
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An overview of metal-free synthetic routes to isoxazoles: the privileged scaffold
Soumyadip Das
and
Kaushik Chanda
 *
*
Department of Chemistry, School of Advanced Sciences, Vellore Institute of Technology, Vellore-632014, India. E-mail: chandakaushik1@gmail.com
First published on 6th October 2021
Abstract
In the field of drug discovery, isoxazole is a five-membered heterocyclic moiety commonly found in many commercially available drugs. In view of their enormous significance, it is always imperative to unleash new eco-friendly synthetic strategies. Among various novel synthetic techniques in use for isoxazole synthesis, most synthetic methods employ Cu(I) or Ru(II) as catalysts for (3 + 2) cycloaddition reaction. The particular disadvantages associated with metal-catalyzed reactions are high costs, low abundance, toxicity, a significant generation of waste, and difficulty to separate from the reaction mixtures. In view of these drawbacks, it is always imperative to develop alternate metal-free synthetic routes. This review article highlights a comprehensive overview on the potential application of metal-free synthetic routes for the synthesis of isoxazoles with significant biological interests.
 Soumyadip Das | Soumyadip Das received his BSc degree from University of Calcutta, India, in 2017 and MSc degree from Vellore Institute of Technology, Vellore, India, in 2019. He is currently pursuing his PhD degree from Vellore Institute of Technology, Vellore, India, under the supervision of Dr Kaushik Chanda. His research interest includes diversity-oriented synthesis, anticancer drug design, and nanocatalysis. |
 Kaushik Chanda | Dr Kaushik Chanda, obtained his MSc in Organic Chemistry from Gauhati University, India in 2001. Subsequently, he worked as a Senior Research Fellow in an ICAR-NATP funded project in St Anthonys College, Shillong, India from 2002 to 2005. In 2006, he moved to Taiwan to pursue a PhD in Applied Chemistry from National Chiao Tung University under the guidance of Prof Chung Ming Sun on the topic of Combinatorial Chemistry. In 2010, he moved to the Department of Chemistry, National Tsing Hua University, Taiwan for an NSC-postdoctoral fellowship in facet dependent organic catalysis with Prof Michael H. Y. Huang. Now, he is working as a Senior Assistant Professor in Department of Chemistry, Vellore Institute of Technology, Vellore. His research interest includes diversity-oriented synthesis, anticancer drug design, drug delivery, sensing applications, and nanocatalysis. |
Introduction
A wide variety of functionalized heterocyclic scaffolds and their synthesis are significant to medicinal chemists as they provide the ability to expand the available drug-like chemical space, which bind to the biological targets based on their chemical diversity.1a,b Furthermore, it is highly desirable to develop robust synthetic methods for the generation of a diverse collection of heterocyclic molecules to accelerate the drug discovery programme. Isoxazole, a five-membered heterocyclic pharmacophore is widely used as a crucial moiety in drug discovery research.1c Functionalized isoxazole scaffolds show different biological activities such as anticancer, as potential HDAC inhibitors, antioxidant, antibacterial, and antimicrobial activity.2 Evidently, the core structure of isoxazole has been found in many drugs such as sulfamethoxazole3a A that acts as an antibiotic, muscimol3b B that acts as GABAA, ibotenic acid3c C that acts as a neurotoxin, parecoxib3d D that acts as a COX2 inhibitor, and leflunomide3e E that acts as an immunosuppressant agent (Fig. 1). Moreover, isoxazole derivatives are an important part of small chemical entities, which exists in synthetic goods of day-to-day use.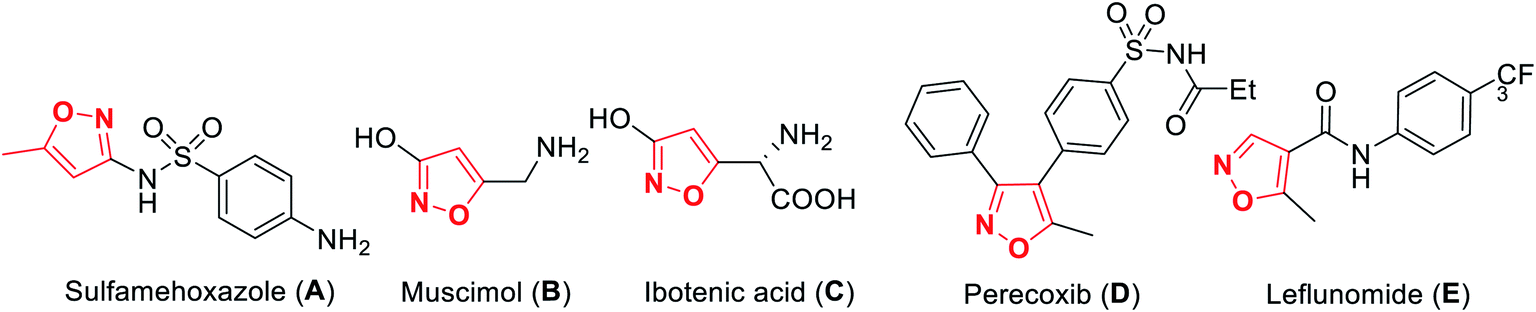 | ||
| Fig. 1 Biologically-active isoxazole-based drug molecules. | ||
Isoxazoles can be synthesized via different pathways using both homogeneous as well as heterogeneous catalysts. Nevertheless, the most broadly researched and reported synthesis of isoxazole derivative is through the (3 + 2) cycloaddition reaction of an alkyne that acts as a dipolarophile and nitrile oxide as the dipole.4 Two predicted mechanisms have been reported for the 1,3-dipolar cycloaddition reaction—firstly, pericyclic cycloaddition reaction via concerted mechanism5a and secondly, via a step-by-step mechanism through diradical intermediate formation.5b Subsequently, the first proposed idea has been accepted, i.e., concerted pathway, via the reaction of the dipole and the dipolarophile (Fig. 2). In 2001, Sharpless and his co-workers described this kind of cycloaddition reaction as ‘Click Chemistry’ for the regioselective synthesis of disubstituted triazoles.6 In 2005, Fokin et al. demonstrated the synthesis of azoles via copper(I) catalysis and the DFT study was performed to predict the extraordinary reactivity.7 Subsequently, in 2008, Müller et al. reported the Pd catalyzed Sonogashira coupling of acid chlorides with terminal alkynes, followed by the 1,3-dipolar cycloaddition with in situ prepared nitrile oxides, which resulted in isoxazoles in good yields via a one-pot three-component reaction.8 Later on, Fokin and his group developed the ruthenium-catalyzed synthetic strategy for the synthesis of 3,4-disubstituted isoxazoles via the (3 + 2) cycloaddition reaction.9 In 2010, DeKorver et al. reported various 1,3-dipolar cycloaddition reactions using copper(II) catalysts such as copper acetate and copper sulfate pentahydrate.10 In the same year, Evano and his group described the copper(II)-catalyzed 1,3-dipolar cycloaddition reaction using ynamides as a new building block for the synthesis of isoxazoles.11 In 2013, we studied the facet-dependent catalytic potential of Cu2O nanocrystals as heterogeneous catalysts for the synthesis of isoxazoles.12 In the year 2015, Szostak and his co-workers reported different synthetic strategies for the synthesis of substituted isoxazoles using metal catalysts.13 In 2018, Nakamura and his group also reported the recent progresses in the synthesis of functionalized isoxazoles via 1,3-dipolar cycloaddition, condensation, cycloisomerization, and direct functionalization reactions.14 Very recently, Li and his group reported the synthetic sequence leading to isoxazoles via the metal-catalyzed cyclization/functionalization of alkynes.15 Subsequently, our group demonstrated the microwave-assisted green synthetic pathway for the synthesis of 3,5-disubstituted isoxazoles using a Cu(I) catalyst.16
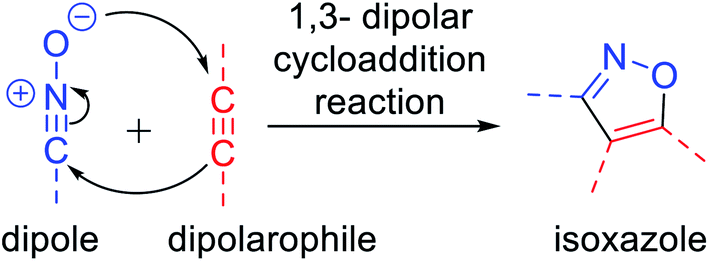 | ||
| Fig. 2 Mechanism of 1,3-dipolar cycloaddition reaction. | ||
It is well known that metal catalysts and organometallic reagents are widely used for academic research as well as industrial purposes. The metal-mediated synthesis of heterocyclic moieties increases day by day. From the physiological point of view, metals can be divided into important classes, which are required compulsorily in the body for some purpose, and the other ones are non-essential, which are absent in organisms. The second class is different from the previous class as metals that execute toxic effects. However, it has been accepted that essential as well as non-essential metals are simultaneously dangerous, if received in excess.17 Homogeneous catalysts are often transition metal complexes and organometallic complexes with high efficiency as well as high selectivity under any reaction environment. However, in reality, the separation of such a metal catalyst from the reaction mixture is very difficult with the generation of waste. Hence, heterogeneous catalysts are generally measured to be technically beneficial over homogeneous catalysts.18 However, a considerable interest has been generated for the synthesis of heterocyclic moieties in the metal-free pathway. The synthetic methodology proceeds very well at room temperature conditions with a good range of products in moderate to excellent yields having excellent chemo- and regioselectivity. Further, the metal-free synthetic reactions avoid the requirement of inert gas, cryogenics, as well as toxic metal contamination, making them useful for the generation of diverse molecular architectures.
In continuation of our efforts to explore the potential application of synthesized bioactive compounds in our laboratory,19 herein, we report metal-free synthetic routes to isoxazoles and challenges to the future design of isoxazoles as the drug molecule in this review. The synthetic protocols reported in this review provide detailed information of the protocols initiated from the metal-free solid phase and green synthetic processes. Furthermore, assembling all synthetic manipulations on one platform will provide key solutions to resolve the current synthetic problems and also promote knowledge about the current synthetic progress in the synthesis of isoxazole analogs. Subsequently, it may be possible to develop new concepts and increase the diversity of synthetic routes to isoxazole moieties.
Metal-free solid phase synthesis of isoxazoles
In 2010, Leonetti and his co-workers developed five-membered heterocyclic ligands having a potential binding feature for a therapeutically interesting enzyme. The final aim was to find new compounds having excellent inhibitory activity toward PKTs or other enzymes such as COXs. All the molecules were synthesized through the solid support using Rink amide resin. Initially, polymer-bound 3-hydroxybenzamide 3 was synthesized with the reaction of Rink amide resin 1 and 3-hydroxybenzoic acid 2 using EDC·HCl as the coupling agent in the presence of dry DMF as the solvent under room temperature condition for 12 h. 91% yield was obtained for intermediate 3.In the coupling step, the use of HOBt and DICI as the coupling agents produced a low yield of 78%. Next, different bromomethyl ketones were reacted with solid-supported 3-hydroxybenzamide 3 using 10% mixture of HMPA in DMF with DIPEA as the base under microwave irradiation for excellent yields of the polymer-bound intermediate 4. However, the use of the base DBU in this step reduced the reaction yield to 67%. The next step involved the synthesis of the polymer-bound enaminoketones 5 by the reaction of polymer conjugates 4 with DMFDMA. For the best result, dry DMF was used as the solvent under microwave irradiation at 120 °C for 1 h, leading to quantitative yields. In the final step, five-membered heterocyclization was done by reacting intermediate 5 with hydroxylamine hydrochloride using a solvent mixture (DMF/i-PrOH, 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) under microwaves at 90 °C for 30 min, followed by cleavage. Finally, the five-membered isoxazoles 6 were obtained in 50–70% yield (Scheme 1).20
:1) under microwaves at 90 °C for 30 min, followed by cleavage. Finally, the five-membered isoxazoles 6 were obtained in 50–70% yield (Scheme 1).20
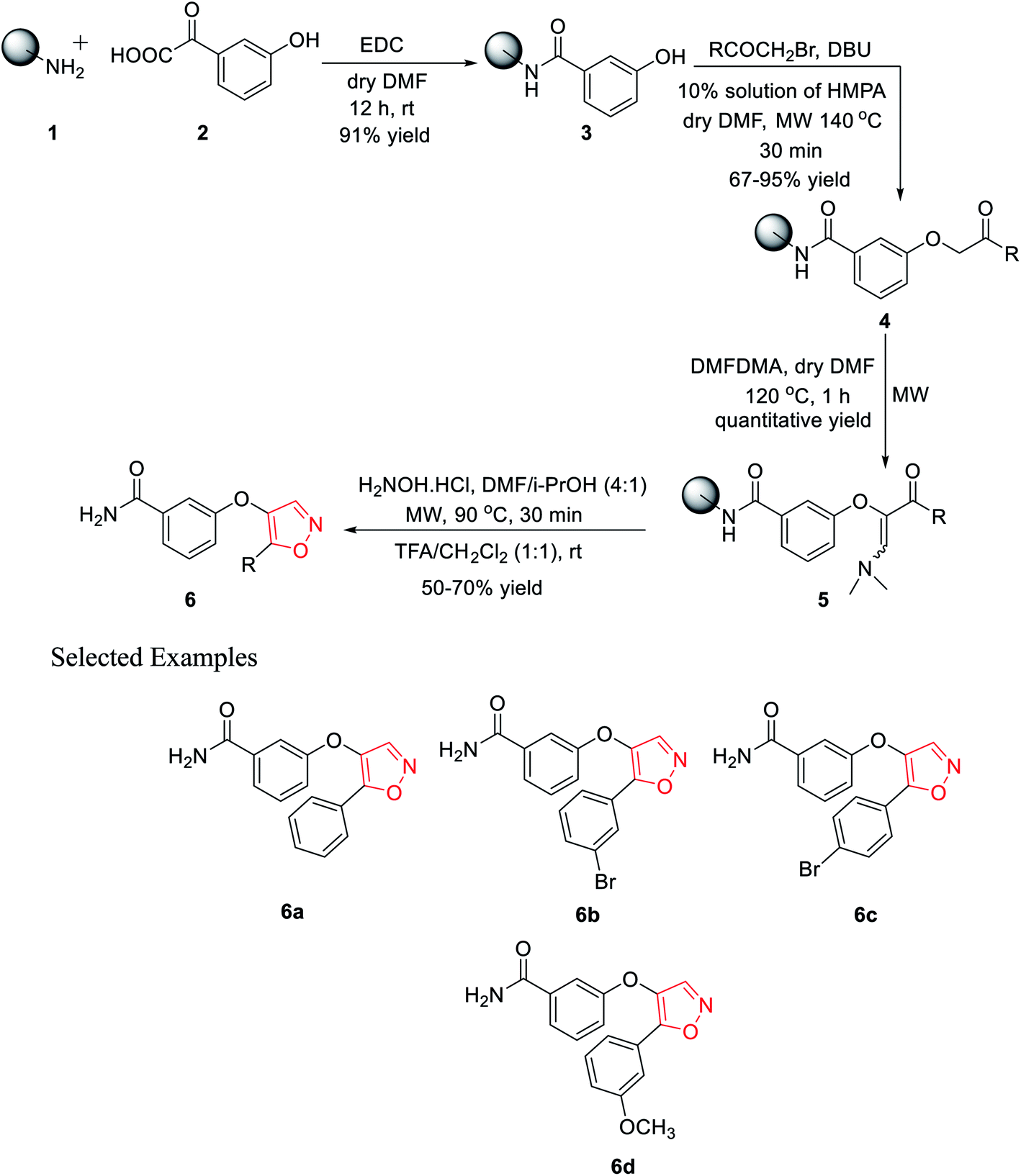 | ||
| Scheme 1 Microwave-assisted solid phase synthesis of 4,5-disubstituted isoxazoles. | ||
In the year 2012, Nefzi and his group introduced the diversity-oriented synthesis of isoxazole derivatives by the 1,3-dipolar cycloaddition reaction of alkyne and in situ developed nitrile oxides.21 The necessary alkyne precursors were developed from the different derivatives of resin-supported carboxylic acids. The solid phase synthesis of all the molecules was done using Houghten's tea-bag approach as the resin was supported with a closed polypropylene mesh.
The first diversity was presented using different carboxylic acid derivatives (examples: cyclopentanecarboxylic acid, benzoic acid, 1-phenyl-1-cyclopentanecarboxylic acid, 4-biphenylcarboxylic acid, 2-nitrobenzoic acid, piperonylic acid, 1-napthalenecarboxylic, diphenylacetic acid, syringyl, 1-phenyl-1-cyclopropylcarboxylic acid, and 1-cyclopenteneacetic acid) and second diversity was introduced using different N-hydroxybenzimidoyl chlorides 9 (Fig. 3).
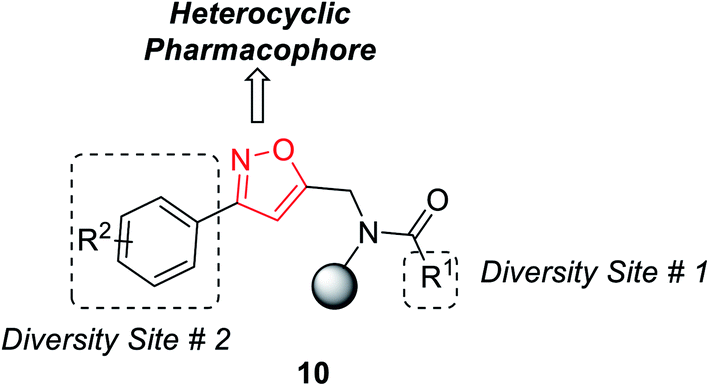 | ||
| Fig. 3 Diversity points of substituted isoxazoles. | ||
Resin-supported carboxylic acid was reacted with propargyl bromide for 5 h and the produced solid-supported secondary amide 8 in the presence of lithium t-butoxide and DMSO as the solvent. Resin-bound isoxazoles 10 were synthesized by the cycloaddition reaction of resin-bound alkynes 8 and in situ generated nitrile oxide of the corresponding N-hydroxybenzimidoyl chloride derivatives 9. Exceptionally, for resin-supported piperonylic acid, original product along with a diol as the side product were obtained. Finally, the cleavage of the resin from resin-bound isoxazoles was achieved in anhydrous HF for 90 min at 0 °C and finally the targeted product 11 was obtained (Scheme 2).21
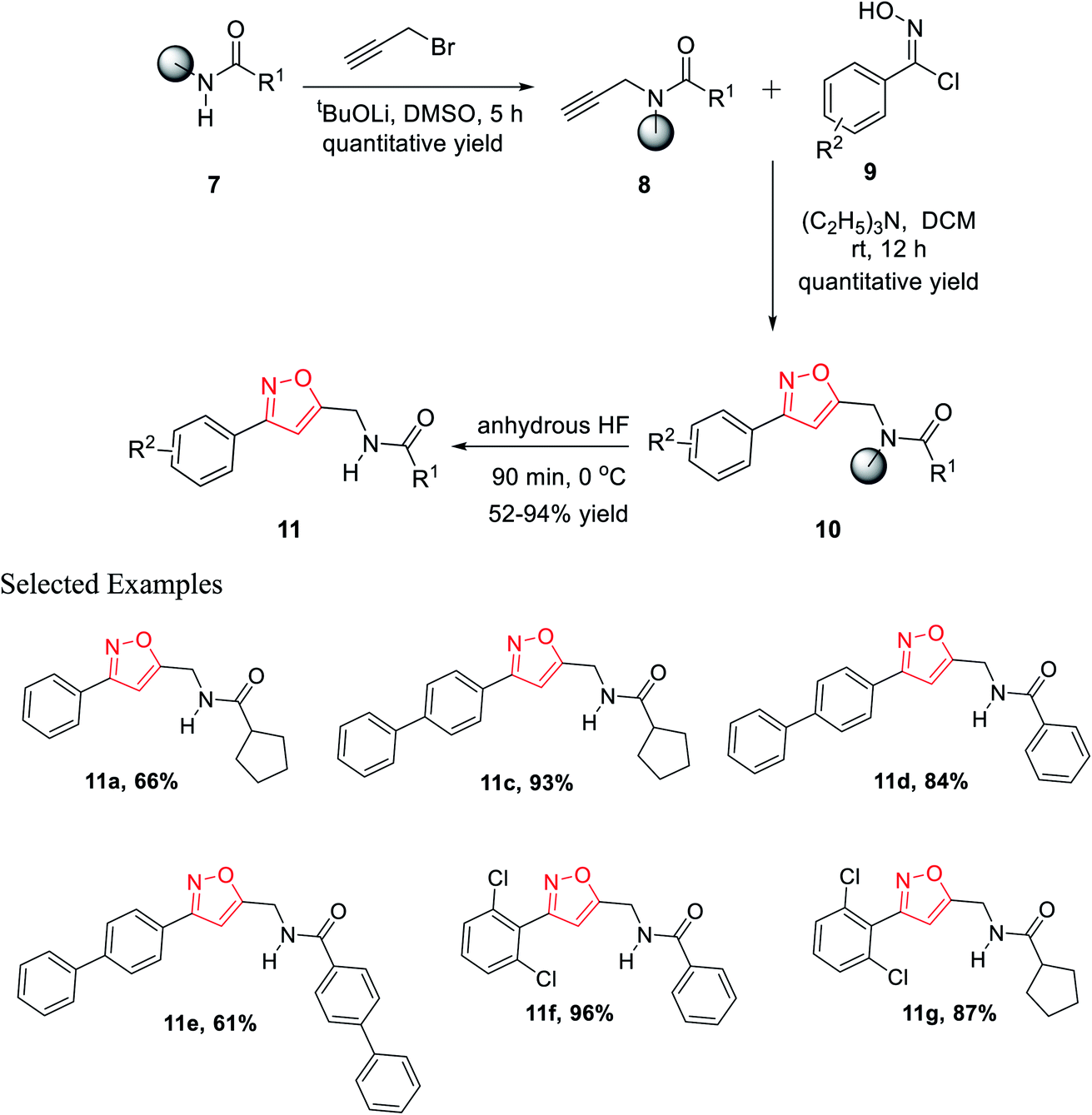 | ||
| Scheme 2 Solid-supported formation of functionalized isoxazoles. | ||
Microwave-assisted metal-free routes to substituted isoxazoles
In 2010, Micheli et al. reported the synthesis of isoxazole derivatives by the 1,3-dipolar cycloaddition reaction. For a successful 1,3-dipolar cycloaddition reaction, one 1,3-dipole and one dipolarophile is required; in this case, 14 and 15 act as the 1,3-dipole and ethyl-2-chloro-2-(hydroxyimino)acetate acts as the dipolarophile.The initial precursor alkynes 14 and 15 were synthesized by the reaction of carboxylic acids 12 and 13 with aniline under conventional coupling conditions using DCC and DMAP as the coupling agents. The in situ formed ethoxycarbonyl formonitrile oxide from ethyl-2-chloro-2-(hydroxyimino)acetate was reacted with dipolarophiles 14 and 15 under microwave condition to obtain ester-functionalized isoxazoles 16 and 17 in quantitative yields. Finally, the ester-functionalized isoxazoles were converted hydroxylamine functionalized isoxazole derivatives 18 and 19 by reacting with hydroxylamine hydrochloride in methanolic KOH solution (Scheme 3). All the synthesized derivatives underwent biological fruition to recognize the molecule furnished with histone deacetylase (HDAC) inhibitory action. HDAC inhibitors are a new class of potential anticancer agents, which play a crucial role in epigenetic or non-epigenetic regulation, apoptosis, cell death, and cell cycle arrest in cancer cells. Compound 19 with an isoxazole skeleton has been elected to have inhibitory activity at various HDAC isoforms. The particular compound exhibited 10–70 folds lower IC50s at HDAC-6 than HDAC 10, 3, 2, and 1.22
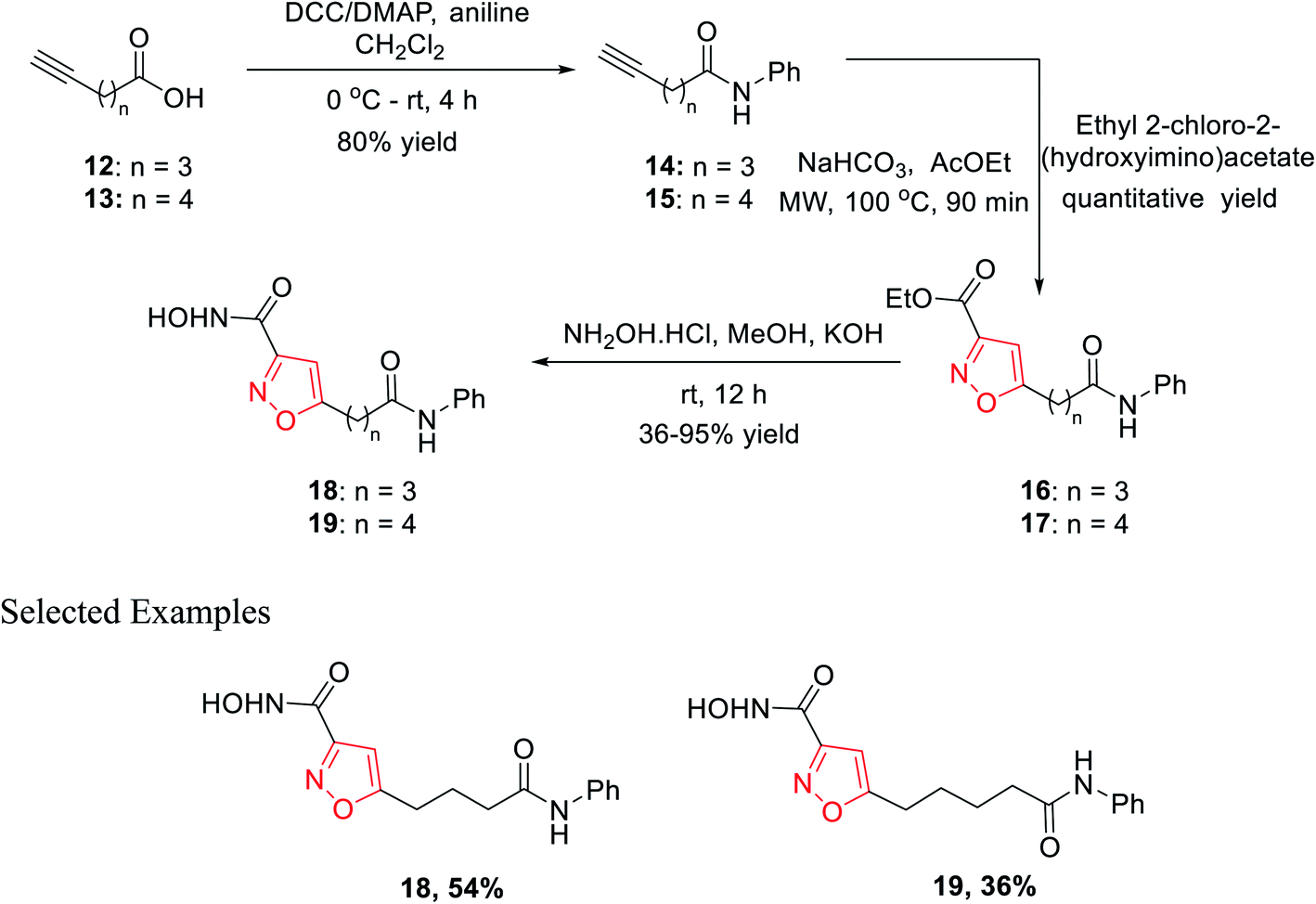 | ||
| Scheme 3 Microwave-assisted metal-free routes to 3,5-disubstituted isoxazoles. | ||
In year 2014, Chondrogianni and co-workers developed a fresh pathway for the synthesis of 3,5-disubstituted isoxazole via microwave irradiation. Initially, aryl aldehydes 20 were reacted with hydroxylamine hydrochloride and furnished the corresponding oximes 21. Subsequently, oxime derivatives 21 were reacted with TsN(Cl)Na·3H2O in the presence of tert-butyl alcohol at room temperature, which yielded the most important nitrile oxide intermediate.
In the presence of microwave irradiation, functionalized alkynes 22 were reacted with nitrile oxides, which afforded the corresponding substituted isoxazoles 23, while no copper catalysts were used in the reaction medium (Scheme 4). Isoxazole derivatives were tested for the anti-ageing and/or antioxidant properties. The test was performed by following two models—one is the nematode C. elegans (in vivo model) and other one is human primary fibroblasts (in vitro model). Among all the derivatives, compound 23f displayed excellent antioxidant properties compared to the typical antioxidant molecules, for example, quercetin.23
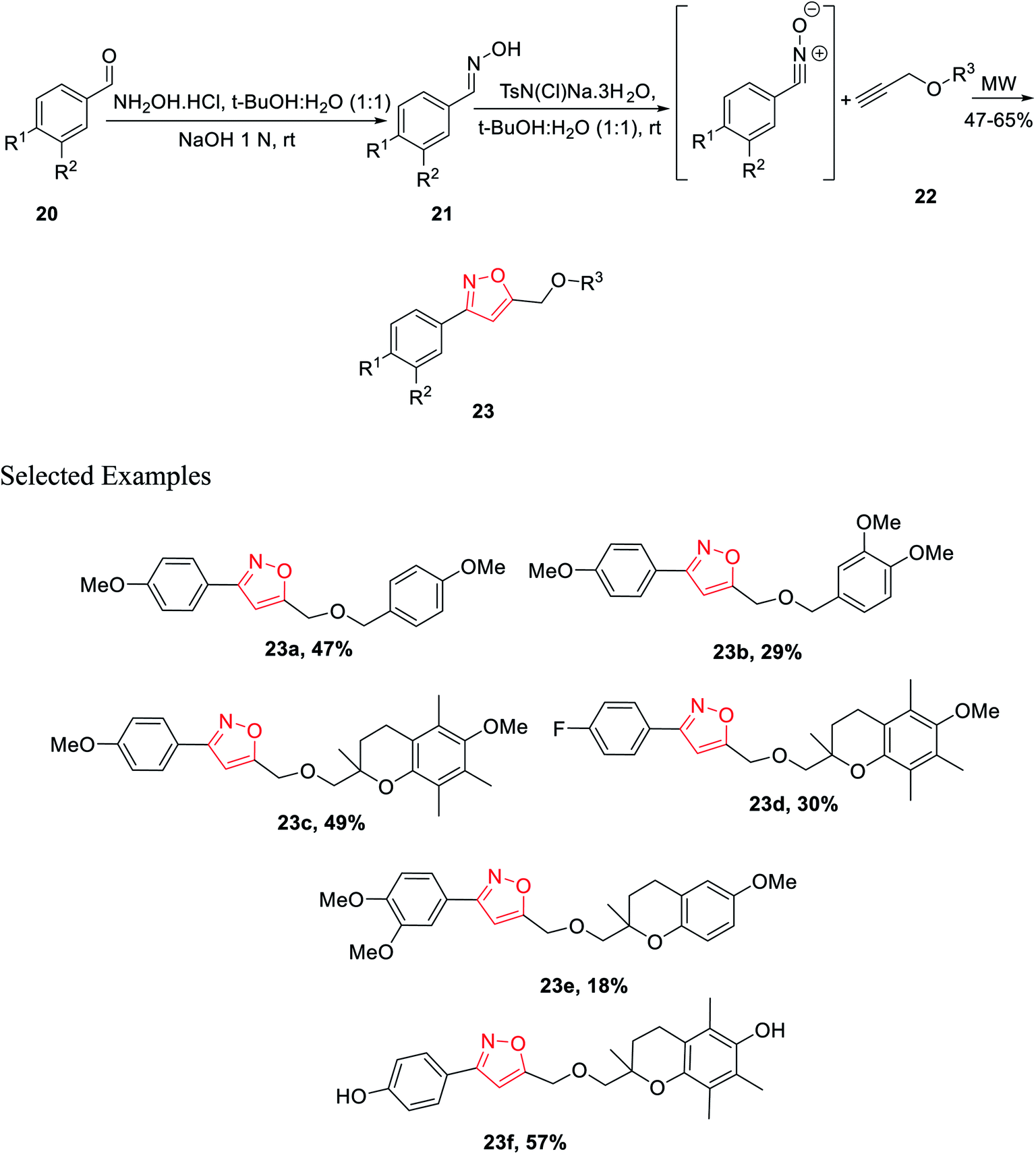 | ||
| Scheme 4 Microwave-assisted synthesis of 3,5-disubstituted isoxazoles. | ||
In the subsequent year, Tiwari et al. reported an extremely regioselective superficial synthesis of unique 3,5-disubstituted isoxazole-linked glyco-conjugates 26 by exploiting the (3 + 2) cycloaddition reaction. The procedure benefited with a prompt and modest way for the initiation of the isoxazole ring at various places of the sugar derivatives with seeming ease. This is a capable method that has been introduced to synthesize the isoxazole-linked glyco-conjugate in the area of carbohydrate chemistry. To synthesize the desired molecule isoxazole-linked glycoconjugate 26, a retrosynthetic study was performed (Scheme 5). D-Glucose 24 was converted to the corresponding glycosyl-β-olefinic ester 25 via a series of reactions, followed by the reaction with nitromethane to obtain intermediate 26, which underwent the (3 + 2) cycloaddition reaction with alkynes to obtain the target derivatives 27.
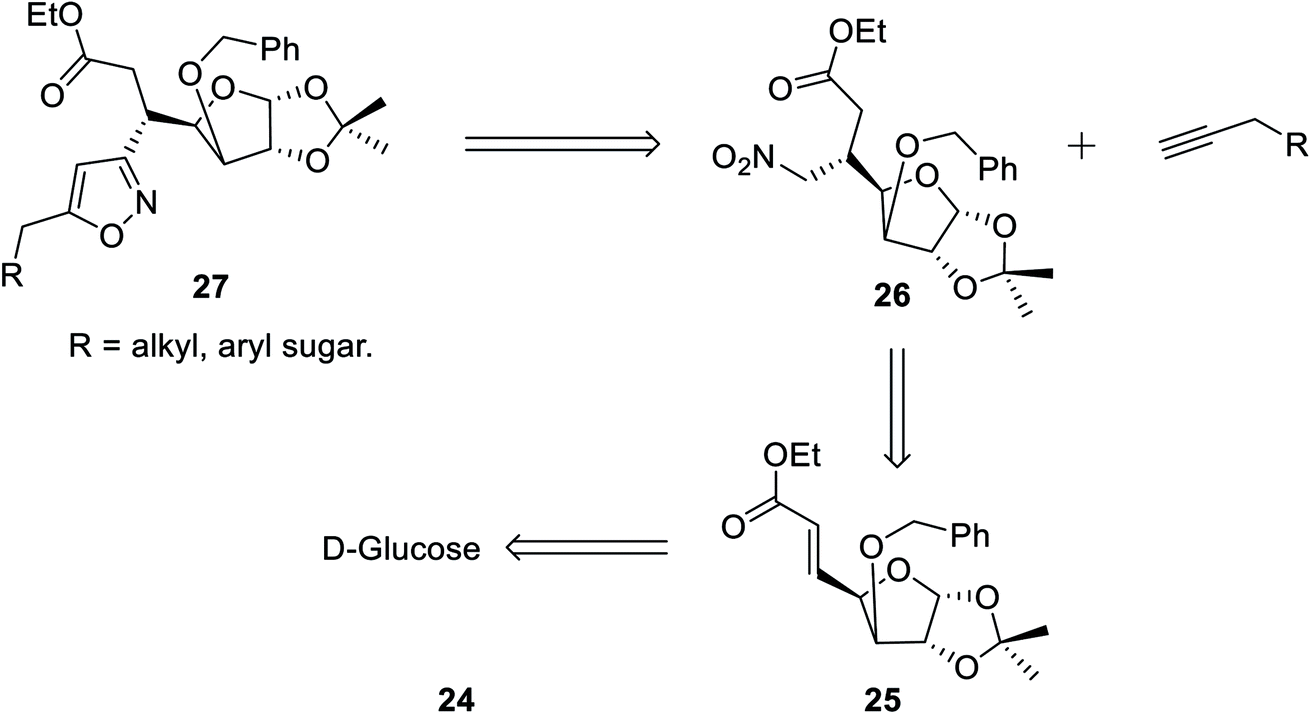 | ||
| Scheme 5 Retrosynthetic analysis for the synthesis of isoxazole-linked glyco-conjugates 27. | ||
The synthetic procedure was initiated with inexpensive and easily affordable D-glucose 24, which subsequently underwent a sequence of reactions, such as the protection of isopropylidene, protection of 3-OH using benzyl group, deprotection of 5,6-isopropylidene, oxidation by NaIO4, and lastly, olefination via the HEW-Wittig method, which yielded glycosyl olefinic ester (1R,2R,3S,4R)-ethyl-[3-O-benzyl-5,6-dideoxy-1,2-O-isopropylidene-D-gluco]-heptfuran-5 enuronate 25.24a,b Compound 25 was refluxed with nitromethane in anhydrous ethanolic medium with K2CO3 as the base for 6 h, which yielded (1R,2R,3S,4R,5R)-ethyl-[3-O-benzyl-5,6-dideoxy-1,2-O-isopropylidene-5-nitromethyl]-b-L-idoheptofuranurnate 26 (Scheme 6). Compound 26 was reacted with alkynes in the presence of 18-crown-6 catalyst, K2CO3, and 4-toluenesulfonyl chloride at 80 °C for 8–10 h resulted the isoxazole-linked glyco-conjugates 27. However, the application of microwave irradiation reduced the reaction time to 15–20 min at 110 °C to obtain isoxazole-linked glycol-conjugates 27 in good yield. The diversity was introduced in compounds 27 by varying the R groups, i.e., varying different sugars, such as monosaccharides like D-mannose, D-galactose, D-glucose, D-fructose, and D-ribose.24c
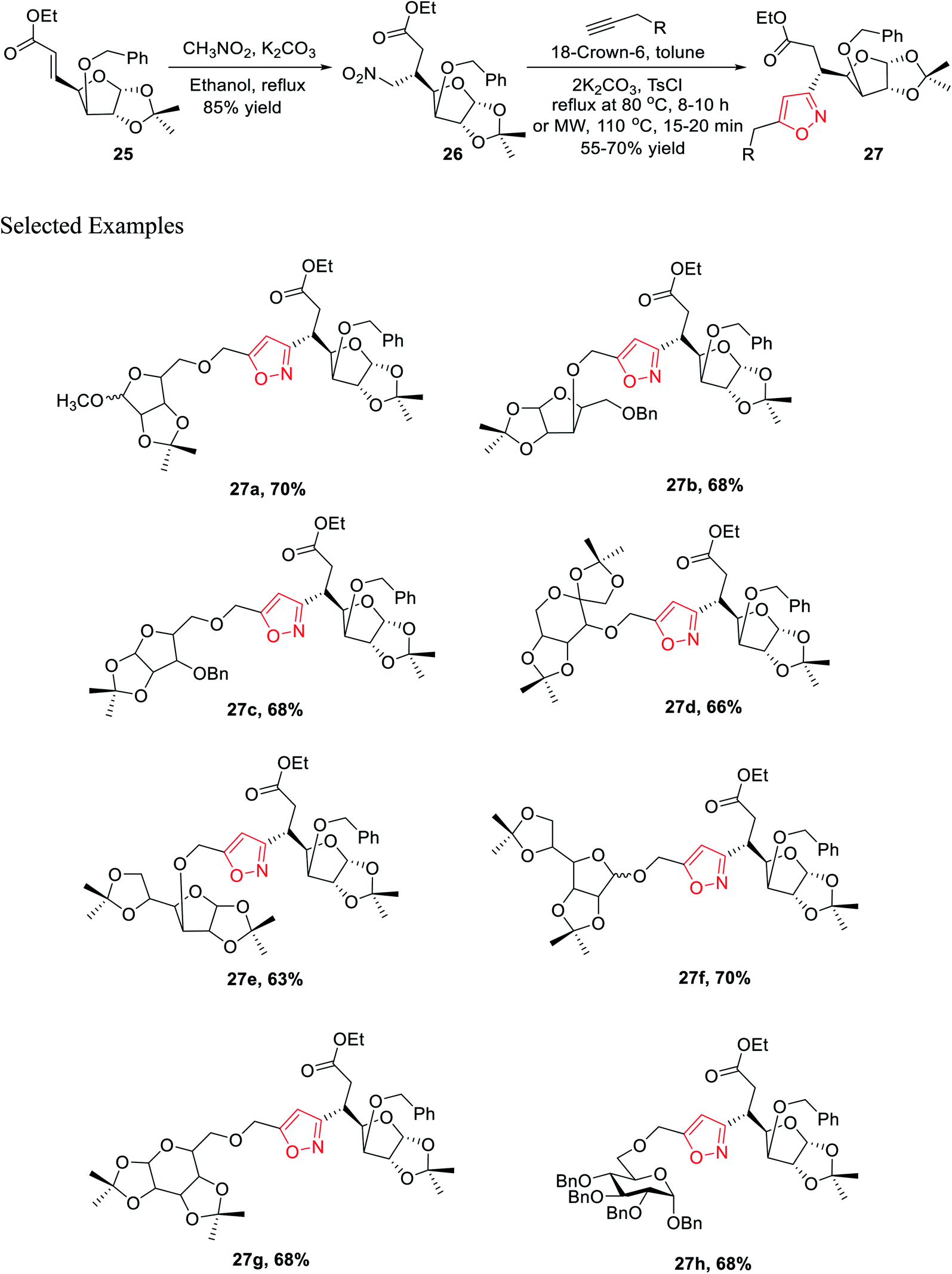 | ||
| Scheme 6 Microwave-assisted regioselective synthesis of unique isoxazole-linked glyco-conjugates. | ||
Metal-free synthesis of isoxazoles via miscellaneous methods
In 2016, Tanyeli's group accomplished a novel method for the formation of isoxazole derivatives by a one-pot cascade reaction via the ultrasonication method.25 To synthesize the isoxazole derivatives 30, ethyl nitroacetate 29 and aromatic aldehyde 28 were reacted in water using 20 mol% DABCO as the catalyst at 80 °C for 24 h under ultrasonication (Scheme 7).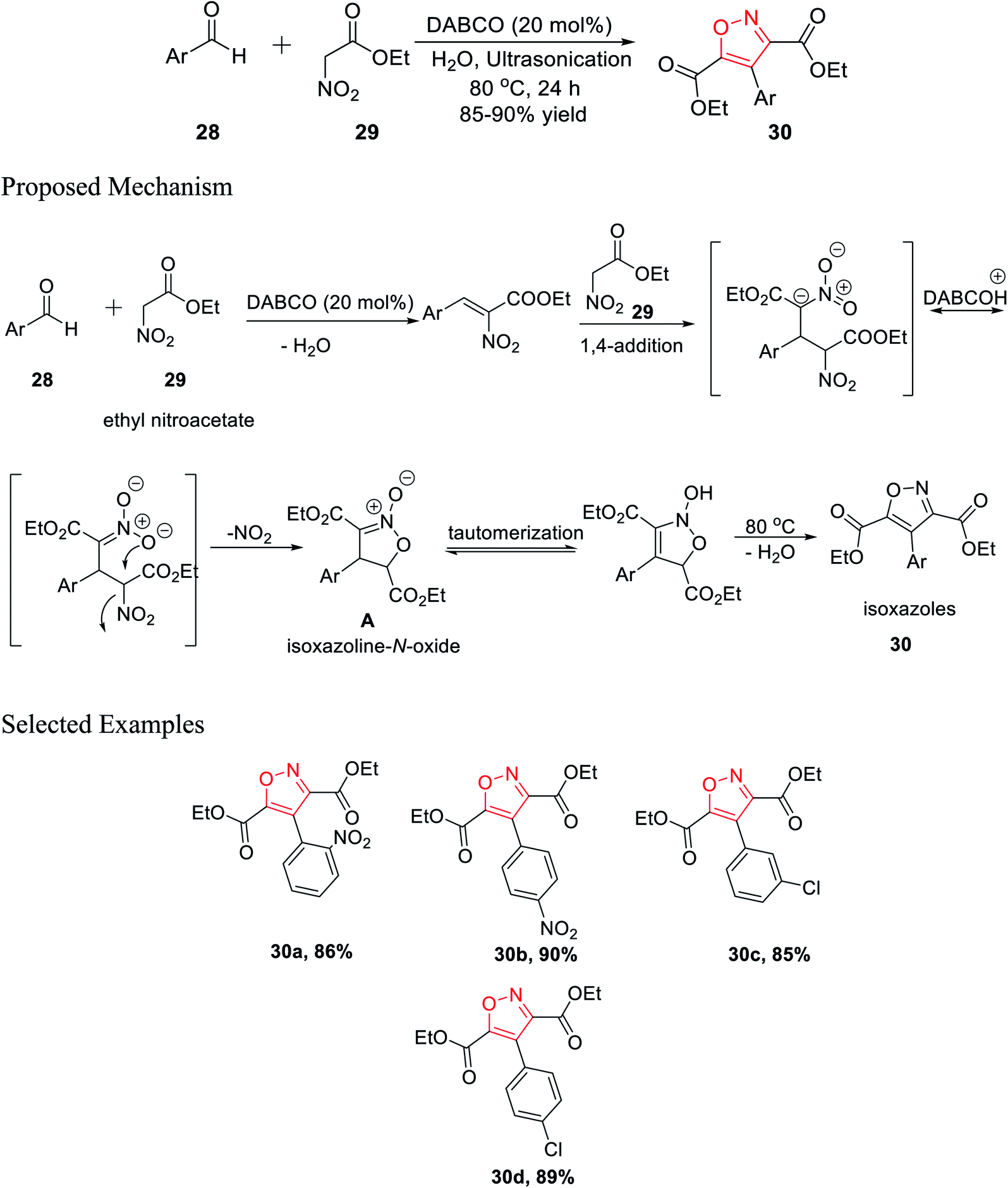 | ||
| Scheme 7 Synthesis of poly substituted isoxazoles via a one-pot cascade reaction. | ||
Different kinds of solvent were used such as toluene, hexane, methanol, ethanol, DCM, chloroform, DMF, DMSO, and, THF but the best result was obtained using water as the solvent. Mechanistically, during the reaction, the corresponding isoxazoline N-oxide intermediate A was isolated, which leads to the final product isoxazoles 30 via tautomerism and dehydration. Diversity has been initiated by changing the Ar group on compound 28.25
Subsequently, in 2011, Rai et al. showed a novel method for the synthesis of isoxazole derivatives and to evaluate their biological activity. To synthesize the privileged scaffold, malonodihydrazide 33 was taken as the starting material, which was synthesized by reacting diethylmalonate 32 with hydrazine hydrate in 1:2 molar ratio under reflux condition for 1.5 h using ethanol as the solvent. The product malonodihydrazide 33 was reacted with different aromatic aldehydes 34 in 1:2 molar ratio under refluxing condition for 30 min in ethanol solvent, which resulted in diaroylhydrazones 35. Further, diaroylhydrazones 35 were reacted with hydroxylamine hydrochloride under refluxing condition for 3 h in ethanol solvent, which led to the final product isoxazoles 36 (Scheme 8). Next, in vitro studies of compounds 36 were carried out for antibacterial and antioxidant activities. Compound 36 with N,N-dimethyl substituents at the para position showed dominant anti-lipid peroxidation, antioxidant, and higher antibacterial activity as compared to standard drugs.26
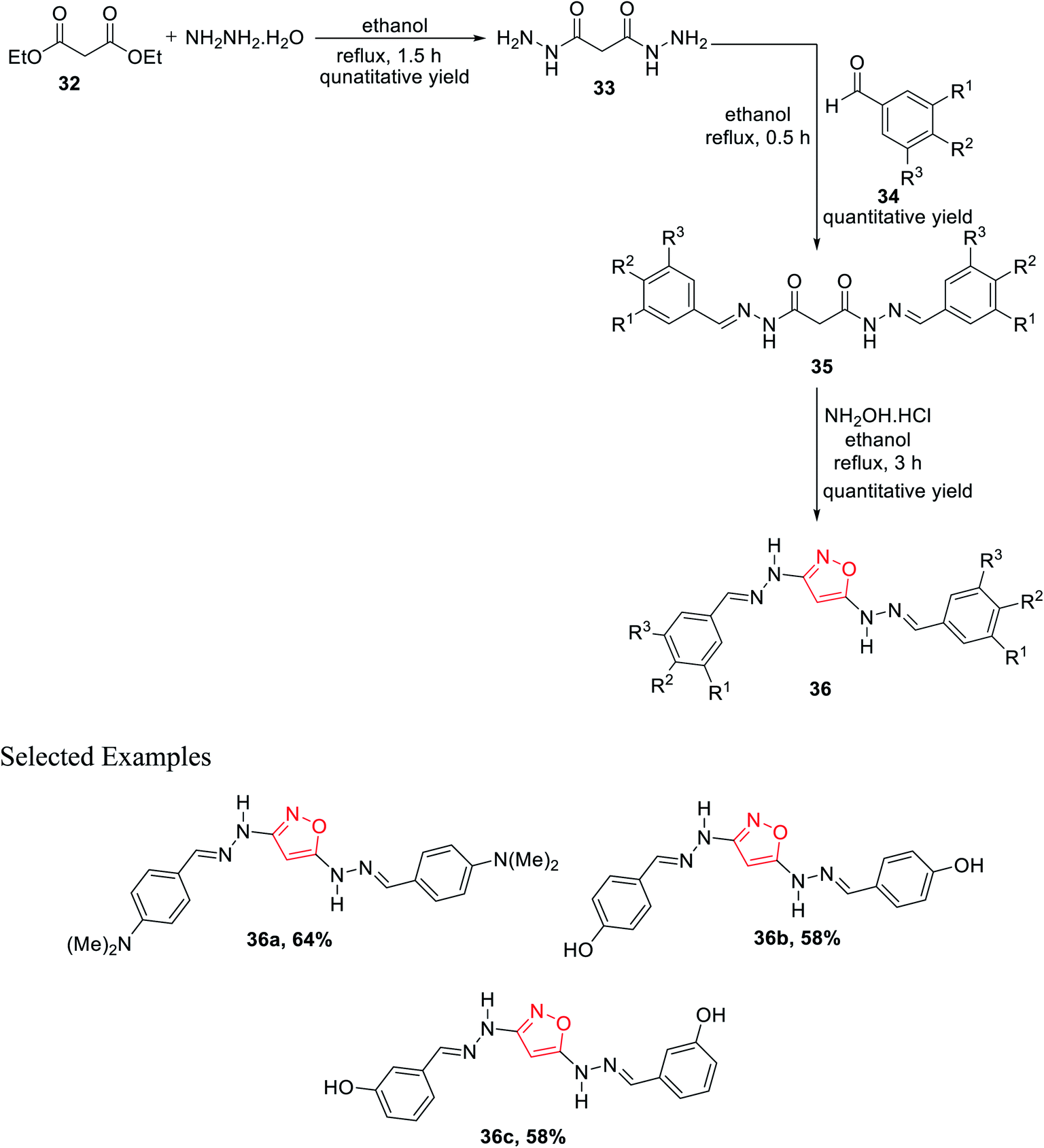 | ||
| Scheme 8 Preparation of 3,5-disubstituted isoxazole derivatives. | ||
In the year 2013, Astudillo and his co-workers introduced a unique synthetic methodology for the generation of 3,5-disubstituted isoxazoles via the 1,3-dipolar cycloaddition reaction of an alkyne and a nitrile oxide. The nitrile oxide was obtained in situ by the conventional method of the reaction of an oxime and an oxidant.
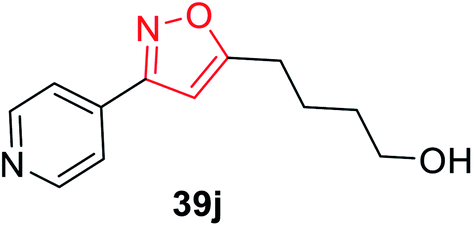
The synthesis of oxime 38 derivatives was achieved by the reaction of hydroxylamine and carbonyl compound 37 in the presence of sodium acetate and ethanol as the solvent for 40 min at room temperature (Scheme 9). The 3,5-disubstituted isoxazoles 39 were prepared by the reaction of oxime 38 and an alkyne in the presence of an oxidizing agent such as sodium hypochlorite and 5% triethylamine via the in situ generated nitrile oxide intermediate. The biological activity showed that the isoxazole derivative 39j (Fig. 4) acts as the acetylcholinesterase (AChE) inhibitor and the docking of the ligand-AchE complex indicates that derivative 39j was placed on the edge of the AChE active site. The main purpose of the AChE inhibitors is prohibiting the enzyme acetylcholinesterase from breaking down the neurotransmitter acetylcholine into choline and acetate, which resulted in an increase in both the level and duration of action of acetylcholine in the central nervous system. Compounds 39g, 39h, and 39i were scrutinized for the biological activity. They displayed the highest biological activity with the lowest IC50 values and they are lower than 200 μM, which are better than that of the other derivatives.27
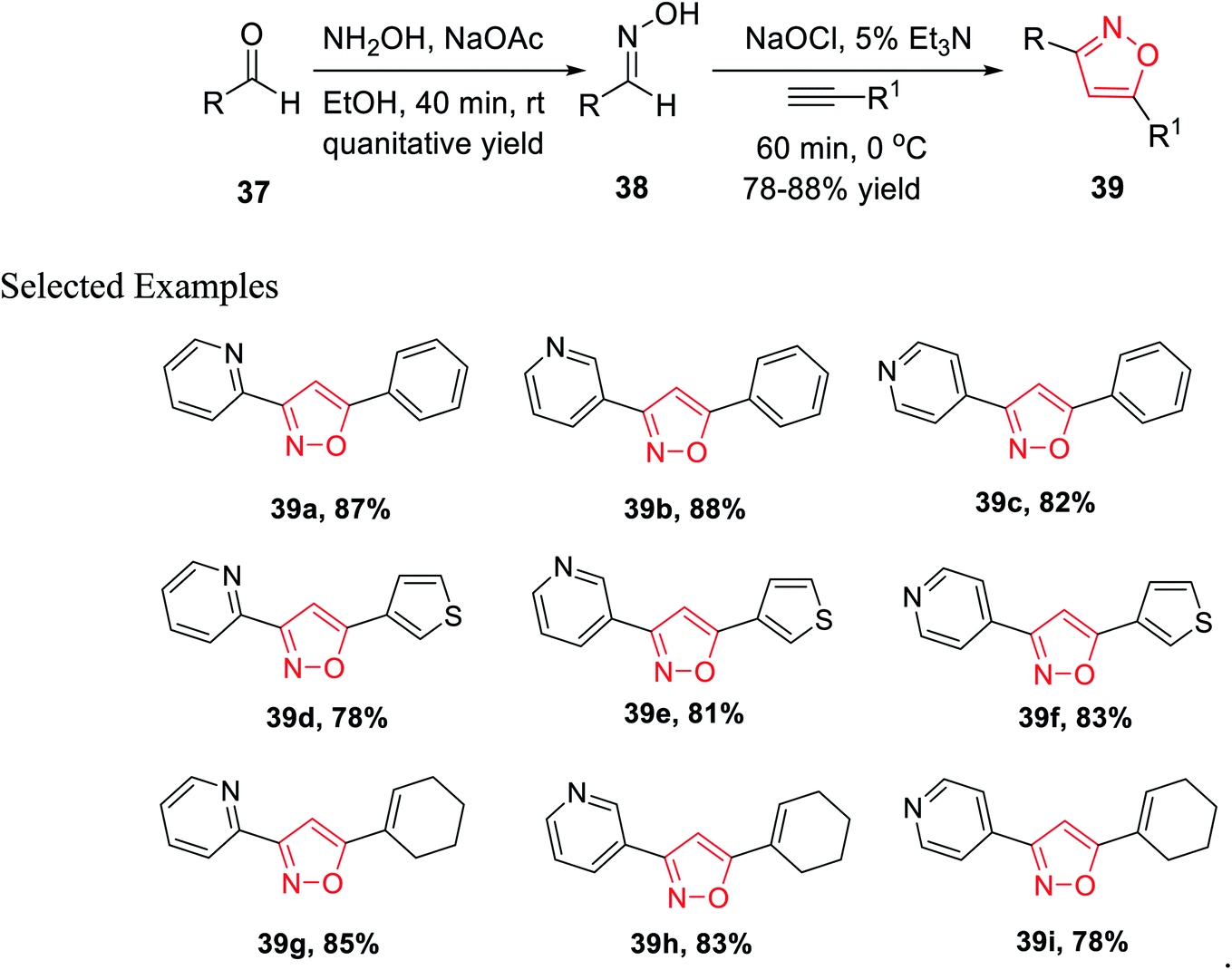 | ||
| Scheme 9 Synthesis of 3,5-disubstituted isoxazole. | ||
 | ||
| Fig. 4 3,5-Disubstituted isoxazole derivative. | ||
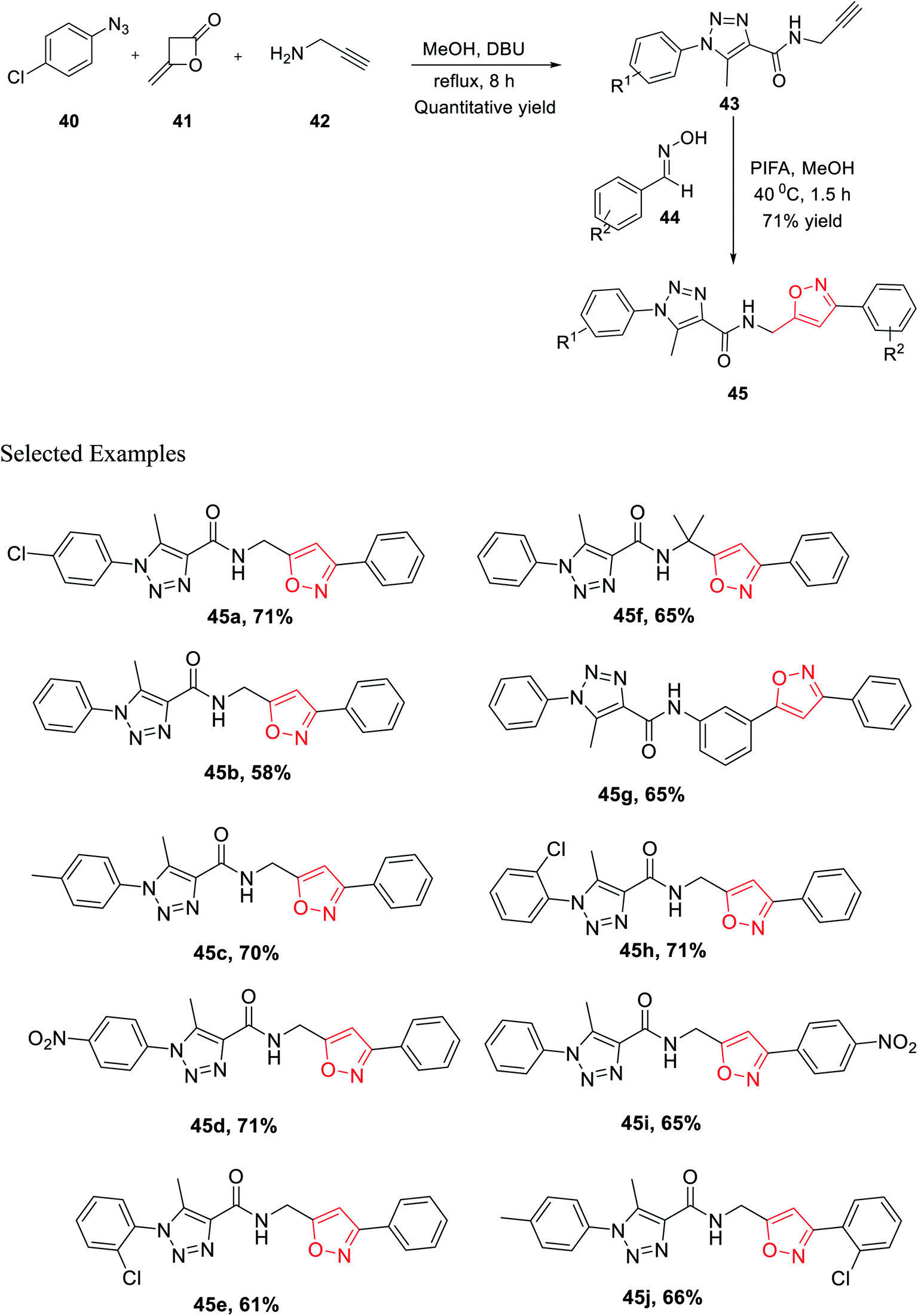
In the same year, Cai and his co-worker demonstrated a novel methodology for the generation of bisfunctionalized 1,2,3-triazole-isoxazole hybrid derivatives having peptide linkage. The methodology started with the synthesis of intermediate 43 by reacting azide 40, diketene 41, and propargyl amine 42 in a sealed tube under refluxing methanol solvent for 8 h. To get the final 1,2,3-triazole-isoxazole derivatives 45, the methanolic solution of intermediate 43 reacts with oxime 44 in the presence of phenyliodine bis(trifluoroacetate) for 1.5 h at 40 °C (Scheme 10). An impressive chemoselective pathway has been introduced to synthesize 1,2,3-triazole-isoxazole derivatives having peptide linkage using a completely metal-free catalyst (particularly not using Cu for triazole synthesis), maintaining the rule of “Green Chemistry” and atom economy up to the mark. Lipinski's rule-of-five study was executed on all the isoxazole derivatives 45(a–j) to assess the drug-likeness or to check the biological or pharmacological activity. It has been found that all the derivatives 45(a–j) follow all the parameters established by Lipinski. Subsequently, the outcomes propose that compounds 45(a–j) were very useful for to discover biological probes and drug leads.28
 | ||
| Scheme 10 Chemoselective formation of bisfunctionalized hybrid 1,2,3-triazole-isoxazole molecules. | ||
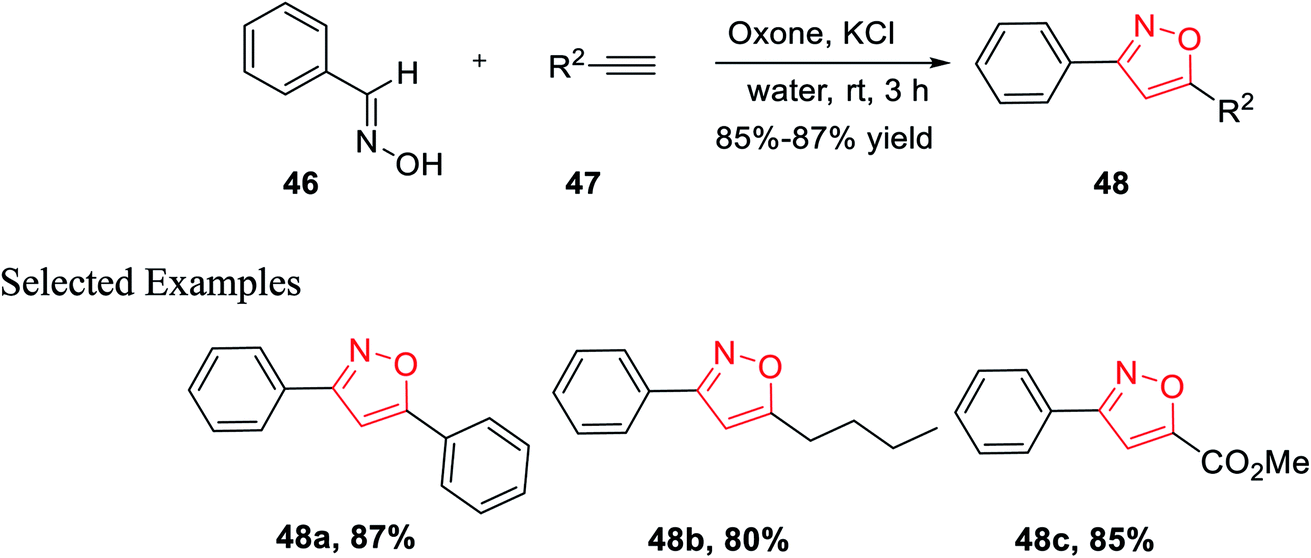
In the year 2014, Yan and his co-workers developed an environmentally benign synthesis of isoxazoles using oxone (an oxidizing agent) in water medium. Oxone (2KHSO5-KHSO4-K2SO4) was used as an efficient oxidizing agent because it has excellent stability, high water solubility, easily transportable, non-toxic in nature, and less costly. The cycloaddition products 48 have been obtained by the reaction of aldoximes 46, alkenes 47 in presence of oxone in aqueous medium at room temperature for 3 h (Scheme 11). The mechanism of this reaction was involved as potassium chloride is oxidized into chlorine in water with the help of oxone, followed by the formation of nitrile oxide from the oxidation of aldoxime by the in situ developed hypochlorous acid. At last, 1,3-dipolar cycloaddition reaction occurred as nitrile oxide reacts with alkyne and resulted in the corresponding 3,5-disubstituted isoxazoles.29
 | ||
| Scheme 11 The cycloaddition of nitrile oxide from aldoxime to alkynes mediated by KCl and oxone. | ||
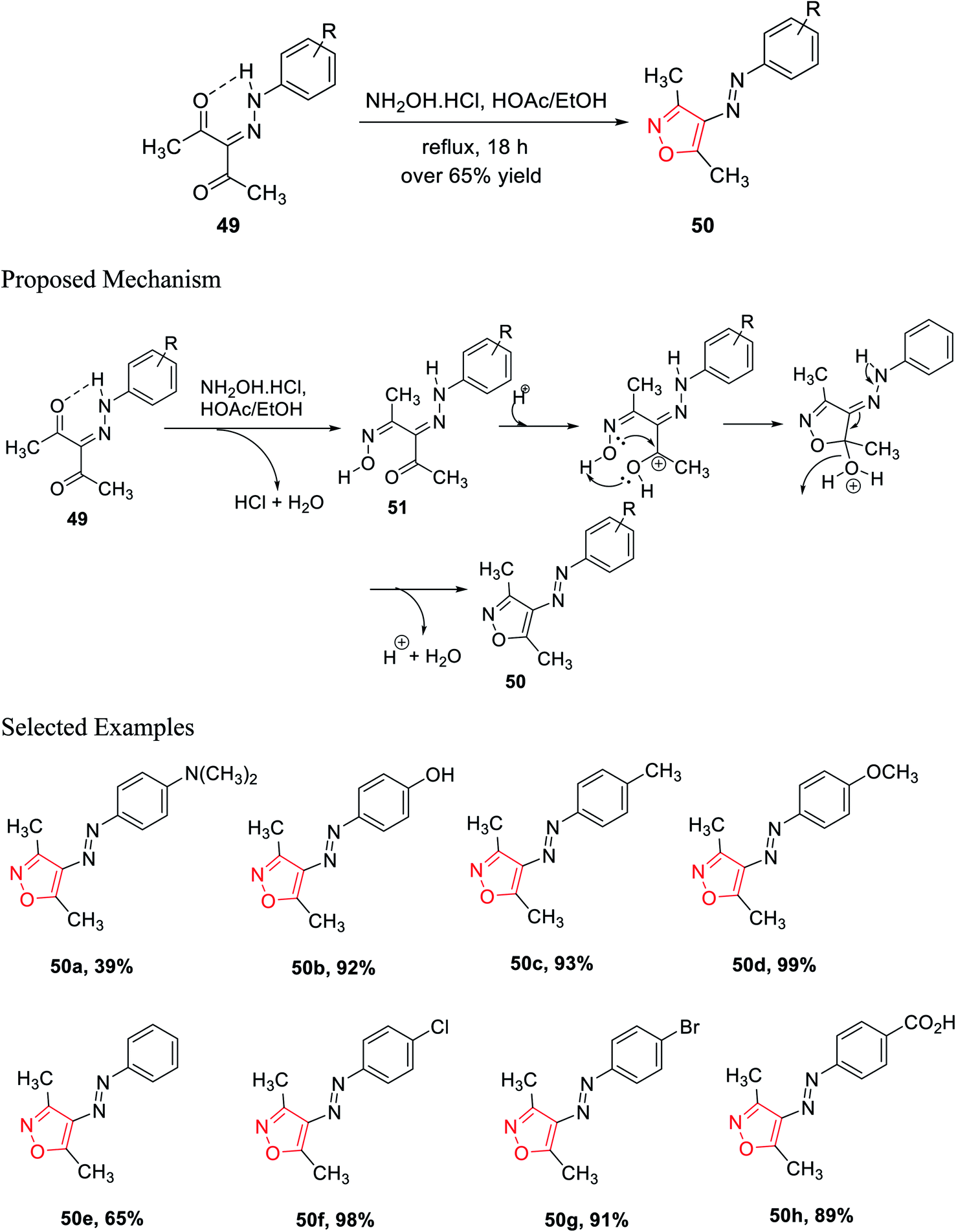
In the subsequent year, Scott and his group established a new method for the preparation of 3,4,5-trisubstituted isoxazole 50 derivatives from β-diketohydrazone 49. The carbonyl group of β-diketohydrazone 49 was stabilized by the existence of the hydrogen bond and its quasi-aromaticity30a (Scheme 12).
 | ||
| Scheme 12 Synthesis of 3,4,5-trisubstituted isoxazole derivatives. | ||
Hence, the electrophilicity on the carbonyl group increased, which created outstanding reactivity difference between the two carbonyl groups; thus, regioselectivity was observed. By maintaining the reaction stoichiometry, water elimination from one carbonyl group was allowed; thus, hydroxylamine hydrochloride reacts with only one carbonyl group and gives the corresponding oxime as the intermediate. After that, the unreacted ketone group reacts with mono oxime intermediate 51, giving the corresponding isoxazole via the addition and displacement reactions. A series of isoxazole molecules have been synthesized using different R groups of 3-(2-(4-R-phenyl)hydrazinylidene)pentane-2,4-diones 49.
3-(2-(4-R-phenyl)hydrazinylidene)pentane-2,4-diones 49 was refluxed for 18 h with NH2OH·HCl, with a stoichiometric ratio of 1:1, in the presence of acetic acid, where ethanol was used as the solvent. Over 65% yields were obtained for each of the (E)-3,5-dimethyl-4-(R-phenyldiazenyl)isoxazole derivatives 50 and were recrystallized by the ethanol–water solution of varying compositions. The biological activity such as the cytotoxic effect of 3,4,5-trisubstituted isoxazoles 50 and the effect of 3,4,5-trisubstituted isoxazoles 50 on the interpretation of p21WAF-1, Bax, and Bcl-2 were observed. The biological study was performed using all the isoxazole derivatives and integrating them into leukemia HL-60 cells culture. Interestingly, compounds 50c and 50f displayed lower IC50 values that are 95.4 μM and 85.6 μM, respectively. Both the molecules exhibit the maximum cytotoxic action toward HL-60 cells.30b
In 2015, Kamal and his co-workers have accomplished a new synthetic method for isoxazole connected with the arylcinnamide moiety.31 Isoxazole-linked arylcinnamide derivatives are an important scaffold; hence, an effort was made to unravel their cytotoxic potential (Scheme 13). The synthesis was started with the condensation of diethyl oxalate 52 and different acetophenones 53 in the presence of sodium ethoxide to obtain compound 54. The reaction of intermediate 54 with hydroxylamine hydrochloride in refluxing ethanol solution resulted in isoxazole-linked esters 55 in good yield. The further reduction of the ester moiety in compound 55 using lithium aluminium hydride yielded 56. Subsequently, the oxidation of isoxazole-linked alcohols 56 by 2-iodoxybenzoic acid (IBX) in dry DMSO introduced the ketone moiety in isoxazoles 57. The ketone-linked isoxazoles 57 were reacted with Ph3PCHCO2C2H5 (C2-Wittig reagent) 58 in the presence of toluene solvent to afford α,β-unsaturated esters 59. Next α,β-unsaturated esters 59 underwent base-mediated hydrolysis and yielded the parallel carboxylic acids 60. The synthesized isoxazole containing carboxylic acids 60 were coupled with different anilines 61 using EDCI/HOBt as the coupling agents to obtain the corresponding isoxazole connected with arylcinnamide moiety 62. These molecules 62(a–i) were assessed for their capability to prevent the progress of several human cancer cell lines, for example A549, HeLa, MDA-MB231, and DU-145, among which some displayed considerable cytotoxic effects. Compounds 62a, 62g, 62h, and 62i displayed moderate cytotoxic activity. These compounds showed the lowest IC50 values compared to the other derivatives, with IC50 values in the range of 2.9–5.4 μM for the HeLa cells. Similarly, they exhibit the lowest IC50 values for DU-145, MDA-MB231, and A549.31
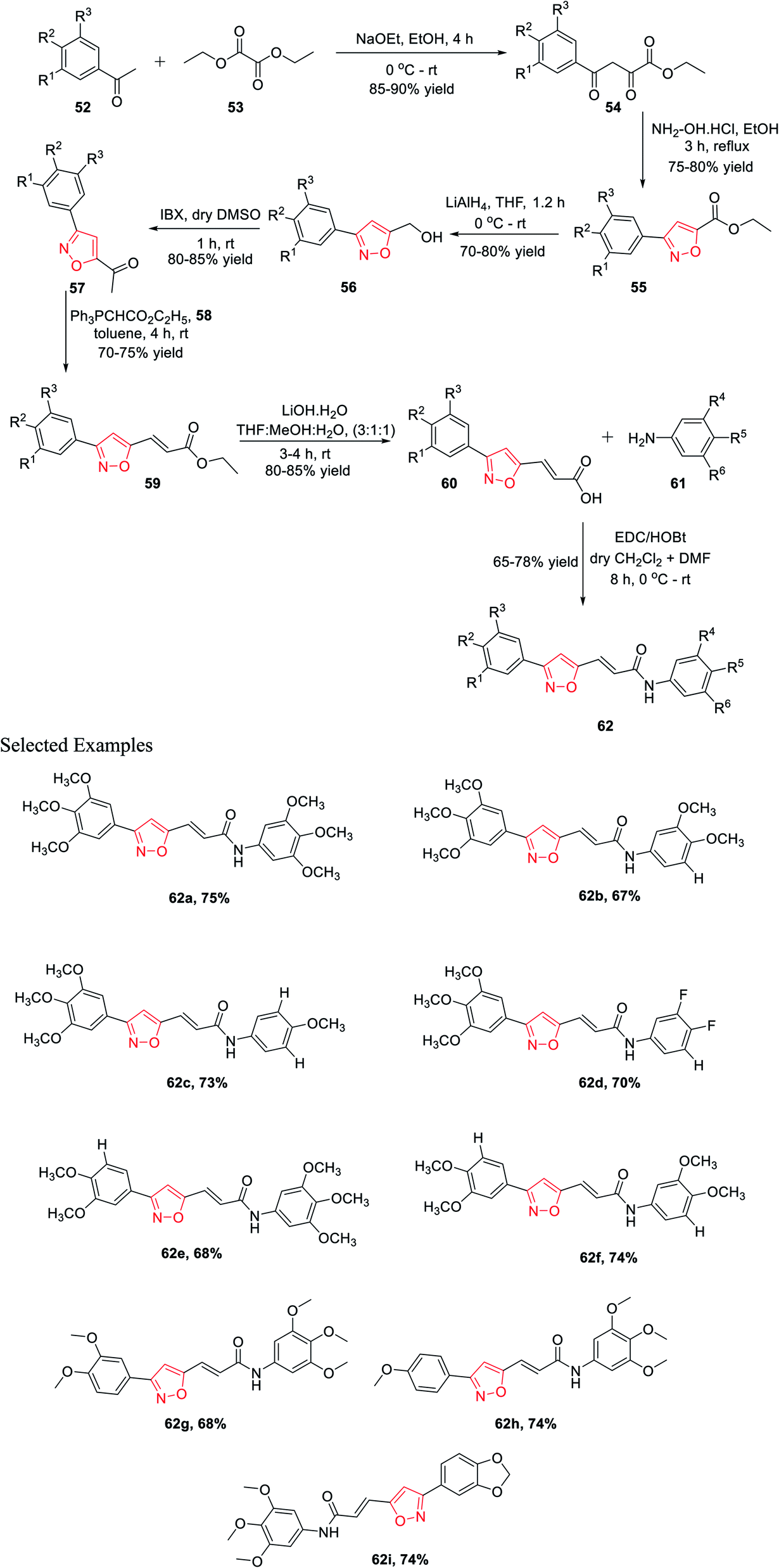 | ||
| Scheme 13 Synthesis of isoxazole-linked arylcinnamides moieties. | ||
In 2016, Mykhailiuk and his group introduced the base-mediated metal-free synthesis of aminoisoxazoles at the multigram scale. Aminoisoxazoles are very useful for peptidomimetics and drug discovery. The (3 + 2)-cycloaddition reaction was the main reaction where the in situ produced nitrile oxide reacted with alkyne/enamines regioselectively (Scheme 14).
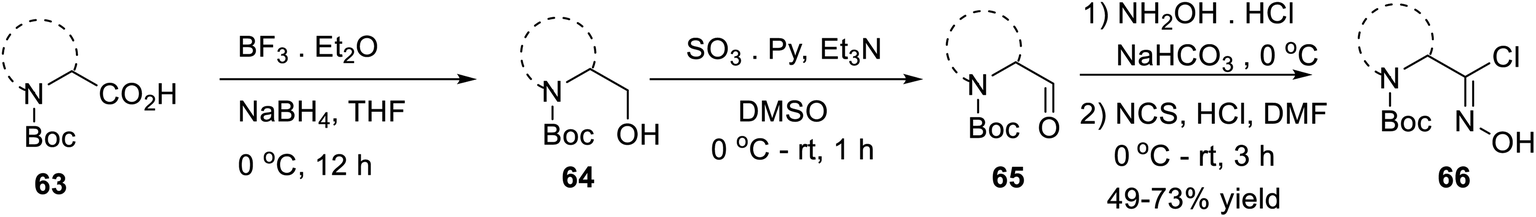 | ||
| Scheme 14 Synthesis of N-Boc-masked chloroxime. | ||
The first step of the synthetic methodology was started with the reduction of the carboxylic group of N-Boc-masked amino acid 63 using NaBH4 from 0 °C to rt for 12 h and yielded the corresponding N-Boc amino alcohol 64. The obtained N-Boc amino alcohol 64 was oxidized by SO3·Et2O in the presence of DMSO solvent from 0 °C to rt, which led to the corresponding aldehyde 65. The resulting aldehyde furnished the oxime by the initial treatment of hydroxylamine hydrochloride in the presence of moderately basic NaHCO3. The so-formed oxime underwent chlorination with N-chlorosuccinimide in DMF from 0 °C to rt for 3 h, which yielded N-Boc-masked chloroxime 66.
Isoxazoles 67 were synthesized from the reaction of alkynes/enamines and in situ created nitrile oxides (Scheme 15). The reaction of N-Boc-masked chloroxime 66 with a mild base such as NaHCO3 or Et3N at room temperature or 0 °C was obtained corresponding to the in situ nitrile oxide in ethyl acetate medium. Then, in situ generated nitrile oxide was reacted with alkyne/enamine in the same ethyl acetate medium at the room temperature for 12 h, which yielded an intermediate that leads to the final product isoxazole 67 at the multigram scale. Compound 67i is commercially available as the ABT-418 drug, which is recognized as a nootropic agent along with both neuroprotective and anxiolytic effects. Compound 67i was also investigated for the treatment of both Alzheimer's disease and attention deficit hyperactivity disorder (ADHD) which is a mental and behavioral disorder characterized by inattention. Previously, the reported synthesized protocol furnished only 7.3% yield of ABT-418. However, herein the potential drug candidate 67i was synthesized with excellent overall yield of 55%.32
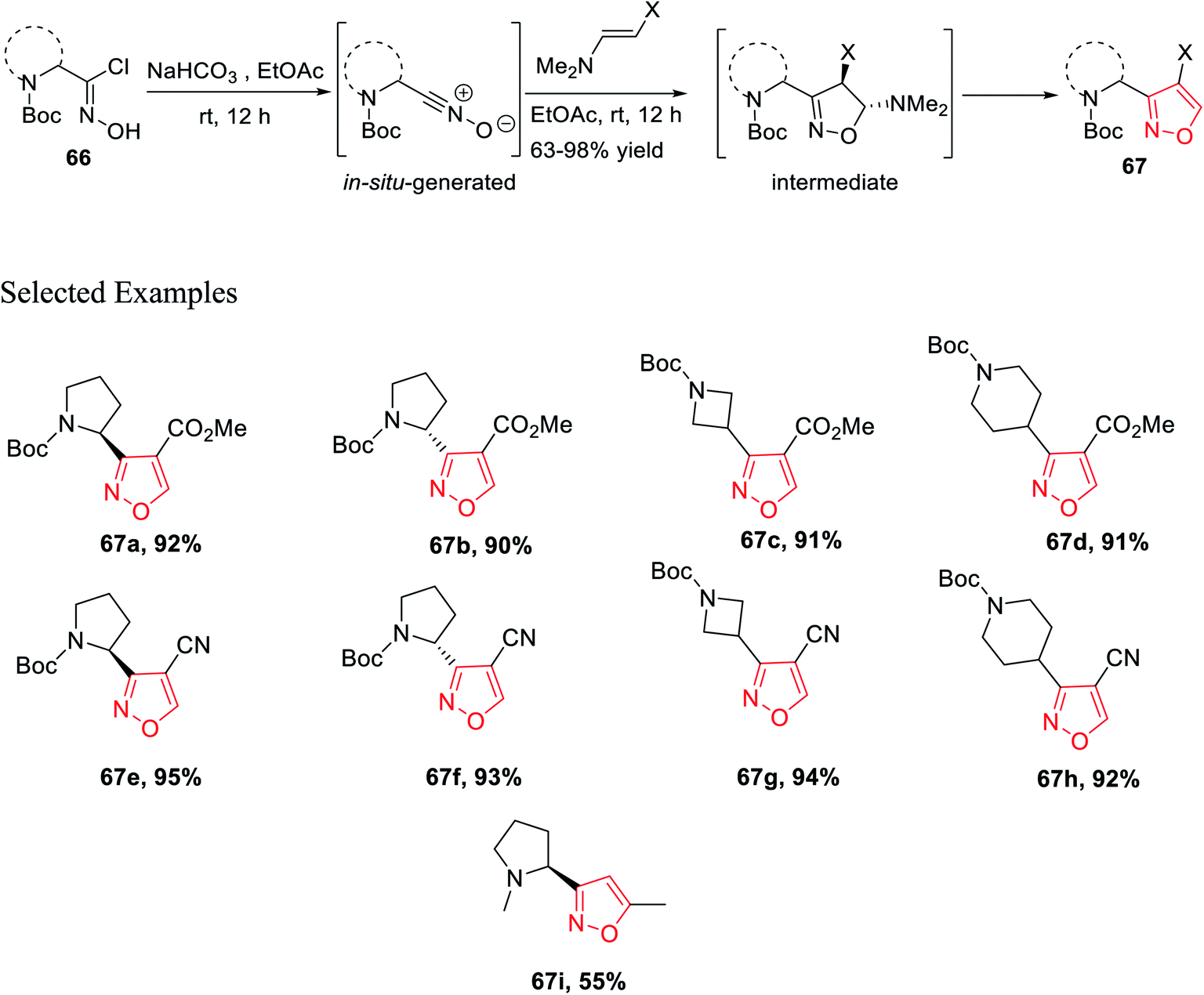 | ||
| Scheme 15 Synthesis of N-Boc-masked isoxazole derivatives. | ||
Again, in the same year, Pan and his co-workers introduced the unique and effective synthesis of 5-substituted isoxazole from TMSN3 and propargylic ketones using TEMPO as the catalyst. This synthetic procedure provided different valuable 5-substituted isoxazoles from easily accessible TMSN3 and propargylic ketones with outstanding yields. Initially, propargylic ketones 68 were reacted with TMSN3 in the presence of TEMPO and methanol as the solvent at room temperature for 12 h. Subsequently, PPh3 was added and the reaction mixture was stirred for another 1 h at room temperature, which resulted in 5-substituted isoxazoles 69 (Scheme 16).33
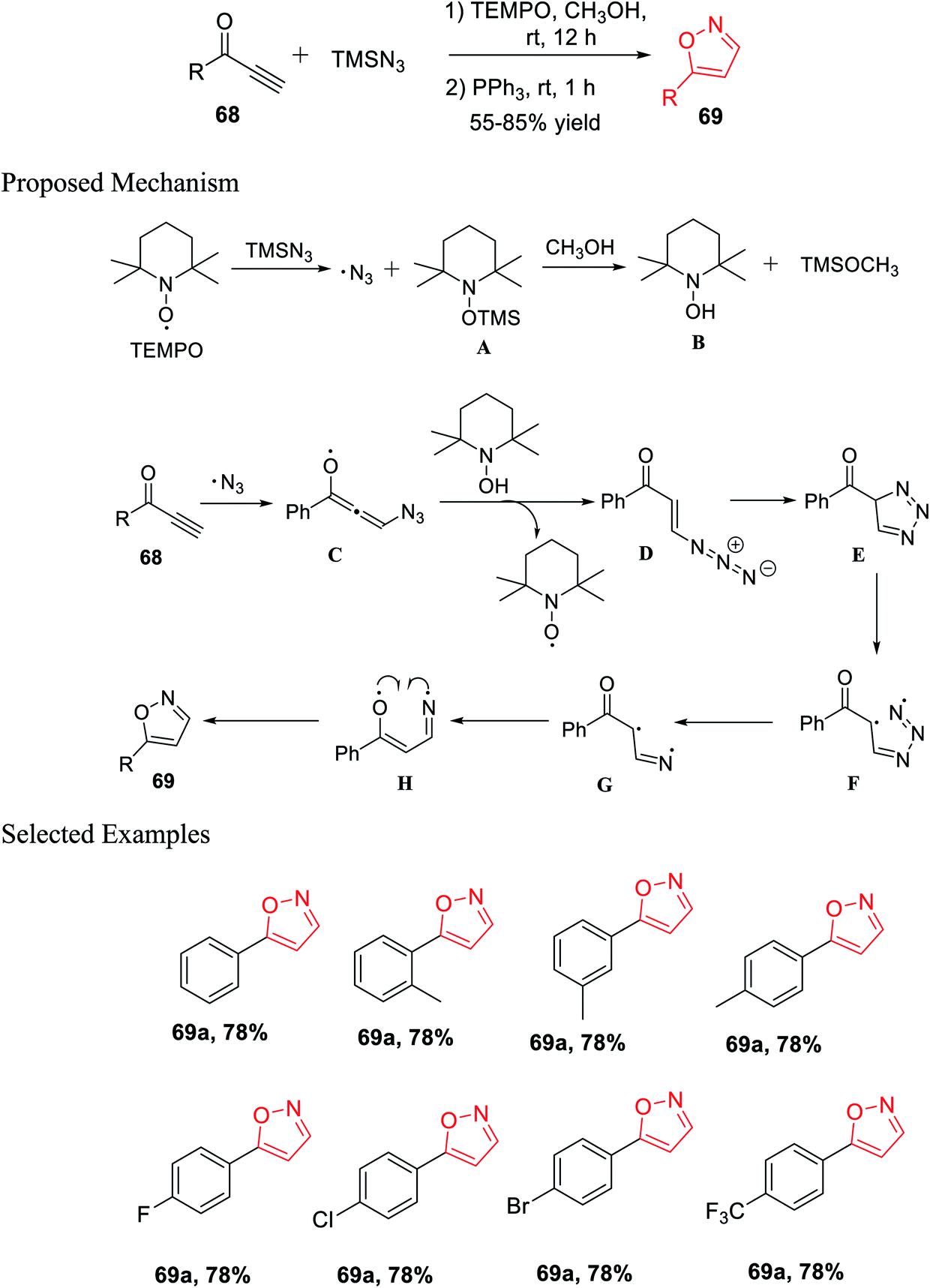 | ||
| Scheme 16 Synthesis of 5-substituted isoxazole using the TEMPO catalyst. | ||
A possible mechanism was proposed for the preparation of 5-substituted isoxazoles. Initially, TEMPO reacted with TMSN3, which gave the azido free radical along with the creation of intermediate A. The TEMPO-coupled TMS compound A was reacted with methanol and produced TEMPOH B and CH3O-TMS. The azido free radical afterward attacked the alkyne and produced radical intermediate C. After radical C reacted totally with the TEMPOH compound B, intermediate D was obtained, which was confirmed by the HRMS study, with the revival of the catalyst. Hence, the intramolecular cycloaddition reaction of azide-alkene took place and yielded triazole E. The obtained triazole E experienced homolytic cleavage to produce the radical intermediate F, which freed N2 and generated the intermediate radical G. Intermediate radical G undergoes one-electron transfer and gave radical compound H, which finally underwent intramolecular radical coupling to furnish 5-substituted isoxazole 69.
Subsequently, Singh's group reported a unique method for the regioselective synthesis of a scaffold containing both β-carboline and isoxazole together via the 1,3-dipolar cycloaddition reaction. To synthesize the desired product, L-tryptophan ester 70 was chosen as the primary precursor. The L-tryptophan ester 70 underwent the sequence of reactions to obtain 1-formyl-9H-β-carbolines 74 and its N-alkylated analogue 76 (Scheme 17).
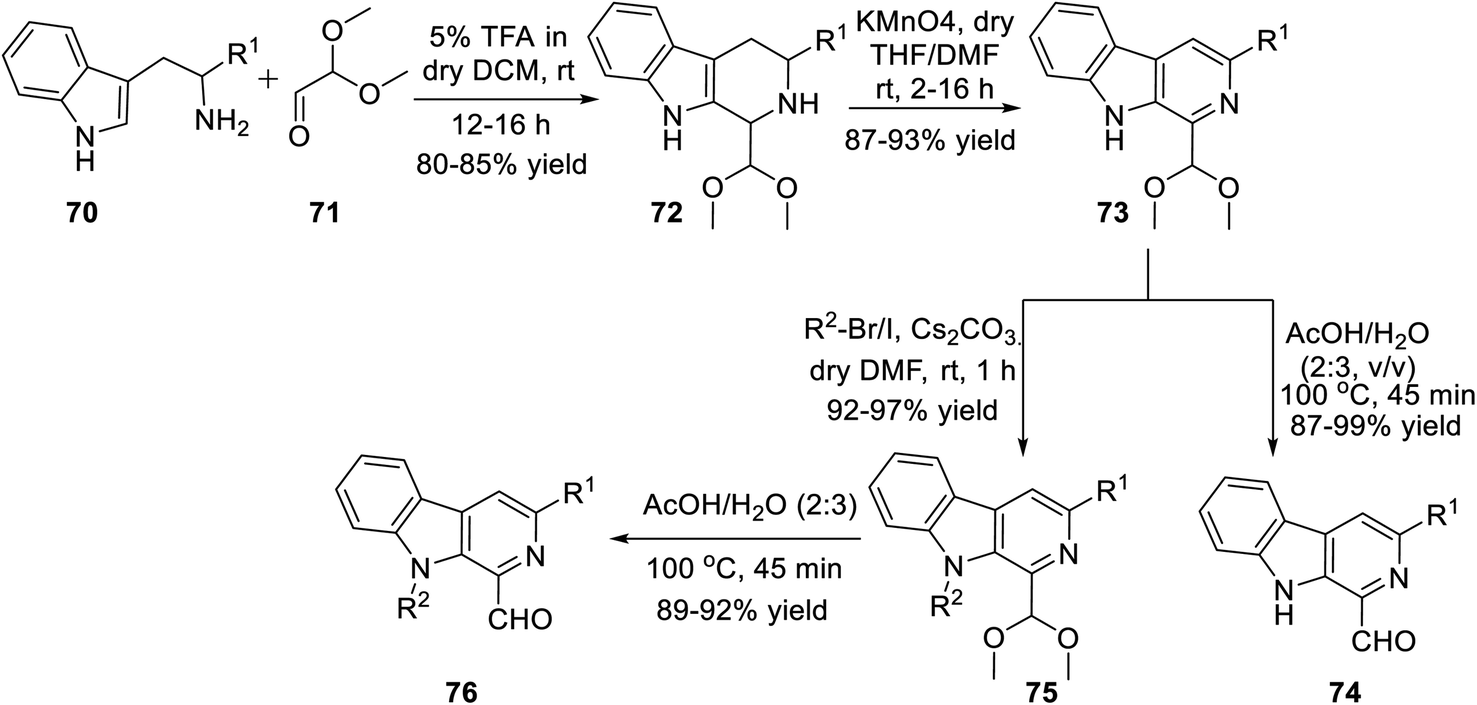 | ||
| Scheme 17 Synthetic procedure of 1-formyl-9H-pyrido[3,4-b]indoles and its N-substituted analogue. | ||
The next sequence of reaction involved the formation of the isoxazole moiety onto the β-carboline scaffold. Initially, N-substituted-β-carbolines 76 were reacted with triethyl phosphonoacetate in dry THF and resulted in the Wittig34a product 77. After that, the nitrile oxides produced in situ were reacted with the Wittig product 77 in the presence of Et3N as the base in anhydrous THF solvent, finally producing the corresponding isoxazoline products 78. Finally, isoxazoline derivatives 78 were oxidized by KMnO4 in DMF and dry THF mixture (1:4) at room temperature, which furnished β-carboline-based isoxazoles 79 (Scheme 18).
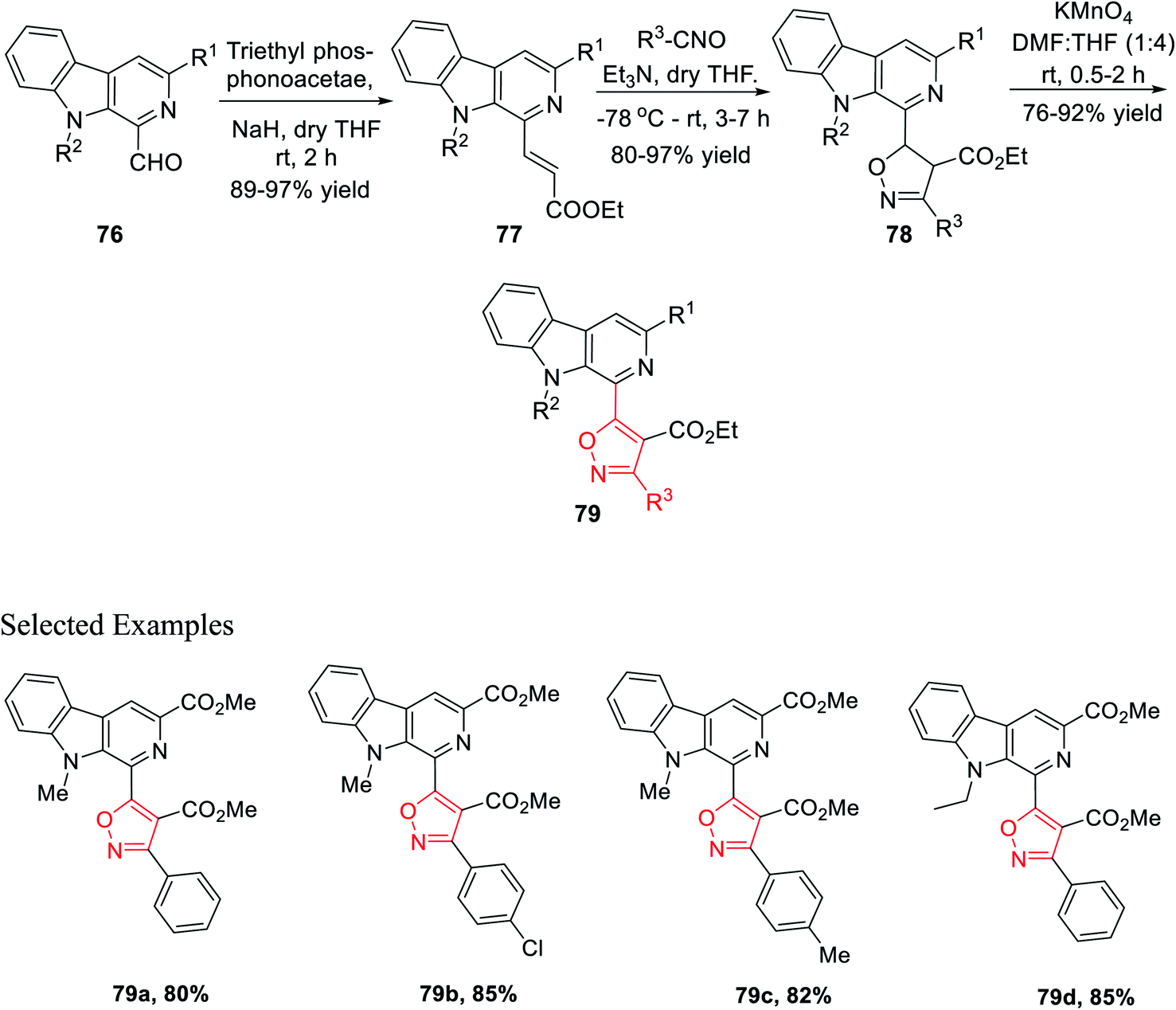 | ||
| Scheme 18 Metal-free multi-step synthesis of β-carboline-based 3,4,5-trisubstituted isoxazoles. | ||
Again, another β-carboline-based chalcone derivatives 77 was synthesized by the Claisen–Schmidt condensation from 1-formyl-pyrido[3,4-b]indole molecules 76 with various acetophenone derivatives containing both electron donating clusters and electron withdrawing clusters. Fascinatingly, β-carboline-linked chalcones 77 (dipolarophile) were reacted effortlessly with different nitrile oxides in the presence of dry THF and Et3N at −78 °C, which yielded the desired isoxazoline derivatives 78. Hence, the isooxazoline derivatives 78 were underwent the same oxidation reaction in the presence of KMnO4 in dry THF and DMF mixture (4:1) at room temperature and furnished the β-carboline-linked isoxazoles 79 with decent to outstanding productivity (Scheme 19).
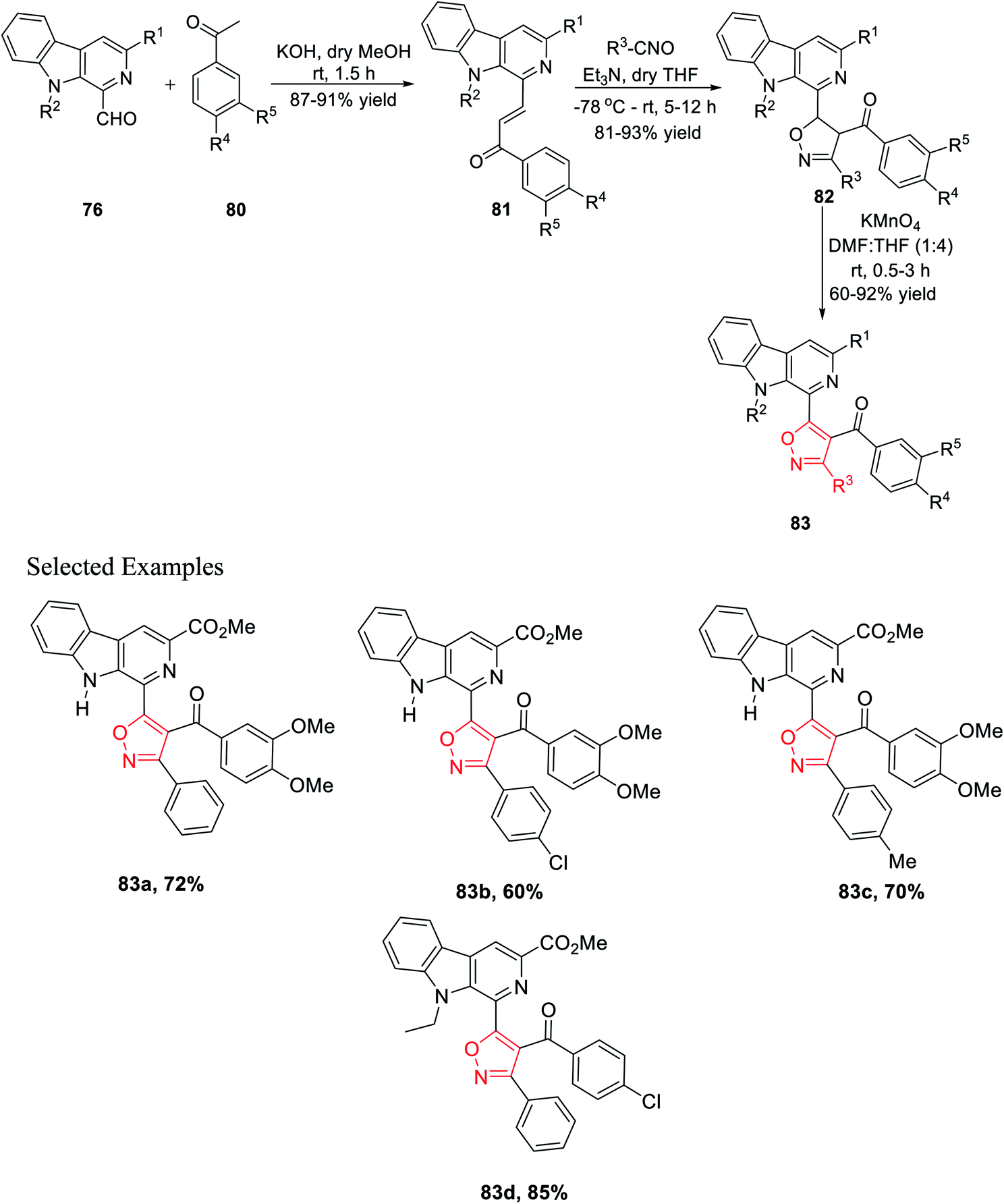 | ||
| Scheme 19 Formation of β-carboline-linked isoxazole derivatives with chalcone as the prototype. | ||
After successfully synthesizing β-carboline-linked isoxazole derivatives, another different kind of β-carboline-linked isoxazole derivatives was synthesized via the formation of the oxime intermediate. The N-alkylated analogue 76 was reacted with NH2OH·HCl in dry methanol to furnish the corresponding oximes 84. Further, oximes 84 were reacted with N-chlorosuccinimide (NCS) in dry DMF at ambient temperature, which yielded the corresponding hydroxyimoyl chlorides 85. Finally, hydroxyimoyl chlorides 85 were reacted with various alkynes 47 (dipolarophiles) via the in situ formation of nitrile oxide using Et3N as the base in anhydrous THF at −78 °C, which afforded the desired products β-carboline-linked isoxazoles 86 (Scheme 20).34b
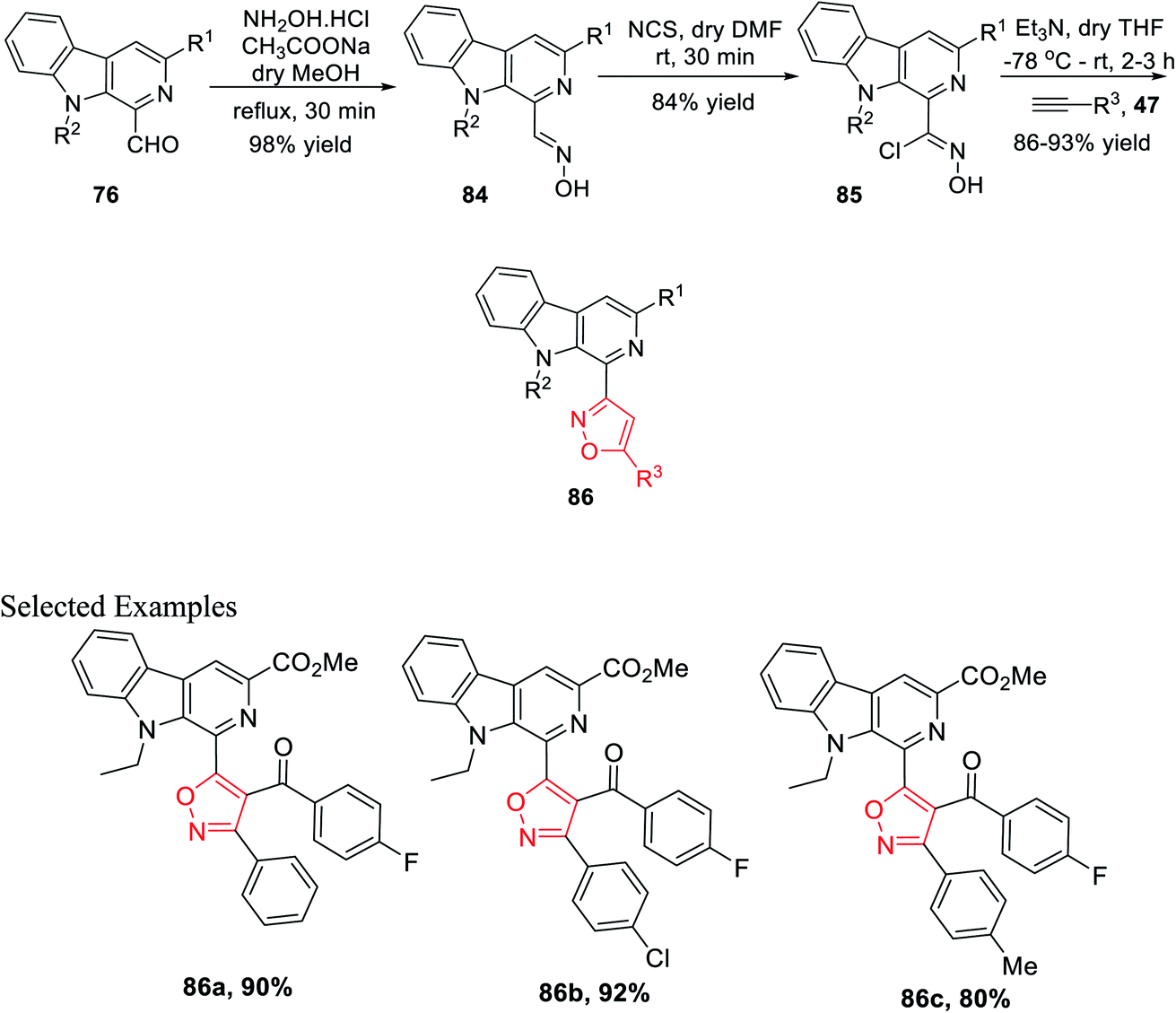 | ||
| Scheme 20 Formation of the β-carboline-(C-3) isoxazole derivative via the oxime intermediate. | ||
In the year 2017, Padmavathi et al. reported a unique method to synthesize isoxazole derivatives having sulphonamide linkage with thiophene. The reaction took place via the routine 1,3-dipolar cycloaddition reaction employing the green approach. To synthesize isoxazole scaffolds having amide linkages with thiophene moiety 93, synthetic intermediate thiophenylamidosulfonyl styrenes 90 were required to be synthesized. Initially, styrene 87 was reacted with sulfuryl chloride in DMF solvent, followed by treatment with aqueous ammonia in THF medium to get arylethenesulfonamide 88. Subsequently, compound 88 was reacted with thiophene-2-carboxylic acid 89 using 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDCI) as the coupling agent along with N,N-dimethylaminopyridine (DMAP) and Et3N as the base, which yielded (E)-N-(arylethenesulfonyl)thiophene-2-carboxamide 90 (Scheme 21).
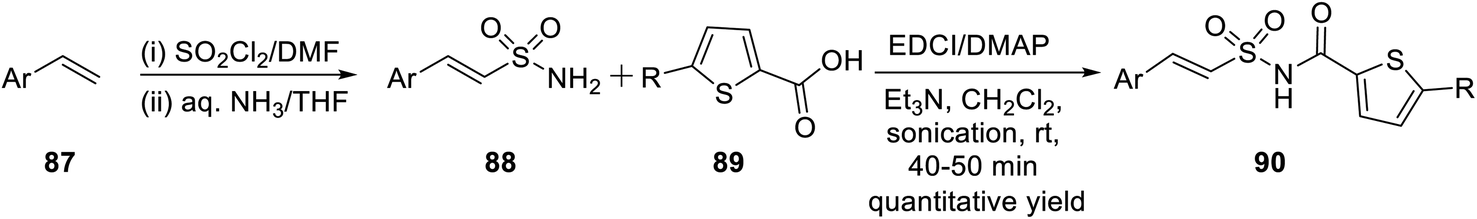 | ||
| Scheme 21 Synthetic procedure for the synthesis of arylethenesulfonyl thiophene-2-carboxamides. | ||
Subsequently, the synthesized (E)-N-(arylethenesulfonyl)thiophene-2-carboxamide 90 was reacted with araldoxime 91 in the presence of CTAB and iodosobenzene in water medium at ambient temperature, which furnished N-((5-aryl-3-phenyl-4,5-dihydroisoxazol-4-yl)sulfonyl)thiophene-2-carboxamide 92. Finally, molecule 92 was oxidized as well as aromatized to N-((5-aryl-3-phenylisoxazol-4-yl)sulfonyl)thiophene-2-carboxamide 93 with the help of I2 in DMSO (Scheme 22).35
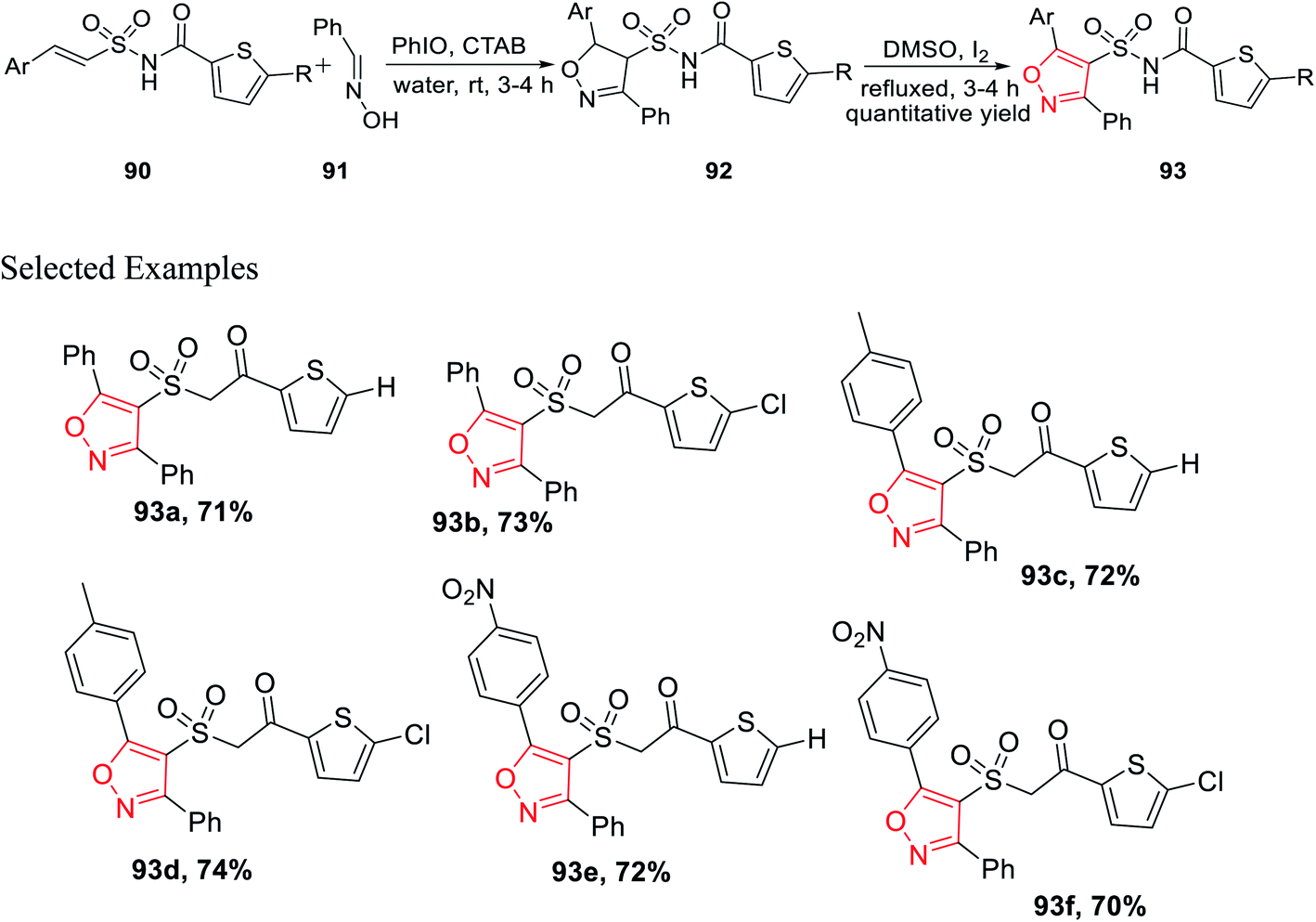 | ||
| Scheme 22 Synthetic pathway for isoxazole derivatives having the sulphonamide linkage with thiophene. | ||
Compounds 93e and 93f demonstrated outstanding antibacterial action on B. subtilis, which was higher than the typical drug chloramphenicol. Subsequently, the same two derivatives exhibit excellent antifungal action, which is better than that of the typical drug, ketoconazole. Again, in vitro study suggests that 93e and 93f act as efficient antibacterial agents toward B. subtilis and antifungal agents toward A. niger. The existence of electron withdrawing groups on the aromatic ring increased the activity.
In 2018, Grygorenko et al. accomplished a new method to synthesize phosphonate-linked isoxazole derivatives through the (3 + 2) cycloaddition reaction. The (3 + 2) cycloaddition reaction took place regioselectively via various types of nitrile oxides and different types of dipolarophile. Initially, the regioselective synthesis of 3,5-disubstituted isoxazoles was attempted under mild basic condition (NaHCO3) at ambient temperature via the reaction of hydroxyimoyl halides and dipolarophile. Unfortunately, a mixture of 3,5-disubstituted isoxazoles and 3,4-disubstituted isoxazoles was obtained. The deciding factor of the regioselectivity of the substituted isoxazole is the leaving group of dipolarophile in the (3 + 2) cycloaddition reaction. Thus, diethyl-1-bromovinyl phosphonate 95 as the dipolarophile was designed to obtain only 3,5-disubstituted isoxazoles 96 regiospecifically via the reaction of oxime 94 with compound 95 under moderately basic condition (NaHCO3) at room temperature (Scheme 23).
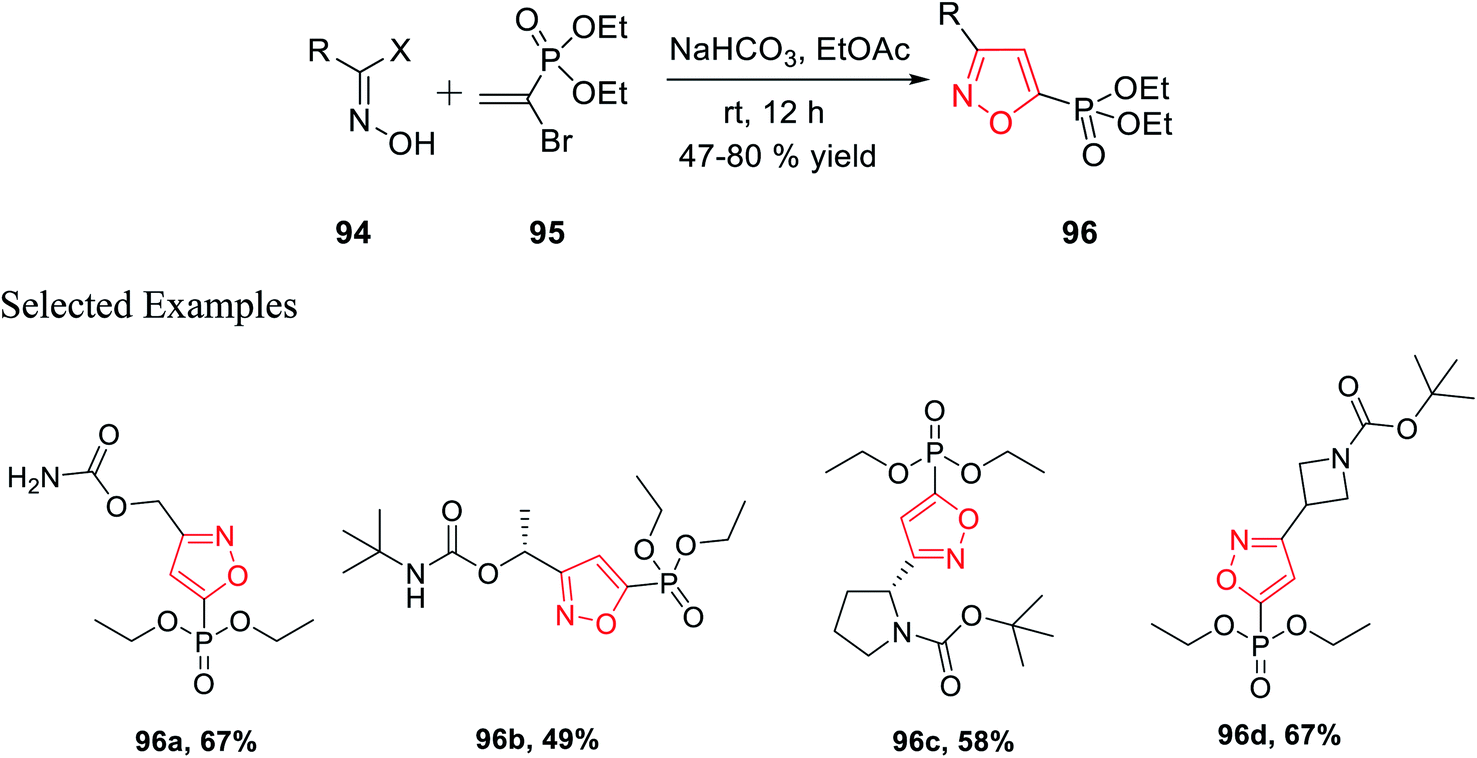 | ||
| Scheme 23 Regioselective synthesis of 3,5-disubstituted isoxazoles. | ||
Similarly, diethyl[2-pyrrolidin-1-ylethenyl]phosphonate 97 was prepared as the dipolarophile to obtain 3,4-disubstituted isoxazoles 98 via the reaction of oxime 94 with compound 97 in the presence of moderately basic condition (NaHCO3) at room temperature (Scheme 24).
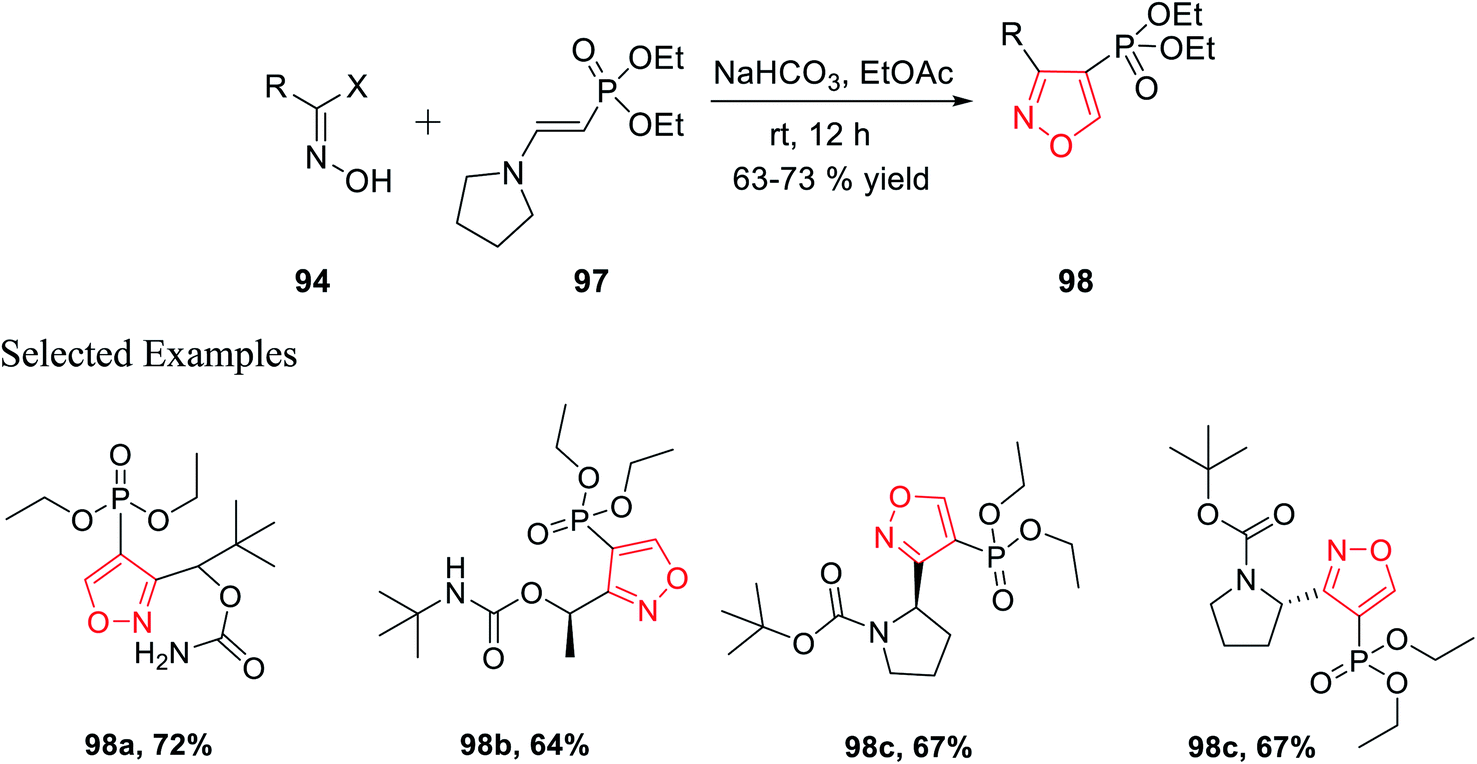 | ||
| Scheme 24 Regioselective preparation of 3,4-disubstituted isoxazoles. | ||
Both the reactions took place via the in situ formation of nitrile oxide. The synthesized phosphonate-linked isoxazole derivatives functioned as a suitable synthetic NDMD receptor.36
In 2019, Mishra and his group introduced a synthetic method for novel 3,5-disubstituted isoxazole scaffolds, which can be used as a possible anti-Parkinson agent. Initially, acetophenone 99 was reacted with diethyl oxalate in the presence of methanol and sodium methoxide at low temperature, which furnished methyl 2,4-dioxo-4-phenylbutanoate 100. Subsequently, intermediate 100 was cyclized with the help of NH2OH·HCl in refluxing methanolic condition for 2–3 h and yielded methyl 5-phenylisoxazole-3-carboxylate 101. Afterward, isoxazole 101 was reacted with hydrazine hydrate in refluxing methanolic condition for 3–5 h, which yielded 5-phenylisoxazole-3-carbohydrazide 102. Finally, the dehydration reaction took place with the reaction of 102 and functionalized benzaldehydes 103 in refluxing methanol solvent for 3–4 h and afforded functionalized N′-benzylidene-5-phenylisoxazole-3-carbohydrazide derivatives 104 (Scheme 25).
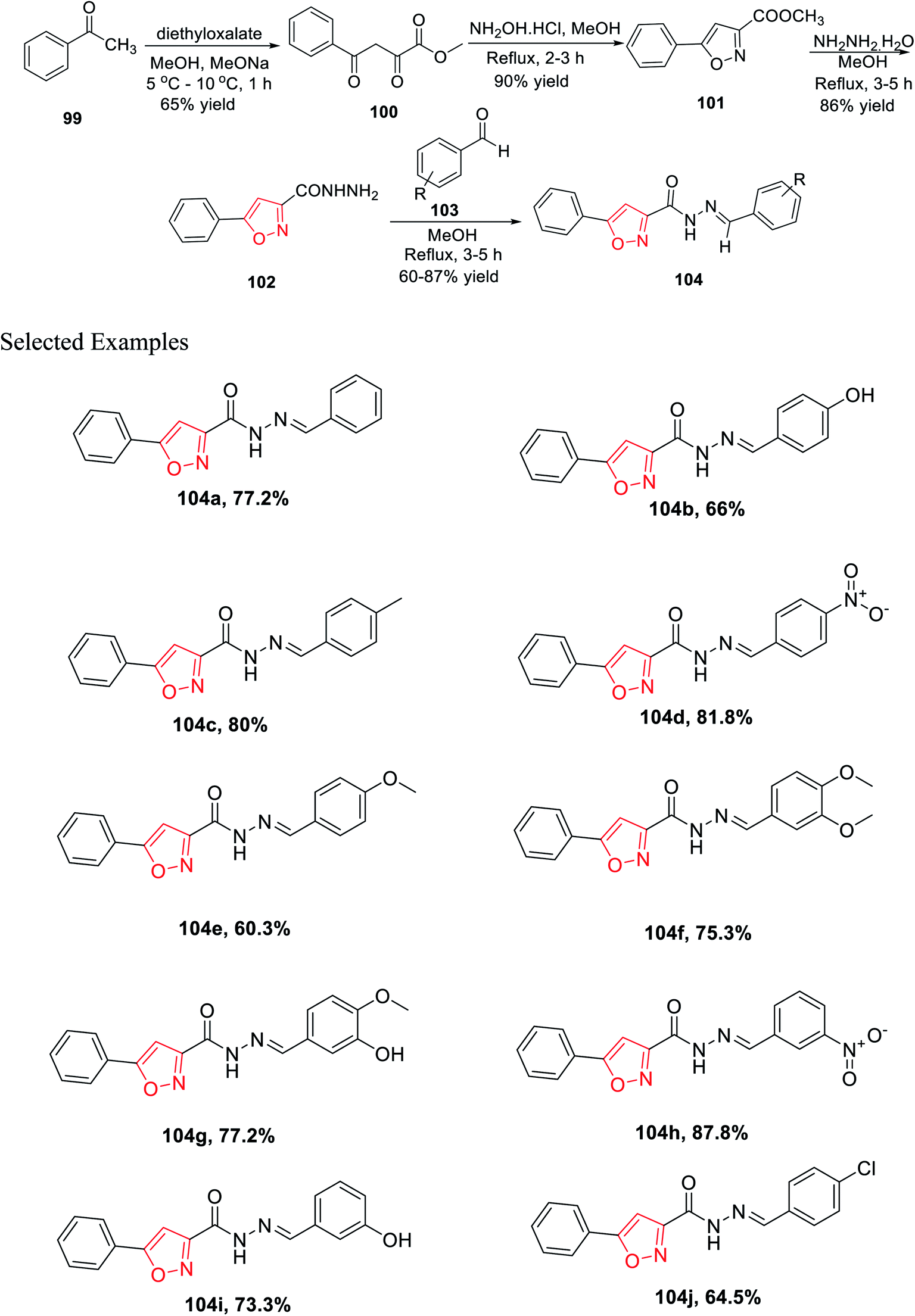 | ||
| Scheme 25 Synthesis of 3,5-disubstituted isoxazole scaffolds. | ||
Intriguingly, monoamine oxidase (MAO) enzymes play a critical role in neurological disorders. Hence, functionalized N′-benzylidene-5-phenylisoxazole-3-carbohydrazide derivatives 104(a–j) were planned, synthesized, and utilized equally for MAO-A and MAO-B inhibition with the help of Amplex Red assays. The effect of the electron withdrawing groups and donating groups at the 4-position and/or the 3-position of the phenyl ring result in the difference in the case of MAO-B inhibition. Among all the derivatives, 104c displayed excellent MAO-B inhibition activity with an IC50 value of 0.0053 ± 0.0003 μM. Compounds 104g and 104b exhibit lower IC50 values than other derivatives. Compounds such as 104e and 104f having the methoxy group and 104j having the chloro group at the 4-position as the electron donating group showed less activity.37
In 2019, Praveen et al. demonstrated a new unique method of TEMPO-catalyzed synthesis of substituted isoxazole derivatives via air oxidation. The use of water as the solvent along with TEMPO as the green oxidant fulfills the criteria of green synthetic procedures. Initially, gram-scale synthesis has performed by reacting ethyl nitroacetate 105 with phenylacetylene 47 in the presence of TEMPO, water, and in open air to give 3,5-disubstituted isoxazole 106 with an excellent yield of 93%. Afterward, the library synthesis was performed by introducing different functionalized nitroacetate 105 and different types of alkyne 107. Functionalized nitroacetate molecules 105 were reacted with alkyne molecules 107, which resulted in 3,5-disubstituted isoxazoles 108 under the same condition using water as the solvent in open air with the TEMPO catalyst (Scheme 26).38
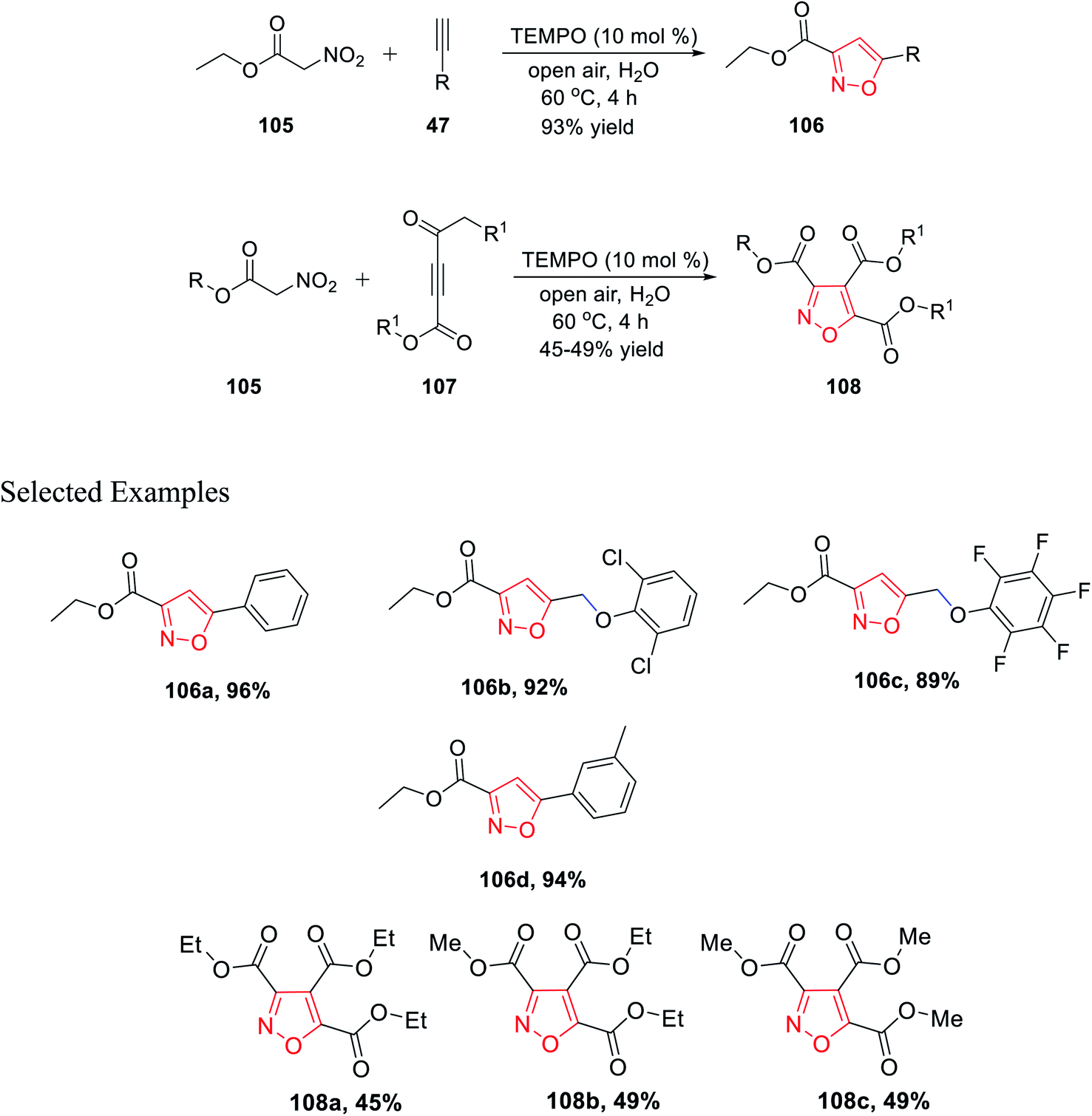 | ||
| Scheme 26 Preparation of highly substituted isoxazoles via TEMPO catalysis in air oxidation. | ||
Further, in the same year, Baranov and his group reported a new method to synthesize 3,4,5-trisubstituted isoxazole derivatives from aromatic aldehyde and nitroacetic esters via the intermediate alkyl-5-hydroxy-6-oxo-4-aryl-6H-1,2-oxazine-3-carboxylate derivatives.
Initially, aromatic aldehydes 110 were reacted with nitroacetic esters 109 in the presence of diethyl amine while using acetonitrile as the solvent for 48 h from 25 °C to 80 °C. Subsequently, the reaction mixture was treated with HCl and hydrated chloroform, and yielded the intermediate alkyl 5-hydroxy-6-oxo-4-aryl-6H-1,2-oxazine-3-carboxylate derivatives 111. Subsequently, intermediates 111 were reacted with various functionalized amines in the presence of chloroform at 70 °C for 12 h, which furnished the 3,4,5-trisubstituted isoxazole derivatives 112 (Scheme 27).39
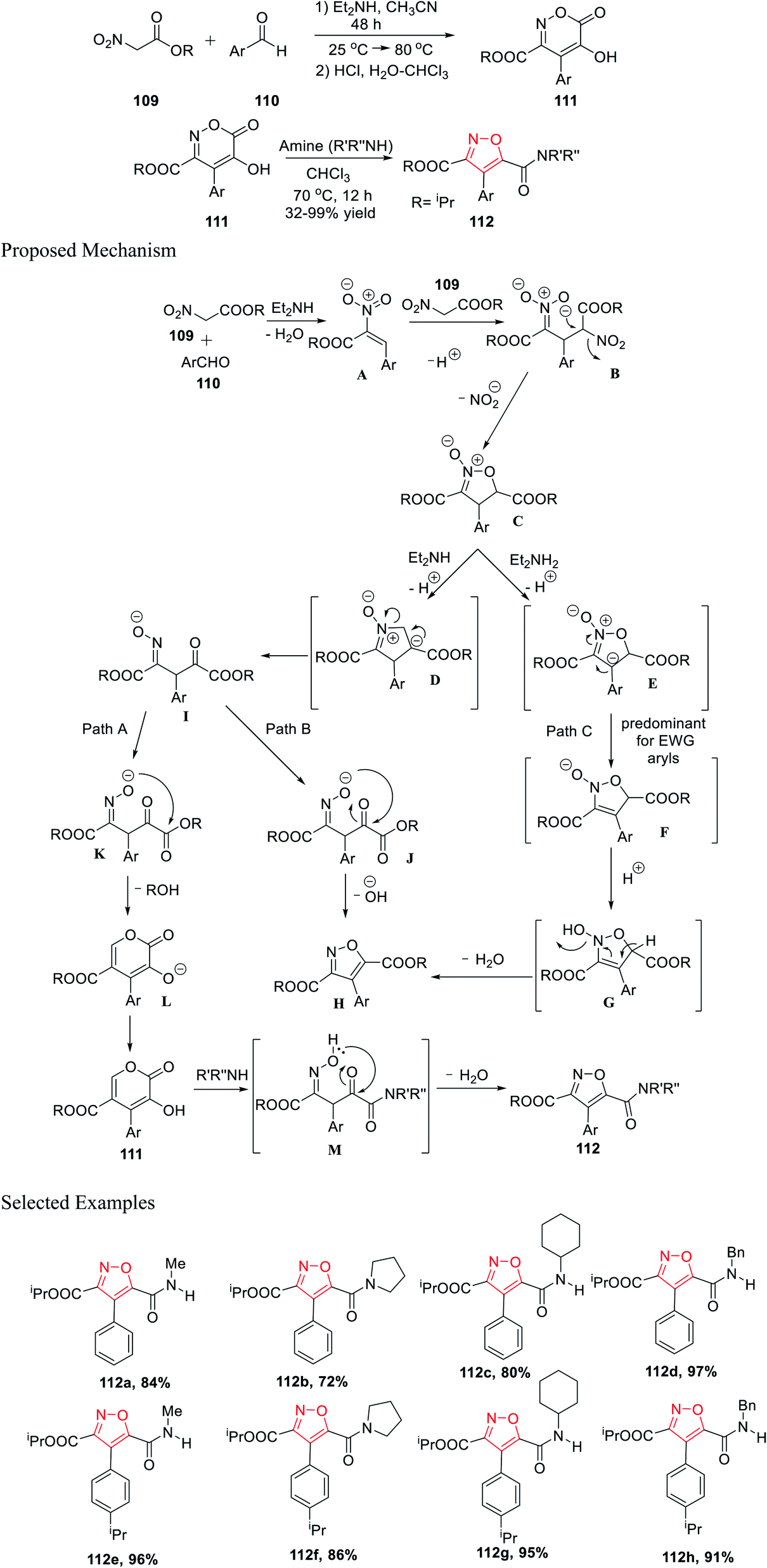 | ||
| Scheme 27 Preparation of 3,4,5-trisubstituted isoxazole derivatives from aromatic aldehyde and nitroacetic esters. | ||
Mechanistically, the synthesis of isoxazole began with the formation of the N-oxide intermediate C through intermediate B. Isoxazoline C was deprotonated in the presence of Et2NH and produced two different types of intermediates E and D, while the formation of E depends on the Ar group. Subsequently, the oxime I underwent cyclization via Path A and Path B, and furnished L (six-membered ring) and H (five-membered ring), respectively.
The transformation to species L and H from I is extremely sensitive and depends on the medium properties. Hence, compound 111 was formed via protonation from L. Finally, compound 112 was formed from compound 111 through the formation of intermediate M, followed by water elimination. Afterward, substituted isoxazole H was formed by water elimination from E (Path C).
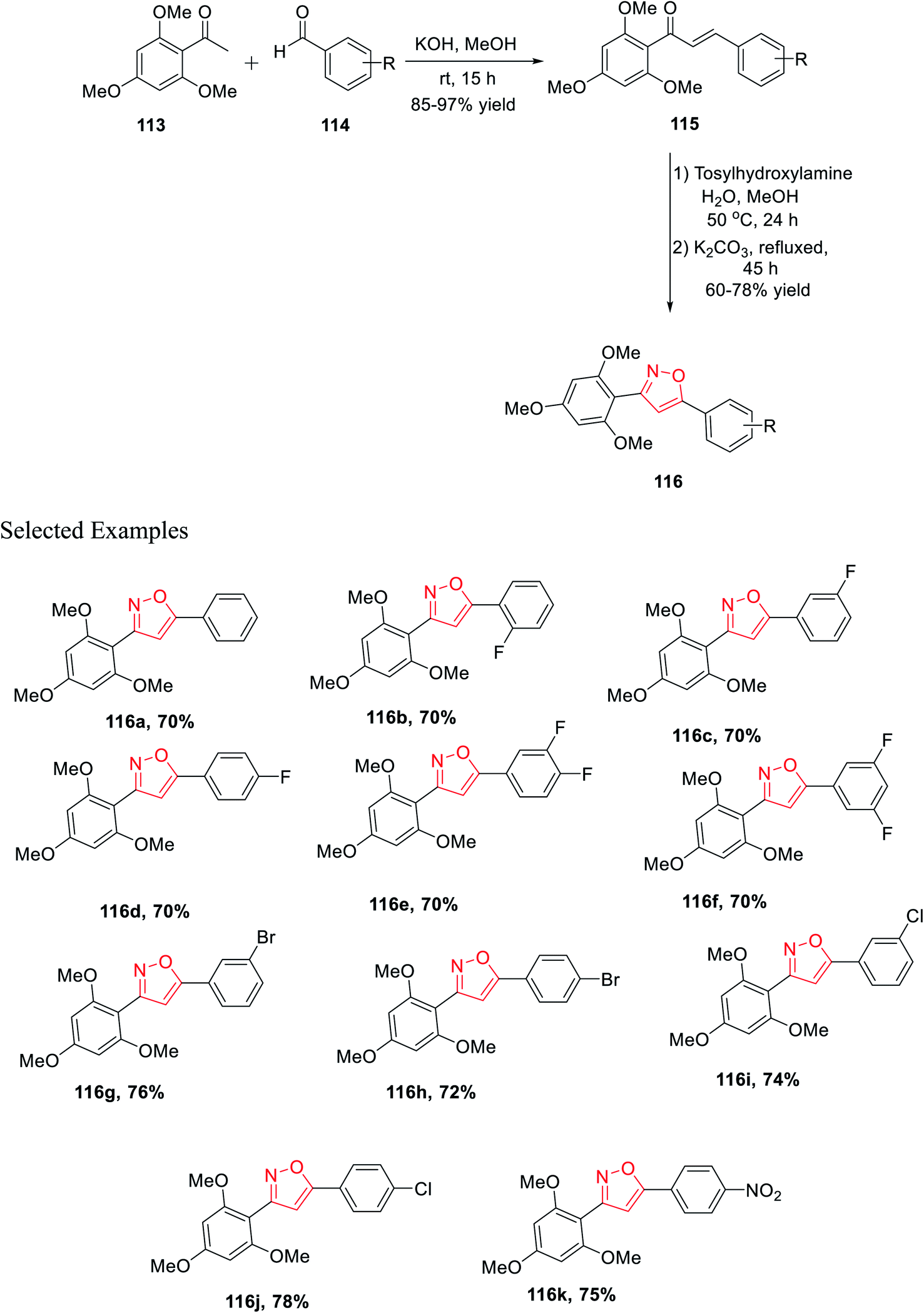
In the last year, Burmaoglu and his co-workers demonstrated a synthetic strategy and biological study of 3,5-diaryl isoxazole scaffolds as effective anticancer mediators. Initially, 2,4,6-trimethoxy acetophenone 113 was reacted with functionalized arylaldehydes 114 in the presence of methanolic KOH at room temperature for 15 h, which afforded functionalized chalcone derivatives 115. Subsequently, in the next step, chalcones 115 were reacted with tosylhydroxylamine in aqueous methanol solvent for 24 h at 50 °C and later K2CO3 was added and refluxed for 45 h, which furnished 3,5-diaryl isoxazole moieties 116 (Scheme 28).
 | ||
| Scheme 28 Preparation of 3,5-diaryl isoxazole scaffolds via the chalcone intermediate. | ||
The biological potential (selectivity and activity) was tested via cancer PC3 cells and non-tumorigenic PNT1a cells for prostate cancer using all the derivatives 116(a–k). Interestingly, compounds (according to their selectivity in the decreasing order) 116c, 116g, 116d, 116f, 116b, 116k, 116e, and 116a were highly selective as well as active toward the cancer cells, which is comparatively better than that of typical epithelial cells. Inadequately, compounds 116h, 116i, and 116j did not exhibit such selectivity toward the cancer cells. Surprisingly, compound 116c showed the maximum selectivity value, which is comparable with the value of 5-FU. Remarkably, compounds 116a displayed the lowest IC50 value with the value of 3.40 ± 0.52 and 2.66 ± 0.22 for PNT1a and PC3, respectively.40
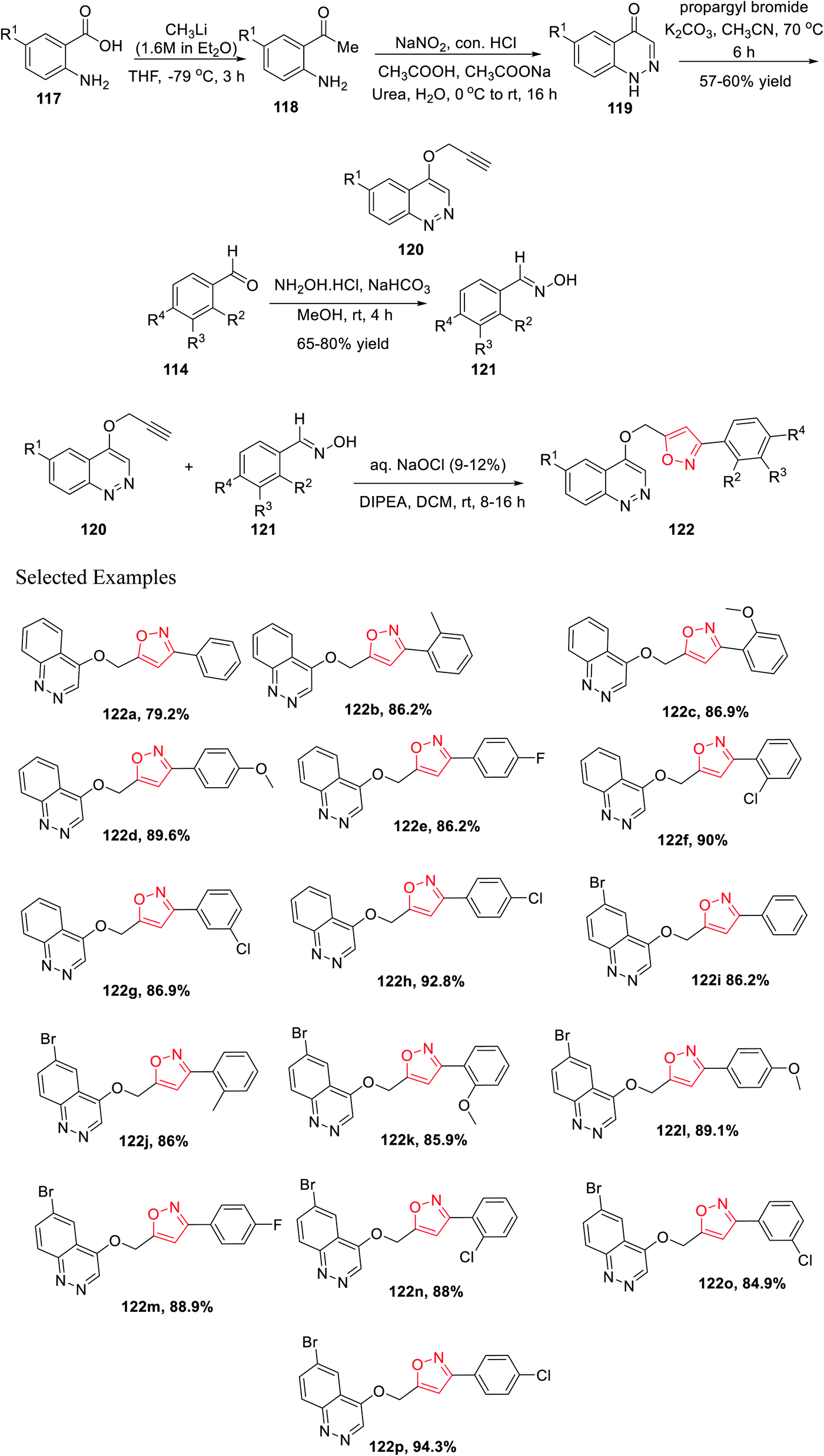
Later, Chitneni and his group accomplished the synthetic methodology as well as the antibacterial activity of unique cinnoline-isoxazole hybrid scaffolds. Initially, anthranilic acid derivatives 117 were reacted with methyl lithium in anhydrous THF solvent for 3 h at −78 °C to produce the corresponding o-amino acetophenones 118. Afterward, the o-amino acetophenones 118 were treated with NaNO2, conc. HCl, urea, and sodium acetate in aqueous medium to produce the corresponding cinnolin-4(1H)-one derivatives 119 in excellent yields. Subsequently, the cinnolin-4(1H)-one derivatives 119 were reacted with propargyl bromide in the presence of K2CO3 in refluxing acetonitrile medium for 8 h to obtain the crucial intermediate cinnoline scaffolds 120.
Again, other main intermediates were synthesized via the reaction of functionalized aromatic aldehydes 114 and hydroxylamine hydrochloride in mild basic condition under methanol medium at room temperature for 4 h to provide the corresponding oxime derivatives 121. Finally, cinnoline scaffolds 120 were reacted with the synthesized oxime derivatives 121 in the presence of aqueous sodium hypochlorite and DIPEA in dichloromethane solvent at ambient temperature for 8–16 h, which furnished the corresponding cinnoline-isoxazole scaffolds 122 with an excellent yield of 65–80% (Scheme 29). All the compounds of 122(a–p) were tested for the antibacterial activity toward the human pathogens, Gram-positive bacteria such as Staphylococcus aureus and Bacillus subtilis, and Gram-negative bacteria such as Pseudomonas aeruginosa and Escherichia coli. Compounds 122h, 122i, 122j, 122m, 122n, 122o, and 122p displayed good inhibition activity and compounds 122f, 122g, 122k, and 122l displayed excellent inhibition activity toward all the abovementioned bacteria. Interestingly, among all the derivatives, compound 122k showed the best result against Pseudomonas aeruginosa.41
 | ||
| Scheme 29 Synthesis of unique cinnoline-isoxazole derivatives. | ||
Summary
Considering the extensive pharmaceutical usage, isoxazole and its derivatives can be synthesized via several synthetic methodologies, which have been established from time to time using microwave, green, solid support, and conventional methods. Herein, we have attempted to explore these methodologies engaged in the formation of isoxazole derivatives due to the easily available low-cost stable starting materials, mild reaction condition, and metal-free reaction condition. The main approach is to synthesize the isoxazole via the 1,3-dipolar cycloaddition reaction of dipolarophile and dipole. In this review article, we have attempted to focus on the latest data available on the synthesis and application of isoxazole derivatives from 2010 to 2020 using metal-free synthetic conditions, which include solid support, microwave-assisted, as well as ultrasonication methods. Herein, we have covered the latest and vital biological activities of isoxazole derivatives such as anti-cancer, HDAC inhibitors, antibiotic, COX2 selective inhibitors, neurotoxin, and anti-rheumatic properties. In conclusion, this isoxazole scaffold can play a major role in drug discovery research and these metal-free synthetic methods will encourage the scientific world to engage more and more ‘metal-free syntheses’ in the drug discovery programme.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Chancellor and Vice Chancellor of VIT for providing opportunity to carry out this study. Further authors are thankful to the management of this institute for providing “SEED MONEY” as research grant.References
- (a) D. C. Swinney and J. Anthony, Nat. Rev. Drug Discovery, 2011, 10, 507–519 CrossRef CAS PubMed; (b) V. Azzarito, K. Long, N. S. Murphy and A. J. Wilson, Nat. Chem., 2013, 5, 161–173 CrossRef CAS PubMed; (c) J. Zhu, J. Mo, H. Z. Lin, Y. Chen and H. P. Sun, Bioorg. Med. Chem., 2018, 23, 3065–3075 CrossRef PubMed.
- (a) P. Kumari, V. S. Mishra, C. Narayana, A. Khanna, A. Chakrabarty and R. Sagar, Sci. Rep., 2020, 10, 6660 CrossRef CAS PubMed; (b) S. Shen, M. Hadley, K. Ustinova, J. Pavlicek, T. Knox, S. Noonepalle, M. T. Tavares, C. A. Zimprich, G. Zhang, M. B. Robers, C. Bařinka, A. P. Kozikowski and A. Villagra, J. Med. Chem., 2019, 62, 8557–8577 CrossRef CAS PubMed; (c) Y. K. Kang, K. J. Shin, K. H. Yoo, K. J. Seo, C. Y. Hong, C. S. Lee, S. Y. Park, D. J. Kim and S. W. Park, Bioorg. Med. Chem. Lett., 2000, 10, 95–99 CrossRef CAS PubMed; (d) D. Garella, E. Borretto, A. Di Stilo, K. Martina, G. Cravotto and P. Cintas, MedChemComm, 2013, 4, 1323–1343 RSC.
- (a) S. Rostamizadeh, Z. Daneshfar and H. Moghimi, Eur. J. Med. Chem., 2019, 171, 364–371 CrossRef CAS PubMed; (b) G. A. R. Johnston, Neurochem. Res., 2014, 39, 1942–1947 CrossRef CAS PubMed; (c) S. Obermeier and M. Muller, Angew. Chem., Int. Ed., 2020, 59, 12432–12435 CrossRef PubMed; (d) R. J. Noveck and R. C. Hubbard, J. Clin. Pharmacol., 2004, 44, 474–480 CrossRef CAS PubMed; (e) A. Prakash and B. Jarvis, Drugs, 1999, 58, 1137–1164 CrossRef CAS PubMed.
- L. W. Page, M. Bailey, P. J. Beswick, S. Frydrych and R. J. Gleave, Tetrahedron Lett., 2010, 51, 3388–3391 CrossRef CAS.
- (a) R. Huisgen, J. Org. Chem., 1968, 33, 2291–2297 CrossRef; (b) R. A. Firestone, J. Org. Chem., 1968, 33, 2285–2290 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- F. Himo, T. Lovell, R. Hilgraf, V. V. Rostovtsev, L. Noodleman, K. B. Sharpless and V. V. Fokin, J. Am. Chem. Soc., 2005, 127, 210–216 CrossRef CAS PubMed.
- B. Willy, F. Rominger and T. J. J. Müller, Synthesis, 2008, 293–303 CAS.
- S. Grecian and V. V. Fokin, Angew. Chem., Int. Ed., 2008, 47, 8285–8287 CrossRef CAS PubMed.
- K. A. DeKorver, H. Li, A. G. Lohse, R. Hayashi, Z. Lu, Y. Zhang and R. P. Hsung, Chem. Rev., 2010, 110, 5064–5106 CrossRef CAS PubMed.
- G. Evano, A. Coste and K. Jouvin, Angew. Chem., Int. Ed., 2010, 49, 2840–2859 CrossRef CAS PubMed.
- K. Chanda, S. Rej and M. H. Huang, Nanoscale, 2013, 5, 12494–12501 RSC.
- F. Hu and M. Szostak, Adv. Synth. Catal., 2015, 357, 2583–2614 CrossRef CAS.
- T. Morita, S. Yugandar, S. Fuse and H. Nakamura, Tetrahedron Lett., 2018, 59, 1159–1171 CrossRef CAS.
- J. Li, Z. Lin, W. Wu and H. Jiang, Org. Chem. Front., 2020, 7, 2325–2348 RSC.
- D. R. Meena, B. Maiti and K. Chanda, Tetrahedron Lett., 2016, 57, 5514–5517 CrossRef CAS.
- K. S. Egorova and V. P. Ananikov, Organometallics, 2017, 36, 4071–4090 CrossRef CAS.
- M. C. Román-Martínez and C. Salinas-Martínez de Lecea, Heterogenization of Homogeneous Catalysts on Carbon Materials, in New and Future Developments in Catalysis, ed. S. L. Suib, Elsevier, Amsterdam, 2013, pp. 55−78 Search PubMed.
- (a) R. D. Padmaja, M. M. Balamurali and K. Chanda, J. Org. Chem., 2019, 84, 11382–11390 CrossRef CAS PubMed; (b) R. L. Panchangam, V. Manickam and K. Chanda, ChemMedChem, 2019, 14, 262–272 CrossRef CAS PubMed; (c) R. N. Rao, R. L. Panchangam, V. Manickam, M. M. Balamurali and K. Chanda, ChemPlusChem, 2020, 85, 1800–1812 CrossRef CAS PubMed; (d) R. N. Rao and K. Chanda, New J. Chem., 2021, 45, 3280–3289 RSC.
- G. Pellegrino, F. Leonetti, A. Carotti, O. Nicolotti, L. Pisani, A. Stefanachi and M. Catto, Tetrahedron Lett., 2010, 51, 1702–1705 CrossRef CAS.
- S. Dadiboyena and A. Nefzi, Tetrahedron Lett., 2012, 53, 2096–2099 CrossRef CAS PubMed.
- P. Conti, L. Tamborini, A. Pinto, L. Sola, R. Ettari, C. Mercurio and C. De Micheli, Eur. J. Med. Chem., 2010, 45, 4331–4338 CrossRef CAS PubMed.
- M. Koufaki, T. Fotopoulou, M. Kapetanou, G. A. Heropoulos, E. S. Gonos and N. Chondrogianni, Eur. J. Med. Chem., 2014, 83, 508–515 CrossRef CAS PubMed.
- (a) R. P. Tripathi, R. Tripathi, V. K. Tiwari, L. Bala, S. Sinha, A. Srivastava, R. Srivastava and B. S. Srivastava, Eur. J. Med. Chem., 2002, 37, 773–781 CrossRef CAS PubMed; (b) V. Prasad, D. Kumar and V. K. Tiwari, RSC Adv., 2013, 3, 5794–5797 RSC; (c) A. Mishra, B. B. Mishra and V. K. Tiwari, RSC Adv., 2015, 5, 41520–41535 RSC.
- A. Rouf, E. Şahin and C. Tanyeli, Tetrahedron, 2017, 73, 331–337 CrossRef CAS.
- E. A. Musad, R. Mohamed, B. Ali Saeed, B. S. Vishwanath and K. M. Lokanatha Rai, Bioorg. Med. Chem. Lett., 2011, 21, 3536–3540 CrossRef CAS PubMed.
- M. Gutiérrez, M. F. Matus, T. Poblete, J. Amigo, G. Vallejos and L. Astudillo, J. Pharm. Pharmacol., 2013, 65, 1796–1804 CrossRef PubMed.
- T. Niu, M. Lv, L. wang, W. Yi and C. Cai, Org. Biomol. Chem., 2013, 11, 1040–1048 RSC.
- L. Han, B. Zhang, M. Zhu and J. Yan, Tetrahedron Lett., 2014, 55, 2308–2311 CrossRef CAS.
- (a) A. Martyniak, I. Majerz and A. Filarowski, RSC Adv., 2012, 2, 8135–8144 RSC; (b) C. Bustos, E. Molins, J.-G. Cárcamo, M. N. Aguilar, C. Sánchez, I. Moreno-Villoslada, H. Nishide, A. Mesías-Salazar, X. Zarate and E. Schott, New J. Chem., 2015, 39, 4295–4307 RSC.
- A. Kamal, A. B. Shaik, B. B. Rao, I. Khan, G. Bharath Kumar and N. Jain, Org. Biomol. Chem., 2015, 13, 10162–10178 RSC.
- B. A. Chalyk, I. Y. Kandaurova, K. V. Hrebeniuk, O. V. Manoilenko, I. B. Kulik, R. T. Iminov, V. Kubyshkin, A. V. Tverdokhlebov, O. K. Ablialimov and P. K. Mykhailiuk, RSC Adv., 2016, 6, 25713–25723 RSC.
- Y. He, Y. Xie, Y. Wang, X. Bin, D. Hu, H. Wang and Y. Pan, RSC Adv., 2016, 6, 58988–58993 RSC.
- (a) G. C. Condie and J. Bergman, Eur. J. Org. Chem., 2004, 2004, 1286–1297 CrossRef; (b) D. Singh, N. Devi, V. Kumar, C. C. Malakar, S. Mehra, R. K. Rawal, B. S. Kaith and V. Singh, RSC Adv., 2016, 6, 88066–88076 RSC.
- D. V. Sowmya, G. Lakshmi Teja, A. Padmaja, V. Kamala Prasad and V. Padmavathi, Eur. J. Med. Chem., 2018, 143, 891–898 CrossRef CAS PubMed.
- B. A. Chalyk, A. S. Sosedko, D. M. Volochnyuk, A. A. Tolmachev, K. S. Gavrilenko, O. S. Liashuk and O. O. Grygorenko, Org. Biomol. Chem., 2018, 16, 9152–9164 RSC.
- N. Agrawal and P. Mishra, Comput. Biol. Chem., 2019, 79, 63–72 CrossRef CAS PubMed.
- M. Vadivelu, S. Sampath, K. Muthu, K. Karthikeyan and C. Praveen, J. Org. Chem., 2019, 84, 13636–13645 CrossRef CAS PubMed.
- A. Y. Smirnov, E. R. Zaitseva, O. A. Belozerova, R. S. Alekseyev, N. S. Baleeva, M. B. Zagudaylova, A. A. Mikhaylov and M. S. Baranov, J. Org. Chem., 2019, 84, 15417–15428 CrossRef CAS PubMed.
- D. A. Aktaş, G. Akinalp, F. Sanli, M. A. Yucel, N. Gambacorta, O. Nicolotti, O. F. Karatas, O. Algul and S. Burmaoglu, Bioorg. Med. Chem. Lett., 2020, 30, 127427 CrossRef PubMed.
- M. B. Bommagani, J. R. Yerrabelly, M. Chitneni, G. Thalari, N. R. Vadiyala, S. K. Boda and P. R. Chitneni, Chem. Data Collect., 2021, 31, 100629 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |