
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA dual polymer composite of poly(3-hexylthiophene) and poly(3,4-ethylenedioxythiophene) hybrid surface heterojunction with g-C3N4 for enhanced photocatalytic hydrogen evolution†
Hailian Baoa,
Xiaodi Chena,
Rui Yuana,
Chao Zhang *a and
Shiai Xu*ab
*a and
Shiai Xu*ab
aSchool of Chemical Engineering, Qinghai University, Xining 810016, Qinghai, China. E-mail: zhangchaoqhu@126.com
bShanghai Key Laboratory of Advanced Polymeric Materials, Key Laboratory for Ultrafine Materials of Ministry of Education, School of Materials Science and Engineering, East China University of Science and Technology, Shanghai 200237, China. E-mail: saxu@ecust.edu.cn
First published on 5th October 2021
Abstract
A surface heterojunction catalyst of g-C3N4–PEDOT/P3HT with P3HT and PEDOT as the polymer sensitizer and hole transport pathway is successfully prepared. The as constructed g-C3N4–PEDOT/P3HT composite exhibits a photocatalyst H2 evolution rate up to 427703.3 μmol h−1 g−1 which is 1059 times higher than that of g-C3N4, 118 times higher than that of g-C3N4–PEDOT with ascorbic acid as sacrificial reagents. What's more, the g-C3N4–PEDOT/P3HT can even show an obviously enhanced photocatalytic H2 evolution rate which is 6.1 times higher than that of pure g-C3N4 in pure water without any sacrificial reagent. Combining the experimental results and molecular dynamic (MD) simulation results, a possible mechanism can be drawn that the existed PEDOT possesses relatively higher hole mobility and can be used as a hole conductor between g-C3N4 and P3HT. Then, the photogenerated holes migration can be accelerated by PEDOT from the VB of g-C3N4 to the VB of P3HT. All those factors may benefit the synergy among g-C3N4, PEDOT and P3HT, which finally facilitates the rapid migration of photoinduced electron–hole pairs and eventually improves the photocatalytic H2 activity process of g-C3N4–PEDOT/P3HT with visible light. The present work may provide useful insights for designing a surface heterojunction composite photocatalyst with high photocatalytic activity for H2 production.
Introduction
As a metal-free semiconductor photocatalyst, graphite carbon nitride (g-C3N4) has attracted abundant attention as a visible light induced water splitting catalyst for hydrogen production.1 However, the visible light photocatalytic activities of pure g-C3N4 are still restricted by the relatively low conductivity of electrons and strong recombination of photogenerated electrons–holes.2,3 Much effort has been devoted to addressing this issue, like element doping,4–6 constructing heterojunctions with another narrowed band-gap semiconductor,7–9 and sensitization by organic dyes,10–13 to enhance the visible light driven photoactivity of g-C3N4.Within these mentioned strategies, the heterojunction design was a common and effective way, due to the easy integration effect of the different semiconductor components.14–18 However, previous research mainly focused on the inorganic–inorganic heterojunction or inorganic–polymer heterojunction.19–21 Recently, the fully polymer–polymer composite based heterojunction systems, applied in the photocatalytic H2 production, have received much attention due to the encouraging progress results.22
Among which, some typical conducting polymers like poly(3-hexylthiophene) (P3HT) and polypyrrole (PPy), were used as polymer composite photocatalyst to improve the H2 production activity of g-C3N4. For example, Yan et al. developed a P3HT–Pt/g-C3N4 composite heterojunction for H2 production with Na2S–Na2SO3 solution as sacrificial reagent.21 Then, they found that the H2 production activity increased to 300 times when 3 wt% P3HT was added and the quantum yield about 2.9% at 420 nm. After that, the same group prepared a new heterostructure of g-C3N4/Au/P3HT/Pt by a self-assembling method.23 The as-prepared heterojunction g-C3N4/Au/P3HT/Pt promoted visible light hydrogen production activity with optimal amount P3HT at 0.5 wt% and obtained the highest H2 evolution rate 320 μmol h−1. Recently, Peng and coworkers introduced a surface heterojunction (SHJ) photocatalyst of P3HT–Pt/g-C3N4. By majorization the composition of P3HT and photoreaction factors, an impressive apparent quantum yield was achieved.24 Besides, Chen's and coworkers prepared the g-C3N4/PPy heterojunction nanoparticles and found that the H2 evolution from pure water was achieved by loading very low weight ratio dispersed PPy nanoparticles.25
However, as the rapid charge recombination always happens during the diffusion of charges on the photocatalyst particle surface after photo-excitation, which is severely limiting the overall photocatalytic efficiency. To further suppress the charge recombination, one of the effective way is to design photocatalysts with spatially separated oxidative and reductive sites.26
Usually, co-catalysts on semiconductor can collect photogenerated carriers, the maintain active sites for catalytic H2 or O2 evolution, and thus improving the activity and stability of photocatalysts by suppressing the photogenerated carriers recombination.22 Currently, plenty of the developed photocatalytic systems use noble metal co-catalysts, such as Pt, Pd, Ag, as the sink of photoinduced electrons to achieve high H2 production activity.22 However, the behavior of photogenerated hole was nearly ignored. As the photogenerated hole may result in the byproduct like O2, and the sacrificial electron donor was commonly used to eliminate these unfavourable photogenerated hole. In fact, these photogenerated hole may recombine with the photoinduced electrons again before it was consumed by the sacrificial reagent.
Poly(3,4-ethylenedioxythiophene) (PEDOT) has been considered to be a perfect conducting polymer in optoelectronic devices, with its perfect conductivities, thermal stability and transparency in the visible range.26 Specially, because of its high hole mobility, PEDOT is often used as a hole conductor in dye sensitized solar cells (DSSCs).27,28 Moreover, Chen et al. found that the PEDOT also could catalyse water oxidation reaction in the typical photoelectrochemical cells.29 What's more, Zong and Wang et al. constructed a g-C3N4–PEDOT composite and PEDOT acted as a hole transport channel.26 Then, the g-C3N4–PEDOT–Pt system was created, in which PEDOT and Pt acted as oxidation and reduction reaction sites, respectively. In this way, the as-prepared g-C3N4–PEDOT–Pt photocatalyst exhibited 4 times higher hydrogen activity than the conventional photocatalyst without PEDOT. However, the photocatalytic activities of pure g-C3N4–PEDOT was still limited due to its relatively large bandgap (2.7 eV), as the PEDOT it self could not used as the light absorber to excite photocatalytic H2 production, resulting in an insufficient sunlight harvesting.
Based on above analysis, we wonder whether it is possible to construct a g-C3N4–PEDOT/P3HT surface heterojunction (SHJ) catalyst with P3HT and PEDOT as the polymer sensitizer and hole transport pathway, respectively. In this paper, we report a new type of g-C3N4 SHJ photocatalyst by co-loading PEDOT and P3HT onto g-C3N4. Moreover, as Pt is often deposited to g-C3N4 as electron sink and proton reduction sites, we successfully construct a g-C3N4–PEDOT/P3HT/Pt composite, in which PEDOT act as hole transport pathway, P3HT as the polymer sensitizer and Pt as the reduction reaction sites. In this way, the g-C3N4–PEDOT/P3HT composite loaded with Pt exhibits the better visible light harvest character, synergy with spatially separated oxidative and reductive sites, and finally remarkably enhanced the photocatalytic H2 evolution activity.
Results and discussion
Structure and morphology of photocatalyst
As shown in Scheme 1, we present the typical synthetic route of g-C3N4–PEDOT/P3HT composite. This process involves two main steps and the detailed process can be found in the experimental section.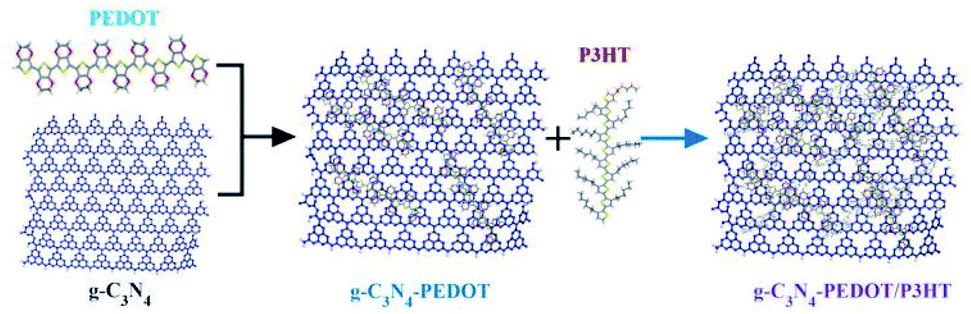 | ||
| Scheme 1 Synthetic route of g-C3N4–PEDOT/P3HT composite. | ||
The morphology of pure g-C3N4 and g-C3N4–PEDOT/P3HT was investigated with transmission electron microscopy (TEM) and high resolution TEM (HRTEM). From Fig. S1,† it was can be observed that g-C3N4 possessed the typical crinkly structure as that reported in our former reports.30 After the PEDOT and P3HT adsorbed to g-C3N4, the main morphologies of g-C3N4–PEDOT/P3HT almost maintain the similar crinkly structure as the pure g-C3N4 (shown in Fig. S2†). Besides, the HRTEM image of the pure g-C3N4 and g-C3N4–PEDOT/P3HT were also exhibited in Fig. S3(a and b).† It can be seen some small nanoparticles with sizes of 5–10 nm are located on the nanoplates of g-C3N4, which could be ascribed to the existence of PEDOT/P3HT. As the unobvious image contrast and amorphous structure between P3HT and PEDOT may lead us hard to distinguish the two polymersemiconductors under the TEM or even under the HRTEM. This kind of similar phenomenon or problem also reported in other literature.23,26 In order to further determine the presence of the PEDOT and P3HT in g-C3N4 and to confirm the distribution of the polymer, element mapping analysis was implemented by the energy dispersive X-ray spectroscopy (EDS). As shown in Fig. 1a and b, all C, N, S and O elements are evenly dispersed at the surface of g-C3N4–PEDOT/P3HT. The clearly appearing hyperintense S and O elements signals only come from the P3HT and PEDOT fragment, while the O elements signals only come from PEDOT fragment. As the contrast sample, the S and O elements disappeared in pure g-C3N4 which further indicated that the g-C3N4–PEDOT/P3HT composite was formed successfully.
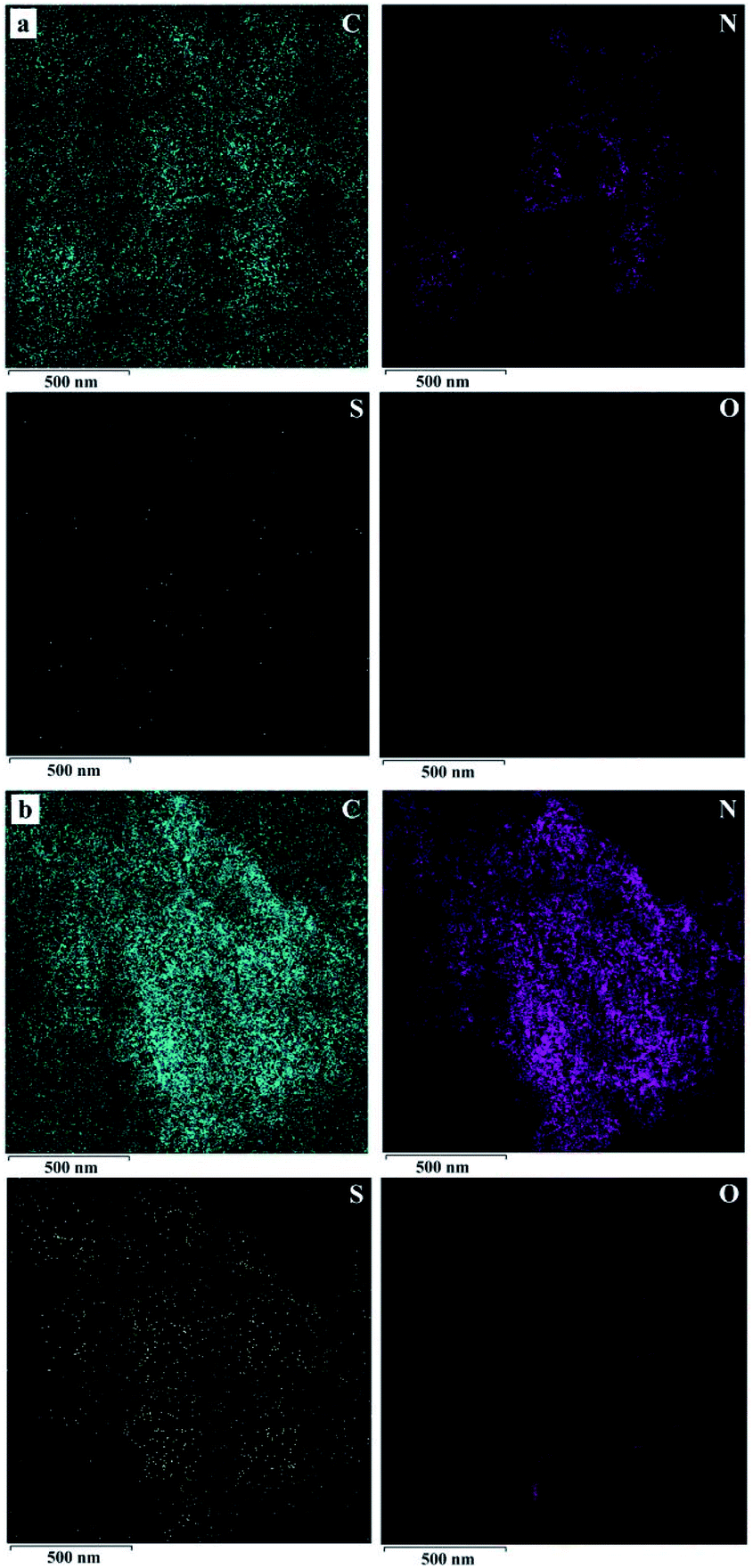 | ||
| Fig. 1 Typical TEM images of the elemental mapping C (green), N (purple), S (yellow) and O (red) (a) g-C3N4, (b) g-C3N4–PEDOT/P3HT. | ||
The XRD pattern of the prepared samples are shown in Fig. 2. All the samples have the similar diffraction peaks at about 12.9° and 27.7°, corresponding to the typical characteristic diffraction peak of the pure g-C3N4.23,24 The diffraction peaks of the different g-C3N4–polymer samples are very similar to those of g-C3N4, indicating that the addition of PEDOT and P3HT would not change the crystal structure of g-C3N4.
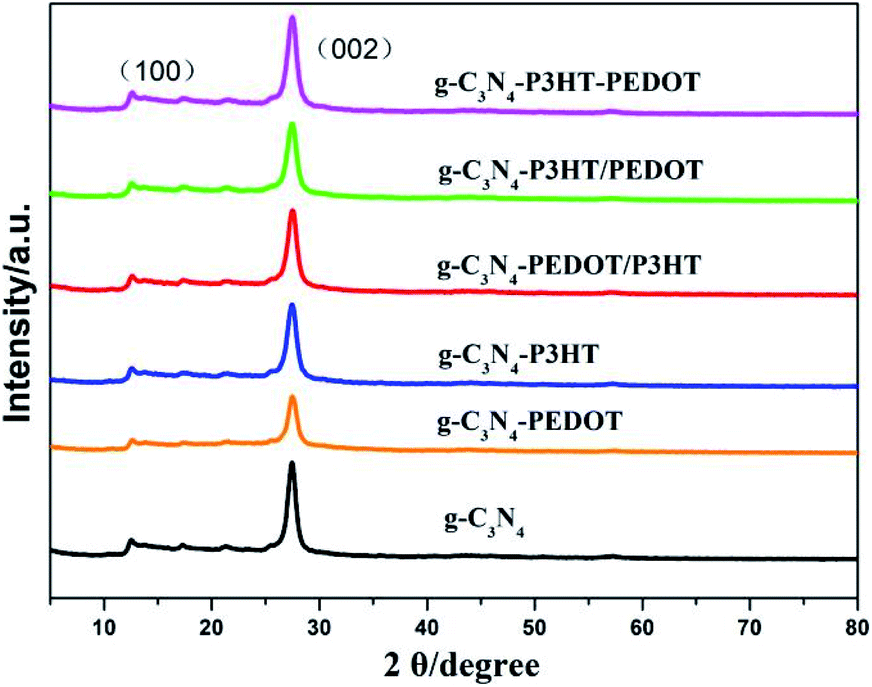 | ||
| Fig. 2 XRD patterns of the as-prepared g-C3N4–polymer samples. | ||
Fig. 3 shows the FTIR spectra of g-C3N4, g-C3N4–P3HT, g-C3N4–PEDOT and g-C3N4–PEDOT/P3HT. As shown in Fig. 2, the obtained composite samples show the typical absorption peak of pure g-C3N4. The typical absorption peak at 807 cm−1 is belonged to the breathing mode of the triazine units.31 In the scope of 1240–1650 cm−1, these peaks are corresponded to the stretching vibration of C–N.32 Additionally, the peaks at around 3150–3450 cm−1 are allocated to stretching vibration modes of the –NH2 and –NH group of g-C3N4. Therefore, the composite photocatalysts g-C3N4–P3HT, g-C3N4–PEDOT and g-C3N4–PEDOT/P3HT show the similar characteristic peaks of pure g-C3N4 and no new peak appear in the spectrum, which means that the relatively low quantity of PEDOT or P3HT in the g-C3N4–polymer composites or without chemical bond formed between g-C3N4 and PEDOT (or P3HT).
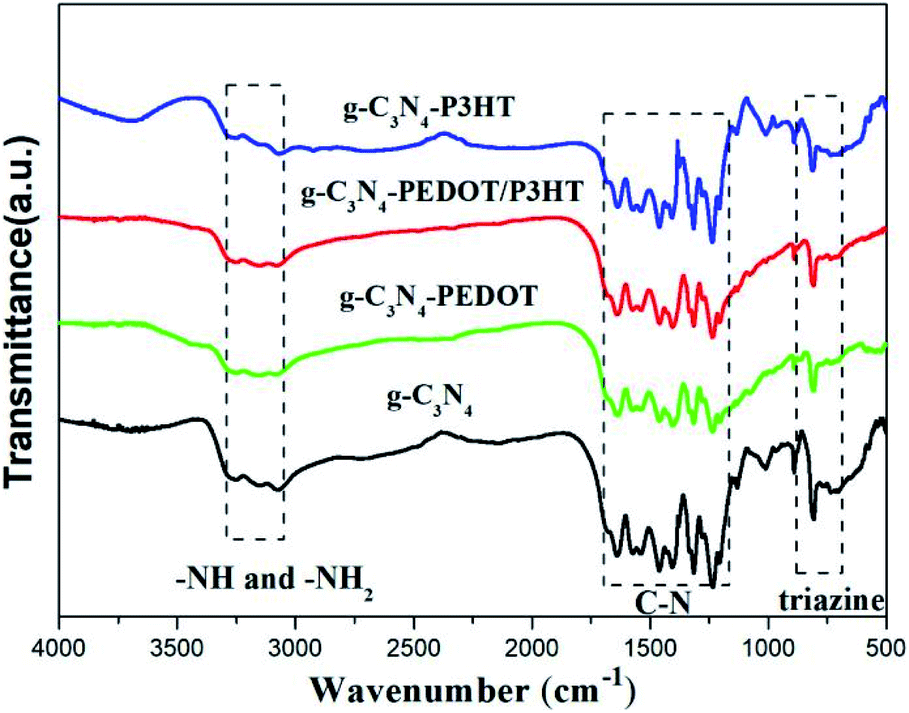 | ||
| Fig. 3 The FTIR spectral of g-C3N4, g-C3N4–P3HT, g-C3N4–PEDOT and g-C3N4–PEDOT/P3HT. | ||
Optical and photoelectrochemical properties analysis
The UV-Vis diffuse reflectance spectra (DRS) of different polymer sensitized g-C3N4 samples are analyzed to show the optical absorption properties. From the Fig. 4, pure g-C3N4 only can absorb visible light with the absorption edge that is shorter than 460 nm, corresponding to a bandgap of ∼2.7 eV. When P3HT is loaded to g-C3N4, the g-C3N4–P3HT shows a distinct spectral absorption band in the scope of 400–700 nm which is belong to the typical absorption area of the pure P3HT. However, the absorption edge of g-C3N4 or g-C3N4–P3HT remained almost unchanged after the impregnation of PEDOT. The similar phenomenon can also be seen in g-C3N4–PEDOT/P3HT, g-C3N4–P3HT/PEDOT and g-C3N4–P3HT–PEDOT (Fig. S4†). These results reveal that PEDOT did not change the electronic structure of g-C3N4 or g-C3N4–P3HT. The little increased baseline of the absorption by PEDOT could be due to the light blue color of PEDOT it self. Besides, the obvious colour change can be found in the ordinary optical photos of the as-prepared g-C3N4–polymer samples also shown in Fig. S5.†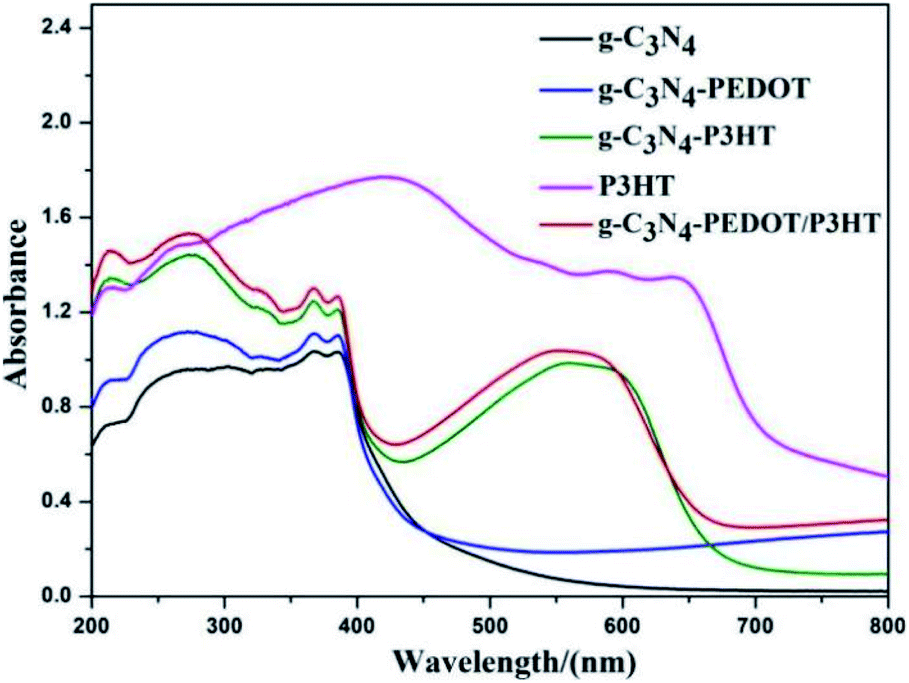 | ||
| Fig. 4 UV-Vis diffuse reflectance spectra of the g-C3N4–polymer samples. | ||
In order to investigate the role of PEDOT, P3HT and photo induced electron–hole separation abilities of obtained samples, the photoluminescence (PL) spectra analysis was performed and illustrated in Fig. 5. As can be seen, pure g-C3N4 shown a broad PL emission peak, corresponding to the fast carrier recombination of pure g-C3N4. When the PEDOT or P3HT was loaded to g-C3N4, the PL peak appeared gradually quenched for g-C3N4–P3HT and g-C3N4–PEDOT. Significant PL quenching was observed in the polymer composite surface heterojunction (SHJ) catalysts. For the PL spectra of g-C3N4–PEDOT/P3HT, g-C3N4–P3HT/PEDOT and g-C3N4–P3HT–PEDOT, the intensity was much lower than that of pure g-C3N4 or g-C3N4–PEDOT. These results indicated that the efficient charge transfer or separation from both PEDOT and P3HT to g-C3N4 could be achieved successfully by the proposed strategy, which was benefit for the enhanced photocatalytic performance.
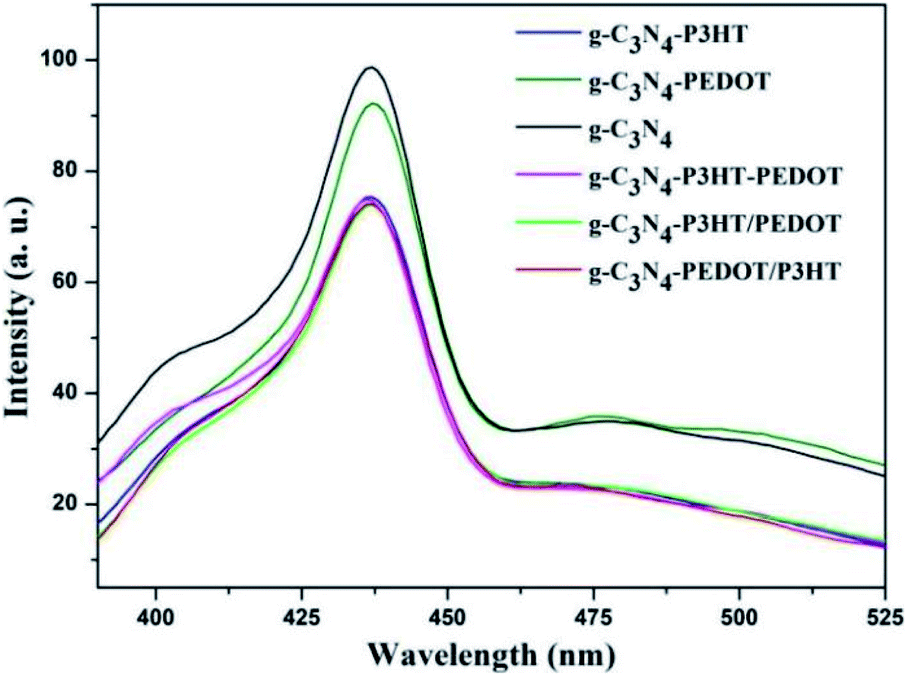 | ||
| Fig. 5 Photoluminescence emission spectra of pure g-C3N4 and all the g-C3N4–polymer composite samples. | ||
Fig. 6 shows the transient photo-current behavior of the obtained g-C3N4, g-C3N4–P3HT, g-C3N4–PEDOT, g-C3N4–P3HT–PEDOT, g-C3N4–P3HT/PEDOT and g-C3N4–PEDOT/P3HT. For the pure g-C3N4, it appears the lower transient photocurrent response, while the photocurrent of g-C3N4–P3HT, g-C3N4–PEDOT, g-C3N4–P3HT/PEDOT or g-C3N4–P3HT–PEDOT is relatively higher than that of the pure g-C3N4. It is obviously observe that the photocurrent of g-C3N4–PEDOT/P3HT is much higher than that of g-C3N4–P3HT and g-C3N4–PEDOT. The much improved photocurrent over g-C3N4–PEDOT/P3HT further intimates the synergy among g-C3N4, PEDOT and P3HT. The highest photocurrent density also demonstrates that g-C3N4–PEDOT/P3HT is superior to g-C3N4–P3HT and g-C3N4–PEDOT in separating photo induced electron–hole pairs.
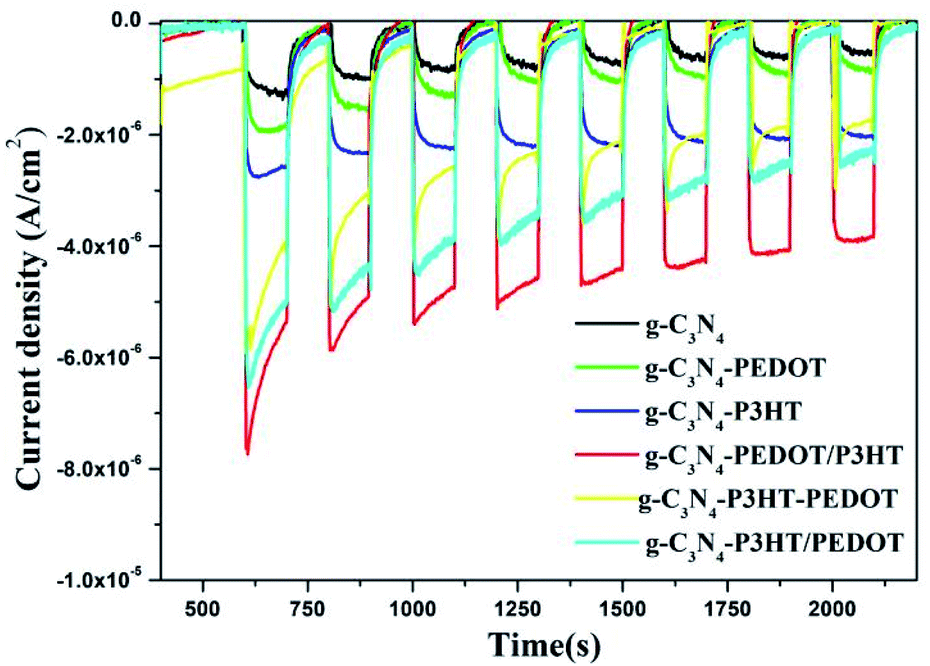 | ||
| Fig. 6 Transient photocurrent performance of the obtained g-C3N4 and the g-C3N4–polymer composite samples in Na2SO4 solution (0.1 M). | ||
Photocatalytic activity of the g-C3N4 composite samples
The photocatalytic H2 evolution performance of the obtained photocatalysts were assessed under visible light using ascorbic acid (AA) as sacrificial reagent. As shown in the Fig. 7a, when loaded with Pt as a cocatalyst, g-C3N4 and g-C3N4–PEDOT shows a photocatalyst H2 evolution rate of 403.7 μmol h−1 g−1 and 3633.9 μmol h−1 g−1, respectively. However, after P3HT added to g-C3N4, the H2 evolution rate of g-C3N4–P3HT reaches at 353233.3 μmol h−1 g−1, which is over 875 times higher than that of g-C3N4 or 97 times higher than that of g-C3N4–PEDOT. As reported, the wide light absorption and efficient charge migration between P3HT and g-C3N4 was responsible for the magnificent photoactivity of the polymer/polymer surface heterojunction catalyst. Based on the aforementioned assumptions, we want to construct a g-C3N4–PEDOT/P3HT surface heterojunction catalyst with P3HT and PEDOT as a polymer sensitizer and hole transport pathway to improve the photocatalytic activity, respectively. In order to verify the above hypothesis, the photocatalyst H2 evolution rate of g-C3N4–PEDOT/P3HT, g-C3N4–P3HT/PEDOT and g-C3N4–P3HT–PEDOT are also tested. When the PEDOT is added to g-C3N4–P3HT, the H2 evolution rate is enhanced obviously, which is of 427703.3 μmol h−1 g−1, 421030.0 μmol h−1 g−1 and 407256.7 μmol h−1 g−1 for g-C3N4–PEDOT/P3HT, g-C3N4–P3HT/PEDOT and g-C3N4–P3HT–PEDOT, respectively. The results also confirm that the order of adding P3HT and PEDOT to g-C3N4 has little effect on the properties of the composite sample. As the best sample g-C3N4–PEDOT/P3HT, the photocatalyst H2 evolution rate is up to 427703.3 μmol h−1 g−1 which is 1059 times higher than that of g-C3N4, 118 times higher than that of g-C3N4–PEDOT or even 1.2 times higher than that of g-C3N4–P3HT. Besides, the apparent quantum yields (AQY) of g-C3N4–PEDOT/P3HT were also tested, the AQY values were 4.4%, 10.6% and 14.9% under 420 nm, 520 nm and 600 nm monochromatic light, respectively. In addition, the comparison of the H2 evolution activity and AQY between this work and other similar polymer composite photocatalysts reported in the literature was added which was shown in Table S1 (ESI†).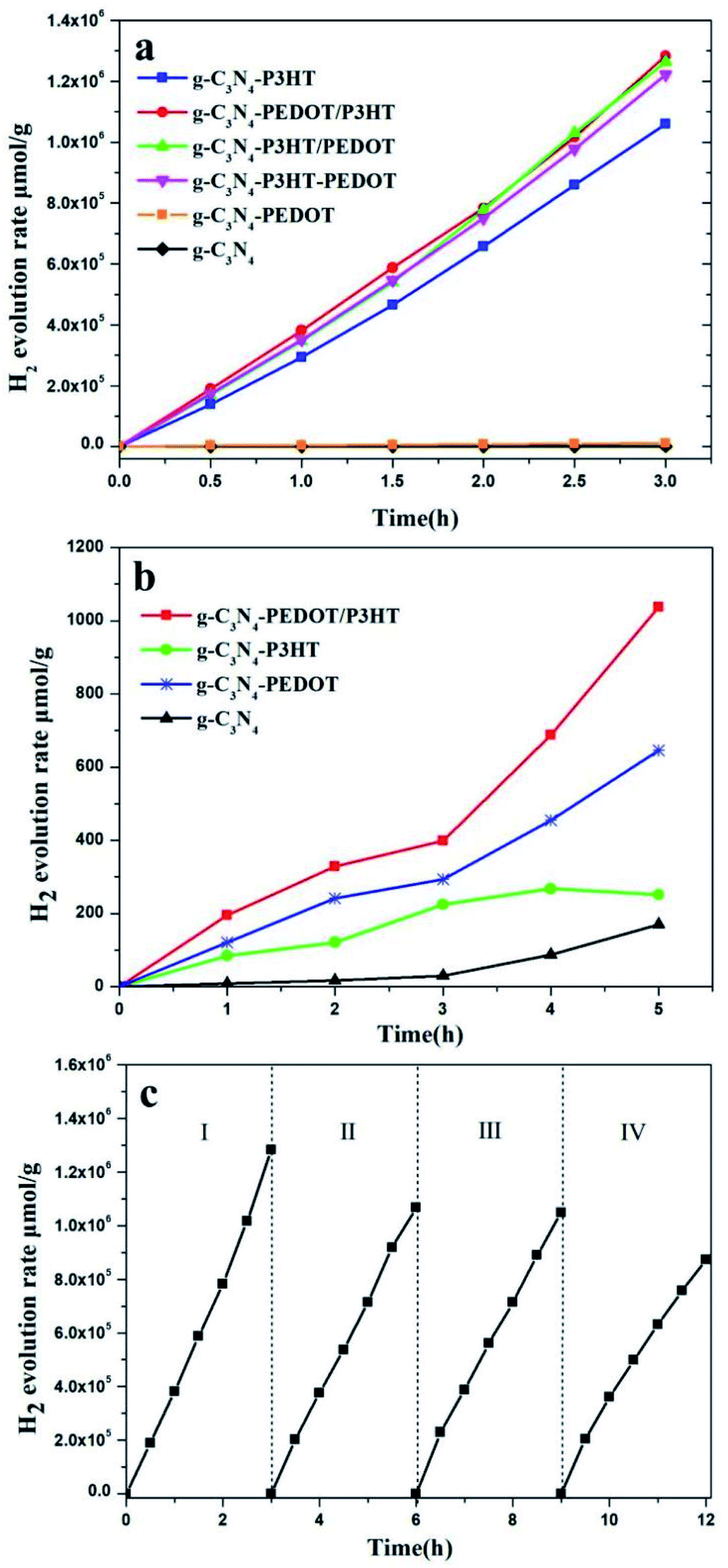 | ||
| Fig. 7 The photocatalytic H2 evolution as a function of reaction time. (a) The comprehensive comparison with AA as sacrificial reagent, (b) the comprehensive comparison from pure water, (c) the recyclable stability of H2 evolution for g-C3N4–PEDOT/P3HT under visible light (λ ≥ 400 nm). | ||
In order to further confirm the separation situation of the photo generated electron–hole, the photocatalytic H2 evolution from pure water is investigated too. The results in Fig. 7b show that the amount of H2 evolution rate of g-C3N4, g-C3N4–P3HT, g-C3N4–PEDOT and g-C3N4–PEDOT/P3HT is 56.9 μmol h−1 g−1, 83.8 μmol h−1 g−1, 215.4 μmol h−1 g−1 and 345.7 μmol h−1 g−1. The g-C3N4 and g-C3N4–P3HT exhibit the lower photocatalytic H2 evolution rate, while the g-C3N4–PEDOT and g-C3N4–PEDOT/P3HT show the obviously enhanced photocatalytic H2 evolution rate which is 3.7 and 6.1 times higher than that of g-C3N4. Besides, the photocatalytic evolved gas is only hydrogen, no oxygen is detected. And these kind of similar phenomenon were also observed in other work, like g-C3N4–Pt or PPy/g-C3N4 in pure water, and the possible product of water oxidation is hydrogen peroxide.25
The encouraging result indeed confirms the synergy between the P3HT and PEDOT when both of them are binding with g-C3N4, which is beneficial for the separation of the photo induced electron–hole. The recyclable stability of g-C3N4–PEDOT/P3HT is also evaluated by the cycling experiments under λ ≥ 400 nm light irradiation conditions (shown Fig. 7c). After four cycles, H2 evolution activity still maintains about 75.0% of the initial one. Besides, the additional characterization of the recycled g-C3N4–PEDOT/P3HT/Pt composite after stability test were performed to further demonstrate the stability of the target photocatalyst. From Fig. 8a and b, it can be seen that the typical crystal structure and main elements characteristic peaks(the specific element of C, N, O and Pt shown in Fig. S6†) appeared no obvious change after four cycles, which further confirmed the photocatalyst was quite stable under the reaction condition.
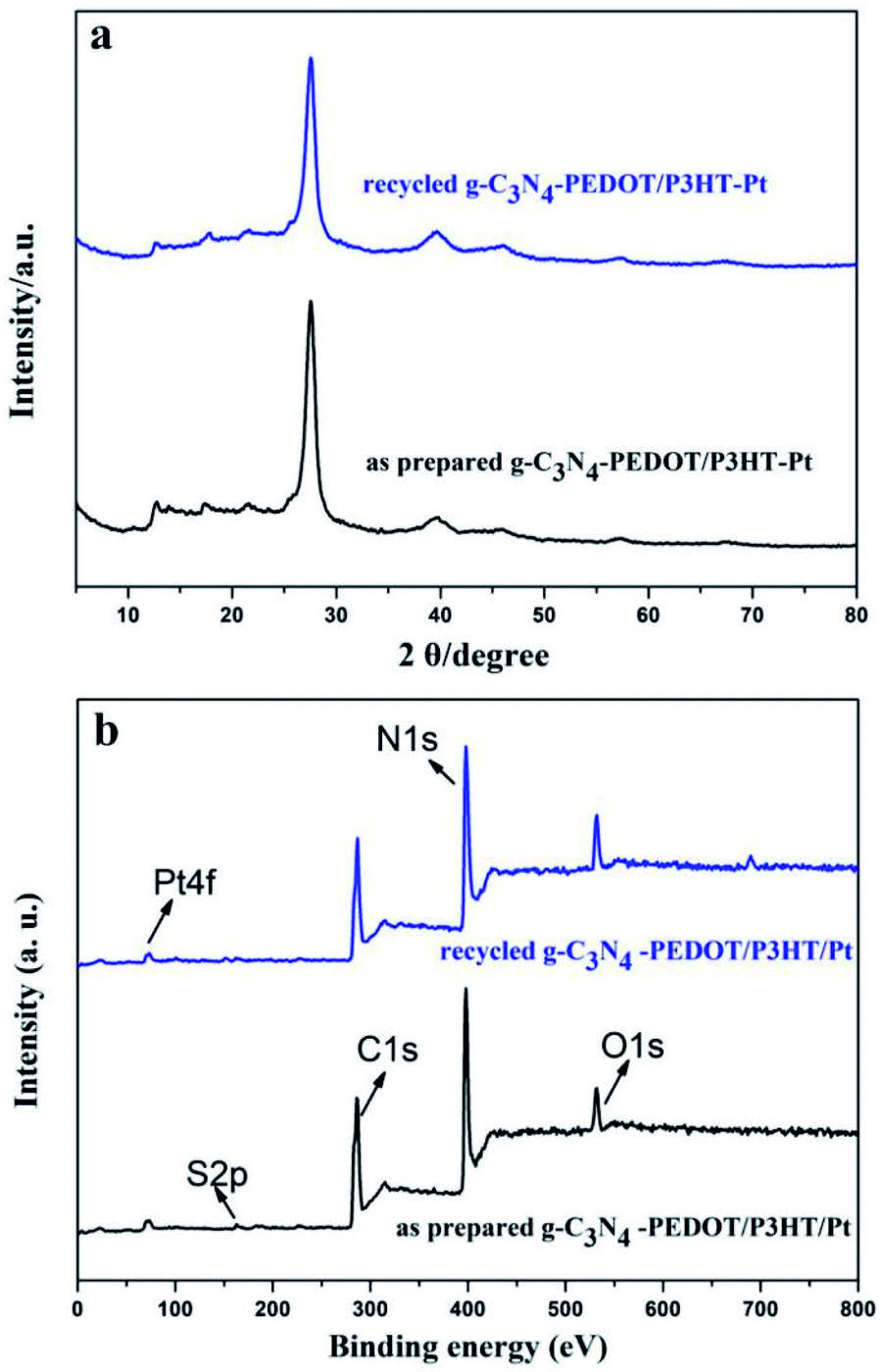 | ||
| Fig. 8 (a) The XRD patterns of the g-C3N4–PEDOT/P3HT/Pt composite before and after stability test in AA solution, (b) the total XPS element spectrum of g-C3N4–PEDOT/P3HT/Pt composite before and after stability test in AA solution. | ||
Photocatalytic mechanism of the g-C3N4–polymer catalyst
In this part, MD simulations were conducted to investigate the possible interaction between g-C3N4 and PEDOT or P3HT polymer. In order to better clarify the microstructure of g-C3N4 hybride with PEDOT or P3HT, the radial distribution functions (RDF) and mean square displacement (MSD) were calculated. Fig. 9(I) and (III) showed the equilibrated configurations and partial enlarged view of g-C3N4–polymer. As can be seen, the g-C3N4 structure was nearly unchanged, and only weak van der Waals forces presented between g-C3N4 and PEDOT or P3HT. From the RDF results in Fig. 9(II), the distance between C and N from g-C3N4 with S from PEDOT was mainly centered at the range about 3.9 Å ∼ 4.1 Å, while the distance between C and N from g-C3N4 with S from P3HT was disperse with a relatively large range about 3.8 Å ∼ 5.0 Å which meant a relatively stronger interaction between g-C3N4 with PEDOT. The similar results were also confirmed by MSD and shown in Fig. 9(IV). For g-C3N4–PEDOT, the diffusion coefficient of g-C3N4 and PEDOT were kept the same level about 5.18 × 10−14 m2 s−1 (g-C3N4) and 7.28 × 10−14 m2 s−1 (PEDOT). However, the diffusion coefficient of g-C3N4 and P3HT appeared a larger difference, about 5.47 × 10−14 m2 s−1 (g-C3N4) and 1.04 × 10−12 m2 s−1 (P3HT). The smaller difference of the diffusion coefficient between g-C3N4 and PEDOT illustrated a relatively stronger interaction between the two compared with that of g-C3N4 and P3HT, which may attributed to the relatively small molecular volume of PEDOT and without any side chain resistance. Then we may imagine that the g-C3N4–PEDOT/P3HT SHJ catalyst could be formed with P3HT as a polymer sensitizer is the upper layer, while the PEDOT as hole transport pathway is interlayer between P3HT and g-C3N4, and the three different part can form an effective mutual synergy system eventually.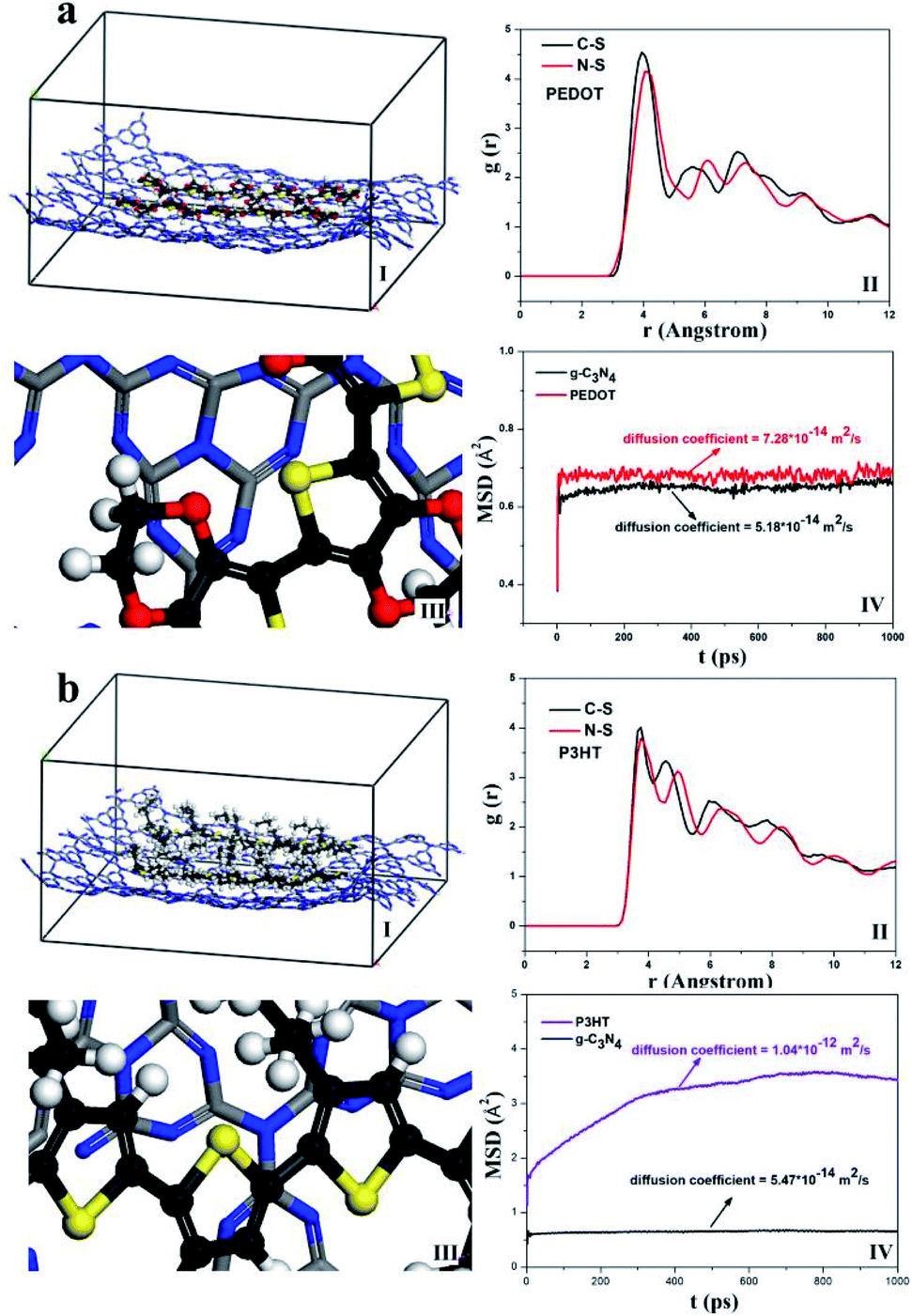 | ||
| Fig. 9 MD simulated equilibrated configurations (I) g-C3N4–polymer (II) RDF (III) partial enlarged view of g-C3N4–polymer and (IV) MSD (a) g-C3N4–PEDOT, (b) g-C3N4–P3HT. | ||
The possible mechanism of photocatalysts for the superior photocatalytic performance of H2 evolution is shown in the Fig. 10. As formerly reported, the LUMO level of P3HT is more negative than the conduction band (CB) of g-C3N4 which is favorable for the photo induced electrons transferring from P3HT to g-C3N4, and then trapped by the Pt for H2 production. Besides, as the valence band (VB) of g-C3N4 is more positive than that of P3HT, the holes may have the trend of spontaneous migration from the VB of g-C3N4 to the VB of P3HT. More importantly, the existing PEDOT possesses the relatively higher hole mobility and can be used as a hole conductor between g-C3N4 and P3HT. Then, the photogenerated holes migration can be accelerated by PEDOT from the VB of g-C3N4 to the VB of P3HT. Finally, the holes on the VB of P3HT are depleted by AA. All those factors may benefit the rapid migration of photo generated electron–hole pairs and eventually facilitate the photocatalytic H2 activity process of g-C3N4–PEDOT/P3HT under visible light.
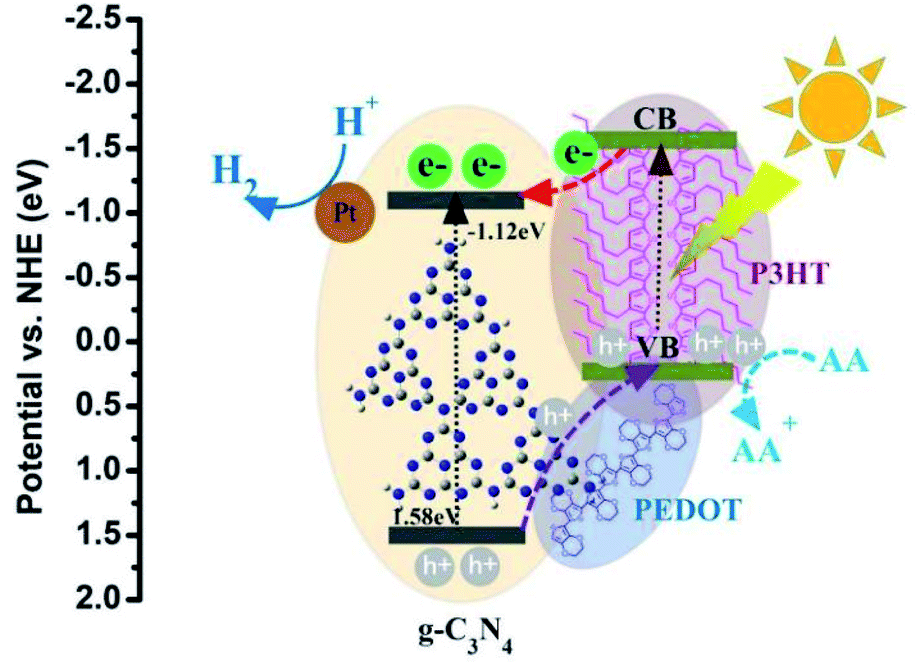 | ||
| Fig. 10 A proposed photocatalytic H2 production mechanism of g-C3N4–PEDOT/P3HT/Pt SHJ catalyst structure. | ||
Conclusions
In conclusion, the g-C3N4–PEDOT/P3HT surface heterojunction (SHJ) catalyst with P3HT and PEDOT as a polymer sensitizer and hole transport pathway was successfully constructed. Moreover, as Pt particles are often deposited to g-C3N4 as electron sink and proton reduction sites, we successfully construct a g-C3N4–PEDOT/P3HT/Pt composite, in which PEDOT act as hole transport pathway, P3HT as the polymer sensitizer and Pt as the reduction reaction sites. In this way, the g-C3N4–PEDOT/P3HT/Pt composite exhibits the photocatalyst H2 evolution rate is up to 427703.3 μmol h−1 g−1 which is 1059 times higher than that of g-C3N4, 118 times higher than that of g-C3N4–PEDOT. The dramatically enhanced photocatalytic performance can be assigned to the synergy among g-C3N4, PEDOT and P3HT, which finally facilitate the separated rapid migration of photoinduced electron–hole pairs. This work may provide a useful insights for designing the surface heterojunction (SHJ) composites photocatalyst with high photocatalytic activity for H2 production.Experimental
Materials
The poly(3-hexylthiophene-2,5-diyl) (regioregular) was acquired from Sigma-Aldrich, poly(3,4-ethylenedioxythiophene)–poly(styrenesulfonate) (1.5% in water) was purchased from Macklin, and both used without any further treatment. The rest reagents and solvents were used as received from commercial suppliers without additional treatment.Synthesis of g-C3N4–polymer composite photocatalysts
g-C3N4 was synthesized according to our previous reported work.32 Briefly, melamine (3 g), urea (7.5 g) and ammonium chloride (2 g) were mixed in an alumina crucible with a cover and the precursor was heated at 600 °C for 4 hours in air with the rate of 5 °C min−1. After cooling to the room temperature, the product was gathered for later use.Photocatalysts characterization
TEM and HRTEM characterization were characterized on a JEOL-2010 electron microscope at an acceleration voltage of 200 kV. The crystal phases of the samples were analyzed by the Rigaku D/MAX 2500X X-ray powder diffractometer with Cu Kα radiation. XPS measurements were performed on a Thermo Scientific ESCALAB Xi+ (monochromatic Mg Kα, 1253.6 eV). The UV-vis spectrometer was carried out on an Agilent Cary 5000 UV-vis-NIR spectrophotometer with BaSO4 as a reference. Photoluminescent (PL) spectra were recorded by a spectrofluorophotometer (Cary Eclipse) with excitation slit widths of 5 nm and emission slit widths of 5 nm. The infrared pattern was analysed by FTIR spectrometer (PE Spectrum Two).Photocatalytic activity analyse
Photocatalytic activity tests were carried out on a CEL-SPH2N-S9 photocatalytic water splitting device (Ceaulight Beijing). The Pt content with 3 wt% was deposited to the catalyst according to the method in our previous work.32 Consequently, the various g-C3N4–polymer composites photocatalysts were prepared first and then the Pt was loaded to composite g-C3N4–polymer catalyst. For specific photocatalytic H2 experiment, 10 mg catalyst loaded with 3 wt% Pt sample was added into 10 mL deionized water containing 1.8 M ascorbic acid as the sacrificial reagent aqueous solution by ultrasonication for 10 min. Prior to the light irradiation, the system was eliminated for 30 min to remove air. Next, the solution temperature was maintained at 3 °C. The measurements were performed under illumination of a Xenon lamp (300 W, Ceaulight Beijing) with a 400 nm cutoff filter and the corresponding incident light intensity was 100 mW cm−2. The photocatalytic H2 evolution rate was detected by a gas chromatography (GC-7920, Ceaulight Beijing) with high purity N2 as carrier gas. For apparent quantum yields (AQY) test, 20 mg as prepared catalyst sample was added into 45 mL 0.6 M H2SO4 solution containing 13.3 g ascorbic acid as sacrificial reagent by ultrasonication for 10 min. Before irradiation, the system was evacuated for 30 min to remove air and the temperature of the solution was maintained at 3 °C. A 300 W Xenon lamp (Ceaulight Beijing) with a 400 nm cutoff filter was used for irradiation the mixture for 60 min and evacuated for another 30 min, then various band-pass filters (420 nm, 520 nm and 600 nm) was used as the irradiation source to measure the monochromatic AQY for the H2. Besides, the specific AQY processing may reference our former work.32Photo electrochemical measurements
The carbon cloth (CeTech Co. Ltd, WO1002) was cleaned by sonication sequentially in acetone, ethanol and deionized water for 30 min each, followed by drying in vacuum at 100 °C. Then, chemical activation was operated by submerging carbon cloth into a mixed solution with concentrated sulfuric acid (H2SO4, 98%) and concentrated nitric acid (HNO3, 68%) in a volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 (H2SO4/HNO3) for 12 hours. Afterwords, the activated carbon cloth was washed with deionized water until neutral, and then placed for standby. Subsequently, 5 mg obtained photocatalysts sample were mixed with 990 μL ethanol, 10 μL Nafion solution (5 wt%, Aladdin). Then, the sample mixture solution was ultrasonic treatment for 30 min. The obtained suspension (about 100 μL) was dropped onto the activated carbon cloth electrode and dried naturally in the air. A three-electrode system was employed where the as prepared carbon cloth electrode, a platinum piece, and a saturated calomel electrode (SCE) were utilized as a working electrode, a counter electrode, and a reference electrode, respectively. Besides, the Na2SO4 solution (0.1 M) was used as electrolyte for the transient photocurrent I–T curve testing with above three-electrode system. The measurements were performed under illumination of a Xenon lamp (300 W, Ceaulight Beijing) with a 400 nm cutoff filter and the corresponding incident light intensity was 100 mW cm−2.
:3 (H2SO4/HNO3) for 12 hours. Afterwords, the activated carbon cloth was washed with deionized water until neutral, and then placed for standby. Subsequently, 5 mg obtained photocatalysts sample were mixed with 990 μL ethanol, 10 μL Nafion solution (5 wt%, Aladdin). Then, the sample mixture solution was ultrasonic treatment for 30 min. The obtained suspension (about 100 μL) was dropped onto the activated carbon cloth electrode and dried naturally in the air. A three-electrode system was employed where the as prepared carbon cloth electrode, a platinum piece, and a saturated calomel electrode (SCE) were utilized as a working electrode, a counter electrode, and a reference electrode, respectively. Besides, the Na2SO4 solution (0.1 M) was used as electrolyte for the transient photocurrent I–T curve testing with above three-electrode system. The measurements were performed under illumination of a Xenon lamp (300 W, Ceaulight Beijing) with a 400 nm cutoff filter and the corresponding incident light intensity was 100 mW cm−2.
Computational methods
In this work, MD simulation with a full atomistic force field was conducted to investigate the possible interation between g-C3N4 and PEDOT or P3HT polymer.33 The COMPASS module from Materials Studio developed by Accelrys Inc was used to conduct the computations. For the target system, the initial simulation system was optimized with 50000 steps. Then, the NVT dynamic equilibration was running for 3 ns with time step 1 fs. Besides, the mean square displacement (MSD) and radial distribution functions (RDF) was obtained by analyzing the data collected at 1000 steps intervals during the last 1 ns. The molecular structure of monolayer g-C3N4, the chain of PEDOT, the chain of P3HT and g-C3N4–polymer composites were depicted in Fig. S7.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the CAS “Light of West China” Program, the Project of Qinghai Science & Technology Department (Grant No. 2021-ZJ-754).Notes and references
- G. Zhang, G. Li, T. Heil, S. Zafeiratos, A. Savateev and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 3433–3437 CrossRef CAS PubMed.
- Y. Sui, J. Liu, Y. Zhang, X. Tian and W. Chen, Nanoscale, 2013, 5, 9150–9155 RSC.
- M. Luo, H. Gong, W. Yang, F. He, Y. Cao, Y. Zhang, Y. Zhang, K. Liu, H. Cao and H. Yan, Int. J. Hydrogen Energ., 2019, 44, 7108–7117 CrossRef CAS.
- Y. Ma, X. L. Wang, Y. S. Jia, X. B. Chen, H. X. Han and C. Li, Chem. Rev., 2014, 114, 9987–10043 CrossRef CAS PubMed.
- X. B. Chen, S. H. Shen, L. J. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- Y. Jiang, Z. Lin, Y. Zhang, Y. Lai and C. Yang, New J. Chem., 2020, 44, 17891–17898 RSC.
- K. Li, B. Chai, T. Y. Peng, J. Mao and L. Zan, ACS Catal., 2013, 3, 170–177 CrossRef CAS.
- J. H. Yang, H. J. Yan, X. L. Wang, F. Y. Wen, Z. J. Wang, D. Y. Fan, J. Y. Shi and C. Li, J. Catal., 2012, 290, 151–157 CrossRef CAS.
- X. Liu, L. Yan, L. He, Z. Yao, L. You, L. Liu, G. Zhou and H. Huang, New J. Chem., 2020, 44, 10739–10746 RSC.
- D. Kim, K. K. Sakimoto, D. Hong and P. D. Yang, Angew. Chem., Int. Ed., 2015, 54, 3259–3266 CrossRef CAS PubMed.
- A. J. Bard and M. A. Fox, Acc. Chem. Res., 1995, 28, 141–145 CrossRef CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- P. Zeng, Y. Zheng, S. Chen, H. Liu, R. Li and T. Peng, Chem. New J. Chem., 2020, 44, 11237–11247 RSC.
- Y. H. Chiu, T. Chang, C. Y. Chen, M. Sone and Y. J. Hsu, Catalysts, 2019, 9, 430 CrossRef CAS.
- Y. H. Chiu, T. H. Lai, M. Y. Kuo, P. Y. Hsieh and Y. J. Hsu, APL Mater., 2019, 7, 080901–080911 CrossRef.
- M. J. Fang, C. W. Tsao and Y. J. Hsu, J. Phys. D: Appl. Phys., 2020, 53, 143001 CrossRef CAS.
- Y. A. Chen, Y. T. Wang, H. S. Moon, K. Yong and Y. J. Hsu, RSC Adv., 2021, 11, 12288 RSC.
- C. W. Tsao, M. J. Fang and Y. J. Hsu, Coordin. Chem.Rev., 2021, 438, 213876 CrossRef CAS.
- L. Li, M. Z. Zhang, C. Ding and J. Xu, Chem. Eng. J., 2021, 419, 129543 CrossRef CAS.
- T. Zheng, J. Xu, Z. Zhang and H. Zeng, RSC Adv., 2015, 5, 99658–99663 RSC.
- H. Yan and Y. Huang, Chem. Commun., 2011, 47, 4168–4170 RSC.
- X. Zhang, T. Peng and S. Song, J. Mater. Chem. A, 2016, 4, 2365–2402 RSC.
- Y. F. Zhang, F. Mao, H. J. Yan, K. W. Liu, H. M. Cao, J. G. Wu and D. Q. Xiao, J. Mater. Chem. A, 2015, 3, 109–115 RSC.
- X. H. Zhang, B. Peng, S. Zhang and T. Y. Peng, ACS Sustain. Chem. Eng., 2015, 3, 1501–1509 CrossRef CAS.
- Y. Sui, J. H. Liu, Y. W. Zhang, X. K. Tian and W. Chen, Nanoscale, 2013, 5, 9150–9155 RSC.
- X. Zheng, Z. Chen, Z. Xu and L. Wang, Chem. Commun., 2014, 50, 6762–6764 RSC.
- A. J. Mozer, D. K. Panda, S. Gambhir, B. W. Jensen and G. G. Wallace, J. Am. Chem. Soc., 2010, 132, 9543–9545 CrossRef CAS PubMed.
- X. Liu, Y. Cheng, L. Wang, L. Cai and B. Liu, Phys. Chem. Chem. Phys., 2012, 14, 7098–7103 RSC.
- X. Li, W. Lu, W. Dong, Q. Chen, D. Wu, W. Zhou and L. Chen, Nanoscale, 2013, 5, 5257–5261 RSC.
- C. Zhang, J. Liu, X. Huang, D. Chen and S. Xu, ACS Omega, 2019, 4, 17148–17159 CrossRef CAS PubMed.
- W. Iqbal, B. Qiu, J. Lei, L. Wang, J. Zhang and M. Anpo, Dalton Trans., 2017, 46, 10678–10684 RSC.
- W. Iqbal, C. Dong, M. Xing, X. Tan and J. Zhang, Catal. Sci. Technol., 2017, 7, 1726–1734 RSC.
- P. Steve, J. Comput. Phys., 1995, 117, 1–19 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05527e |
| This journal is © The Royal Society of Chemistry 2021 |