. 6c).
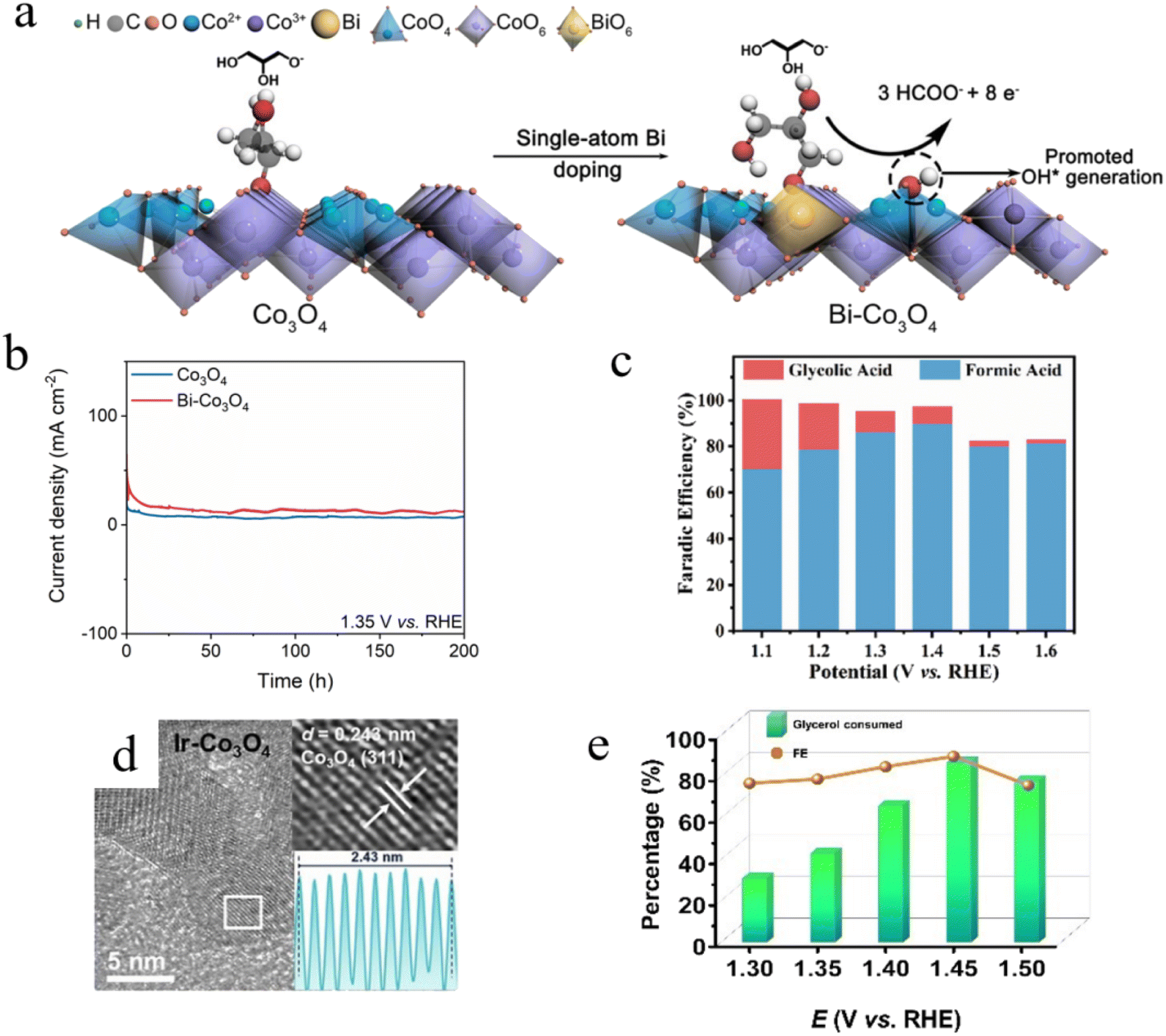 | ||
| Fig. 6 (a) Schematic of the promoting effects of single-atom Bi doping on co-adsorption of OH* and glycerol.44 (b) Chronoamperometry curves of Co3O4 and Bi–Co3O4 in 1.0 M KOH with 0.1 M glycerol at 1.35 V vs. RHE.44 (c) Faradaic efficiency of glycolic acid (FEGCA) and formic acid (FEFA) at varying potentials for NiCo2O4/NF.45 (d) HRTEM images of Ir–Co3O4.46 (e) Glycerol conversion and formate FE at different potentials for Ir–Co3O4/NF tested in a 1 M KOH solution containing 0.1 M glycerol.46 Reproduced with permission from ref. 44–46. Copyright 2022, American Chemical Society, Copyright 2023, Wiley-VCH, Copyright 2023, Royal Society of Chemistry. | ||
Although non-precious metal doping has demonstrated remarkable efficacy in enhancing the GEOR performance, the incorporation of noble metals can further optimize catalytic behavior through strain engineering-induced lattice distortion in the host oxides, thereby modulating their electronic structures to achieve superior GEOR activity. Xu et al.46 doped Co3O4 with Ir, which caused lattice expansion in Co3O4 and modulated its electronic structure due to the larger atomic radius of Ir compared to Co (Fig. 6d). The Ir–Co3O4 catalyst grown on a nickel foam substrate (Ir–Co3O4/NF) exhibited lattice-expansion-induced strain effects and demonstrated an excellent performance in both electrocatalytic glycerol-to-formic acid conversion and hydrogen evolution reactions. The results showed that Ir–Co3O4/NF achieved the highest glycerol conversion efficiency (87%) and formic acid FE (92%) at 1.45 V vs. RHE (Fig. 6e). Furthermore, the LSV curves of Ir–Co3O4/NF before and after 5000 CV measurements were nearly identical, indicating its good stability.
Non-metal doping (N, P, S, B) represents a paradigm-shifting approach in the design of GEOR catalysts, where tailored electronic structures and surface functionalities overcome conventional activity–stability trade-offs. Chen et al.47 demonstrated this via a Co vacancy-rich, P-doped nanosheet (P3–Co), which in an MEA electrolyzer achieved the co-production of NH3 (98.2% FE) and formate (93.4% FE) at an industrially relevant current density (100 mA cm−2) for 300 h, a record for nitrate–glycerol valorization. According to operando spectroscopy and DFT calculations, this breakthrough was attributed to the P-induced charge redistribution and Co defect-mediated kinetic acceleration, establishing a dual-doping-defect design principle for next-generation electrocatalysts.
2.3 Defect engineering
Defect engineering can alter the coordination number and electronic structure of the active sites in catalysts, enriching the number of active sites, effectively modulating the electronic density of the active centers, and optimizing the adsorption and activation of molecules, which are crucial for improving the electrocatalytic conversion efficiency. Vacancies and other defects on metal catalytic surfaces can influence the electronic structure of neighboring atoms, alter the reaction barriers, and thereby effectively modify the reaction pathways and product distribution. Xia et al.48 reported a chromium ion leaching strategy to optimize the metal coordination through vacancies, enhancing the co-adsorption process and intrinsic GEOR activity of nickel-based catalysts. The NiCrO catalyst with vacancy structures (NiCrO–VCr,O) achieved a current density of 10 mA cm−2 at only 1.37 V vs. RHE. The leaching of Cr resulted in the creation of cation vacancies, accompanied by the partial loss of lattice oxygen linked to Cr, generating abundant oxygen vacancies (Fig. 7a). The optimized coordination environment improved the adsorption of OH− and optimized its electronic structure, facilitating the rapid formation of NiOOH. At 1.45 V vs. RHE, the glycerol conversion rate, formic acid selectivity, and FE reached 99%, 98%, and 96%, respectively. Moreover, NiCrO–VCr,O exhibited outstanding stability with the conversion rate and selectivity in a flow electrolyzer remaining above 90% during long-term continuous electrolysis. DFT calculations were performed to construct (200) facet models of NiO and NiCrO–VCr,O, and the adsorption energies of OH−, glycerol, and co-adsorption of OH−/glycerol were calculated (Fig. 7b and c–h) On the NiO (200) surface, all three adsorption processes exhibited positive adsorption energies, indicating that the adsorption of OH− and glycerol during the formation of NiOOH species and the glycerol oxidation reaction (GOR) process is thermodynamically unfavorable. In contrast, the NiCrO–VCr,O (200) surface demonstrated negative adsorption energies for all three processes, exhibiting a significant adsorption advantage for both OH− and glycerol molecules. These results theoretically elucidate the origin of the superior capability for the formation of NiOOH species and the highly efficient GOR activity of the NiCrO–VCr,O surface. | ||
| Fig. 7 (a) Schematic of the activation process.48 (b) Adsorption energies of OH−, glycerol, and the co-adsorption of OH− and glycerol on the NiO (200) surface and the NiCrO–VCr,O (200) surface.48 Adsorption configurations on NiO and NiCrO–VCr, O for (c and d) OH−, (e and f) glycerol, and (g and h) co-adsorption of OH− and glycerol.48 Reproduced with permission from ref. 48. Copyright 2024, American Chemical Society. | ||
Wu et al.49 used closely packed defect sites with synergistic effects to regulate the GEOR activity by controlling the oxygen vacancy density in Cu–Co oxide nanosheets (CuCo2O4 NS). The systems with multiple closely spaced oxygen vacancies synergistically promoted C–C bond cleavage, and CuCo2O4−x NS with the highest oxygen vacancy density (CuCo2O4−x−2) oxidized C3 molecules to C1 molecules with nearly 100% selectivity and an FE of approximately 99%. Fang et al.50 reported the synthesis of sulfur-doped manganese hydroxide nanosheets (Mn–Co–S/NF) on a nickel foam substrate. SEM images revealed that Mn–Co–S exhibited a typical nanosheet morphology (Fig. 10h). During GEOR, the doped sulfur in Mn–Co–S/NF was partially replaced by oxygen, forming a layered porous structure with abundant oxygen vacancies and strong charge transfer capability. Low-temperature electron paramagnetic resonance spectra also confirmed the formation of oxygen vacancies in the reconstructed Mn–Co–S/NF (Fig. 10i). Mn–Co–S/NF exhibited a low potential of 1.347 V vs. RHE at 100 mA cm−2, with the FE for FA reaching 96.6% at 1.35 V vs. RHE. Time-potential tests showed that Mn–Co–S/NF maintained GEOR stability for up to 60 h at 10 mA cm−2. Feng et al.51 constructed a 3D antler-like VC-Co3O4/CoSe2 array with abundant O and Se dual vacancies on nickel foam. The synergistic effect of the dual vacancies endowed the VC-Co3O4/CoSe2 catalysts with abundant active sites and optimized charge distribution, resulting in excellent catalytic activity and stability in GEOR.
2.4 Heterostructure construction
Heterostructure design is an important strategy for constructing electrocatalytic active interfaces, and new catalytic sites can be created by adjusting the electronic structure and lattice strain at the interface to improve the interface charge transfer kinetics.52 The construction of heterostructures between two components usually results in structural distortion and charge redistribution at the interface, providing new active sites for the reaction intermediates. Importantly, the rational design of heterogeneous interfaces can maximize the synergistic effect, and the formation of heterogeneous structures is a classic strategy to effectively improve the performance of GEOR catalysts. Xu et al.53 synthesized an NiOOH/Ni3S2/NF model catalyst using a two-step method. The unique electronic structure of Ni3S2 enhanced its electron transport capability, while NiOOH provided excellent glycerol adsorption properties, and the synergy of the two significantly improved the GEOR activity (Fig. 8a and b). This catalyst achieved a current density of 10 mA cm−2 at 1.227 V vs. RHE and FE for formic acid as high as 97.7% at 1.4 V vs. RHE. The authors further inferred the reaction pathway of glycerol based on NMR and in situ infrared data (Fig. 8c). The terminal hydroxyl group of glycerol first loses 2 electrons to form GLAD, which then loses 2 electrons to produce GLA. GA loses electrons and undergoes bond cleavage to form GA and FA. GA is further oxidized to OA. The hydroxyl group at the intermediate position of glycerol is oxidized to form DHA, which can interconvert with GLAD under alkaline conditions. DHA further loses 2 electrons to be oxidized to hydroxy-pyruvic acid, and ultimately oxidized to OA and FA. The final product of both pathways is FA, corresponding to the high FE of 97.7% for FA observed with NiOOH/Ni3S2/NF. Feng et al.54 prepared NiSe–Ni3Se2/NF using an electrodeposition method to construct heterogeneous interfaces and developed a theoretical structural model of NiSe–Ni3Se2 heterojunctions for glycerol adsorption (Fig. 8d). The construction of the NiSe–Ni3Se2 heterojunction structure resulted in the distortion of the NiSe structure, exposing highly active adsorption sites (Ni sites) (Fig. 8e and f). Theoretical calculations confirmed this observation, showing that the adsorption energy of glycerol on the heterojunction NiSe (ΔEgly = −1.64 eV) was higher than on Ni3Se2 (−1.47 eV) or single NiSe (−1.40 eV), indicating that glycerol molecules linger longer on the heterojunction surface, facilitating subsequent reactions. Additionally, the d-band center of NiSe–Ni3Se2 was closer to the Fermi level compared to Ni3Se2 and NiSe, further indicating that the construction of NiSe–Ni3Se2 heterojunctions enhanced the glycerol adsorption capability and promoted GOR catalysis. Liu et al.55 fabricated a freestanding Ni3N–Ni0.2Mo0.8N nanowire array with a heterostructure on a carbon cloth (CC) electrode. The unique composition leveraging the nanowire array structure and the synergistic effects of the heterointerface optimized the adsorption energy of the catalyst for reactants and intermediates, resulting in excellent GEOR activity. The results showed that at the optimal potential of 1.35 V vs. RHE, the FE for glycerol conversion to formic acid exceeded 96%. The results showed that at the optimal potential of 1.35 V vs. RHE, the FE for glycerol conversion to formic acid exceeded 96% (Fig. 8g). All the above-mentioned studies show that the synergistic effect of heterojunction catalysts provides more possibilities for the electrocatalyzed oxidation of glycerol to prepare high-value-added chemicals, laying the foundation for exploring the reaction paths and active sites for the electrocatalyzed oxidation of glycerol. | ||
| Fig. 8 (a) Raman spectra of Ni3S2/NF and NiOOH/Ni3S2/NF.53 (b) LSV curves of NF, NOOH/NF, Ni3S2/NF and NiOOH/Ni3S2/NF.53 (c) Proposed GEOR pathway in alkaline media based on the NiOOH/Ni3S2/NF electrode.53 (d) Theoretical structure models of adsorption glycerol on NiSe–Ni3Se2 heterojunction. The yellow, green, purple, blue, pink, and grey balls represent the Ni, Se, Mo, H, O, and C atoms, respectively.54 (e) XRD pattern of NiSe–Ni3S2/NF.54 (f) HRTEM analysis of NiSe–Ni3Se2/NF.54 (g) Calculated faradaic efficiencies for formic acid products in a sequence of five successive electrocatalytic cycles.55 (h) LSV curves of NiV LDH and E-NiV LDH in 1 mol L−1 KOH and 1 mol L−1 KOH with 0.1 mol L−1 glycerol.56 (i) FE of formate production and conversion from glycerol to formate with time in chronoamperometric test at 1.46 V vs. RHE.56 (j) FE of formate production and conversion from glycerol to formate for 5 electrolysis cycles at 1.46 V (vs. RHE).56 Reproduced with permission from ref. 53–56. Copyright 2023 Elsevier, Copyright 2023, Wiley-VCH, Copyright 2022, Elsevier, Copyright 2022, Springer Nature. | ||
In addition, nickel-based double layer hydroxides have a two-dimensional nanolayer structure, which have strong structural tunability and abundant active sites and show excellent performance in GEOR. Dong et al.56 reported the activation of Ni sites in an NiV layered double hydroxide (LDH) using electrochemical and N2/H2 plasma regulation, which enhanced GEOR and HER activity. The electrochemically regulated NiV LDH featured abundant Ni(III) sites, exhibiting an excellent electrocatalytic performance for glycerol oxidation (Fig. 8h), achieving a current density of 10 mA cm−2 at only 1.23 V vs. RHE. The E-NiV LDH achieved a high FE (94%) and high conversion rate for formates (98%), while also demonstrating good reusability after five cycles of glycerol electrolysis (Fig. 8i and j). In addition to catalyst regulation, the electrochemical interfacial microenvironment (e.g., cation effects) is another factor influencing GEOR activity. Wu et al.57 discovered that electrolyte cations can regulate the formate selectivity. The best FE of FA can be achieved at 81.3% using LiOH electrolyte on NiOOH catalyst, which was higher than that using NaOH, KOH and CsOH as electrolytes. In situ infrared reflection absorption spectroscopy (IRRAS) and DFT calculations revealed that Li+ cations significantly stabilized the aldehyde intermediates (glyceraldehyde and glycolaldehyde) and promoted C–C cleavage to form formates, thereby enhancing the selectivity for formate.
2.5 Crystal phase regulation
Due to the varying charge distributions of different atomic configurations, the electronic properties of materials are highly dependent on their crystal phase.58 Adjusting the crystal phase can alter surface adsorption properties and charge states, resulting in excellent conductivity and higher surface activity.59 Chiang's team60 used crystal facet engineering to adjust the proportion of reaction facets and control facets conducive to the reaction. Using spinel Co3O4 with dominant {001} and {111} crystal facets as a model catalyst, and they comprehensively investigated its facet dependence and catalytic performance in GEOR (Fig. 9a and b). Co3O4 micro-octahedra with highly exposed {111} facets exhibited over 60% selectivity for DHA compared to micro-cubic Co3O4 dominated by {001} facets in 0.1 M NaBi electrolyte. This may be attributed to its higher density of active cobalt ions, enabling better the adsorption and transfer of reactants. Variations in atomic arrangement can also influence the charge transfer efficiency on the electrocatalyst, and the synergy between enhanced charge transfer and preferential adsorption of glycerol favors selective production of DHA. Depending on the position of the oxidized hydroxyl group in glycerol (Fig. 9c), it can bind to active sites and undergo partial oxidation through C–H cleavage to form intermediates such as GLAD or DHA, both of which can be oxidized to GLA. Glycerol can also directly undergo C–C bond cleavage to form FA. | ||
| Fig. 9 HRTEM analysis of (a) C–Co3O4 and (b) O–Co3O4.60 (c) Proposed reaction pathway of the GEOR over O–Co3O4.60 Reproduced with permission from ref. 60. Copyright 2023, Elsevier. | ||
Chiang's team61 regulated the selectivity of glycerol oxidation products through the crystal phase modulation of oxides. The authors62 also used MnO4 as a GEOR catalyst and found that at high potentials, the Raman spectroscopy-guided transformation of α-MnO2 to δ-MnO2 in nanoneedle electrodes reduced C–C bond cleavage (Fig. 10a and b), thereby enhancing the selectivity for DHA (Fig. 10c). Building upon this foundation, Tran et al.63 further investigated the correlation between the MnO2 crystalline phases and the resulting reaction products using Raman spectroscopy. They examined three polymorphs, α-MnO2, β-MnO2, γ-MnO2, and found that all exhibited comparable selectivity toward dihydroxyacetone (DHA, ∼50%) and glyceraldehyde (GLYD, ∼40%) in 0.1 M Na2B4O7 solution (as shown in Fig. 10d). Among them, γ-MnO2 demonstrated a superior catalytic performance, which was attributed to the preferential adsorption of secondary hydroxyl groups on its surface, leading to a lower onset potential (Fig. 10e). The yield of C3 products on γ-MnO2 was 1.4-times higher than that on α-MnO2 and 2.5-times higher than that on β-MnO2 (Fig. 10f). This study revealed that the catalyst surface exhibits a stronger affinity for secondary hydroxyl groups (–OH) compared to primary hydroxyls, thereby consistently favoring the formation of DHA over GLYD. Under high anodic potentials, all the MnO2 polymorphs were observed to undergo structural transformation into low-crystallinity δ-MnO2. This phase transition is believed to suppress C–C bond cleavage, thereby enhancing the selectivity toward C3 products.
 | ||
| Fig. 10 (a) Operando Raman spectra of the catalyst at different applied potentials.62 (b) Lorentz fitting peak of the Raman spectrum of the catalyst at 1.85 V vs. RHE.62 (c) Product selectivity of the GEOR over MnO2 after running the GEOR for 3 h and its corresponding DHA yield at different applied potentials. Electrolyte: 0.1 M Na2B4O7 and 0.1 M glycerol.62 (d) Distribution of the liquid product for α-MnO2, β-MnO2, and γ-MnO2.63 (e) LSV of α-, β-, and γ-MnO2 in a 0.1 M Na2B4O7 solution with and without 0.1 M glycerol.63 (f) ECSA-normalized yield of liquid products after the 3 h GEOR using α-, β-, and γ-MnO2 at 1.85 V vs. RHE.63 Reproduced with permission from ref. 62 and 63. Copyright 2021, Elsevier, Copyright 2023, American Chemical Society. | ||
2.6 Morphology control engineering
Morphology control engineering is mainly aimed at increasing the electrochemical active area and exposing more active sites in catalysts, thus effectively improving their GEOR activity. Zhu et al.64 synthesized three-dimensional flower-like CoCu-MOF and leaf-like polyaniline (CoCu-MOF/PANI) hybrid materials using hydrothermal and in situ polymerization methods, forming a unique and stable 3D nanoflower structure. The three-dimensional flower-like CoCu-MOF featured a distinct mesoporous structure and a large specific surface area (Fig. 11a and b), which provided low-resistance pathways, increased the number of electroactive sites, and accelerated ion diffusion. The leaf-like polyaniline acted as a separation layer to prevent the agglomeration of CoCu-MOF, exhibiting excellent electrocatalytic activity, high stability, and good tolerance to poisoning in glycerol oxidation. The authors also found that when the CoCu-MOF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PANI mass ratio was 1:1, the material exhibited the highest catalytic activity for glycerol oxidation in a 1.0 mol L−1 C3H8O3 and 1.0 mol L−1 NaOH solution (Fig. 11c).
:PANI mass ratio was 1:1, the material exhibited the highest catalytic activity for glycerol oxidation in a 1.0 mol L−1 C3H8O3 and 1.0 mol L−1 NaOH solution (Fig. 11c).
 | ||
| Fig. 11 (a) N2 adsorption–desorption isotherms and (inset) partially enlarged plot and (b) corresponding pore size distribution of CoCu-MOF, PANI and CoCu-MOF/PANI.64 (c) CV curves of CoCu-MOF/PANI with different mass ratios in 0.5 mol L−1 KOH + 0.5 mol L−1 C3H8O3.64 Copyright (2020), with permission from Elsevier. Reproduced with permission from ref. 64. Copyright 2020, Elsevier. | ||
2.7 Compositing strategies
The rational integration of multiple components through composite design harnesses interfacial synergy, simultaneously boosting the catalytic activity, steering reaction pathways for selective product formation, and reinforcing structural robustness under operational conditions. Xi et al.65 prepared spherical CuO–Co3O4/NF catalysts via a solvothermal method (Fig. 12a), and electrochemical tests showed that the catalyst exhibited the best electrocatalytic activity at a Cu:Co molar ratio of 1:1. When copper ions were incorporated in the cobalt oxide lattice, they disrupted the ordered structure, causing lattice distortion; the more copper ions incorporated, the more oxygen vacancies may be generated. Compared to Co3O4/NF, the Raman spectrum of CuO–Co3O4/NF exhibited red-shifted peaks, reduced peak intensity, and broadened peak shapes, indicating possible lattice distortion or defects (Fig. 12b and c). The abundant oxygen vacancies in CuO–Co3O4/NF improved the electron transport efficiency, exposed more active sites, and reduced the binding energy of adsorbed intermediates, thereby accelerating the mass transfer of the reactants. CuO–Co3O4/NF achieved a current density of 10 mA cm−2 at just 1.13 V vs. RHE, outperforming other single-metal catalysts such as CuO and Co3O4. The electrolysis products consisted of formic acid, glycolic acid, and glyceric acid, with formic acid showing the highest selectivity (90.3%). Du et al.66 used a low-temperature sulfidation method to in situ grow Cu nanoflake arrays on copper mesh, uniformly decorating Cu nanoparticles on the Cu nanoflakes via disproportionation reactions. The prepared Cu–CuS/BM catalyst exhibited good GEOR activity and demonstrated excellent stability and catalytic performance in the direct oxidation of glycerol to formates (Fig. 12d and e). At the optimal potential of 1.45 V vs. RHE, the selectivity for formates reached as high as 86.0%, with an FE of 90.4%. Density functional theory (DFT) calculations and X-ray photoelectron spectroscopy (XPS) analysis confirmed that the combination of Cu nanoparticles and CuS reduced the electron cloud density of Cu, facilitating the adsorption of glycerol molecules and accelerating the GEOR process. Rizk et al.67 deposited dendritic Cu/Cu2O foam onto smooth Cu electrodes and used them as efficient catalysts for GEOR. After incorporating Cl− into the porous Cu/Cu2O film, its structure and shape underwent significant changes, resulting in cubic-structured copper particles (Fig. 12f). The results showed that the electrochemical active surface area (ECSA) of the catalyst increased by 2.9 times, effectively enhancing the stability and glycerol oxidation kinetics of the catalyst. Huang et al.68 developed a MnO2–CuO/CF composite electrode for GEOR via in situ hydrothermal growth on copper foam. This catalyst achieved a current density of 10.7 mA cm−2 at 1.3 V vs. RHE, with an average DHA selectivity of up to 60% during a 5 h test. This was attributed to the adsorption energy of DHA being lower on MnO2(100) than on CuO(111), confirming that DHA was more easily desorbed from MnO2(100) (Fig. 12g). Additionally, the overlapping configuration formed by combining CuO with MnO2 helps regulate the electronic density of MnO2 (Fig. 12h). MnO2 was affected by the inner CuO, resulting in a decrease in electron density and an increase in the average oxidation state (AOS) value, which prevented the further oxidation of the reaction and improved the selectivity for DHA.
 | ||
| Fig. 12 (a) SEM images of CuO–Co3O4.65 (b) Raman spectra of CuO–Co3O4/NF.65 XPS spectra of (c) O 1s for CuO–Co3O4/NF before and after reaction, Co3O4/NF and CuO/NF.65 (d) LSV curves of Cu–CuS/BM in 0.1 M KOH, the BM, CuS/BM, Cu–BM, and Cu–CuS/BM in 0.1 M KOH + 0.1 M GLY solution.66 (e) Linear relationship between the current density and the scan rate to determine the Cdl values.66 (f) SEM image of the film at high chloride concentration showing the cubic structure.67 (g) Adsorption energy of CuO(111) by DHA.68 (h) EDS element mapping images of MnO2–CuO/CF.68 (i) Normalized valence-band spectra of NiMoO4/NF and Ni–MoO2/NF.69 Reproduced with permission from ref. 65–69. Copyright 2023, Elsevier, Copyright 2022, American Chemical Society, Copyright 2020, Wiley-VCH, Copyright 2024, Elsevier, Copyright 2025, Elsevier. | ||
Xiong et al.69 successfully synthesized an Ni nanoparticle-anchored MoO2 nanorod catalyst (Ni–MoO2/NF) via a hydrothermal-annealing reduction approach. At a constant current density of 100 mA cm−2, the catalyst achieved a glycerol conversion rate of 57.5% and formate selectivity of approximately 80% after 4 h of continuous electrolysis. The synergistic interaction between Ni and MoO2 effectively modulated the electronic structure, accelerating electron transfer and promoting the in situ formation of the active NiOOH species. Valence band spectroscopy analysis revealed that the d-band center of Ni–MoO2/NF was located at −2.31 eV, which is significantly closer to the Fermi level compared to that of NiMoO4/NF (−3.01 eV) (Fig. 12i). This shift in the d-band center enhanced the adsorption affinity of Ni–MoO2/NF toward glycerol and *OH intermediates, thereby facilitating C–C bond cleavage within the glycerol molecule. Consequently, the selectivity toward formate was markedly improved.
Wolfgang Schuhmann and co-workers developed an integrated electrolysis system comprised of an Ni foam-supported NixB composite anode for glycerol oxidation and a BiOBr-modified gas-diffusion cathode for CO2 reduction (CO2RR).70 This paired reactor achieved a remarkable total formate faradaic efficiency of 141% (45% from anode + 96% from cathode) at an industrially relevant current density of 200 mA cm−2. Notably, the electrode selectivity in the paired configuration remained comparable to half-cell measurements (Fig. 13). This work demonstrates that simultaneous formate production via paired electrolysis at unprecedented current densities represents an effective strategy to enhance the co-production of value-added chemicals through electrochemical CO2RR.
 | ||
| Fig. 13 Schematic representation of an OER‖CO2RR reactor (a) and GOR‖CO2RR paired electrolysis reactor (b), and picture of a used paired electrolyzer (c).70 Reproduced with permission from ref. 70. Copyright 2023, Wiley-VCH. | ||
3 The unique contribution of this review
This review systematically summarizes the recent research advances in non-precious metal catalysts for the GEOR and proposes innovative insights addressing key challenges in this field. The primary contributions are reflected in the following three aspects.Firstly, we establish a comprehensive analytical framework for the “composition–structure–performance” relationship of non-precious metal catalysts. By systematically categorizing electronic structure modulation strategies (e.g., heteroatom doping, defect engineering, heterostructure construction, morphology control, and crystal phase regulation) in Ni-, Co-, Cu-, and Mn-based catalysts, we elucidate their quantitative structure–activity relationship between the electronic density of states of active sites and the energy barrier for C–C bond cleavage. This addresses the current research limitation of focusing predominantly on individual material systems.
Secondly, we introduce the perspective of “dynamic catalytic mechanisms”. Unlike the static active sites of traditional noble metal catalysts, non-precious metal catalysts often undergo in situ oxidative reconstruction during the reaction, forming high-valent active species (e.g., NiOOH) or active hydroxyl groups (OH*). This review integrates experimental characterization and theoretical computational evidence to clarify the decisive role of this dynamic evolution in enhancing the selectivity of glycerol oxidation, providing new design principles for adaptive catalysts.
Finally, we evaluate the performance of non-precious metal catalysts in proton exchange membrane electrolyzers, offering critical guidance for bridging the gap between laboratory research and industrial-scale applications.
4 Summary and outlook
In summary, this paper introduced and reviewed the recent progress in Ni-, Co-, Cu-, and Mn-based catalysts for GEOR, discussing in detail how catalyst synthesis strategies (doping engineering, alloying strategies, defect engineering, heterostructure construction, morphology engineering, and phase regulation) improved the GEOR performance. Currently, alloying remains an effective strategy to tune the physicochemical properties of materials by introducing specific elements, enabling synergistic effects among components to enhance their catalytic activity. Similar to alloying strategies, doping engineering aims to overcome the limitations of single materials by adjusting the electronic structure and coordination environment of compounds, thereby increasing the number of active sites and optimizing the adsorption/desorption of the intermediates to improve the electrocatalytic activity. Defect engineering can expose more active sites by introducing vacancies and modifying the surface morphology, thereby enhancing the utilization of the internal active sites. Moreover, these defect sites facilitate rapid charge transfer and improve the conductivity. However, excessive vacancies may disrupt the crystal structure, negatively affecting the electrocatalytic performance. Additionally, optimizing the structure and enhancing the stability of catalysts through heterostructure construction is another effective approach to improve their GEOR performance. Crystal phase regulation strategies optimize the adsorption performance of catalysts by altering their crystal phase structures, thereby effectively improving the GEOR reaction kinetics. However, achieving precise quantitative control using this strategy remains a major challenge in catalyst preparation. These typical catalyst design strategies have effectively modulated various non-noble metal materials, enhancing their activity, stability, and product selectivity in GEOR. This clearly reflects the progress made in recent years in the optimization of GEOR catalysts and the modulation of their GEOR activity, but there are still many challenges encountered.Thus, we propose the following outlook for addressing these challenges.
(1) Transition metal sulfides, nitrides, etc., often undergo partial or complete reconstruction after electrocatalytic glycerol oxidation. The structural evolution process and identification of the true active sites in GEOR catalysts require further elucidation through in situ spectroscopy and other in situ characterization techniques.
(2) Investigating the electrochemical stability of non-metal catalysts. The current literature primarily focuses on enhancing the GEOR activity of catalysts, while effective strategies for improving their stability remain under explored. The deactivation of catalysts in the GEOR may be caused by the structural collapse of the catalyst or poisoning by the substrate/intermediate products. Therefore, exploring the deactivation mechanism and improving the stability of non-metal catalysts still need further research.
(3) Regulating product selectivity. At present, the main product of non-noble metal GEOR is formic acid, while other products such as glyceraldehyde, glyceric acid, and lactic acid remain insufficiently explored.
(4) Developing non-noble metal electrocatalysts for GEOR in acidic or neutral electrolyte. Currently, most GEOR electrolytes are alkaline, requiring acidification and neutralization during product separation and purification, which increases the acid consumption and incurs additional costs for salt waste treatment.
(5) Coupling GEOR with cathodic reduction reactions. Currently, the cathodic reduction reaction in GEOR is mostly the hydrogen evolution reaction (HER), which can be replaced by carbon dioxide reduction (CO2RR) or nitrogen reduction (NRR). These coupled electrocatalytic systems offer new perspectives for the practical application of GEOR.
Data availability
No primary research results, software or code has been included and no new data were generated or analysed as part of this review.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by the Natural Science Foundation of Liaoning Province (2023-BSBA-251), Natural Science Foundation of Inner Mongolia Autonomous Region of China (2024QN08039) and Basic Scientific Research Project of Higher Education Department of Liaoning Province (LJ212410142153).References
- S. Budžaki, G. Miljić, M. Tišma, S. Sundaram and V. Hessel, Is there a future for enzymatic biodiesel industrial production in microreactors?, Appl. Energy, 2017, 201, 124–134 CrossRef.
- I. Ambat, V. Srivastava and M. Sillanpää, Recent advancement in biodiesel production methodologies using various feedstock: A review, Renewable Sustainable Energy Rev., 2018, 90, 356–369 CrossRef CAS.
- A. Almena, L. Bueno, M. Díez and M. Mariano, Integrated biodiesel facilities: Review of glycerol-based production of fuels and chemicals, Clean Technol. Environ., 2018, 20, 1639–1661 CrossRef CAS.
- X. Zhang, D. Zhou, X. Wang, J. Zhou, J. Li, M. Zhang, Y. Shen, H. Chu and Y. Qu, Overcoming the deactivation of Pt/CNT by introducing CeO2 for selective base-free glycerol-to-glyceric acid oxidation, ACS Catal., 2020, 10, 3832–3837 CrossRef CAS.
- J. Li, Z. Li, Z. Zheng, X. Zhang, H. Zhang, H. Wei and H. Chu, Tuning the product selectivity toward the high yield of glyceric acid in Pt-CeO2/CNT electrocatalyzed oxidation of glycerol, ChemCatChem, 2022, 14, e202200509 CrossRef CAS.
- L. Clarizia, I. D. Somma, L. Onotri, R. Andreozzi and R. Marotta, Kinetic modeling of hydrogen generation over nano-Cu (s)/TiO2 catalyst through photoreforming of alcohols, Catal. Today, 2017, 281, 117–123 CrossRef CAS.
- E. A. Kozlova, A. Y. Kurenkova, E. Y. Gerasimov, N. V. Gromov, T. B. Medvedeva, A. A. Saraev and V. V. Kaichev, Comparative study of photoreforming of glycerol on Pt/TiO2 and CuOx/TiO2 photocatalysts under UV light, Mater. Lett., 2021, 283, 128901 CrossRef CAS.
- M. Deng, W. Yang, K. Xiong, H. Zhang, J. Chen, M. Yang and X. Gan, High-valence molybdenum doped Ni(OH)2 nanosheets enhancing hydrogen production and glycerol oxidation to formic acid, Int. J. Hydrogen Energy, 2024, 59, 128901 Search PubMed.
- M. H. Haider, N. F. Dummer, D. W. Knight, R. L. Jenkins, M. Howard, J. Moulijn, S. H. Taylor and G. J. Hutchings, Efficient green methanol synthesis from glycerol, Nat. Chem., 2015, 7, 1028–1032 CrossRef CAS PubMed.
- Y. Fernández, A. Arenillas, M. A. Díez, J. J. Pis and J. A. Menéndez, Pyrolysis of glycerol over activated carbons for syngas production, J. Anal. Appl. Pyrolysis, 2009, 84, 145–150 CrossRef.
- Y. Gu, A. Azzouzi, Y. Pouilloux, F. Jérôme and J. Barrault, Heterogeneously catalyzed etherification of glycerol: new pathways for transformation of glycerol to more valuable chemicals, Green Chem., 2008, 10, 164–167 RSC.
- M. Malyaadri, K. Jagadeeswaraiah, P. S. S. Prasad and N. Lingaiah, Synthesis of glycerol carbonate by transesterification of glycerol with dimethyl carbonate over Mg/Al/Zr catalysts, Appl. Catal., A, 2011, 401, 153–157 CrossRef CAS.
- M. Guschakowski and U. Schröder, Direct and indirect electrooxidation of glycerol to value-added products, ChemSusChem, 2021, 14, 5216–5225 CrossRef CAS PubMed.
- Z. An, H. Ma, H. Han, Z. Huang, Y. Jiang, W. Wang, Y. Zhu, H. Song, X. Shu, X. Xiang and J. He, Insights into the multiple synergies of supports in the selective oxidation of glycerol to dihydroxyacetone: layered double hydroxide supported Au, ACS Catal., 2020, 10, 12437–12453 CrossRef CAS.
- H. Sheng, A. N. Janes, R. D. Ross, H. Hofstetter, K. Lee, J. R. Schmidt and S. Jin, Linear paired electrochemical valorization of glycerol enabled by the electro-Fenton process using a stable NiSe2 cathode, Nat. Catal., 2022, 5, 716–725 CrossRef CAS.
- A. M. Verma, L. Laverdure, M. M. Melander and K. Honkala, Mechanistic origins of the pH dependency in Au-catalyzed glycerol electro-oxidation: insight from first-principles calculations, ACS Catal., 2021, 12, 662–675 CrossRef.
- J. Li, K. Jiang, S. Bai, C. Guan, H. Wei and H. Chu, High productivity of tartronate from electrocatalytic oxidation of high concentration glycerol through facilitating the intermediate conversion, Appl. Catal., B, 2022, 317, 121784 CrossRef CAS.
- D. Kim, L. Oh, Y. Tan, H. Song, H. Kim and J. Oh, Enhancing glycerol conversion and selectivity toward glycolic acid via precise nanostructuring of electrocatalysts, ACS Catal., 2021, 11, 14926–14931 CrossRef CAS.
- I. Terekhina, J. White, A. Cornell and M. Johnsson, Electrocatalytic oxidation of glycerol to value-added compounds on Pd nanocrystals, ACS Appl. Nano Mater., 2023, 6, 11211–11220 CrossRef CAS.
- T. Yang and Y. Shen, Coupling glycerol conversion with hydrogen production using alloyed electrocatalysts, Langmuir, 2023, 39, 12855–12864 CrossRef CAS PubMed.
- B. Katryniok, H. Kimura, E. Skrzyńska, J. Girardon, P. Fongarland, M. Capron, R. Ducoulombier, N. Mimura, S. Paul and F. Dumeignil, Selective catalytic oxidation of glycerol: perspectives for high value chemicals, Green Chem., 2011, 13, 1960–1979 RSC.
- D. M. Morales, D. Jambrec, M. A. Kazakova, M. Braun, N. Sikdar, A. Koul, A. Brix, S. Seisel, C. Andronescu and W. Schuhmann, Electrocatalytic conversion of glycerol to oxalate on Ni oxide nanoparticles-modified oxidized multiwalled carbon nanotubes, ACS Catal., 2022, 12, 982–992 CrossRef CAS.
- X. Han, H. Sheng, C. Yu, T. W. Walker, G. W. Huber, J. Qiu and S. Jin, Electrocatalytic oxidation of glycerol to formic acid by CuCo2O4 spinel oxide nanostructure catalysts, ACS Catal., 2020, 10, 6741–6752 CrossRef CAS.
- T. Vo, P. Ho and C. Chiang, Operando mechanistic studies of selective oxidation of glycerol to dihydroxyacetone over amorphous cobalt oxide, Appl. Catal., B, 2022, 300, 120723 CrossRef CAS.
- X. Huang, Y. Zou and J. Jiang, Electrochemical oxidation of glycerol to dihydroxyacetone in borate buffer: enhancing activity and selectivity by borate-polyol coordination chemistry, ACS Sustainable Chem. Eng., 2021, 9, 14470–14479 CrossRef CAS.
- J. Zhang, Y. Shen and H. Li, Electrolysis of glycerol by non-noble metal hydroxides and oxides, ACS Appl. Energy Mater., 2023, 6, 5508–5518 CrossRef CAS.
- Z. Xia, C. Ma, Y. Fan, Y. Lu, Y. Huang, Y. Pan, Y. Wu, Q. Luo, Y. He, C. Dong, S. Wang and Y. Zou, Vacancy optimized coordination on nickel oxide for selective electrocatalytic oxidation of glycerol, ACS Catal., 2024, 14, 1930–1938 CrossRef CAS.
- L. Wu, Q. Wu, Y. Han, D. Zhang, R. Zhang, N. Song, X. Wu, J. Zeng, P. Yuan, J. Chen, A. Du, K. Huang and X. Yao, Strengthening the synergy between oxygen vacancies in electrocatalysts for efficient glycerol electrooxidation, Adv. Mater., 2024, 36, 2401857 CrossRef CAS PubMed.
- Q. Yin, X. Xie, L. Xu and S. Geng, Boosting electrocatalytic glycerol oxidation reaction to formate based on Mn-doped Ni7P3, J. Alloys Compd., 2025, 1022, 179867 CrossRef CAS.
- R. K. P. Purushothaman, J. van Haveren, D. S. van Es, I. Melián-Cabrera, J. D. Meeldijk and H. J. Heeres, An efficient one pot conversion of glycerol to lactic acid using bimetallic gold-platinum catalysts on a nanocrystalline CeO2 support, Appl. Catal., B, 2014, 147, 92–100 CrossRef CAS.
- C. Zhang, T. Wang, X. Liu and Y. Ding, Selective oxidation of glycerol to lactic acid over activated carbon supported Pt catalyst in alkaline solution, Chin. J. Catal., 2016, 37, 502–509 CrossRef CAS.
- C. H. Lam, A. J. Bloomfield and P. T. Anastas, A switchable route to valuable commodity chemicals from glycerol via electrocatalytic oxidation with an earth abundant metal oxidation catalyst, Green Chem., 2017, 19, 1958–1968 RSC.
- V. L. Oliveira, C. Morais, K. Servat, T. W. Napporn, G. Tremiliosi-Filho and K. B. Kokoh, Glycerol oxidation on nickel based nanocatalysts in alkaline medium-Identification of the reaction products, J. Electroanal. Chem., 2013, 703, 56–62 CrossRef CAS.
- V. L. Oliveira, C. Morais, K. Servat, T. W. Napporn, G. Tremiliosi-Filho and K. B. Kokoh, Studies of the reaction products resulted from glycerol electrooxidation on Ni-based materials in alkaline medium, Electrochim. Acta, 2014, 117, 255–262 CrossRef CAS.
- B. Habibi and N. Delnavaz, Electrooxidation of glycerol on nickel and nickel alloy(Ni-Cu and Ni-Co) nanoparticles in alkaline media, RSC Adv., 2016, 6, 31797–31806 RSC.
- M. E. Ghaith, M. G. A. El-Moghny, G. A. El-Nagar, H. H. Alalawy, M. E. El-Shakre and M. S. El-Deab, Tailor-designed binary Ni-Cu nano dendrites decorated 3D-carbon felts for efficient glycerol electrooxidation, RSC Adv., 2023, 13, 895–905 RSC.
- Q. Zhang, X. Zhang, B. Liu, P. Jing, X. Xu, H. Hao, R. Gao and J. Zhang, Active hydroxyl-mediated preferential cleavage of carbon-carbon bonds in electrocatalytic glycerol oxidation, Angew. Chem., Int. Ed., 2025, 64, e202420942 CrossRef CAS PubMed.
- L. Shen, L. Sun, M. Douthwaite, O. Akdim, S. Taylor and G. J. Hutchings, Hollow Au1Cu1 (111) bimetallic catalyst promotes the selective electrochemical conversion of glycerol into glycolic acid, ACS Catal., 2024, 14, 11343–11351 CrossRef CAS.
- L. Fan, Y. Ji, G. Wang, J. Chen, K. Chen, X. Liu and Z. Wen, High entropy alloy electrocatalytic electrode toward alkaline glycerol valorization coupling with acidic hydrogen production, J. Am. Chem. Soc., 2022, 144, 7224–7235 CrossRef CAS PubMed.
- H. Yao, Y. Wang, Y. Zheng, X. Yu, J. Ge, Y. Zhu and X. Guo, High-entropy selenides: A new platform for highly selective oxidation of glycerol to formate and energy-saving hydrogen evolution in alkali-acid hybrid electrolytic cell, Nano Res., 2023, 16, 10832–10839 CrossRef CAS.
- M. S. E. Houache, K. Hughes, R. Safari, G. A. Botton and E. A. Baranova, Modification of nickel surfaces by bismuth: effect on electrochemical activity and selectivity toward glycerol, ACS Appl. Mater. Interfaces, 2020, 12, 15095–15107 CrossRef CAS PubMed.
- M. K. Goetz, E. Usman and K. Choi, Understanding and suppressing C-C cleavage during glycerol oxidation for C3 chemical production, ACS Catal., 2023, 13, 15758–15769 CrossRef CAS.
- Y. Wang, R. Ge, X. Liu, J. Li and H. Duan, An anion leaching strategy towards metal oxyhydroxides synthesis for electrocatalytic oxidation of glycerol, Acta Phys.-Chim. Sin., 2024, 40, 48–51 Search PubMed.
- Y. Wang, Y. Zhu, Z. Xie, S. Xu, M. Xu, Z. Li, L. Ma, R. Ge, H. Zhou, Z. Li, X. Kong, L. Zheng, J. Zhou and H. Duan, Efficient electrocatalytic oxidation of glycerol via promoted OH* generation over single-atom-bismuth-doped spinel Co3O4, ACS Catal., 2022, 12, 12432–12443 CrossRef CAS.
- W. Luo, H. Tian, Q. Li, G. Meng, Z. Chang, C. Chen, R. Shen, X. Yu, L. Zhu, F. Kong, X. Cui and J. Shi, Controllable electron distribution reconstruction of spinel NiCo2O4 boosting glycerol oxidation at elevated current density, Adv. Funct. Mater., 2024, 34, 2306995 CrossRef CAS.
- Y. Xu, T. Liu, K. Shi, H. Yu, K. Deng, Z. Wang, X. Li, L. Wang and H. Wang, Iridium-incorporated Co3O4 with lattice expansion for energy-efficient green hydrogen production coupled with glycerol valorization, Chem. Commun., 2023, 59, 1817–1820 RSC.
- Z. Yu, K. Chen, Y. Tong, C. Lin, X. Zheng and P. Chen, Phosphorus-modulated cobalt nanosheets with confined metal defects for enhanced kinetics in nitrite-glycerol co-electrolysis, Adv. Funct. Mater., 2025, 35, 2506735 CrossRef.
- Z. Xia, C. Ma, Y. Fan, Y. Lu, Y. Huang, Y. Pan, Y. Wu, Q. Luo, Y. He, C. Dong, S. Wang and Y. Zou, Vacancy optimized coordination on nickel oxide for selective electrocatalytic oxidation of glycerol, ACS Catal., 2024, 14, 1930–1938 CrossRef CAS.
- L. Wu, Q. Wu, Y. Han, D. Zhang, R. Zhang, N. Song, X. Wu, J. Zeng, P. Yuan, J. Chen, A. Du, K. Huang and X. Yao, Strengthening the synergy between oxygen vacancies in electrocatalysts for efficient glycerol electrooxidation, Adv. Mater., 2024, 2401857 CrossRef CAS PubMed.
- Y. Fang, C. Dai, X. Liu, Y. Wang, C. Ju, S. He, R. Shi, Y. Liu, J. Zhang, Y. Zhu and J. Wang, Sulfur-doped manganese-cobalt hydroxide with promoted surface reconstruction for glycerol electrooxidation assisted hydrogen production, Nano Energy, 2024, 127, 109754 CrossRef CAS.
- X. Feng, Y. Shi, C. Wang, J. Yang, J. Qin and X. Li, Dual vacancies engineering on cobalt-based materials to synergistic enhance electrocatalytic oxidation of urea and glycerol, Int. J. Hydrogen Energy, 2024, 71, 1230–1241 CrossRef CAS.
- Y. Jia, L. Zhang, G. Gao, H. Chen, B. Wang, J. Zhou, M. T. Soo, M. Hong, X. Yan, G. Qian, J. Zou, A. Du and X. Yao, A heterostructure coupling of exfoliated Ni-Fe hydroxide nanosheet and defective graphene as a bifunctional electrocatalyst for overall water splitting, Adv. Mater., 2017, 29, 1700017 CrossRef PubMed.
- L. Xu, Y. Yang, C. Li, R. Ning, J. Ma, M. Yao, S. Geng and F. Liu, Unveiling the mechanism of electrocatalytic oxidation of glycerol by in-situ electrochemical spectroscopy, Chem. Eng. J., 2024, 481, 148304 CrossRef CAS.
- Y. Feng, X. He, M. Cheng, Y. Zhu, W. Wang, Y. Zhang, H. Zhang and G. Zhang, Selective adsorption behavior modulation on nickel selenide by heteroatom implantation and heterointerface construction achieves efficient Co-production of H2 and formate, Small, 2023, 19, 2301986 CrossRef CAS PubMed.
- X. Liu, Z. Fang, X. Teng, Y. Niu, S. Gong, W. Chen, T. J. Meyer and Z. Chen, Paired formate and H2 productions via efficient bifunctional Ni-Mo nitride nanowire electrocatalysts, J. Energy Chem., 2022, 72, 432–441 CrossRef CAS.
- L. Dong, G. Chang, X. Yu, Y. Feng and X. Yao, Regulating Ni site in NiV LDH for efficient electrocatalytic production of formate and hydrogen by glycerol electrolysis, Rare Met., 2022, 41, 1583–1594 CrossRef CAS.
- J. Wu, J. Li, Y. Li, X. Ma, W. Zhang, Y. Hao, W. Cai, Z. Liu and M. Gong, Steering the glycerol electro-reforming selectivity via cation-intermediate interactions, Angew. Chem., Int. Ed., 2022, 61, e202113362 CrossRef CAS PubMed.
- R. S. Berry and B. M. Smirnov, Phase transitions in various kinds of clusters, Phys.-Usp., 2009, 52, 137–164 CrossRef CAS.
- P. Chen, K. Xu, S. Tao, T. Zhou, Y. Tong, H. Ding, L. Zhang, W. Chu, C. Wu and Y. Xie, Phase-transformation engineering in cobalt diselenide realizing enhanced catalytic activity for hydrogen evolution in an alkaline medium, Adv. Mater., 2016, 28, 7527–7532 CrossRef CAS PubMed.
- T. G. Vo, P. Y. Tsai and C. Y. Chiang, Tuning selectivity and activity of the electrochemical glycerol oxidation reaction by manipulating morphology and exposed facet of spinel cobalt oxides, J. Catal., 2023, 424, 64–73 CrossRef CAS.
- T. G. Vo, P. Y Tsai and C. Y. Chiang, Tuning selectivity and activity of the electrochemical glycerol oxidation reaction by manipulating morphology and exposed facet of spinel cobalt oxides, J. Catal., 2023, 424, 64–73 CrossRef CAS.
- G. S. Tran, T. G. Vo and C. Y. Chiang, Earth-abundant manganese oxide nanoneedle as highly efficient electrocatalyst for selective glycerol electro-oxidation to dihydroxyacetone, J. Catal., 2021, 404, 139–148 CrossRef CAS.
- G. S. Tran, T. G Vo and C. Y. Chiang, Operando revealing the crystal phase transformation and electrocatalytic activity correlation of MnO2 toward glycerol electrooxidation, ACS Appl. Mater. Interfaces, 2023, 15, 22662–22671 CrossRef CAS PubMed.
- S. Zhu, A. Xie, X. Tao, J. Zhang, B. Wei, Z. Liu, Y. Tao and S. Luo, Enhanced electrocatalytic performance of CoCu-MOF/polyaniline for glycerol oxidation, Electroanal. Chem., 2020, 857, 113748 CrossRef CAS.
- Z. Xi, H. Zhou, Y. Liu and C. Xu, CuO-Co3O4 nanoparticle catalysts rich in oxygen vacancies toward upgrading anodic electro-oxidation of glycerol, Electrochim. Acta, 2023, 470, 143285 CrossRef CAS.
- J. Du, Y. Qin, T. Dou, J. Ge, Y. Wang, X. Zhao, F. Zhang and X. Lei, Copper nanoparticles dotted on copper sulfide nanosheets for selective electrocatalytic oxidation of glycerol to formate, ACS Appl. Nano Mater., 2022, 5, 10174–10182 CrossRef CAS.
- M. R. Rizk, M. G. Moghny, G. A. Nagar, A. A. Mazhar and M. S. El-Deab, Tailor-designed porous catalysts: nickel-doped Cu/Cu2O foams for efficient glycerol electro-oxidation, ChemElectroChem, 2020, 7, 951–958 CrossRef CAS.
- Z. Huang, H. Ren, J. Guo, Y. Tang, D. Ye, J. Zhang and H. Zhao, High DHA selectivity and low-cost electrode for glycerol oxidation: CuO regulates MnO2 electron density to promote DHA desorption, Appl. Catal., B, 2024, 351, 123986 CrossRef CAS.
- Y. Gao, Z. Li, M. Deng, W. Yang, M. Yang, X. Gan, K. Xiong and H. Zhang, Anchoring Ni nanoparticles on MoO2 nanorods for enhancing electrocatalytic oxidation of glycerol to formate, Colloids Surf., A, 2025, 715, 136661 CrossRef CAS.
- J. R. C. Junqueira, D. Das, A. C. Brix, S. Dieckhöfer, J. Weidner, X. Wang, J. Shi and W. Schuhmann, Simultaneous anodic and cathodic formate production in a paired electrolyzer by CO2 reduction and glycerol oxidation, ChemSusChem, 2023, 16, e202202349 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |