
Oxidative degradation of sequence-defined peptoid oligomers†
Hattie C.
Schunk
 ab,
Mariah J.
Austin
a,
Bradley Z.
Taha
a,
Matthew S.
McClellan
a,
Laura J.
Suggs
b and
Adrianne M.
Rosales‡
*a
ab,
Mariah J.
Austin
a,
Bradley Z.
Taha
a,
Matthew S.
McClellan
a,
Laura J.
Suggs
b and
Adrianne M.
Rosales‡
*a
aMcKetta Department of Chemical Engineering, University of Texas at Austin, Austin, TX 78712, USA. E-mail: arosales@che.utexas.edu
bDepartment of Biomedical Engineering, University of Texas at Austin, Austin, TX 78712, USA
First published on 28th September 2022
Abstract
Due to their N-substitution, peptoids are generally regarded as resistant to biological degradation, such as enzymatic and hydrolytic mechanisms. This stability is an especially attractive feature for therapeutic development and is a selling point of many previous biological studies. However, another key mode of degradation remains to be fully explored, namely oxidative degradation mediated by reactive oxygen and nitrogen species (ROS/RNS). ROS and RNS are biologically relevant in numerous contexts where biomaterials may be present. Thus, improving understanding of peptoid oxidative susceptibility is crucial to exploit their full potential in the biomaterials field, where an oxidatively-labile but enzymatically stable molecule can offer attractive properties. Toward this end, we demonstrate a fundamental characterization of sequence-defined peptoid chains in the presence of chemically generated ROS, as compared to ROS-susceptible peptides such as proline and lysine oligomers. Lysine oligomers showed the fastest degradation rates to ROS and the enzyme trypsin. Peptoids degraded in metal catalyzed oxidation conditions at rates on par with poly(prolines), while maintaining resistance to enzymatic degradation. Furthermore, lysine-containing peptide–peptoid hybrid molecules showed tunability in both ROS-mediated and enzyme-mediated degradation, with rates intermediate to lysine and peptoid oligomers. When lysine-mimetic side-chains were incorporated into a peptoid backbone, the rate of degradation matched that of the lysine peptide oligomers, but remained resistant to enzymatic degradation. These results expand understanding of peptoid degradation to oxidative and enzymatic mechanisms, and demonstrate the potential for peptoid incorporation into materials where selectivity towards oxidative degradation is necessary, or directed enzymatic susceptibility is desired.
Design, System, ApplicationHere, we have explored molecular design principles of a series of peptide, peptoid, and peptide–peptoid hybrid oligomers for oxidative degradation to chemically generated reactive oxygen species (ROS). Taking inspiration from the tertiary amide structure of poly(proline), an established ROS-sensitive molecule, we designed a series of peptoids (N-substituted glycines) with various side chains for oxidative degradation. Specifically, using liquid chromatography and fluorescence tracking, we demonstrate that oxidative degradation rates can be modulated by both monomer identity and configuration. Lysine sidechains proved to be especially oxidatively susceptible. In peptide oligomers of lysine, the molecule was quickly degraded by both ROS and trypsin; however, lysine-mimetic peptoid oligomers enabled comparable oxidative susceptibility while inhibiting enzymatic degradation. This proteolytic resistance of peptoids is advantageous for biorecognition in cases where targeting biological species in complex mixtures is desired, such as developing ROS-selective biomaterials as responsive therapeutic materials or biosensors. Furthermore, oligomers with both peptide and peptoid residues afforded a straightforward way to direct enzymatic degradation by trypsin, thus achieving an adaptable degradative response to multiple biological stimuli. |
Introduction
Reactive oxygen and nitrogen species (ROS/RNS), such as H2O2 and NO, can lead to oxidative degradation of a variety of molecules, including peptides, proteins, and polymers.1–3 Through these processes, ROS contributes to the development of oxidative stress, especially during disease and inflammation.4 As a result, numerous ROS-degradable peptides and proteins have been exploited as responsive therapeutic materials5 or sensing oligomers to report cell phenotype.6,7 While these biomolecules offer a route to ROS-responsive biomaterials, cross-reactivity (i.e., degradation by other biological species such as enzymes) remains an issue. Ideally, ROS-sensitive biomaterials would resist degradation by other biological mechanisms and offer a means of fine-tuning control of degradation rate. For example, poly(prolines) have been explored as ROS-sensitive materials with good enzymatic stability;8–10 however, their loose helical chain structures often complicate synthesis and solubility. Furthermore, their synthetic and structural limitations restrict further functionalization, making it difficult to tune biomaterial properties or add side-chain moieties for specific interactions with biological systems. D-Amino acids, such as poly(D-lysine), have also been proposed as alternative ROS-sensitive biomaterials because they are not recognized by most proteases.11 However, there is literature reporting biodegradation of D-lysine by pancreatic enzyme extracts,12 thus limiting their use in vivo. Here, we propose to address these constraints by using peptoids (N-substituted glycines) as a model system.Peptoids feature an N-substituted polyamide backbone, which eliminates the backbone chirality and hydrogen bonding donors seen in peptides (Fig. 1A). Peptoids can be synthesized using ring-opening polymerizations in solution13 or stepwise submonomer methods, which allow for exact sequence control and monodispersity.14 Their chemical synthesis is even amenable to direct integration with peptide solid-phase methods, allowing for the generation of peptide–peptoid (i.e., “peptomer”)15 hybrid molecules. Additionally, peptoids use commercially available primary amines, which means that they can access a large bio-orthogonal chemical diversity and properties similar to those of other synthetic polymers.16
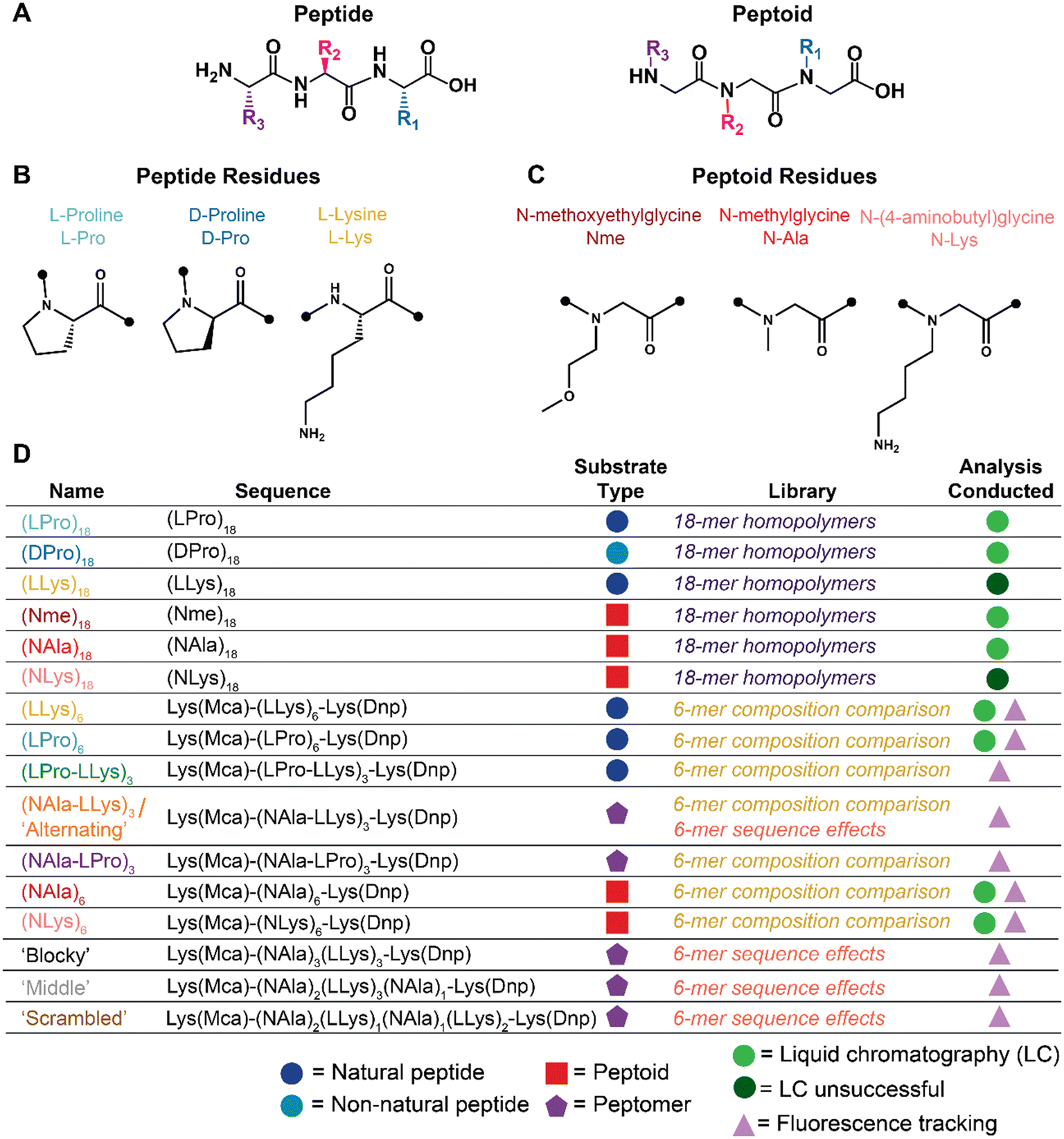 | ||
| Fig. 1 Summary schematic of peptide and peptoid molecules explored for oxidative and enzymatic degradation. A) Peptide vs. peptoid chemical structure depicted with representative trimers. B) Peptide and C) peptoid residues explored for oxidative and enzymatic degradation. D) Name, sequence, substrate classification, applicable library of investigation, and analyses conducted of all oligomers investigated. Full chemical structures, MALDI-TOF spectra, and LC-MS chromatographs for each molecule are included in Fig. S1–S5.† | ||
Given their polyamide backbone and structural similarity to peptides, peptoids are a unique class of materials that lie in a converging area between synthetic polymers and biological materials.17,18 As a result, peptoids have been used in biologically motivated applications,19 including drug discovery, therapeutics, diagnostics, and antifouling surfaces.20 It is well-established that peptoids are resistant to certain modes of biological degradation, such as hydrolytic and enzymatic mechanisms, due to their N-substitution.21,22 As a result, peptoids are generally regarded as being inherently bio-stable. However, stability of biomaterials depends not only on resistance to hydrolysis and proteolysis, but to degradation by other species such as ROS and RNS as well.7 Despite their structural similarity, peptoid reactivity to oxidative stimuli is far less explored than that of peptides. Thus, expanding fundamental characterization of peptoid susceptibility towards species such as ROS is essential to capitalizing on their full potential as bio-responsive materials.
As mentioned above, the oxidative susceptibility of the only N-substituted amino acid, proline, is known to lead to peptide backbone degradation. Poly(proline) degradability has been leveraged for ROS-degradable linkers in tissue engineering scaffolds,10 indicating that oxidation may represent a significant degradation mechanism for other N-substituted molecules in environments where ROS species are elevated.23 However, only one study has systematically explored the oxidative degradation of peptoids: homopolymers of poly(N-ethylglycine) were monitored upon incubation in hydrogen peroxide (H2O2, 0.5–50 μM) with a copper sulfate catalyst (CuSO4, 50 μM), and degradation was compared to polyethylene glycol (PEG) and poly(2-oxazoline).2 This work revealed that polydisperse peptoid backbones were found to be more susceptible to H2O2 with CuSO4 catalyst than PEG or poly(2-oxazolines), as observed through rapid broadening of gel permeation chromatography peaks and shifts to higher elution volumes. Given these previous results, it is feasible that peptoids may degrade via oxidative mechanisms at rates on par with peptides, but offer the benefit of proteolytic stability and expanded chemical functionality.
The goal of this work is to establish a fundamental investigation of the oxidative susceptibility of three types of peptides (Fig. 1B), in comparison to three types of synthetic peptoids (Fig. 1C), and demonstrate how their unique structure has the potential to impact selective interactions with biologically relevant degradative species (i.e., enzymes vs. ROS). First, we developed a library of six homopolymer oligomers of constant chain length (Fig. 1D) to establish a baseline comparison of peptides to structurally similar peptoids for oxidative degradation to chemically-generated ROS. Then, we adapted a Förster resonance energy transfer (FRET) reporter design and synthesized twelve additional fluorescent oligomers (Fig. 1D) to show that side-chain identity, sequence, and peptide content may enable a path for fine-tuning degradation behavior in certain biological contexts. Together, our library demonstrates the potential of peptoids for future applications as biomaterials, given their selective oxidative degradability, hydrolytic stability, enzymatic tunability, and vast chemical space for further functionality as hybrid molecules.
Materials and methods
Materials
Amino acids were all Fmoc-protected and purchased from Chem-Impex International, Inc. (Wood Dale, IL), along with Rink amide resin, O-(1H-6-chlorobenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HCTU, ≥99%), bromoacetic acid (≥99%), and N,N′-diisopropylcarbodiimide (DIC, ≥99%). Peptoid submonomors, sarcosine, and modified lysines were also purchased from Chem-Impex International Inc., namely N-Boc-1,4 butanediamine (N-lysine, ≥99%), Fmoc-N-methylglycine (N-alanine, ≥99%), and the fluorophore and quencher pre-loaded on L-lysines: Nα-Fmoc-Nε-7-methoxycoumarin-4-acetyl-L-lysine (Lys(Mca), ≥97%) and Nα-Fmoc-Nε-2,4-dinitrophenyl-L-lysine (Lys(Dnp), ≥98.5%). Triisopropylsilane (TIPS, 98%) was purchased from Acros Organics (Fir Lawn, NJ). N-Methylmorpholine (NMM, 99%) and piperidine (99.5%) were purchased from Millipore Sigma (Burlington, MA). All other solvents were purchased from Fisher Scientific (Hampton, NH) at the following purity levels: dimethylformamide (DMF, Certified ACS: ≥99.8%), trifluoroacetic acid (TFA, peptide synthesis grade: ≥99.5%), diethyl ether (ether, ACS Reagent: ≥99%), acetonitrile (ACN, HPLC: ≥99.9%), and phosphate buffered saline (PBS, 10×, RNAse free). Trypsin (0.25%, 1 mM EDTA in HBSS) was purchased from Caisson Labs. Inc. (Smithfield, UT). Hydrogen peroxide (H2O2,Certified ACS: 30%) was purchased from Fisher Scientific, and copper(II) sulfate (CuSO4, Reagent Plus®: ≥99%) was purchased from Millipore Sigma.Oligomer synthesis
Peptides, peptoids, and peptomers were all synthesized using Rink amide polystyrene resin (0.43 mmol g−1) on a Prelude X automated peptide synthesizer (Gyros Protein Technologies) at a scale of 50 μmol. Fmoc groups were removed from the resin and subsequent amino acids by washing twice with 20% piperidine in DMF. Peptide residues utilized Fmoc-protected amino acids (250 mM, 5× molar excess) coupled using HCTU activator (250 mM, 5× molar excess) and NMM (500 mM, 10× molar excess). Coupling steps were performed twice. Peptoid residues were installed according to previously published submonomer synthesis methods.24 First, bromoacylation occurs via addition of bromoacetic acid (1.2 M in DMF) and DIC at a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.93.25 The bromine is then displaced by a primary amine (2 M in DMF) to install the entire peptoid residue. Fluorescent oligomers were synthesized with lysines functionalized with a FRET pair: 2,4-dinitropheyl (Dnp) as a quencher on the carboxy terminus and 7-methoxycoumarin-4-acetic acid (Mca) as a fluorophore on the N-terminus. Upon completion of synthesis, oligomers were cleaved from the resin using a cleavage cocktail comprised of 95% TFA/2.5% water/2.5% TIPS for oligomers with side-chain protecting groups (lysine containing residues) or 95% TFA/5% water for oligomers lacking any side-chain protecting groups. The resin was then filtered off and the oligomers were prepared for purification.
:0.93.25 The bromine is then displaced by a primary amine (2 M in DMF) to install the entire peptoid residue. Fluorescent oligomers were synthesized with lysines functionalized with a FRET pair: 2,4-dinitropheyl (Dnp) as a quencher on the carboxy terminus and 7-methoxycoumarin-4-acetic acid (Mca) as a fluorophore on the N-terminus. Upon completion of synthesis, oligomers were cleaved from the resin using a cleavage cocktail comprised of 95% TFA/2.5% water/2.5% TIPS for oligomers with side-chain protecting groups (lysine containing residues) or 95% TFA/5% water for oligomers lacking any side-chain protecting groups. The resin was then filtered off and the oligomers were prepared for purification.
Oligomer purification
Crude purification by ether precipitation was possible for all peptides. A ten-fold volume excess of diethyl ether was chilled and the oligomers dissolved in cleavage cocktail were added dropwise, centrifuged to collect the precipitate, then washed twice with fresh ether. For the all-peptoid oligomers, the cleavage cocktail was evaporated on a rotary evaporator. Oligomers were then dissolved in a mixture of 20% ACN/80% water with 0.01% TFA and purified by high performance liquid chromatography (HPLC) with a semi-prep C18 column on a Dionex UltiMate 3000 UHPLC with a 25 minute gradient (10 minute equilibration period followed by increasing ACN ratio from 20–100% over 25 min) run at 10 mL min−1. Fractions were collected according to their UV signal at 214 nm and separation continued until at least 90% purity was achieved. Purified oligomers were then lyophilized and analyzed on an analytical Thermo BioBasic™ C18 column (250 mm × 4.6 mm, i.d. 5 μm) and via matrix-assisted laser desorption ionization-time of flight (MALDI-TOF, Bruker Autoflex maX) mass spectrometry (MS) (Fig. S1–S5†) to confirm purity and molecular weight, respectively.18-mer degradation studies
To investigate degradation of the 18-mers following synthesis and purification, oligomers were dissolved in 1× PBS at a concentration of 1 mg mL−1 and exposed to either 10 mM H2O2 only, 10 μM–1 M H2O2 + 50 μM CuSO4, or 0.1 μM trypsin. A control containing 1 mg mL−1 substrate in 1× PBS buffer alone was also performed. Samples were then incubated at 37 °C and pH was monitored at physiologically relevant levels (pH 7.4). At defined intervals, aliquots were removed and flash frozen in liquid nitrogen to quench the reaction, and then lyophilized for further analysis using liquid-chromatography/mass spectrometry (LC-MS) with an electrospray ionization (ESI) source. For experiments lasting 2 hours (H2O2 + CuSO4), timepoints were taken every 15 minutes. For experiments lasting seven days (H2O2 and trypsin), timepoints were taken every 24 hours, at which time stimuli were also replenished. For 18-mer oligomers with neutral residues (L-proline, D-proline, N-alanine and N-methoxyethylglycine) lyophilized samples were reconstituted at 0.1 mg mL−1 in methanol and analyzed on an Agilent Technologies 6120 Single Quadropole LC-MS with an Agilent ZORBAX Eclipse Plus C18 column coupled with an ESI source. A 12 minute gradient ramp running methanol and water with 0.1% formic acid spanned from 5% to 100% methanol. Reactions were done in triplicate and degradation was monitored by decreasing main peak areas of chromatogram peaks in comparison to control. Specifically, spectra were extracted at 214 nm and the max absorbance and corresponding retention time of the control peaks were identified using Origin Pro's built-in Peak Analysis/Finder tool with a baseline at y = 0. The absorbance of each sample peak was normalized against its respective control at fixed retention time, and plotted against incubation time. Exponential decay degradation rates were fit using GraphPad Prism's built-in One Phase Decay Analysis according to the equation| y = (y0 − Plateau)e−kx + Plateau |
Degradation monitoring of fluorescent 6-mers via HPLC/LC-MS
For 6-mer fluorescent oligomers with positively charged sides chains (L and N-lysine), degradation was monitored by decreasing main peak areas and shifting retention times of analytical HPLC peaks in comparison to controls (as described above). For these molecules, reverse phase-HPLC was used in place of LC-MS given the greater level of control over chromatography parameters (the LC-MS instrument is a core facility instrument with predetermined settings; the HPLC is our instrument). This allowed us to adjust the solvent gradient and flow rate, enabling separation of oligomer peaks from the solvent peaks. Specifically, lyophilized samples were dissolved at 0.1 mg mL−1 in 15% ACN/85% water with 0.1% TFA. HPLC analysis conditions included a mobile phase containing ACN and H2O with 0.1% (v/v) TFA at a flowrate of 1 mL min−1. A 15 minute gradient was employed (10 minute equilibration period followed by increasing ACN ratio from 15–100% over 15 min) with the eluents measured at 214 nm. Following HPLC, the samples associated with each eluted HPLC peak were collected and individually analyzed by ESI using flow injection analysis to monitor intact substrate masses and confirm degradation.Degradation monitoring using fluorescence assays
Fluorescent reporter oligomer degradation was monitored in real-time by tracking the fluorescence signal of the Mca fluorophore as it was liberated from the Dnp quencher by oxidative and enzymatic degradation. Oligomers were first dissolved in DMSO at a concentration of 1 mM, as measured by the Dnp group's absorbance at 363 nm on a NanoDrop OneC Microvolume UV-Vis spectrophotometer. Oligomers were then diluted to 20 μM with 20% DMSO in 1× PBS. 50 μL of substrate solution was combined with either 50 μL of buffer or stimuli (oxidative or enzymatic) solution in triplicate in a black 96-well plate to get a final substrate concentration of 10 μM. The plate was oscillated for 10 seconds to mix and then read on a BioTek Synergy H1 Multi-Mode Microplate Reader at Ex./Em. 325/392 nm for three hours.Fluorescence assay data analysis
10 μM fluorescence traces were fit to the following built-in exponential plateau function in GraphPad Prism 9:| y = yM − (yM − y0)e−kt |
Degradation product identification
Samples of degraded substrates were measured after 3 hours using LC-MS. Specifically, 10 μM of substrate in 10% DMSO was incubated for 3 hours at 37 °C with the same ROS and enzyme concentrations used for fluorescence assays. Controls without ROS or enzyme were prepared in the same way. After 3 hours, the reactions were quenched in liquid nitrogen, then lyophilized. Lyophilized samples were reconstituted at 50 μM in water with 25% acetonitrile and analyzed by LC-MS, as described above. Spectra were extracted in Agilent's ChemStation software at 210 nm and 400 nm, along with extracted ion chromatographs at each possible fragment mass. The fragments in the 400 nm trace (only those with a Dnp group) were matched to their extracted mass peak at the same retention time to identify degradation products. Three samples were analyzed using LC-MS to confirm degradation.Statistical analysis
Numeric data is reported as the mean ± standard deviation of n = 3 independent runs, unless otherwise indicated. Oxidative and enzymatic half-lives were compared using 95% confidence intervals to individually determine significant differences between mean degradation rates of different oligomers.Results and discussion
Peptide, peptoid and peptomer library development
The N-substituted backbone of both poly(prolines) and peptoids is known to confer proteolytic resistance and makes poly(proline) an especially intriguing comparison for the initial investigation of oxidative degradation of peptoids.23 Furthermore, the known oxidative susceptibility of amino acids beyond proline (e.g., lysine, arginine, histidine)26 also make charged side-chains an interesting selection for studying oxidative susceptibility. While past studies focused on high molecular weight (MW) polymers with degrees of polymerization ranging from approximately 50 to 120,2 we chose to investigate shorter, sequence-defined oligomers given their ease with which to vary side-chain structure and placement. For peptides, we selected L-proline (LPro), D-proline (DPro), and L-lysine (LLys) residues (Fig. 1B). For peptoids, we aimed to select submonomers with translocated sidechains that match the amino acids of the peptides (Fig. 1C). Given that the proline monomer is already N-substituted and therefore does not have an analogous peptoid substitution, we selected sarcosine (NAla) using the N-methylglycine monomer because it is the simplest peptoid residue and enables investigation of backbone effects. We employed N-(4-aminobutyl)glycine to generate a peptoid lysine mimic (NLys) for our study. Finally, we selected the N-methoxyethylglycine (Nme) residue using the N-methoxyethylamine submonomer due to its hydrophilic ether groups and high reaction efficiency.Using solid phase synthesis, we first generated six homopolymer oligomers with chain lengths of 18 residues each (Fig. 1D). We quickly found that analyzing degradation of the highly hydrophilic lysine molecules using LC methods was extremely difficult, so we sought a new design for our oligomers. Our new design consisted of a set of peptoids, peptides, and peptomers six residues in chain length and flanked with lysines functionalized with a Dnp/Mca FRET pair (Fig. 1D).27 In addition to enabling real-time fluorescence tracking of degradation, these bulky, aromatic groups effectively shifted the polarity of our lysine oligomers, thereby facilitating analysis as the increase in hydrophobicity afforded resolution by LC.
Oxidative and enzymatic degradation of peptide vs. peptoid homopolymers
To initially compare the timescales of oxidative degradation, as well as begin to explore side-chain effects for our 18-mer oligomers, we conducted degradation studies in aqueous buffer with H2O2 (10 mM) and a metal catalyzed oxidation (MCO) reaction system using H2O2 (10 mM) and CuSO4 (50 μM). Both of these ROS generators produce highly reactive hydroxyperoxide (˙OOH) and hydroxyl (˙OH) radicals, which oxidize amino acid side-chains and cause backbone cleavage.2 Numerous systems for studying ROS can be found in the literature.10,28–30 While these two model oxidative conditions represent only a portion of the potential sources of oxidative stress encountered in biological systems,2 and the concentration of these ROS radicals in vivo has also long been a matter of debate,31–36 these chemically generated ROS species and concentrations allowed for benchmarking of our peptoid degradation against other polymeric and peptide oxidative degradation studies in the literature. Furthermore, MCO is the most common mechanism of protein oxidation in living cells, therefore making it relevant in vitro and in vivo. In fact, MCO reactions with metals such as Cu+ or Fe2+ interacting with H2O2 are commonly used to mimic cellular production of ˙OOH and ˙OH.2,4,37–40 We also included trypsin (0.1 μM) degradation studies to investigate the relative proteolytic stability of our peptoid vs. peptide oligomers. Similarly, while trypsin does not cover the entire landscape of proteolysis, it is a key digestive enzyme and is frequently used in cell culture and other biotechnology applications.41For degradation studies, each 18-mer oligomer was exposed to one of our three oxidative or enzymatic conditions (H2O2, H2O2 + CuSO4, or trypsin) and prepared for chromatographic analysis. As mentioned, the highly hydrophilic nature of the lysine oligomers ((LLys)18 and (NLys)18) made analysis by LC difficult given the fast elution from the C18 column. Both oligomers eluted at the same retention time as the solvent peaks, thus confounding the degradation study analysis (Fig. S6†). As a result, only (LPro)18, (DPro)18, (Nme)18, and (NAla)18 were tested.
All four oligomers showed decreasing peak areas over seven days for 10 mM H2O2 and over two hours for the MCO condition (Fig. 2A and B and S7†). In both cases, the main peak associated with the intact oligomers decreased, indicating degradation, and the degree of degradation was faster for the MCO condition relative to the H2O2 condition for both oligomers (two hours vs. seven days). In addition to time-dependent degradation, we also found the MCO reaction led to concentration-dependent degradation, with faster degradation occurring at higher ROS concentrations (Fig. S8†). Furthermore, all oligomers maintained stability against proteolysis by trypsin over seven days (Fig. 2A and B and S7†).
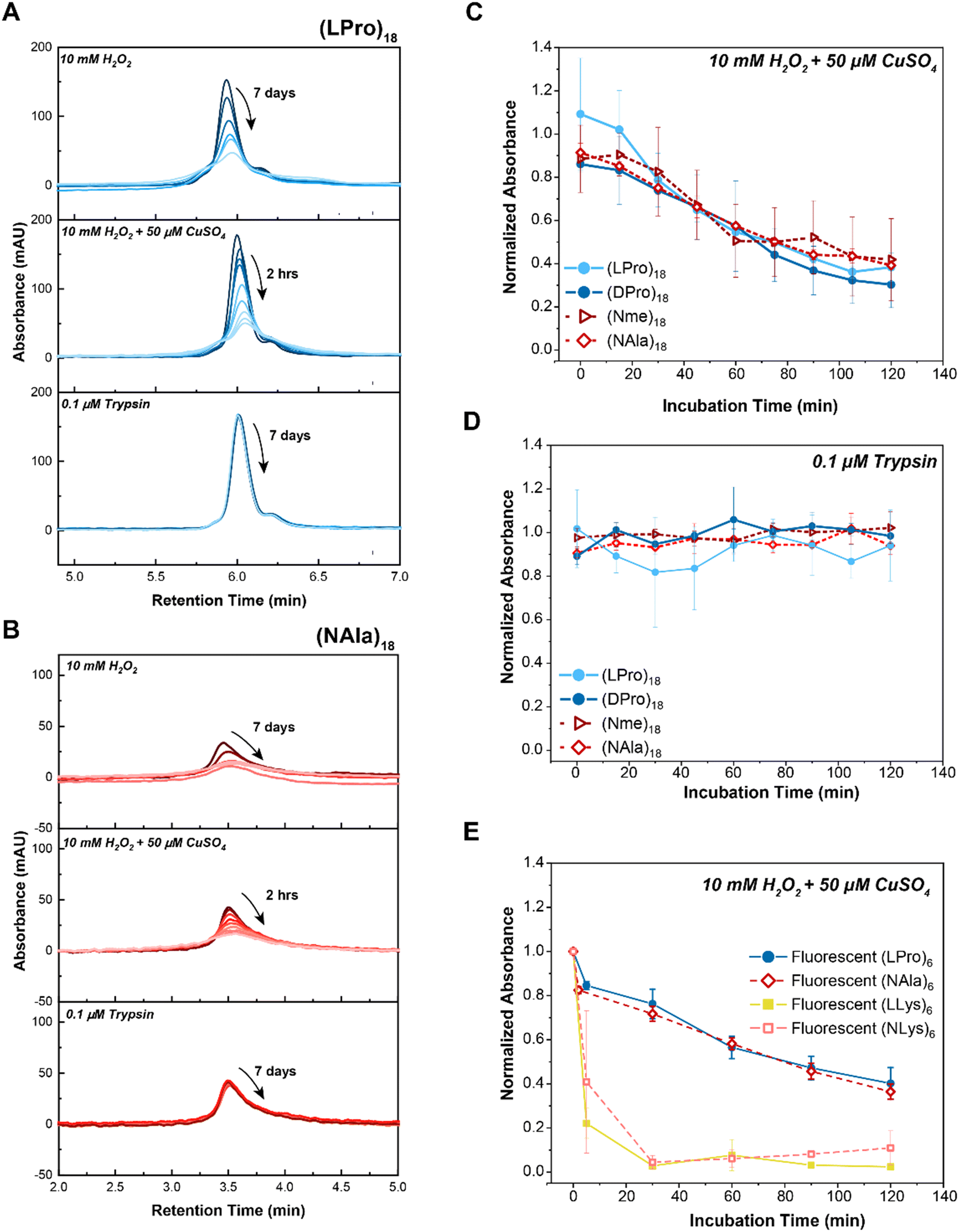 | ||
| Fig. 2 Comparison of peptide vs. peptoid oxidative and enzymatic degradation. A) LC traces of selected 18-mer L-proline peptide, (LPro)18, in comparison to B) 18-mer N-methylglycine peptoid, (NAla)18, upon exposure to 10 mM H2O2 (top panel), 10 mM H2O2 + 50 μM CuSO4 (middle panel) or 10 μM trypsin (bottom panel). As indicated by the arrows, timepoints were taken at 15 minute intervals over the course of 2 hours for H2O2 + 50 μM CuSO4 (MCO) and at 24 hour intervals over the course of 7 days for H2O2 and Trypsin. LC traces of the other 18-mer oligomers can be found in Fig. S7.† C) Comparison of 18-mer oligomer peptide and peptoid degradation rates when exposed to oxidative (10 mM H2O2 + 50 μM CuSO4) and D) enzymatic (1 μM trypsin) stimuli. E) Comparison of 6-mer fluorescent homopolymer peptides and peptoids when exposed to 10 mM H2O2 + 50 μM CuSO4. Peptides are shown with closed markers and solid lines. Peptoids are shown with open markers and dashed lines. Each point represents max absorbance of degraded substrate samples normalized to max absorbance of control (n = 3). Lines only represent a guide for the eye. Error bars represent standard deviation from three experimental replicates. | ||
To quantify degradation rates, the maximum absorbance of each sample was normalized against its respective control (i.e., sample in buffer without H2O2 or trypsin) at fixed retention times. Notably, all peptoid oligomers investigated degraded in response to chemically generated ROS (Fig. 2C), in contrast to the consistent intact LC peaks observed for oligomers exposed to trypsin over the 2 hour course of the study (Fig. 2D). Degradation rates to MCO were further compared by performing an exponential decay fit on the data points (Fig. S9A–D†) and calculating the oxidative half-lives, t0.5, of each substrate (Fig. S9E and F†). While (Nme)18 and (NAla)18 had slightly larger half-lives ((Nme)18: t0.5 = 86 min and (NAla)18: t0.5 = 97 min) than the peptides ((LPro)18: t0.5 = 65 min and (D-Pro)18: t0.5 = 66 min), all peptoids exhibited half-lives on-par with N-substituted peptides. Only (NAla)18 exhibited a half-life significantly greater than (LPro)18 based on the 95% confidence interval.
Potential degradation mechanisms were further investigated for (LPro)18, (DPro)18, (NAla)18 and (Nme)18 using the mass spectra collected throughout the course of LC-MS (Fig. S10–S13†). (LPro)18 (Fig. S10†) and (DPro)18 (Fig. S11†) exhibited analogous LC-MS traces, indicating that the D-amino acid structure does not have an effect on oxidative degradation mechanisms, and thereby concluding our investigation of the D-oriented analog. Current proposed mechanisms for oxidation of proline by MCO state that peptide bond cleavage is a result of the generation of carbonyl derivatives such as glutamic semialdehydes.26,42 However, identifying and tracking carbonylation using MS is known to be technically challenging given the many types of modifications that result in carbonyl residues.38 Carbonyl assays using 2,4-dinitropheylhydrazine have been used to estimate protein and peptide carbonylation.1,42 However, these are optimized for high MW proteins and often result in carbonyl products being missed when used for analyzing peptides due to ion suppression and only a limited number of the most abundant ions (typically the parent peptide species).42 Thus, it was not surprising that the mass spectrometry results provided no clear insight regarding mechanisms of degradation.
Apart from the intact oligomer masses, we were unable to identify additional masses for (LPro)18 and (NAla)18 (Fig. S10 and S12†). However, when analyzing the mass spectrometry of the 60 and 120 min timepoints for (Nme)18, we noticed new masses arising within side peaks of the LC chromatogram (Fig. S13B and C†). Upon further investigation, it was determined that these new masses (specifically, a mass change of −60.06 from the intact structure) corresponded with cleavage of the ether side-chains. After 60 minutes, masses corresponding to one side-chain cleaved were identified (Fig. S13B†), and after 120 minutes, masses corresponding to one and two side-chains cleaved were identified (Fig. S13C†). Interestingly, (Nme)18 had the slowest (and most variable) oxidative half-life of all oligomers, likely a result of the additional side-chain sites for oxidation slowing backbone degradation. Altogether, LC-MS provided a way to monitor the disappearance of intact oligomer, and yielded some mechanistic insight regarding oxidative degradation of (Nme)18 side-chains.
We wanted to further investigate our other peptides and peptoids with charged side-chains (LLys and NLys). Given our previous difficulty analyzing degradation of the highly hydrophilic (LLys)18 and (NLys)18 oligomers using LC methods, we attempted degradation studies of our more hydrophobic 6-mer FRET reporter oligomers: (LLys)6 and (NLys)6. Studies were performed in aqueous buffer using the MCO reaction system as previously described, then separated by HPLC and subsequently analyzed by MS. Excitingly, the bulky Dnp group on the 6-mer lysine oligomers enabled successful separation of oligomer peaks from the solvent peak on the HPLC chromatogram (Fig. S14 and S15†). Fluorescently tagged (LPro)6 and (NAla)6 were also analyzed (Fig. S16 and S17†), and the maximum absorbance of each sample was again normalized against its respective controls at fixed retention times to quantify degradation rates (Fig. 2E). As before, oxidative half-lives of each substrate were calculated by performing an exponential decay fit on the data points (Fig. S18†). The results showed that (LPro)6 and (NAla)6 degraded at rates similar to their 18-mer counterparts, while also revealing rapid degradation by (LLys)6 and (NLys)6.
We were intrigued by the rapid degradation for both (NLys)6 and (LLys)6 compared to (LPro)6 and (NAla)6, so again turned to LC-MS investigation in an attempt to elucidate mechanisms of oxidation. As mentioned, current proposed mechanisms for oxidation of proline state that peptide bond cleavage is a result of the generation of carbonyl derivatives. Carbonyl derivatives are also relevant to the oxidative degradation of lysines, leading to 2-amino-adipic semialdehydes.26,38 Generation of these groups is associated with loss of ammonia groups, which carry a positive charge. This leads to decreased ionization efficiency and reduces the chance of detecting those peptides using positive-ion mode MS.38 Thus, it was not a surprise that mechanisms once again could not be substantiated from the MS data (negative-mode MS also provided no further insight).
Given the widespread difficulty in tracking oxidation degradation mechanisms for proteins and polymers, simulation studies have been used to characterize the attack of ˙OH radicals and their resulting intermediates.43 Radical attack can occur at the backbone, or at the side-chains, depending on the relative stability of the formed intermediates. In simulation studies on the oxidation of lysines caused by ˙OH radicals, the side-chain is expected to be the primary site of attack because it leads to the most stable carbonyl derivative (usually an aldehyde).43 We speculate that side-chain splitting leads to increased reactivity and susceptibility to further oxidative attack, which could accelerate backbone cleavage. This aligns with our observations of decreased half-life for (LLys)6 compared to those oligomers without protruding side-chains ((LPro)6 and (NAla6)). Furthermore, given the very similar chromatogram profiles (Fig. S14 and S15†) of (LLys)6 and (NLys)6, we suspect the peptoid oligomer degrades by the same mechanism (˙OH attack of lysine side-chains increasing reactivity of the molecule and accelerating backbone degradation).
Fluorescent reporter degradation studies
As a result of our difficulty analyzing degradation of the highly hydrophilic lysine-containing molecules using LC methods, we moved forward with our fluorescent reporter design for the remainder of our studies. Protease activity is frequently tracked using short peptides that are equipped with common FRET reporter systems. These systems operate such that while the peptide is intact, fluorescence is quenched. Then, upon cleavage of the peptide, fluorescence is restored (Fig. 3A). This new approach not only increased hydrophobicity of our lysine-containing molecules, but also allowed degradation of our oligomers to be monitored in situ. All of our oligomers were a total of eight residues in length, six of which were the active sequence with fluorophore and quencher moieties attached to modified lysines on each end.27 The length was selected to ensure efficient energy transfer from the fluorophore molecules (distance range recommended for separating the donor and acceptor pair is 10–100 Å).44 Our peptides range from about 28–32 Å based on estimates of average amino acid lengths.45 For these molecules, L-lysine and L-proline were selected as the peptide residues and N-alanine and N-lysine were employed as peptoid residues. These residues were selected given our outstanding interest in the oxidative susceptibility of oligomers with lysine side-chains, LLys and NLys, and our interest in the significantly different oxidative degradation rates of N-substituted residues, NAla and LPro, as revealed in the 18-mer homopolymer degradation study.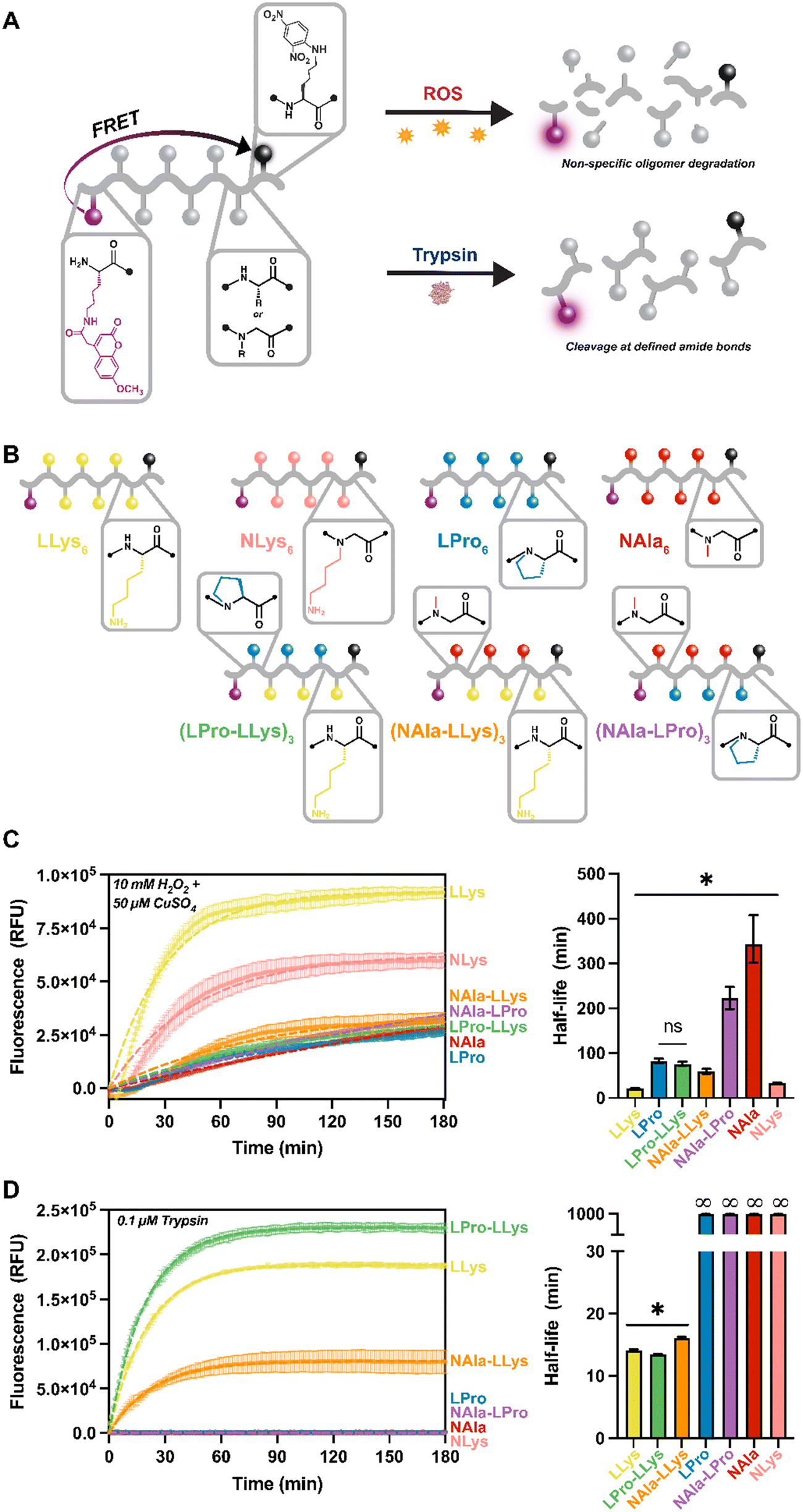 | ||
| Fig. 3 Fluorescent reporter ‘composition comparison’ degradation study. A) Schematic of FRET design and resulting degradation products. Each 6-mer oligomer sequence is flanked with L-lysines functionalized with 2,4-dinitropheyl (Dnp) quencher on the C-terminus and 7-methoxycoumarin (Mca) fluorophore on the N-terminus. B) Schematic of fluorescent 6-mer library structures and sequences. C) Fluorescence tracking of degradation when exposed to 10 mM H2O2 + 50 μM CuSO4 stimuli for 3 hours and comparison of half-lives (min) from exponential plateau fit. D) Fluorescence tracking of degradation when exposed to 0.1 μM trypsin stimuli for 3 hours and comparison of half-lives (min) from exponential plateau fit. All oligomers were at a concentration of 10 μM. For fluorescence tracking, error bars represent standard deviation from three experimental replicates and the dashed line represents exponential plateau model fit. For bar-graphs, error bars represent the 95% confidence intervals. Significance brackets extend over all samples that are significantly different from one another such that *p ≤ 0.05 with exceptions (ns) denoted. | ||
For this study, four homopolymers of each residue: (LLys)6, (LPro)6, (NAla)6, and (NLys)6 were first synthesized to establish a baseline for our new reporter design. We were also curious how changing peptide content and residue type would affect degradation rates, especially considering the fast degradation of (LLys)6. Therefore, we synthesized three peptomers incorporating our fastest degrading residue (LLys) and our slowest degrading peptide and peptoid residues (LPro and NAla) in an alternating sequence: (LPro-LLys)3, (NAla-LLys)3, and (NAla-LPro)3. Following synthesis, a spectral fluorescence scan of all substrates was conducted to ensure the fluorophore was fully quenched (Fig. S19†).
For fluorescence studies, 10 μM of each fluorescent oligomer was exposed to the same MCO and trypsin concentrations used for the 18-mer library. Oligomers were tracked for three hours to establish susceptibility to oxidative and enzymatic degradation as indicated by increasing fluorescence signals (Fig. 3C and D). To ensure the fluorescence increase was not a result of Mca fluorophore instability, Mca was exposed to the same conditions (Fig. S20†). To better visualize variance across oligomers, the three-hour fluorescent traces were fit directly to an exponential plateau function and the k constants were tabulated to determine the half-life of each oligomer (Fig. 3C and D). As expected, all oligomers degraded to MCO as indicated by the increasing fluorescent signals, and the fastest degrading substrate was (LLys)6 (Fig. 3C, yellow). In agreement with our previous LC study (Fig. 2E), we found that (NLys)6 exhibited a heightened sensitivity to MCO compared to (LPro)6 and (NAla)6 (Fig. 3B, pink, blue, and red), and that (NAla)6 had the longest half-life (Fig. 3C, red).
Notably, when LPro and NAla residues were combined in an alternating sequence ((NAla-LPro)3), the half-life was intermediate to each homopolymer (Fig. 3C, purple). Interestingly, the other alternating sequences, (LPro-LLys)3 and (NAla-LLys)3, had significantly shorter half-lives (Fig. 3B, green and orange), again suggesting that the presence of lysine side-chains speeds up the oxidation reactions. Furthermore, because fluorescence degradation tracking is only sensitive to one initial backbone cleavage event, the fast oxidative degradation observed here indicates accelerated backbone cleavage for oligomers containing lysine side-chains, supporting our earlier hypothesis.
When exposed to trypsin, all LLys-containing oligomers were degraded, while all fully N-substituted molecules (containing NAla, LPro, and NLys residues) remained quenched, and therefore intact (Fig. 3D), demonstrating that N-substitution does generate proteolytic resistance, making these molecules selectively degraded by oxidation. NLys is exceptionally notable considering its short oxidative half-life and complete enzymatic resistance, and thus could be leveraged as a highly selective molecule. Interestingly, the LLys-containing oligomers that consisted only of peptide residues ((LLys)6 and (LPro-LLys)3) degraded more quickly (Fig. 3D, yellow green) than (NAla-LLys)3 (Fig. 3D, orange) which consisted of half peptoid residues. The fact that the N-substitution in LPro did not have this effect suggests that the NAla residue specifically alters trypsin degradation behavior. Studies have explored oligomers with non-natural residues to leverage alternative backbone interactions and side-chains that change recognition by proteases,46–48 which could be a reason for this altered cleavage behavior.
Given the results of the fluorescent reporter library, we sought to investigate the effect of changing sequence on oxidative and enzymatic degradation rates. Specifically, we were curious to explore how the significantly different sensitivities of L-lysine and N-alanine residues to MCO and trypsin alter fluorescence response when the sequence of the residues was changed, but the overall molecular composition was kept constant. Using the same fluorescent reporter design as previously, we synthesized three new peptomers with LLys and NAla residues in various combinations to create a ‘sequence effects’ library. This library included the (NAla-LLys)3 alternating substrate used in the previous study, and oligomers designated ‘blocky’, ‘scrambled’, or ‘middle’ (Fig. 4A) according to their distribution of lysine residues. After synthesis and purification, fluorescence assays were conducted as previously described.
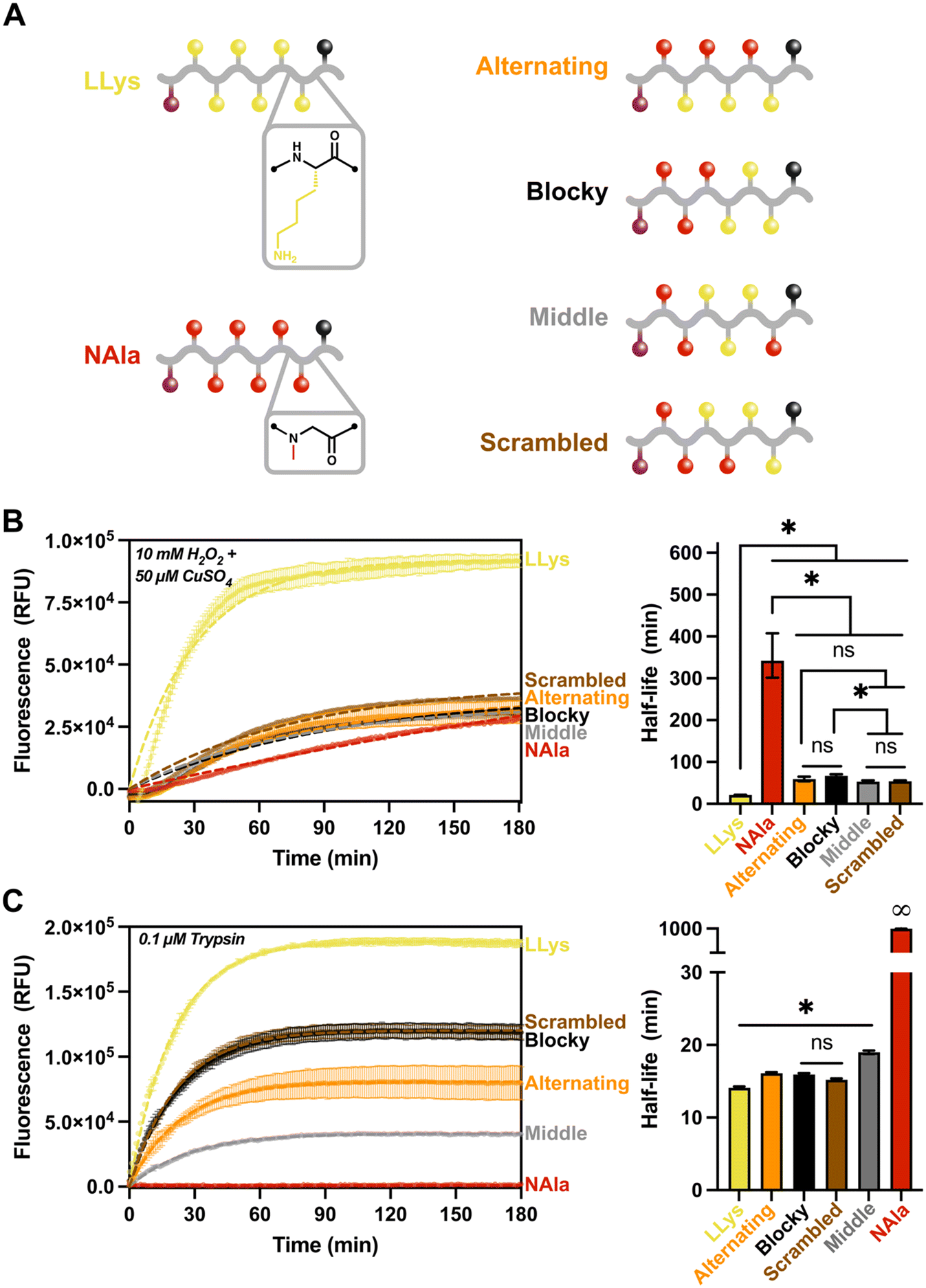 | ||
| Fig. 4 Fluorescent reporter ‘sequence effects’ degradation study. A) Schematic of fluorescent 6-mer sequence effect library. B) Fluorescence tracking of degradation when exposed to 10 mM H2O2 + 50 μM CuSO4 stimuli for 3 hours and comparison of half-lives (min) from exponential plateau fit. C) Fluorescence tracking of degradation when exposed to 0.1 μM trypsin stimuli for 3 hours and comparison of half-lives (min) from exponential plateau fit. All oligomers were at a concentration of 10 μM. For fluorescence tracking, error bars represent standard deviation from three experimental replicates and the dashed line represents exponential plateau model fit. For bar-graphs, error bars represent the 95% confidence intervals. Significance brackets extend over all samples that are significantly different from one another such that *p ≤ 0.05 with exceptions (ns) denoted. | ||
Given the high ROS sensitivity and high trypsin sensitivity of LLys in comparison to NAla, we sought to answer the following questions with our ‘sequence effects’ library: 1) does grouping LLys residues together change enzymatic and oxidative degradation rates? And 2) does changing the location of peptoid (NAla) substitutions alter degradation behavior? As shown (Fig. 4B), grouping LLys residues together (‘Blocky’ and ‘Middle’ oligomers) or altering the location of NAla did not significantly change degradation by ROS. Rather, it appeared that increasing LLys content was most important in shortening the oxidative half-life.
For degradation by trypsin, the location of the NAla residues proved to be the most important factor affecting degradation rates (Fig. 4C). Trypsin is an endopeptidase that cleaves on the C-terminal side of L-lysine and L-arginine amino acid residues. As shown (Fig. 4C), the degradation was fastest for LLys, followed by the ‘blocky’, ‘scrambled’ and ‘alternating’ combinations. Trypsin cleavage is known to be slowed down in the presence of acidic residues49 (i.e., when the pKa of the molecule increases). This likely explains the slower rate for the oligomers with lower LLys content, given that the side-chain of lysines are known to act as bases and are often protonated at physiological pH.43 Another notable observation was that the ‘middle’ sequence appeared to cleave significantly slower, and is the only oligomer that contains an NAla residues adjacent to the C-terminus. This suggests that trypsin preferentially cleaves at the C-terminus. To investigate our hypothesis, we analyzed our control and degraded oligomers using LC-MS (Fig. S21–S25†). The mass spectrometry results revealed that all except the ‘middle’ oligomer (Fig. S24†) did in fact contain the “quencher only” mass as the primary product. The ‘middle’ oligomer was also the only oligomer with detectable amounts of intact substrate after three hours, confirming the slow-degrading behavior observed on the fluorescence trace. We also analyzed (LPro-LLys)3 (Fig. S26†), and the only cleavage product identified was the quencher, further supporting our hypothesis and also agreeing with other studies stating that cleavage does not occur when the L-lysine residue is adjacent to L-proline.49
Trypsin's preference for the C-terminus explains the faster cleavage for LLys6, ‘blocky’, ‘scrambled’, and ‘alternating’ oligomers compared to the ‘middle’ oligomer. However, the slower rate for the ‘alternating’ sequence was intriguing, and suggests trypsin might also prefer a peptide residue on both sides of the cleavage site. Again, LC-MS supported this hypothesis, given that all identified cleavage products occurred at spots in which LLys was located on both sides of the cleavage site (recall that the quencher is functionalized on a lysine residue). Finally, the exceptionally short half-life of (LPro-LLys)3 compared to the (NAla-LLys)3/‘alternating’ oligomer suggests that the rate of cleavage is heavily influenced by the presence and location of NAla residues, perhaps by decreasing the pKa surrounding the cleavage sites or by altering recognition by the trypsin protease. However, it is important to keep in mind that because fluorescence degradation tracking is only sensitive to an initial cleavage event, this means that the rates shown may not reflect the actual proteolytic activity in the case of multiple cleavage sites.
Conclusion
Here, we compared the oxidative and enzymatic degradation of peptoids (N-substituted glycines) to oxidatively and enzymatically labile peptides: poly(prolines) and poly(lysines). Our results present evidence that sequence-defined peptoids and peptide–peptoid hybrids can be used as a tool for developing biomaterials with fine-tuned degradation behavior. Although poly(prolines), another N-substituted polyamide, are a viable ROS-sensitive molecule for certain biological applications, their synthetic and structural limitation make it difficult to tune biomaterial properties for imparting specific interactions with biological systems. Alternatively, the N-substitution of peptoids has major implications for biorecognition and proteolytic resistance in such cases where targeting biological species in complex mixtures is desired. For example, given that chronic oxidative stress is a hallmark of many inflammatory diseases such as cancer, neurodegeneration, and cardiovascular dysfunction,4,50 biomaterials exhibiting selectivity towards oxidative over enzymatic degradation may be a promising route for detecting disease by identifying ROS in vitro and in vivo. Thus, we present sequence-defined peptoids and peptomers as a complementary material to poly(prolines) and poly(lysines), but with the benefit of added enzymatic stability (for peptoids) and ability to tune ROS degradation rates over a span of a few minutes (LLys and NLys) to several hours (NAla). Additionally, we have also demonstrated that NAla substitutions provide a straightforward strategy for tuning enzymatic degradation rates to trypsin, thus opening the door for further functionality. Future work will aim to gain a full picture of peptoids' oxidative susceptibility to other biologically relevant ROS species and concentrations, as well as further explore sequence and side-chain effects on enzymatic degradation behavior. We anticipate these studies will enable a route for fine-tuning degradation behavior of peptoid-based materials for use in a variety of biomedical applications.Author contributions
H. C. S., M. J. A., L. J. S., and A. M. R. conceived the research and designed experiments. H. C. S., M. J. A., B. Z. T., and M. S. M. performed the research and collected data. H. C. S., M. J. A., L. J. S., and A. M. R. analyzed the data. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Science Foundation (DMR-2046746, A. M. R.) and by the National Institutes of Health (R35GM138193 to A. M. R. and R01EB015007 to L. J. S.). In addition, H. C. S and M. J. A. were supported through a National Science Foundation Graduate Research Fellowship (Program Award No. 000392968). We also acknowledge the use of shared facilities at the University of Texas Mass Spectrometry and Proteomics Facilities.References
- B. S. Berlett and E. R. Stadtman, J. Biol. Chem., 1997, 272, 20313–20316 CrossRef CAS.
- J. Ulbricht, R. Jordan and R. Luxenhofer, Biomaterials, 2014, 35, 4848–4861 CrossRef CAS PubMed.
- P. E. Starke-Reed and C. N. Oliver, Arch. Biochem. Biophys., 1989, 275, 559–567 CrossRef CAS.
- M. Mittal, M. R. Siddiqui, K. Tran, S. P. Reddy and A. B. Malik, Antioxid. Redox Signaling, 2014, 20, 1126–1167 CrossRef CAS.
- S. Joshi-Barr, C. de Gracia Lux, E. Mahmoud and A. Almutairi, Antioxid. Redox Signaling, 2014, 21, 730–754 CrossRef CAS.
- D. S. Hernandez, H. C. Schunk, K. M. Shankar, A. M. Rosales and L. J. Suggs, Nanoscale Adv., 2020, 2, 3849–3857 RSC.
- H. C. Schunk, D. S. Hernandez, M. J. Austin, K. S. Dhada, A. M. Rosales and L. J. Suggs, J. Mater. Chem. B, 2020, 8, 3460–3487 RSC.
- S. H. Lee, T. C. Boire, J. B. Lee, M. K. Gupta, A. L. Zachman, R. Rath and H. J. Sung, J. Mater. Chem. B, 2014, 2, 7109–7113 RSC.
- F. El-Mohtadi, R. D'Arcy and N. Tirelli, Macromol. Rapid Commun., 2019, 40, 1800699 CrossRef.
- S. S. Yu, R. L. Koblin, A. L. Zachman, D. S. Perrien, L. H. Hofmeister, T. D. Giorgio and H. Sung, Biomacromolecules, 2011, 12, 4357–4366 CrossRef CAS.
- S. K. Lee, L. J. Mortensen, C. P. Lin and C. H. Tung, Nat. Commun., 2014, 5, 1–8 Search PubMed.
- M. A. Stahmann, E. Tsuyuki and H. Tsuyuki, J. Biol. Chem., 1956, 222, 479–485 CrossRef CAS.
- L. Guo, J. Li, Z. Brown, K. Ghale and D. Zhang, Biopolymers, 2011, 96, 596–603 CrossRef CAS PubMed.
- R. N. Zuckermann, J. M. Kerr, W. H. Moosf and S. B. H. Kent, J. Am. Chem. Soc., 1992, 114, 10646–10647 CrossRef CAS.
- S. Østergaard and A. Holm, Mol. Diversity, 1997, 3, 17–27 CrossRef.
- A. M. Rosales, R. A. Segalman and R. N. Zuckermann, Soft Matter, 2013, 9, 8400–8414 RSC.
- A. S. Knight, E. Y. Zhou, M. B. Francis and R. N. Zuckermann, Adv. Mater., 2015, 27, 5665–5691 CrossRef CAS.
- A. Battigelli, Biopolymers, 2019, 110, e23265 CrossRef PubMed.
- R. Luxenhofer, C. Fetsch and A. Grossmann, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2731–2752 CrossRef CAS.
- K. H. A. Lau, Biomater. Sci., 2014, 2, 627–633 RSC.
- S. M. Miller, R. J. Simon, S. Ng, R. N. Zuckermann, J. M. Kerr and W. H. Moos, Drug Dev. Res., 1995, 35, 20–32 CrossRef CAS.
- S. M. Miller, R. J. Simon, S. Ng, R. N. Zuckermann, J. M. Kerr and W. H. Moos, Bioorg. Med. Chem. Lett., 1994, 4, 2657–2662 CrossRef CAS.
- E. R. Stadtman and R. L. Levine, Amino Acids, 2003, 25, 207–218 CrossRef CAS PubMed.
- A. S. Culf and R. J. Ouellette, Molecules, 2010, 15, 5282–5335 CrossRef CAS PubMed.
- B. C. Lee, R. N. Zuckermann and K. A. Dill, J. Am. Chem. Soc., 2005, 127, 10999–11009 CrossRef CAS PubMed.
- A. Amici, R. L. Levine, L. Tsai and E. R. Stadtman, J. Biol. Chem., 1989, 264, 3341–3346 CrossRef CAS.
- C. G. Knight, F. Willenbrock and G. Murphy, FEBS Lett., 1992, 296, 263–266 CrossRef CAS.
- T. Nguyen, C. de Gracia Lux, S. Joshi-Barr, A. Almutairi, N. Fomina, E. Schopf and E. Mahmoud, J. Am. Chem. Soc., 2012, 134, 15758–15764 CrossRef PubMed.
- D. K. Dempsey, C. Carranza, C. P. Chawla, P. Gray, J. H. Eoh, S. Cereceres and E. M. Cosgriff-Hernandez, J. Biomed. Mater. Res., Part A, 2014, 102, 3649–3665 CrossRef PubMed.
- M. Lange, S. Braune, K. Luetzow, K. Richau, N. Scharnagl, M. Weinhart, A. T. Neffe, F. Jung, R. Haag and A. Lendlein, Macromol. Rapid Commun., 2012, 33, 1487–1492 CrossRef CAS.
- P. Wardman, Free Radical Biol. Med., 2007, 43, 995–1022 CrossRef CAS.
- C. Nathan and A. Cunningham-Bussel, Nat. Rev. Immunol., 2013, 13, 349–361 CrossRef CAS PubMed.
- S. G. Rhee, T. S. Chang, W. Jeong and D. Kang, Mol. Cells, 2010, 29, 539–549 CrossRef CAS PubMed.
- C. C. Winterbourn, Biochim. Biophys. Acta, Gen. Subj., 2014, 1840, 730–738 CrossRef CAS PubMed.
- C. C. Winterbourn, Nat. Chem. Biol., 2008, 4, 278–286 CrossRef CAS.
- M. B. Grisham, Comp. Biochem. Physiol., Part A: Mol. Integr. Physiol., 2013, 165, 429–438 CrossRef CAS PubMed.
- J. R. Requena, C. C. Chao, R. L. Levine and E. R. Stadtman, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 69–74 CrossRef CAS.
- I. M. Møller, A. Rogowska-Wrzesinska and R. S. P. Rao, J. Proteomics, 2011, 74, 2228–2242 CrossRef.
- M. R. McCall and B. Frei, in Developments in Cardiovascular Medicine, 2000, pp. 75–98 Search PubMed.
- T. A. Wynn and K. M. Vannella, Immunity, 2016, 44, 450–462 CrossRef CAS.
- J. M. Chen, E. S. Radisky and C. Férec, Handb. Proteolytic Enzymes, 2013, 3, 2600–2609 Search PubMed.
- C. L. Hawkins and M. J. Davies, J. Biol. Chem., 2019, 294, 19683–19708 CrossRef CAS PubMed.
- J. Uranga, J. I. Mujika, R. Grande-Aztatzi and J. M. Matxain, J. Phys. Chem. B, 2018, 122, 4956–4971 CrossRef CAS PubMed.
- M. Poreba, A. Szalek, W. Rut, P. Kasperkiewicz, I. Rutkowska-Wlodarczyk, S. J. Snipas, Y. Itoh, D. Turk, B. Turk, C. M. Overall, L. Kaczmarek, G. S. Salvesen and M. Drag, Sci. Rep., 2017, 7, 1–13 CrossRef.
- S. R. K. Ainavarapu, J. Brujić, H. H. Huang, A. P. Wiita, H. Lu, L. Li, K. A. Walther, M. Carrion-Vazquez, H. Li and J. M. Fernandez, Biophys. J., 2007, 92, 225–233 CrossRef CAS PubMed.
- B. L. Zerfas, R. A. Coleman, A. F. Salazar-Chaparro, N. J. Macatangay and D. J. Trader, ACS Chem. Biol., 2020, 15, 2588–2596 CrossRef CAS.
- Y. Yamawaki, T. Yufu and T. Kato, Processes, 2021, 9, 242 CrossRef CAS.
- M. J. Austin, H. C. Schunk, C. M. Watkins, N. R. Ling, J. M. Chauvin, L. D. Morton and A. M. Rosales, bioRxiv, 2022, preprint, DOI:10.1101/2022.08.31.506126.
- R. J. Simpson, Cold Spring Harb. Protoc., 2006, 2006, pdb.prot4550 CrossRef PubMed.
- J. Lugrin, N. Rosenblatt-Velin, R. Parapanov and L. Liaudet, Biol. Chem., 2014, 395, 203–230 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2me00179a |
| ‡ McKetta Department of Chemical Engineering, University of Texas at Austin, Austin, TX 78712, USA. |
| This journal is © The Royal Society of Chemistry 2023 |