
Methods and strategies for the synthesis of diverse nanoparticles and their applications: a comprehensive overview
Chetna Dhand
a,
Neeraj Dwivedi
b,
Xian Jun Loh
c,
Alice Ng Jie Ying
a,
Navin Kumar Verma
ad,
Roger W. Beuerman
*ae,
Rajamani Lakshminarayanan
*ae and
Seeram Ramakrishna
*f
aAnti-Infectives Research Group, Singapore Eye Research Institute, Singapore 169856. E-mail: lakshminarayanan.rajamani@seri.com.sg; Tel: +65-6576-7276
bDepartment of Electrical and Computer Engineering, National University of Singapore, Singapore 117582
cInstitute of Materials Research and Engineering, A*STAR (Agency for Science, Technology and Research), 3 Research Link, Singapore 117602
dLee Kong Chian School of Medicine, Nanyang Technological University, Research Techno Plaza, 50 Nanyang Drive, Singapore 635753
eDuke-NUS SRP Neuroscience and Behavioral Disorders, Singapore 169857
fCenter for Nanofibers and Nanotechnology, Department of Mechanical Engineering, National University of Singapore, Singapore 117576. E-mail: seeram@nus.edu.sg; Fax: +65-68725563; Tel: +65-65162216
First published on 26th November 2015
Abstract
Ongoing advances in nanotechnology research have established a variety of methods to synthesize nanoparticles (NPs) from a diverse range of materials, including metals, semiconductors, ceramics, metal oxides, polymers, etc. Depending upon their origin and synthesis methods, NPs possess unique physicochemical, structural and morphological characteristics, which are important in a wide variety of applications concomitant to electronic, optoelectronic, optical, electrochemical, environment and biomedical fields. This review provides a comprehensive overview on various physical, chemical and bio-assisted methods largely employed to synthesize and fabricate NPs of varying size, surface characteristics, functionalities and physicochemical behavior. The key applications of nanoparticles have also been discussed.
 Chetna Dhand | Chetna Dhand is working as a postdoctoral research fellow at Singapore Eye Research Institute, Singapore. She has completed her PhD in Chemistry from University of Delhi in collaboration with National Physical Laboratory, New Delhi, India. Her major research interests include nanomaterials for different biomedical applications including antimicrobial wound dressings and contact lenses, targeted drug delivery, tissue engineering and biosensors. |
 Neeraj Dwivedi | Neeraj Dwivedi obtained his PhD in Physics from the Indian Institute of Technology Delhi, India. Currently, he is working as a Postdoctoral Research Fellow in the Department of Electrical and Computer Engineering, National University of Singapore, Singapore. His research interest includes material science, thin films and coatings, nanomaterials, MEMS, nanobiotechnology. |
 Xian Jun Loh | Dr Xian Jun Loh is a polymer chemist working in the inter-disciplinary field of biomaterials. He is currently a Senior Scientist and the Head of Research Planning at the Institute of Materials Research and Engineering (IMRE) and an Assistant Professor at the National University of Singapore (NUS). He is also an adjunct scientist at the Singapore Eye Research Institute. His main research interests are in the design of supramolecular and stimuli-responsive polymers and hydrogels for biomedical and personal care applications. Currently, he is the author and co-author of 84 journal papers, 11 patents, 10 book chapters and 3 books. |
 Alice Ng Jie Ying | Alice Ng Jie Ying obtained her Bachelor (Hons) degree in Science (Biomedical Sciences) with specialisation in Cancer Biology from University of Bradford, UK. She worked as Research Officer at Anti-Infective Research Group, Singapore Eye Research Institute (SERI), Singapore. Her research focuses is on designing nanoparticles for exploring their applications as drug delivery carriers for eye anti-infectives. |
 Navin Kumar Verma | Navin K. Verma completed his PhD and postdoctoral training in Clinical Medicine at Trinity College Dublin, Ireland. In 2013, Dr Verma joined Lee Kong Chan School of Medicine, Nanyang Technological University Singapore, where he is currently an Assistant Professor of Immunology and Cell Biology. His research is focused on the understanding of signal transduction processes involved in the migration and effector functions of T-lymphocytes in health and diseases. He is also studying the underlying mechanisms by which mammalian cells and tissues interact with biologically responsive nanoparticles, nanostructures and polymeric matrices for their potential therapeutic or diagnostic medical applications. |
 Roger W. Beuerman | Roger Beuerman is currently Senior Scientific Director of the Singapore Eye Research Institute, Professor of Neuroscience and Behavioral Disorders at DUKE-NUS School of Medicine and Adjunct Professor of Ophthalmology, Yong Loo Lin, School of Medicine at the National University of Singapore. Roger and his colleagues have developed two well-known translational programs: one a broad program in infectious disease, including design of new antimicrobials, animal models and biofilms and the other in biomarkers for diseases such as dry eye, and keratoconus that reveal basic biological information about the disease as well as severity. His group has developed new classes of antibiotics effective against resistant forms of Pseudomonas and the CREs. He has more than 300 peer reviewed publications and has co-authored three texts in ophthalmology. |
 Rajamani Lakshminarayanan | R. Lakshminarayanan obtained his PhD from the Department of Chemistry at the National University of Singapore. He was a recipient of the Singapore Millennium Foundation Postdoctoral Fellow and then worked as Research Associate at the University of Southern California. Currently, he is working as a Principal Research Scientist at the Singapore Eye Research Institute. His major interests include antimicrobial peptides design, topical delivery of antimicrobial peptides and protein aggregation diseases. |
 Seeram Ramakrishna | Seeram Ramakrishna, FREng, is the Director of Center for Nanofibers & Nanotechnology at National University of Singapore. He authored 6 books and ∼650 ISI listed journal papers, which attracted ∼46 |
1 Introduction
‘There's Plenty of Room at the Bottom’ a revolutionary lecture by Richard Feynman in 1959 has provided a new thought process to the scientific community to miniaturize and advance prevailing technologies.1 Inspired with this vision and substantiated with tremendous research efforts, a new branch of science called nanotechnology originated.2 Nanotechnology is basically an amalgamation of two words “Nano” and “Technology” that deals with dimensions and tolerances of less than 100 nanometres, especially the engineering of individual atoms and molecules. With a promise to advance and benefit the society through its wide range of applications in medicines, energy, environment, information technology, aerospace science etc., nanotechnology has emerged as a cutting-edge science in this modern era.3–5 Owing to its capability to display improved characteristics at small dimensions, nanomaterial is considered as one of the crucial components of nanotechnology. Enriched with outstanding physical, chemical, biological, optical and electronic properties, nanomaterials helped in improving the existing technologies and also opened new avenues to develop novel scientific/technological fields. Depending upon the desired application, the nanomaterials can be synthesized in diverse shapes and dimensions as described in Table 1.| Type of nanomaterials | Synthesis | Applications |
|---|---|---|
Nanocage: First reported in 2002, this class of nanomaterials has hollow interior and porous walls containing metallic nanoparticles (MNPs) inside. Their size ranges between 10–150 nm6  |
Nanocages can be synthesized by simple template engaged galvanic replacement reaction among the metallic precursor and the nanotemplates of more reactive metals6,7 | As tracers for tracking using multi-photon luminescence, contrasting agents for photoacoustic and multimodal imaging, photothermal agents for the selective destruction of cancerous tissue and drug delivery vehicles for smart release in response to external stimuli such as NIR radiation or high-intensity focused ultrasound8 |
Nanocrystal: Nanocrystal is single or multi-phase polycrystalline solids with grain size of less than 100 nm9  |
Murray et al. have reported high temperature colloidal method to synthesize semiconducting magnetic nanocrystals that are uniform in size to ±one atomic layer, composition, shape, internal structure, and surface chemistry.10 Li et al. have developed an unified liquid–solid–solution phase transfer synthetic strategy to fabricate monodisperse nanocrystals with different chemistries and properties using versatile materials including noble metal, magnetic/dielectric, semiconducting, rare-earth fluorescent, biomedical, organic optoelectronic semiconducting and conducting polymers NPs.11 Hyeon et al. have nicely reviewed the synthesis strategies to design monodisperse nanocrystals of metals, semiconductors and metal oxides12 | Memory devices, solar cells, solid-state displays, photo-detectors and field-effect transistor (FET) detectors13,14 |
Nanobelt: Nanobelt is thin and flat sheets of ribbon-like structures that are typically 30–300 nm in size.15 Nanobelts with rectangular cross-section and well-defined crystalline facets enable to attain unique optical confinement, microcavity, catalysis and piezoelectric effect16  |
Wang et al. have reported the synthesis of 1D ultra-long nanobelts of lanthanum hydroxide using composite-hydroxide-mediated synthesis method16 | Nanobelts have profound impact in the field of self-powered nanodevices and nanosystems. They have applications in FET devices, nanometer-sized ultrasensitive gas and biosensors, resonators, cantilevers, etc.17 |
Nanofiber (NF): These are 2D fibre structures having diameter less than 100 nm  |
Electrospinning is the most explored method for large scale production of NFs of versatile materials including polymers, metal oxides, carbon based materials, supramolecular dipeptides, etc.18–20 Other method to design NFs are emulsion polymerization, self-assembly, melt blowing and phase separation etc.21–23 | Water filtration systems, surgical implants, biosensors, drug delivery systems, electronic devices, tissue engineering, etc.24–26 |
Nanoparticle (NP): According to IUPAC, particle of any shape with dimensions in the 1 × 10−9 and 1 × 10−7 m range is known as nanoparticle (NP)  |
Different chemical, physical and biological strategies are available to design NPs of various material types (polymers, inorganic oxides, metals, etc.) of diverse sizes, surface characteristics and topography.27 Our review is an attempt to compile all the synthesis methods for the NP design | NPs are having a wide range of applications that includes their role in biomedical devices (biosensors, tissue engineering, drug delivery, bioactuators, bio-imaging devices, etc.), electronics and optoelectronic devices, food industry, construction industries, etc.28 |
Nanotube (NT) and Nanorod (NR): NT is microscopic tube whose diameter is measured in nanometers (usually <100 nm).29 NT are mostly hollow. On the contrary NR is solid structure with aspect ratio of ∼3–5 and each of their dimension from 1–100 nm30  |
Arc-discharge, laser ablation, and chemical vapour deposition (CVD) are some of the methods used to prepare metallic/semiconducting nanotubes and nanorods.31 Emulsion polymerization method is another chemical strategy to design soft nanotubular structures32 | Nanotubes specifically carbon nanotubes (CNT) has the potential to design wonder technologies. CNT yarns and sheets have already been known to have promising applications in supercapacitors, actuators and electromagnetic shields.29,33 Continuous efforts are under progress to use these nanostructures in biomedical fields and nanomedicines34 |
| Nanowire (NW): Nanowire is smart 1D nanostructural material with dimensions of the order of 10−9 meters | Nanowires can be synthesized both by top-down approach or bottom-up approaches. The commonly used techniques for the synthesis of nanowires are chemical, electrochemical, photochemical, electrophoresis, CVD, physical vapour deposition (PVD), plasma assisted chemical vapour deposition (PACVD), etc.35,36 | Since semiconductor nanowires show tremendous electronic and optoelectronic properties, they can be used for fabrication of p–n junctions, transistors, solar cells, sensors, etc.37,38 |
Quantum dot (QD): Quantum dots is nanocrystal of semiconducting material, small enough to exhibit quantum mechanical properties and their excitons are confined in all three dimensions. These exhibits strong size dependent optical and electronic properties. QD can contain as few as 100 to 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 atoms within the quantum dot volume, with a diameter of 10 to 50 atoms39 000 atoms within the quantum dot volume, with a diameter of 10 to 50 atoms39 |
Three routes to design QD are: (i) organometallic method that consists of three components viz., precursors, organic surfactants and solvents, (ii) aqueous synthesis using short-chain thiols as stabilising agents, (iii) biological method using microorganisms40,41 | Due to their superb optical and electronic properties, they can be extensively applied to light-emitting diodes, lasers, biomarkers, biosensor devices and biomedical imaging41–43 |
Nanocomposites: It is multiphase material where at least one of the constituent phases has one dimension less than 100 nm.44 The promise of nanocomposites lies in their multifunctionality, the possibility of realizing unique combinations of properties unachievable with traditional pristine materials45  |
Different chemical, physical and biological methods can be employed to derive different nanocomposites depending upon the materials concerned | Applications of nanocomposites are in microelectronic industry, aerospace, electronic packaging and also in catalysis46–48 |
Due to its small dimension and exceptional surface properties, NPs have shown great potential for diverse applications. In general, NPs are the most fundamental element of nanomaterials and act as bridging link between the atomic/molecular structures and nanomaterials. The NPs demonstrate size and shape dependent properties, which can be tailored over wide range. Dendrimers, liposomes, polymer micelles, quantum dots are the other structural analogues of NPs, used for various applications.49,50 Depending upon the type of the material, NPs are used for broad spectrum applications including drug delivery,51,52 biosensors,53 bioimaging,54 molecular tagging,55 food technology,56 textile manufacturing,57 antimicrobial coatings,58 quantum computers,59 quantum lasers,60 energy and environment, etc. (discussed in Section 2.4).
To enfold such a broad range of applications, one of the most essential aspects is to understand various possible synthesis methods to design desired NPs. Selection of the synthesis method to realize required NPs largely depends on the desired size, appropriate surface properties and the type of concerned material such as metals, semiconductors, ceramics, polymers, etc. In present article, we understand and review the key synthesis methods for the preparation of various types of NPs. Synthesis methods are broadly divided into three parts viz., physical methods, chemical methods and bio-assisted methods. In addition, attempts have also been made to provide a brief discussion on the key applications of different types of nanoparticles.
2 Methods for the synthesis of nanoparticles
High-throughput NPs with good and controlled quality are desirable for their commercialization in various fields of applications. There are two basic approaches commonly employed to prepare NPs; (1) top-down approach, where synthesis is initialized with the bulk counterpart that leaches out systematically bit-after-bit leading to the generation of fine NPs. Photolithography, electron beam lithography, milling techniques, anodization, ion and plasma etching are some of the commonly used top-down methods for the mass production of NPs; (2) bottom-up approach, which involves the coalescence or assembling of atoms and molecules to generate diverse range of NPs. Examples of bottom-up approach include self-assembly of monomer/polymer molecules, chemical or electrochemical nanostructural precipitation, sol–gel processing, laser pyrolysis, chemical vapour deposition (CVD), plasma or flame spraying synthesis and bio-assisted synthesis.61 In general, NP synthesis methods can be divided in three groups – (1) physical methods, (2) chemical methods, and (3) bio-assisted methods (Fig. 1).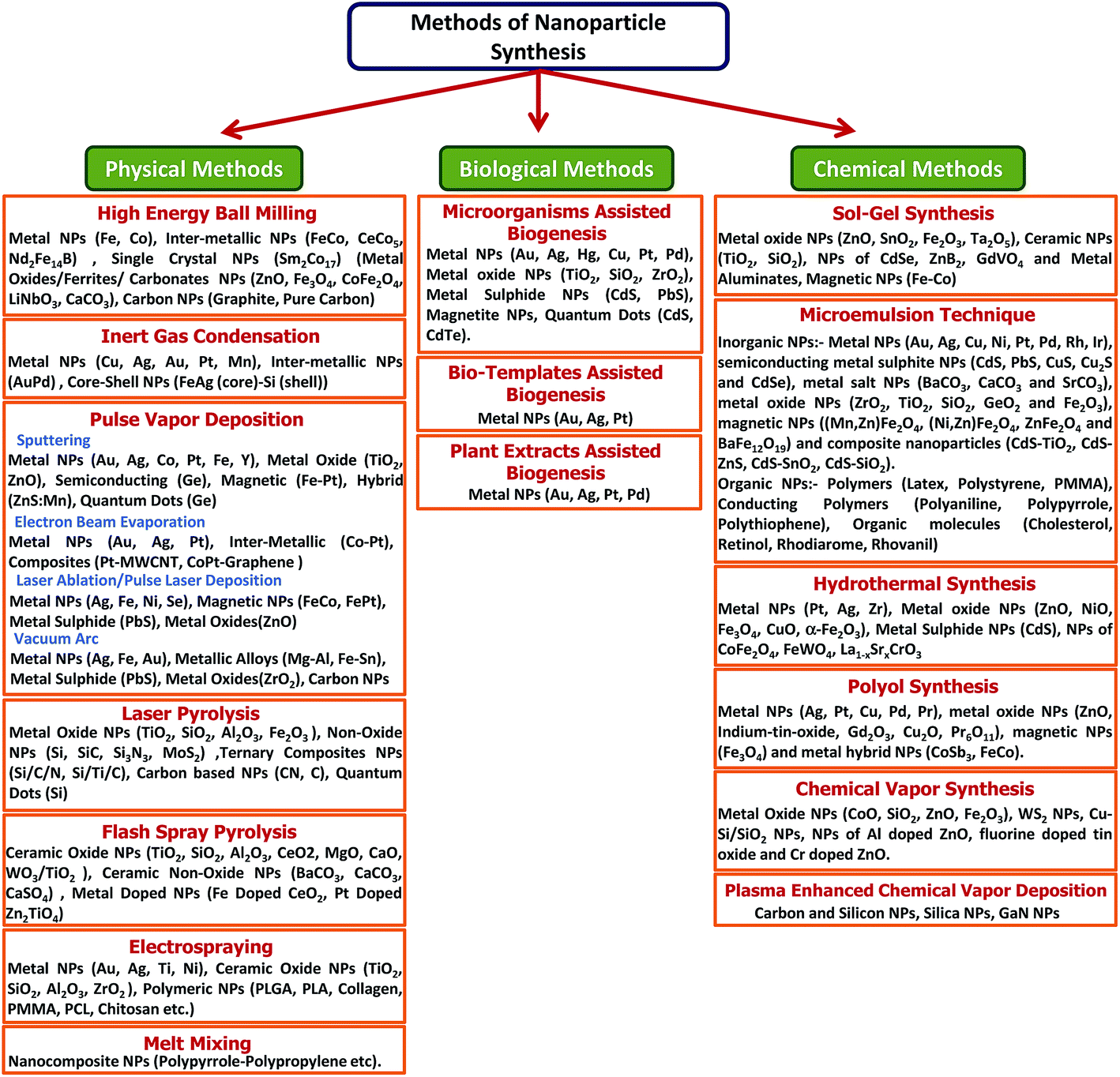 | ||
| Fig. 1 Overview on different type of synthesis methods to produce variety of nanoparticles. | ||
2.1 Physical methods for the synthesis of nanoparticles
Physical methods apply mechanical pressure, high energy radiations, thermal energy or electrical energy to cause material abrasion, melting, evaporation or condensation to generate NPs. These methods mainly operate on top-down strategy and are advantageous as they are free of solvent contamination and produce uniform monodisperse NPs. At the same time, the abundant waste produced during the synthesis makes physical processes less economical. High energy ball milling, laser ablation, electrospraying, inert gas condensation, physical vapour deposition, laser pyrolysis, flash spray pyrolysis, melt mixing are some of the most commonly used physical methods to generate NPs.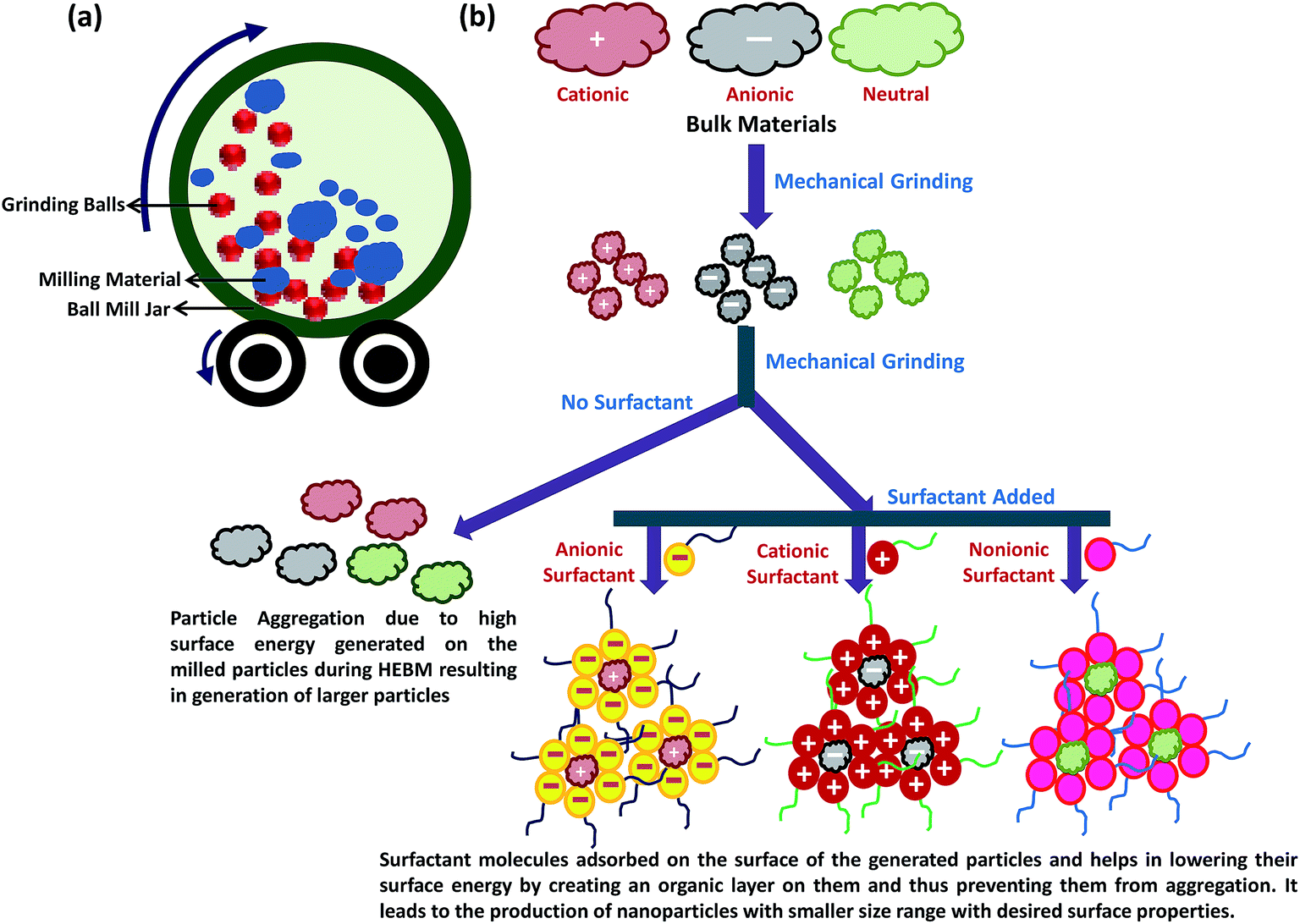 | ||
| Fig. 2 (a) High energy ball milling (HEBM) system and (b) schematic representation of the NPs synthesis using HEBM method with and without surfactant. | ||
Using metallic iron powder as the precursor material, Carvalho et al. synthesized superparamagnetic magnetite NPs with size ranging from 12 nm to 20 nm by HEBM process.63 Bartwal and co-workers optimized various milling parameters (milling time, rotation per minute; rpm, ball size, etc.) to prepare uniform LiNbO3 NPs with particle size ranging from ∼30 to 60 nm.64 Salah and co-workers synthesized ZnO NPs of ∼30 nm size from ZnO microcrystalline powder using HEBM process.65 Synthesized ZnO NPs showed antibacterial activities due to their desirable lattice constant. Chen et al. utilized microwave assisted HEBM method to prepare pure and well-crystallized cobalt ferrite NPs with mean size of 20 nm, and high saturation magnetization (82.9 emu g−1), at low temperature (≤100 °C) without subsequent calcination.66 Cobalt oxalate hydrate and Fe powder were used as the raw materials for this synthesis and stainless steel or pure iron balls with diameter of 1.5 mm were used for milling. Xing et al. recently established HEBM as the powerful green synthesis method for large-scale production of nitrogen doped carbon NPs for catalytic applications.62
Currently, surfactant assisted high energy ball milling is used as an efficient strategy for the synthesis of NPs with precise size and specific surface characteristics (Fig. 2b). Surfactants are the surface active agents containing both hydrophobic and hydrophilic properties and can be classified as anionic, cationic, zwitterionic and nonionic depending upon the surface charge characteristics of their hydrophilic group.67 Following adsorption on the material surface, the surfactant molecules generate electrostatic/steric forces which stabilize the milling particles, and thus minimize the uncontrolled fracturing of particles. Surfactant can also lower the surface energy of the freshly generated fine particles by forming thin organic layer and introducing long range capillary forces that lower the energy for crack propagation. This prevents the particles from agglomeration and cold welding that may lead to enhancement of particle size. Nature and amount of the surfactant used during the HEBM tremendously affect the physical characteristics of the NPs. Normally, increasing the surfactant volume can decrease the particle size by second to third order of magnitude. Zheng et al. designed rare-earth-based magnetically hard single-crystal NPs of Sm2Co17 with average diameter of 9 nm and 26 nm by HEBM using oleic acid and oleylamine, respectively, as the surfactant with SmCo5 as precursor.68 Islam et al. used this surfactant assisted HEBM method to prepare pure CaCO3 NPs with diameter of 30 nm from cockle shells using dodecyl dimethyl betaine (BS-12).69 Surfactant assisted HEBM has also been used to synthesize graphite NPs of the size ranging from 1–30 nm,70 where the carbon black was used as a precursor and phosphate ester as a surfactant in aqueous media.
Benelmekki et al. reported the synthesis of metallic-dielectric multi-core–shell NPs by IGC method.76 The FeAg NPs were deposited in Si shell (Fig. 3a–c), where Si shell protected the NPs against oxidation and restrained their aggregation. The basic principle for the synthesis of such hybrid nanoparticles (HNPs) was based on the condensation of atomic vapor produced from sputtering of multiple targets (Fe, Ag and Si) simultaneously at high pressure. Argon working pressure was kept at 10 mbar during the deposition. Using DC sputtering, vapours of metal atoms were produced near the target surface and condensed into nanoclusters along its propagation path through aggregation zone as shown in Fig. 3a. These nanoclusters were then extracted and accelerated due to difference in pressure between aggregation zone (10 mbar) and deposition unit (10−5 mbar) and then deposited onto the substrate (Fig. 3b and c). The mechanism responsible for the fabrication of multi-core–shell HNPs is illustrated in Fig. 3d. The mechanism was divided into three steps. Step 1 was associated with the nucleation and growth of the NPs. Plasma density was found to be an important parameter, which influences the nucleation, growth and crystallinity of the NPs. The plasma density and sputtering yield (∼1.2) were maximum in case of Ag. This led to the nucleation and growth of the highly crystalline Ag NPs before leaving the plasma zone, which then quenched into mono-crystalline NPs in aggregation zone. Since the plasma density and sputtering yield (0.45) was lower in case of Fe than Ag, their nucleation and growth occurred when the atoms reached the aggregation zone. This transportation process caused loss of energy of the atoms resulting in the creation of amorphous nanoclusters/NPs. The plasma density and sputtering yield (0.29) was lowest in case of Si leading to the formation of amorphous Si nanoclusters/NPs. Step 2 involve cluster–cluster collisions, where the nanoclusters collide and coalescence with each other in aggregation zone, leading to the formation of larger NPs. However, formation of core/shell or dumbbell-like structures was most probable due to the large positive free energy of Ag and Fe mixing. In other words, during collision in aggregation zone, Ag and Fe nanoclusters could not completely coalescence due to lack of energy. This led to the formation of dumbbell-like structures in amorphous Si matrix. Finally, during their movement through aggregation zone, AgFe/Si core–shell NPs were found to collide and partially coalescence with each other resulting in the formation of multi-core–shell HNPs in step 3. Fabricated HNPs were found to be the promising candidates for magneto-optic bio-applications.
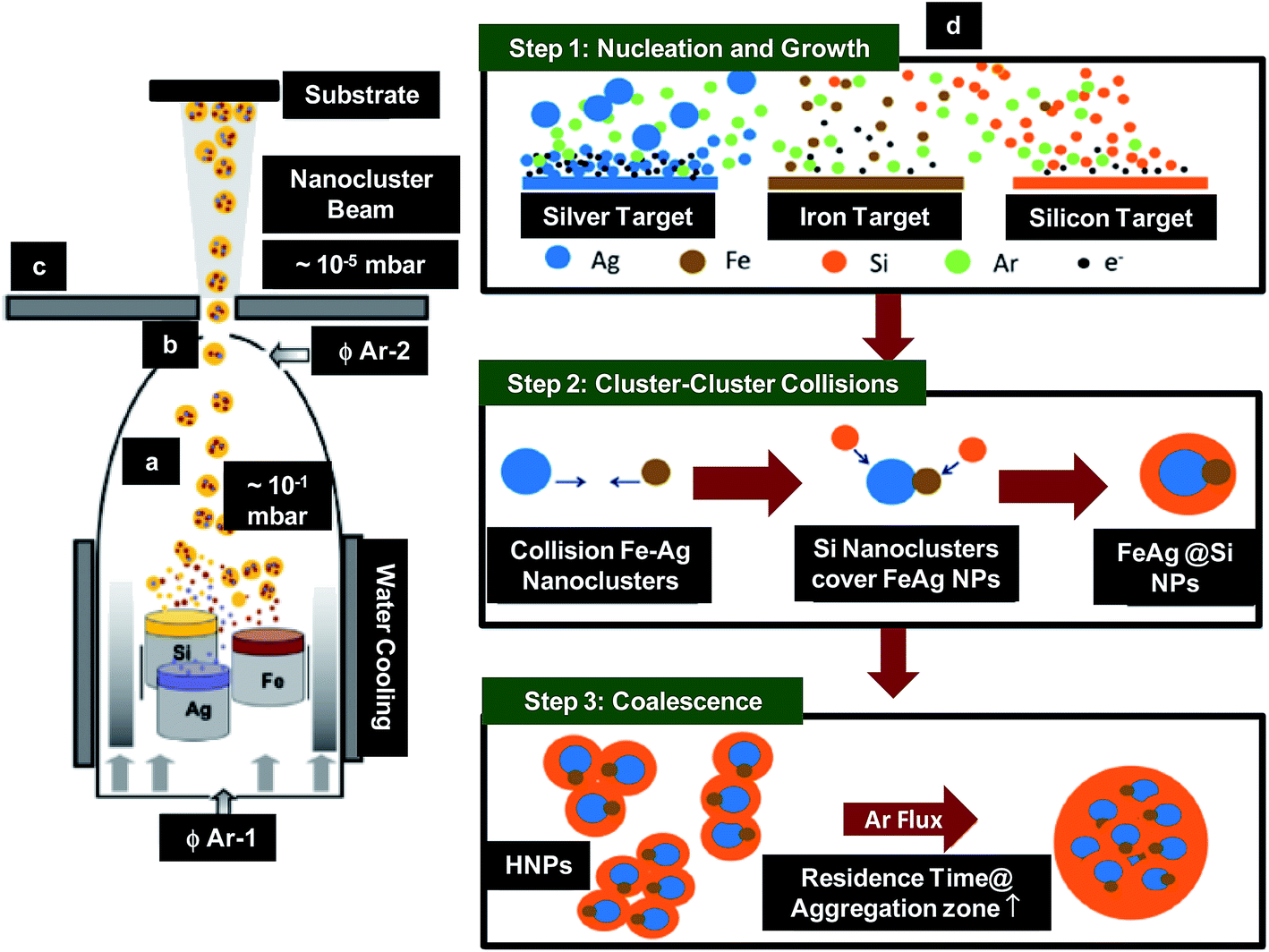 | ||
| Fig. 3 Schematic showing (a)–(c) IGC system used for synthesis of HNPs; (a) aggregation zone, (b) aperture through which formed nanoclusters moved and (c) deposition section and (d) mechanism responsible for the synthesis of HNPs. Reproduced with permission from ref. 76. | ||
(i) Sputtering
(ii) Electron beam evaporation
(iii) Pulsed laser deposition
(iv) Vacuum arc
Fig. 4 shows the schematics representations of different PVD techniques.
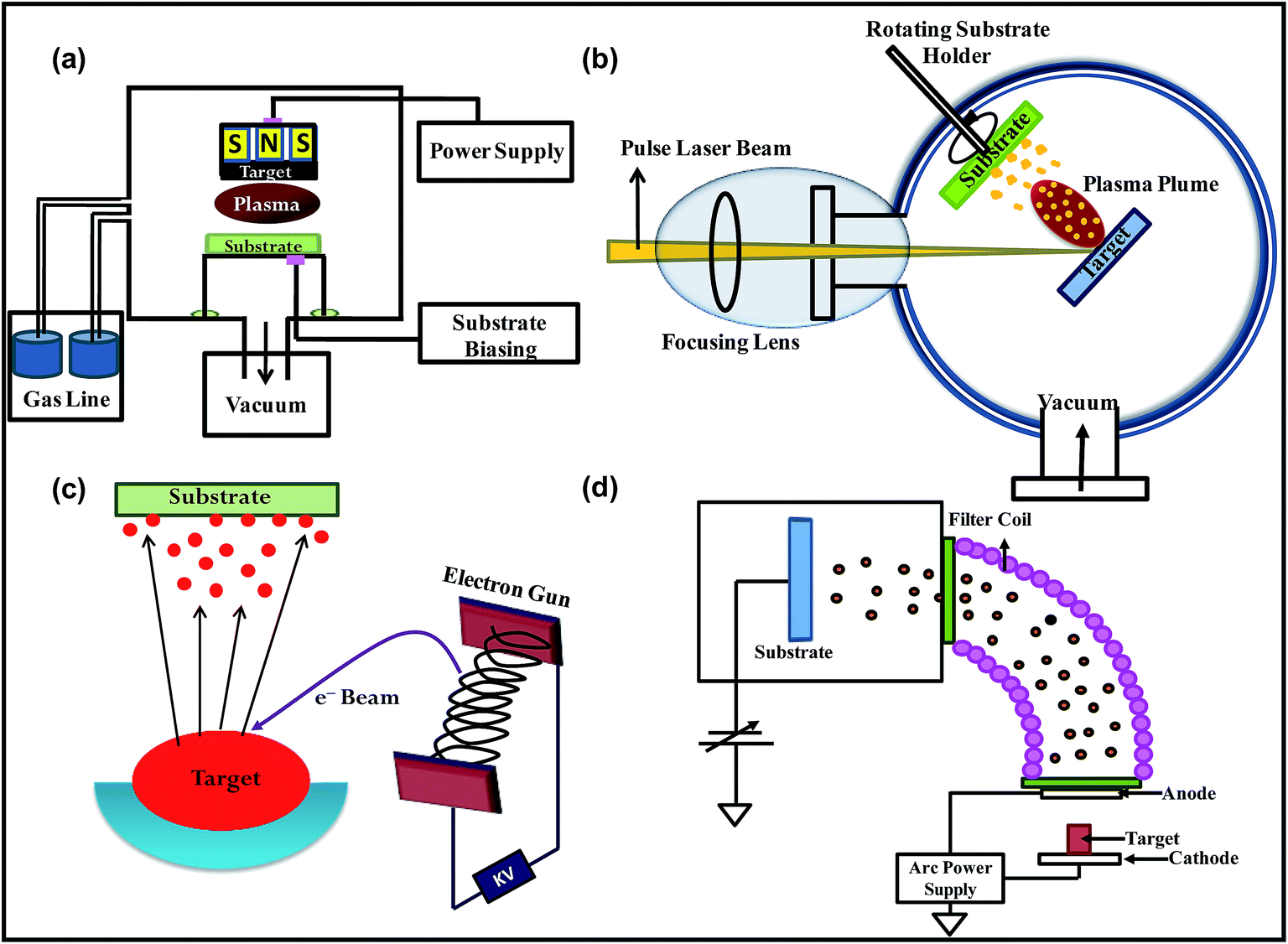 | ||
| Fig. 4 Schematic representation of (a) plasma sputtering, (b) electron beam evaporation, (c) pulsed laser deposition and (d) vacuum arc technique. | ||
2.1.3.1 Sputtering. Sputtering is a vacuum-based PVD process, which is often used to deposit films and NPs (Fig. 4a). Sputtering works on the principle of momentum transfer in which the atoms from the target (which is made up of material to be deposited) are ejected by the ion bombardment. The deposition of material by sputtering can be achieved using DC, pulsed DC and radio frequency (RF) powers. The sputter deposition takes place in following steps:
(i) The plasma of neutral gases, commonly Ar, is generated between the two electrodes by the collisions of electrons to gaseous molecules
(ii) Ions present in the plasma are then accelerated toward the target by applying potential between the two electrodes
(iii) These ions with appropriate energy thus hit the target, leading to ejection of the material from the target
(iv) The ejected material are then transported and deposited onto the substrate
Simple sputtering and magnetron sputtering are two types of sputtering processes. The difference between the two processes arises due to the fact that simple sputtering does not contain magnets behind the target, while magnets are always present in case of magnetron sputtering. The magnetron sputtering is upgraded version of simple sputtering and has following two advantages over former process:
• Higher deposition rate
• Prevent target overheating and damage
Higher deposition rate in magnetron sputtering is obtained due to the fact that under the influence of magnetic field the path of electrons become curved i.e. they follow helical path. This leads to more ionization of gases, causing more ions hit to the target, and hence results in higher deposition rate. In sputtering, sputtering yield is an important parameter, which describes the number of atoms ejected per incident ions. It should be noted that DC sputtering can be used to deposit nanostructures of conducting materials, while the deposition of insulating material by DC sputtering is difficult. However, RF sputtering can be used for the deposition of insulating materials also. Further, when reactive gases are introduced into the sputtering process chamber then sputtering process can be called as reactive sputtering. Reactive sputtering is used to deposit variety of compound/hybrid materials.
In all types of sputter deposition processes the NPs of ideal size can be synthesized by selecting the appropriate deposition conditions. For example, Hatakeyama and Nishikawa have synthesized Au NPs by analytically varying the sputtering conditions that influences the particle size and its distributions.78 Veith et al. used magnetron sputtering for the deposition of Au NPs on WO3 and activated carbon surfaces.79 Using magnetron sputtering and selecting the appropriate deposition parameters, Bouchat et al. reported the synthesis of variety of metallic and non-metallic NPs including titanium oxide (TiO2), silver (Ag), gold (Au), yttrium (Y), carbon (C), cobalt (Co) and iron (Fe).80 Asanithi et al. optimized the process conditions for the deposition of uniform sized, <10 nm, silver NPs by DC magnetron sputtering.81 While varying target-to-substrate (TTS) distance from 10 to 20 cm, they demonstrated significant tailoring of particle size with the formation of highly uniform small grains (3.8 ± 0.7 nm) at optimized TTS distance of 20 cm. Moreover, TTS was also reported to influence the shape and distribution of the Ag NPs. They further tuned the sputtering current (50 mA) at optimized TTS to design monodisperse Ag NPs. However, at higher sputtering current worm-like morphology with increased grain size was obtained. Ichida et al. synthesized germanium (Ge) NPs and Ge quantum dots (Ge QDs), both used in electronic and optoelectronic industry, by high pressure RF magnetron sputtering.82
Ghosh et al. fabricated Mn doped ZnS NPs (ZnS:Mn) by RF magnetron sputtering and studied their photoluminescence and field emission properties.83 At an optimum Mn concentration, the excellent photoluminescence and field emission properties in ZnS:Mn NPs were realized. In another work, Veith et al. fabricated 2 nm size Au NPs on γ-Al2O3 using one step magnetron sputtering process which was comparable to those obtained by traditional chemical methods.84 Ramalingam et al. employed tilted-target sputtering in order to grow sub-2 nm sizes and density tuneable Pt NPs at room temperature.85 They showed that tilting of target angle and variation of growth time significantly altered the mean diameter and density of Pt NPs (Fig. 5).
 | ||
| Fig. 5 High resolution transmission electron microscopy images of Pt NPs synthesized by sputtering process. (a)–(f) Pt NPs grown at constant target angle of 23.8° but varied growth time of (a) 5 s, (b) 10 s, (c) 20 s, (d) 30 s, (e) 45 s and (f) 55 s. (g)–(i) Pt NPs grown at constant growth time of 2 min but varied target angle of (g) 16.2°, (h) 23.8° and (i) 38.8°. Reproduced with permission from ref. 85. | ||
Sputtering process is also widely used for the synthesis of magnetic NPs for numerous applications. For example, Zhang et al. demonstrated the synthesis of L10 ordered FePt NPs on MgO substrate by DC magnetron sputtering.86 The FePt NPs were grown at base pressure of 5 × 10−7 Torr and low argon pressure of 1.5 × 10−3 Torr at 750 °C. During the deposition of FePt NPs the sputtering rate was 1.2 Å s−1. Takahashi et al. synthesized FePt NPs by sputtering method and studied the effect of size on ordering of the NPs.87 They suggested that there is a critical value of size of NPs for ordering i.e. FCC FePt NPs with size <4 nm diameters do not display L10 ordering. Further, Dmitrieva et al. fabricated FePt NPs by sputtering process and suggested that inclusion of nitrogen during NP deposition can enhance the L10 phase of FePt NPs.88
2.1.3.2 Electron beam evaporation (EBE). Electron beam evaporation is a vacuum-based PVD process which is used to fabricate thin films and NPs. EBE system consists of vacuum unit, electron beam source and target materials (Fig. 4b). Electron beam source consists of a filament, which is heated by passing current through it resulting in the generation of electron beam. The generated electron beam is focused and then directed onto the target material using magnets. The deposition of films by EBE takes place in the following steps:
(i) Electron beam hits the target and heat the target material
(ii) Target material (atoms) evaporates when temperature reaches above its boiling point
(iii) Evaporated material then transported and
(iv) Condensed onto the substrate
Advantage of EBE methods are
(i) High deposition rate
(ii) Can be used to deposit materials ranging from conducting to insulating
(iii) Unlike thermal evaporation, EBE can be used to deposit materials of higher boiling point
By selecting optimum process parameters and conditions during EBE process, NPs of different sizes and shapes can be deposited. Perry et al. have reported the use of EBE in the development of NPs for both 2D and 3D metal patterning.89 Hsieh et al. synthesized Au NPs and Pt NPs on multiwall carbon nanotubes (MWCNTs) to form composite electrodes for sensors and energy storage applications.90 They observed that the distribution of Au NPs was better on MWCNTs than Pt NPs. Uhm et al. demonstrated the fabrication of antibacterial Ag NPs on diameter-controlled TiO2 nanotubes by facile EBE process.91 Further, Castaldi et al. prepared CoPt NPs of size between 5 nm and 20 nm on thermally oxidized silicon using EBE process and found that the size of NPs was dependent on the thickness of the CoPt layer.92
Using EBE process, Bello et al. fabricated Ag NPs decorated three dimensional graphene (GR) scaffold for electrochemical application.93 In comparison to pure GR electrode, the GR/Ag NPs electrode showed higher capacitive performance and better cyclability for supercapacitor applications.
2.1.3.3 Laser ablation (LA) and pulse laser deposition (PLD). Laser ablation method utilizes a high power laser beam that evaporates particles from a solid source.94 In normal LA process, the laser can be either continuous laser or pulsed laser. LA offers a flexible approach in the production of micro- and nano-structures of polymeric materials. Kris et al. reported a micro-lens assembly with the utilization of LA technique by scanning the polymer surface to achieve a lens shape with the optimal focal distance and diameter.95 Gopal et al. have synthesized colloidal zinc metallic NPs using the LA technique.96
PLD is another vacuum-based PVD process that employs laser energy to remove the material from the target. The high power laser pulses hit the surfaces of target, leading to melting, evaporation and ionization of the material (Fig. 4c). Finally the ablated materials deposit onto the substrate. PLD utilize pulsed laser beam, mainly coming from either excimer laser or Nd:YAG laser, for the ablation of the material. PLD is used for the preparation of variety of materials including polymers, oxides, metallic systems, fullerenes, carbides, nitrides, etc.
In context to NPs production, Lee et al. reported the synthesis of FeCo NPs in an inert gas atmosphere by PLD.97 Ong et al. examined the role of process parameters such as number of pulses, ambient gas pressure and temperature gradient on the morphology and size of FeCo NPs synthesized by PLD.98 The morphology of FeCo was changed from linear interconnected chains to fibrous when the number of pulses were increased. At low pressure, NPs tend to form floccules-like nano-networks while chain-like network was observed at higher pressure.98 In addition, the average size of NPs was increased with increasing gas pressure. Andrea et al. used PLD for the synthesis Ag NPs arrays for surface enhanced Raman scattering (SERS).99 They controlled the size and morphology of Ag NPs by optimizing the Ar pressure and number of pulses. In another work, Jing et al. deposited Ag NPs on nickel hydroxide nanosheet arrays using PLD for application to SERS.100 They found that deposition time influenced the size and inter-particle gap of Ag NPs and hence could control the SERS enhancement. Kumar et al. synthesized self-assembled magnetic NPs by PLD and demonstrated the embedment of Fe NPs and Ni NPs of sizes 5–10 nm in amorphous alumina and crystalline TiN materials.101 Quintana et al. performed an interesting work involving the fabrication of selenium (Se) NPs on three different substrates: (1) metallic Au film, (2) Si wafers and (3) glass using PLD.102 While analyzing the morphology of the deposited NPs, they found that size, shape and population density of Se NPs were strongly dependent on the experimental process parameters and underlying base such as energy density, number of pulses and substrate. Lin et al. studied the effect of target–substrate geometry and ambient gas pressure on the synthesis of FePt NPs by PLD.103 They suggested that backward plume, which is a novel target–substrate geometry can produce FePt NPs with high uniformity, better phase formation than conventional PLD process. Amoruso et al. fabricated Ni NPs by femtosecond PLD, which was confirmed by atomic force microscopy (AFM).104 Dhlamini et al. reported the synthesis of lead sulphide (PbS) NPs in amorphous SiO2 matrix on Si substrate using PLD and realized excellent photoluminescence properties.105 Table 2 covers different NPs synthesized by the PLD method with the process details.
| Type of nanoparticles | Deposition medium | Deposition parameters | Size of nanoparticles | Laser type | References |
|---|---|---|---|---|---|
| a Abbreviations: AD – average diameter, BP – basic pressure, CTAB – cetyltrimethylammonium bromide, IT – incubation time, LED – laser energy density, LI – laser intensity, LIA – laser incident angle, MS – mean size, NLS – number of laser shots, PW – pulse width, RP – residual pressure, RR – repetition time, SDS – dodecylsulfuric acid sodium salt, ST – substrate temperature, TTS distance – target-substrate distance. | |||||
| Ag NPs | Aqueous SDS solution | LI: 120 mJ per pulse, RR: 10 Hz, PW: 5 ns, TTS distance: 4.5 mm | 4.2 ± 1.9 nm in SDS (anionic surfactant) | Nd:YAG laser, 532 nm | 94 |
| Aqueous CTAB solution | LI: 120 mJ per pulse, RR: 10 Hz, PW: 5 ns, TTS distance: 4.5 mm | 7.8 ± 4.5 nm in CTAB (cationic surfactant) | |||
| Aqueous SDS solution | LI: 60 mJ per pulse, RR: 10 Hz, PW: 5 ns, TTS distance: 4.5 mm | 6.8 ± 2.7 nm in SDS | |||
| Aqueous CTAB solution | LI: 60 mJ per pulse, RR: 10 Hz, PW: 5 ns, TTS distance: 4.5 mm | 9.4 ± 5.9 nm in CTAB | |||
| Ag NPs | Ar gas, 70 Pa pressure | BP: 2 × 10−4 Pa, LED: 2 J cm−2, LIA: 45°, TTS distance: 35 mm, NLS: 15000 & 30000, RR: 10 Hz |
1.5–4.5 nm | 248 nm KrF laser | 99 |
| Ni NPs | Vacuum | RP: 10−5 Pa, LI: 1 mJ per pulse, LIA: 45°, TTS distance: 30 mm, NLS: 200, LED: 0.3 J cm−2 | 5–100 nm, MS ≈ 40 nm | 1 kHz Ti:sapphire oscillator–amplifier system emitting at 780 nm | 104 |
| Ni NPs | High vacuum | BP: 5 × 10−7 Torr, RR: 10 Hz, PW: 10 ns, LED: 2 J cm−2, ST: 500 °C | ∼10 nm | — | 101 |
| Fe NPs | 4.5–9 nm | ||||
| Se NPs | High vacuum | BP: 8 × 10−4 Torr, PD: 108 W cm−2, PW: 10 ns, LIA: 45°, TTS distance: 35 mm | 75–100 nm | Nd:YAG laser, 532 nm | 102 |
| ZnO NPs | Aqueous SDS solution | RR: 10 Hz, LI: 100 mJ per pulse, IT: 60 min | 9.4–32.1 nm, AD: 14.7 nm | Nd:YAG laser, 532 nm | 96 |
| PbS | O2 gas, 300 mTorr pressure | RR 10 Hz, PW: 30 ns, TTS distance ∼ 50 mm, LI: 183 mJ per pulse | 10–50 nm, AD: 17 nm | 248 nm KrF laser | 105 |
| FeCo NPs | Ar gas, pressure varying from 10–90 kPa | BP: 10−4 Pa, RR: 10 Hz, PW: 10 ns, ST: 150 °C | 4.8–8.6 nm | Nd:YAG laser, 532 nm | 98 |
| FePt NPs | Ar gas | BP: 5 × 10−5 mbar, LI: 75 mJ per pulse, LED: 955 J cm−2, TTS distance: 6 mm | 4.8–22 nm | Nd:YAG laser, 532 nm | 103 |
2.1.3.4 Vacuum arc (VA). Vacuum arc is a vacuum-based PVD process in which the arc is used to vaporize the material for the synthesis of metallic, ceramic and composite NPs and films (Fig. 4d). Akbari et al. synthesized the Mg–Al alloy NPs by varying Mg and Al concentrations using plasma arc process.106 Lei et al. used vacuum arc discharge process for the synthesis of inter-metallic Fe–Sn NPs.107 Amaratunga demonstrated the synthesis of thin film of linked carbon NPs using localized high pressure arc discharge process and realized excellent mechanical properties in deposited material.108 Chhowalla and Amaratunga used a similar approach of localized high pressure arc discharge for the synthesis of thin film of fullerene-like MoS2 NPs, which showed extremely low friction.109 Fig. 6a and b show the high resolution electron microscope images of thin films of cross-linked carbon NPs and fullerene-like MoS2 NPs, respectively.
 | ||
| Fig. 6 (a) High resolution electron microscopic image of thin film of carbon cross-linked NPs and (b) high resolution transmission electron microscopic image of thin film of fullerene-like MoS2 NPs. Reproduced with permission from ref. 108 and 109. | ||
 | ||
| Fig. 7 Schematic illustration of (a) laser pyrolysis, (b) flame spray pyrolysis and (c) electrospraying technique for the synthesis of NPs. | ||
Rosaria D'Amato et al. utilized laser pyrolysis technique to synthesize variety of ceramic NPs (e.g. SiC, SiO2 and TiO2) for energy applications and cultural heritage preservation using silane (SiH4), tetraethyl orthosilicate (Si(OC2H5)4) and titanium(IV) isopropoxide respectively as precursor materials.110 Owing to the advantage of single step doping in this method, Scarisoreanu et al. synthesized carbon coated TiO2 NPs for photocatalysis applications.111 Marino et al. synthesized carbon nitride NPs with varying nitrogen incorporation between 2% and 20% using laser pyrolysis.112 Jansa et al. fabricated carbon NPs using pulsed laser pyrolysis of hydrocarbons and found that at same experimental parameters, the carbon NPs produced using benzene showed higher conductivity than those produced using either acetylene or ethylene.113 They also found the variation of size of NPs with varying laser power.
Silicon carbide (SiC), silicon and molybdenum disulphide (MoS2) NPs have also been produced by the laser pyrolysis technique using the same technique.114 Veintemillas-Verdaguer et al. synthesized magnetic composites Fe-based NPs encapsulated in carbon/silica (C/SiO2@Fe) or carbon (C@Fe) with average diameter of 29 ± 3 nm and 56 ± 3 nm, rerspectively.115 Laser pyrolysis of the carbonyl precursors is emerging as an excellent technique for the direct synthesis of iron NPs with smaller particle size, narrow size distribution without much noticeable aggregation. Following the same lines, Bomat et al. have prepared the iron–iron oxide core–shell NPs (10–30 nm) by laser pyrolysis of Fe(CO)5 vapours and in situ passivation by controlled oxidation process.116 Erogbogbo et al. have reported the synthesis of biocompatible Si QD by laser-driven pyrolysis of silane using phospholipid micelles for the imaging of cancer cells.117
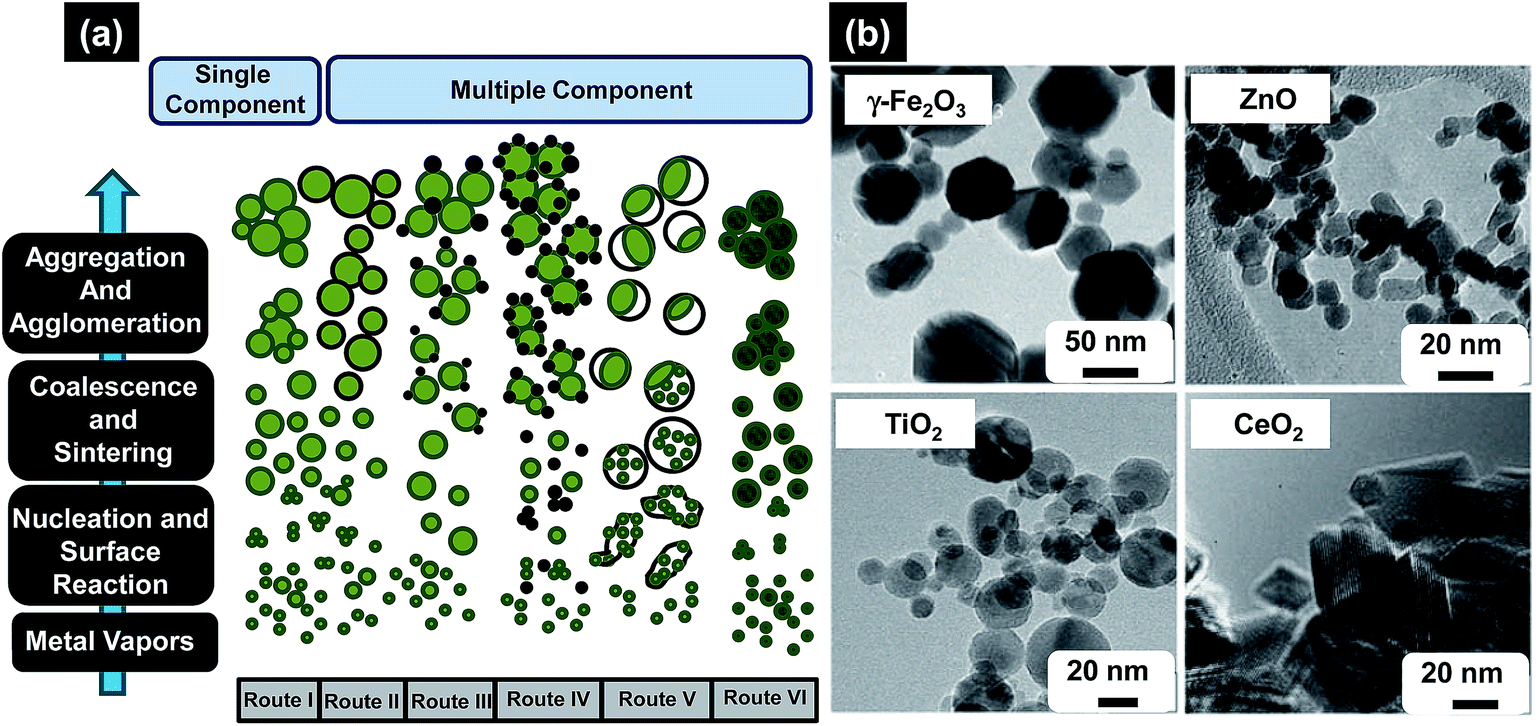 | ||
| Fig. 8 (a) Formation of different particle configurations via the gas-to-particle mechanism for single and multicomponent systems, (b) as-prepared metal oxides NPs of different morphologies made by FSP, varying from hexagonal/octagonal platelet g-Fe2O3, lightly oblongated ZnO, spherical TiO2, and rhomboid-shaped CeO2 with sharp edges. Reproduced with permission from ref. 118. | ||
Sokolowski et al. first utilized this strategy to synthesize Al2O3 NPs by combusting an ultrasonically-dispersed spray of aluminium acetylacetonate in benzene–ethanol solvent mixture.119 Later, FSP process was used tremendously to design various oxide and non-oxide ceramic NPs e.g. CeO2, SiO2, Al2O3, MgO, C–Cu, BaCO3, CaO, CaCO3, CaSO4, etc. Madler et al. adequately reviewed the research articles concerning the design and fabrication of NPs by FSP method and also tabulated very nicely all different oxide and non-oxide ceramic NPs synthesized using this method.118 Some of the most notable applications of the FSP derived NPs are in the areas of catalysis optics and photonics, sensors, health care, magnetic materials, etc.120–123 For example, Arutanti et al. fabricated WO3/TiO2 NPs by flame-assisted spray pyrolysis and explored them for photocatalysis.124 They studied the role of amount of WO3 and TiO2 in the range 0–100 wt% on size, shape, crystallinity, and photocatalytic performance. While analyzing the results they found that photocatalytic performance of composite WO3/TiO2 NPs was superior compared to 100 wt% TiO2 catalyst. Channei et al. synthesized Fe-doped CeO2 NPs with varying amount of Fe using flame-assisted spray pyrolysis for photocatalysis application and found that photocatalytic efficiency was 100% when 2 mol% Fe was introduced in CeO2.125 Similarly Siriwong et al. synthesized Pt-doped Zn2TiO4 NPs with varying amount of Pt from 0.25–1.0 mol% using flame-assisted spray pyrolysis and suggested promising use of this process for the synthesis of high quality and controlled sized NPs.126
The electrospraying technique can also be utilized to transform water vapour present in the atmosphere into engineered water nano-structures (EWNS). The EWNS has a size of 25 nm that is equipped with an extraordinary set of physical, chemical, biological and morphological properties with low toxicity level that possess a remarkable mechanism of action as EWNS is loaded with reactive oxygen species (ROS) that is able to interact with and to stop the activity of harmful airborne bacteria.132 With the electrospraying method, NPs of various type and sizes can be synthesized based on the use of a variety of solvents (Table 3).
| Type of nanoparticles | Solvent | Size | Application | References |
|---|---|---|---|---|
| a Abbreviations: PLGA – poly(lactic-co-glycolic acid), PLA – poly-DL-lactic-acid, PMSQ – polymethylsilsesquioxane, PMMA – poly(methyl methacrylate), PCL – polycaprolactone, COLL – collagen. | ||||
| Au NPs | Water + methanol | 20–100 nm | Biomedical field, cancer nanotechnology, molecular imaging, molecular diagnosis, targeted therapy and bioinformatics | 133 and 134 |
| Ag NPs | Toluene | 3–7 nm | Biosensor, anti-reflection coatings, artificial joint replacement, cancer diagnosis, anti-bacterial activity | 135 and 136 |
| Sn NPs | Isopropyl alcohol | 3 nm | Humidity sensor, microelectronics and batteries | 137 and 138 |
| SiO2 NPs | Ethylene glycol | 20 nm | Photodynamic therapy, magnetic resonance imaging, optical imaging, drug delivery, gene delivery and protein delivery | 139 and 140 |
| Si NPs | 1-Octanol | 3 nm | Biological interface devices, anti-cancer drug delivery and cancer detection | 139 and 141 |
| Al2O3 NPs | Ethanol | 500 nm | Catalysis, waste water treatment, heat transfer fluidics, biosensors, drug delivery and nanocomposites | 139 and 142 |
| Ni NPs | Ethylene glycol monoethyl ether acetate + alkylnaphthalene + polyamine | 2–3 nm | Magnetism, energy technology and biomedicine | 139 and 143 |
| TiO2 NPs | 1-Butanol | 10–40 nm | Drug delivery, cosmetics, anti-bacterial materials, sun-screens and electronics | 139 and 144 |
| ZrO2 NPs | 1-Butanol | 10–40 nm | Catalysis, anti-corrosion coating in surgical appliances, explosive primers, dental implant material, cell and tissue engineering | 139,145 and 146 |
| Pt NPs | Ethanol | 8 nm | Chemotherapy, cellular imaging, MRI, sun-screen protection and anti-septic products | 147 and 148 |
| PLGA NPs | Acetonitrile and dimethyl sulfoxide | 165 nm to 1.2 μm | Nanocarriers for drug delivery, cancer diagnosis and imaging | 131 and 149 |
| PLA NPs | Butanol/methylene chloride mixture | 100 nm | Drug encapsulation, thin film deposition and biomedical engineering | 150 and 151 |
| PMSQ NPs | Ethanol | 62 ± 20 nm | Organic thin film insulator and printed electronics | 152 and 153 |
| PMMA NPs | Acetone | 300 nm | Drug/SiRNA systems, artificial enzymes and photostable bio-imaging agents | 154 and 155 |
| PCL NPs | Chloroform | 4.8 ± 0.2–17.7 ± 0.5 μm | Pharmaceutics, drug delivery, cancer treatment and cell culture | 156–158 |
| COLL/NaCl NPs, COLL/CaCl2 NPs | Acetic acid | 900 nm (COLL/NaCl) or 630 nm (COLL/CaCl2) | Biomedical and therapeutics, nanomedicine | 159 and 160 |
| Chitosan NPs | Acetic acid | 520 nm | Anti-cancer treatment, drug delivery, gene delivery and growth factor carriers | 161 and 162 |
Zuhal et al. have utilized the melt mixing technique to prepare nanocomposite NPs of conducting polymer polypyrrole with polypropylene and the resultant NPs have been then processed further using injection moulding technique.165 Weiss et al. have reported in situ formation of hybrid NPs during melt blending of hydroxybenzoate (HBA) and 27% hydroxynaphthanoate liquid polymer polyesters with zinc salts of sulfonated polystyrene ionomers at elevated temperatures due to chemical reaction that involved the liquid crystalline polyester and residual zinc acetate from the neutralization of the ionomer.166
2.2 Chemical methods for the synthesis of nanoparticles
Sol–gel method, microemulsion technique, hydrothermal synthesis, polyol synthesis, chemical vapour synthesis and plasma enhanced chemical vapour deposition technique are some of the most commonly used chemical methods for the NPs synthesis.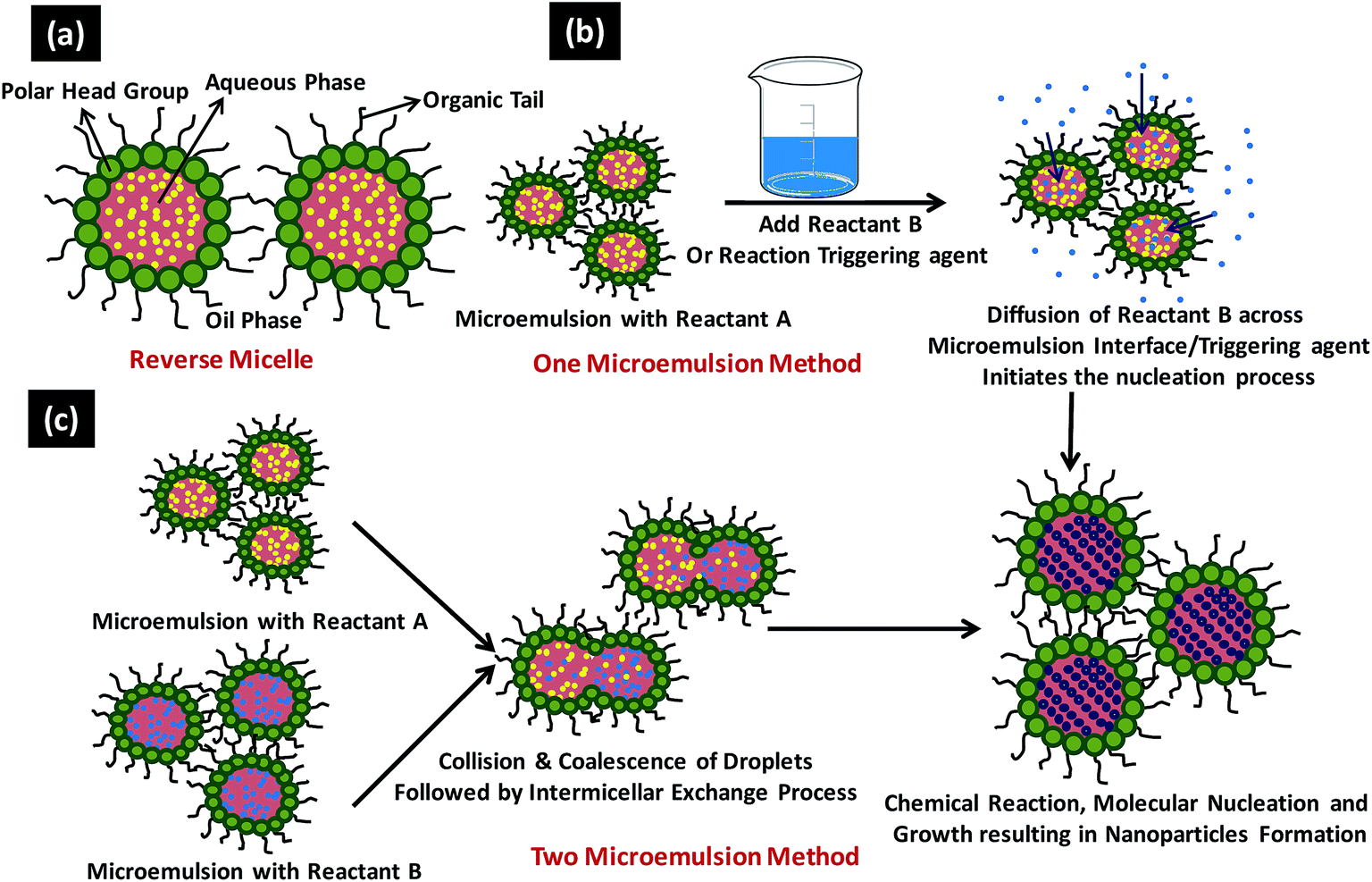 | ||
| Fig. 9 Shows (a) typical reverse micelle system, (b) various steps involved in one microemulsion process and (c) reaction sequence involved in the two microemulsion nanoparticles synthesis. | ||
In general there are two microemulsion routes to synthesize the NPs namely (1) one microemulsion method and the (2) two microemulsion method (Fig. 9b and c).178 One microemulsion method can be further divided into two types i.e., energy triggering method that needs a triggering agent to initiate the nucleation reaction within the single microemulsion containing the precursor and other is one microemulsion plus reactant method which is initiated by adding one of the reactant directly into microemulsion already carrying the second reactant (Fig. 9b). Malik et al. have reported the laser and radiolytically induced colloidal Au NPs formation in w/o microemulsion system with diameter ranging from 2.5–10 nm.178 One microemulsion processes are diffusion controlled since the second trigger/reactant has to diffuse through the interfacial wall of the microemulsion encapsulating the first reactant to accomplish the NPs synthesis. In two microemulsion method, the two microemulsions carrying the separate reactants are mixed together in appropriate ratios (Fig. 9c). Brownian motion of the micelles helps them to approach each other resulting in intermicellar collisions and sufficiently energetic collisions leads to the mixing of the micellar components. Once both the reactant comes in a same micellar compartment, the chemical reaction takes place in this nanoreactor. As the critical number of molecules (Ncrit) attained inside the micelle, it initiates the nucleation process and results in NPs formation. Numerous intermicellar collisions are needed for the sufficient reactant exchange, their mixing and finally their reaction to terminate at the end product.
Microemulsion technique was used most commonly for the synthesis of the inorganic nanomaterials including metal NPs (Au, Pt, Pd), semiconducting metal sulphite NPs (CdS, PbS, CuS, Cu2S and CdSe), metal salt NPs (BaCO3, CaCO3 and SrCO3), metal oxide NPs (ZrO2, TiO2, SiO2, GeO2 and Fe2O3), magnetic NPs ((Mn,Zn)Fe2O4, (Ni,Zn)Fe2O4, ZnFe2O4 and BaFe12O19) and composite NPs (CdS–TiO2, CdS–ZnS, CdS–SnO2). Metal NPs were easily prepared using microemulsion technique using the reduction strategy. Water-to-surfactant molar ratio (ω), type of continuous phase, metal ion concentration, type and concentration of the reducing agent, structure and amount of the surfactant used are some of the key parameters that control the metal NPs synthesis effectively.177 Boutonnet et al. have used the reverse micelle microemulsion strategy to synthesize Pt, Pd, Rh and Ir NPs employing H2PtCl6, Pd(NH2)4Cl2, RhCl2 and IrCl3, respectively as the precursors with hydrazine (N2H4) and active hydrogen as the reducing agent.179 Solla-Gullon et al. have recently reported fast, easy and scalable production of Pt cubic NPs by reducing H2PtCl6 with sodium borohydride using w/o microemulsion process.180 They revealed significant effect of introducing varying amount of HCl in the H2PtCl6/NaBH4 microemulsion system in controlling the shape/surface of the Pt NPs. Zhang et al. have studied the synthesis of Ag NPs and also investigated the effect of ω (water/surfactant ratio) on the particle size and their size distribution.181 Results revealed that there is continuous decrease in the particle size from ∼5 nm to ∼1.5 nm with reduction in the ω value from 15 to 2.5. They also investigated the effect of reducing agent concentration on the Ag NPs synthesis and leads to the fact that both less and excessive amount of reducing agent can hinder the synthesis process by affecting the stability of the colloids.181 The same group have investigated the effect of increasing surfactant (sodium bis(2-ethylhexyl) sulfosuccinate; AOT) concentration on the Ag NP synthesis and observed reduction in the particle size with increasing surfactant amount.182 Pileni et al. have introduced the synthesis of Cu NPs by reverse micelles using hydrazine as the reducing agent and revealed the effect of increasing water content in controlling the NPs size and the oxide content within the NPs structure.183 CdS NPs were synthesized in AOT and triton reverse micelles with cadmium lauryl sulphate and cadmium AOT as the functional surfactant.184 The diameter of the NPs is reported to be dependent on the relative composition of Cd2+ and S2− ions. Chen et al. have accounted the synthesis of Ni NPs with average diameter of 4.7 nm using cationic w/o microemulsion of water/CTAB (cetyltrimethylammonium bromide)/n-hexanol system by the reduction of nickel chloride with hydrazine at an elevated temperature.185 Subrata Kundu reported a novel methodology to synthesize shape-selective Au NPs by reduction of Au(III) ions with alkaline 2,7-dihydroxynaphthalene (DNP) in CTAB micellar media under 30 min continuous UV-irradiation.186 The size and shape of the NPs were reported to be tuned by varying the Au(III):CTAB molar ratio. Similar synthetic pathway was accounted to synthesize catalytically enhanced shape selective rhodium nanoparticles (Rh NPs) in the presence of DNP (reducing agent) and CTAB (surfactant) using 6 h UV-irradiation.187 The growth of the NPs with different shapes (spherical, flower-like or cubic) was found to be directed by the surfactant-to-metal ion molar ratio and the concentration of DHN. Praharaj et al. also reported the size selective synthesis of Au NPs with average diameter from 9 nm to 15 nm in toluene employing cationic surfactant of variable chain length.188 Asher and co-workers developed and demonstrated a novel strategy to synthesize silica–CdS nanocomposites that allocate tailoring the size and morphology of the resultant NPs.189 Gold–silver bimetallic NPs with varying mole fractions were synthesized in water-in-oil microemulsions of water/aerosol OT/isooctane system by co-reduction of HAuCl4 and AgNO3 with hydrazine.190 Voucher et al. reported the preparation of crystalline NPs of hexacyanoferrate, cobalt pentacyanonitrosylferrate and chromium hexacyanochromate coordination polymers in water-in-oil microemulsions.191 Mandal et al. reported the synthesis of bimetallic core–shell Pd–Au and Pd–Ag NPs of the size range of 10–30 nm using TX-100 micellar system by seed-mediated method.192
Microemulsion technique was not progressed much in term of designing organic NPs due to phase separation constraint and thus only few reports are available.178 This method is known to be microemulsion polymerization when it comes to organic structures. Atik and Thomas reported first successful microemulsion polymerization in 1981 of latex NPs (average diameter: 20–35 nm) using CTAB/styrene/hexanol/water oil–water microemulsion coordination.193 Three component microemulsion method was described to synthesize polystyrene NPs within size range of 20–30 nm using dodecyl trimethyl ammonium bromide (DTAB) and potassium persulphate (KPS) initiator.194 Holdcroft and Guillet synthesized polystyrene NPs using pulse UV-triggered microemulsion polymerization.195 Palani et al. studied the polymerization of MMA using MMA/ethylene glycol dimethacrylate/water system with acrylamide as amphiphile.196 Organic NPs of cholesterol, retinol, Rhodiarome, Rhovanil were also been reported using different microemulsion systems including AOT/heptane/water, Triton/decanol/water, and CTABr/hexanol/water.197 Microemulsion method was also been used to synthesize NPs of conducting polymers including polypyrrole, polyaniline, polythiophene, etc.198–200
In a chemical solution, NPs are produced from a colloidal system that consists of two or more phases (solid, liquid or gas states) of matter (e.g. gels and foams) mixed together under controlled pressure and temperature. The advantage of using this method includes the capability to synthesize a huge amount of NPs with an optimized size, morphology, composition and surface chemistry that is rationally inexpensive.202 Du et al. reported the facile one-pot synthesis of Pt NPs using hydrothermal method and observed excellent electrocatalytic activity.203 Ma et al. fabricated hydrophilic NaYF4:Yb,Er NPs using this method and observed good bright upconversion luminescence. They suggested that these hydrophilic NaYF4:Yb,Er NPs have great potential as luminescent labelling materials for biological applications.204 This method was also used for the synthesis of NPs of oxides of iron, nickel and copper such as Fe3O4, α-Fe2O3, NiO, CuO, etc.205–207 Hydrothermal is a facile and fast process for the synthesis of NPs of various other materials such as CoFe2O4, Ag, FeWO4, La1−xSrxCrO3, CdS, Zr, ZnO, etc.208–214
Xia et al. reported the synthesis of polycrystalline NPs of Cu2O using copper nitrate, ethylene glycol and poly(vinyl pyrrolidone) as the precursor, reducing agent and the stabilizing agent, respectively.219 Morphological transformation of these spherical Cu2O NPs to nanocubes was observed by introducing NaCl in the reaction mixture which is explained by the fact that the added Cl− ions play a role in retarding the reduction kinetics, stabilizing the {100} plane of the Cu2O crystals that inducing the formation of single-crystal nanocubes, which can grow in size via Oswald ripening. Low temperature synthesis of ITO NPs was carried out by Jeyadevan and coworkers in polyols and alcohols including ethylene glycol, trimethylene glycol and 1-heptanol.220 The NPs characteristics were found to be influenced by the type of polyol used as the reaction medium. In another work, Moon et al. reported the preparation of ZnO NPs by polyol method and showed that the amount of water and way of precursor addition affects the characteristic of the NPs immensely.221 Similarly, the polyol synthesis method was also used to synthesize NPs of Gd2O3 and Pr6O11.222,223
Polyol process was also employed for developing low temperature strategies to synthesize magnetic NPs of magnetite, Fe3O4, etc.224,225 Cheng et al. have very nicely described the effect of two different polyols (ethylene glycol; EG and 1,2-propylene glycol; PG) having different reduction capability on the morphology evolution of the Fe3O4 magnetic NPs (Fig. 10).225 Due to the higher reductive ability of PG, there is faster formation and growth rate for the NPs leads to Fe3O4 aggregation and generation of more misaligned mesocrystals. In the mesocrystal, the neighbouring NPs merge together and finally integrate into a single crystal with a porous structure during the Ostwald ripening process. However, due to relatively lower NP formation rate in EG, most of the particles got enough time to self-assemble along the same orientation in the cluster. Each cluster, with its highly ordered arrangement of NPs, can be regarded as a single mesocrystal. And this mesocrystal can retain its secondary structure owing to the lower surface energy. This one-pot facile chemical process can also be used to design hybrid metal NPs including CoSb3, FeCo, etc.226,227
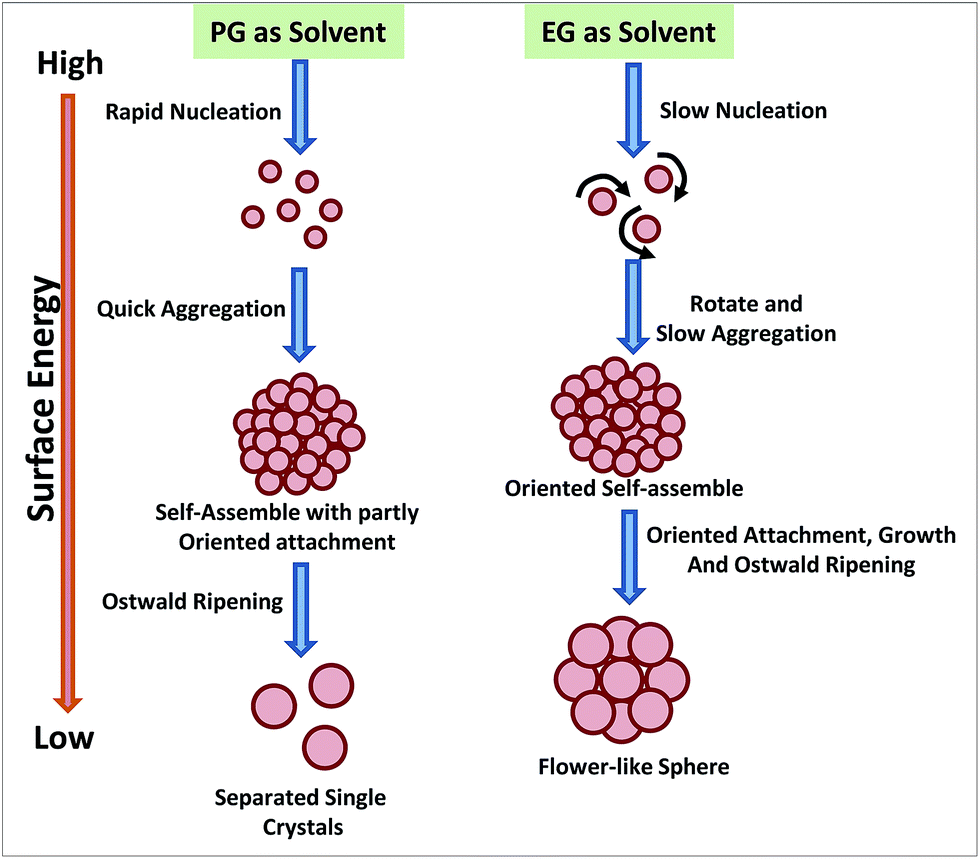 | ||
| Fig. 10 The morphology evolution of iron oxide NPs in polyol processes using two different polyols. Adapted from ref. 225. | ||
(i) High temperatures (in hot wall reactors)
(ii) High supersaturations (high partial pressure of monomers at a low vapor pressure of the bulk solid)
(iii) Long residence times (low gas flows or long reactors)
(iv) Small substrates
Hence, when the process conditions adjusted in such a way that CVD process produces NPs instead of thin solid films, then the modified process is called as CVS. The process CVS also termed as chemical vapor reaction (CVR), chemical vapor precipitation (CVP) and chemical vapor condensation (CVC). In this synthesis method, the precursors that exist in the three different states (solid, liquid, gas) are produced in the form of vapour in the reactor under an environment that requires particles to undergo the process of nucleation. In addition, CVS forms multi-component or doped NPs by the use of a variety of precursors. For instance, erbium (Er) had been incorporated into Si NPs by the use of precursors such as the organometallic erbium and disilane compound, and composite NPs are produced by encapsulating a material within the other (e.g. silicon tetrachloride in reaction with sodium chloride that result in the formation of sodium chloride-encapsulated silicon particles).228 CVS method has been extensively utilized for the synthesis of NPs using a variety of materials including ZnO, WS2, iron oxide, copper and silicon oxides, Al doped ZnO, fluorine doped tin oxide, Cr doped ZnO, etc.229–236 Polarz et al. synthesized size selected ZnO NPs (6–30 nm) by CVS process using the tetrameric alkyl-alkoxy zinc compound [CH3ZnOCH(CH3)2]4 as the precursor, which was chemically transformed into ZnO through gas-phase reactions.229 Hartner et al. synthesized Al doped ZnO (AZO) NPs by CVS process using diethylzinc (DEZn) and triethylaluminum (TEAl) as precursors for Zn and Al, respectively, while He gas was chosen as carrier gas.230 Oxygen gas with constant flow rate of 1000 sccm was inserted in alumina tube with an inner diameter of 19 mm, and temperature and pressure of the system was kept at 1073 K and 20 mbar, respectively. With these conditions, high quality AZO NPs were synthesized and their particle size was found to decrease with increasing the Al concentration. In comparison to other processes, CVS process has following advantages for the synthesis of AZO NPs:
(i) In contrast to wet synthesis, CVS process allows high percentage of Al doping in ZnO
(ii) CVS process can produce NPs at higher growth rate, leading to reduction of the cost of the material
(iii) CVS produced AZO NPs with high crystallinity
(iv) Unlike other chemical process, CVS process avoids the use of additional step which is the removal of organic contaminant after the synthesis
Jin et al. reported the synthesis of Cr doped ZnO (CZO) NPs using CVS process.231 Their CVS system was splitted into two zones for evaporation and subsequently pyrolysis of the precursors. Zinc acetylacetonate (Zn(acac)2) and chromium acetylacetonate (Cr(acac)3) were used as precursors for the synthesis of CZO NPs. These precursors were evaporated from alumina boat at low temperature first zone and then transported through He carrier gas into high temperature second zone for pyrolysis and oxidizing at 1173 K. It should be noted that the temperature in first zone was kept at 493 K for pure ZnO NPs, and changed to 423 K for CZO NPs. The crystallographic and microscopic analysis revealed that the size of pure ZnO NPs was ∼19 nm, which was decreased to ∼6 nm in case of CZO NPs with Cr concentration of 6 at%. Suffner et al. demonstrated the synthesis of fluorine doped SnO2 (FTO) NPs using CVS method.232 Tetramethyltin (TMT) and difluoromethane (DFM) were used as the precursors, while oxygen and helium were used as the reactive and carrier gases, respectively, for the synthesis of FTO NPs. The flow rate of TMT, oxygen and helium was kept constant at 25 sccm, 1500 sccm and 150 sccm, respectively, while it was changed from 0 to 1.5 sccm for DFM in order to synthesize pure tin oxide (TO) NPs and FTO NPs. The chemical reactions for the synthesis of these NPs occurred at temperature and pressure of 1200 °C and 15 mbar, respectively. When synthesized at DFM flow rate of 1.5 sccm, size of FTO NPs were found to be in the range of 3 to 10 nm. The synthesized ZnO, AZO, CZO and FTO NPs can find wide electronic and optoelectronic applications.
Using atmospheric pressure CVS (APCVS) process, Lahde et al. reported the synthesis of Cu NP composite in an amorphous silicon dioxide (a-SiO2), where Cu NPs were either coated with a-SiO2 or embedded in a-SiO2 matrix.233 In order to prepare NP composite structure using APCVS, first step was to synthesize metal–organic precursor. They synthesized metal–organic precursor [CuN(SiMe3)2]4, also called as Cu(I), using standard process. The ARCVS system, which was used for synthesis of NP composite, included different zones such as:
(i) Precursor feeding zone
(ii) Heating zone
(iii) Dilution and cooling zone
(iv) Sampling and particle collection zone
Using a heat bath, the synthesized Cu(I) precursor was vaporized at temperature between 100 and 250 °C. The pre-heated (100–250 °C) pure nitrogen or a mixture of hydrogen (10% v) and nitrogen was used as carriers, while the flow rate was kept to be 0.3 L min−1. After this, precursor vapor was transported to heating zone of system and mixed with nitrogen at flow rate of 1.8 L min−1 at temperature 800 °C. However, at the exit point of the heating zone the aerosol was diluted with nitrogen at the flow rate of 10.4 L min−1 to quench the particle. Finally, the particle sampling and collection was done from the gas phase. Inclusion of hydrogen in nitrogen gas significantly affected the structure of NP composite. In pure nitrogen atmosphere the Si/SiO2 protective coating was formed around the Cu NPs. While in mixed hydrogen/nitrogen environment, the Cu NPs were embedded in SiO2 matrix. Schematic showing the mechanism of formation of NP composite in pure nitrogen and mixed nitrogen/hydrogen atmosphere is depicted in Fig. 11.
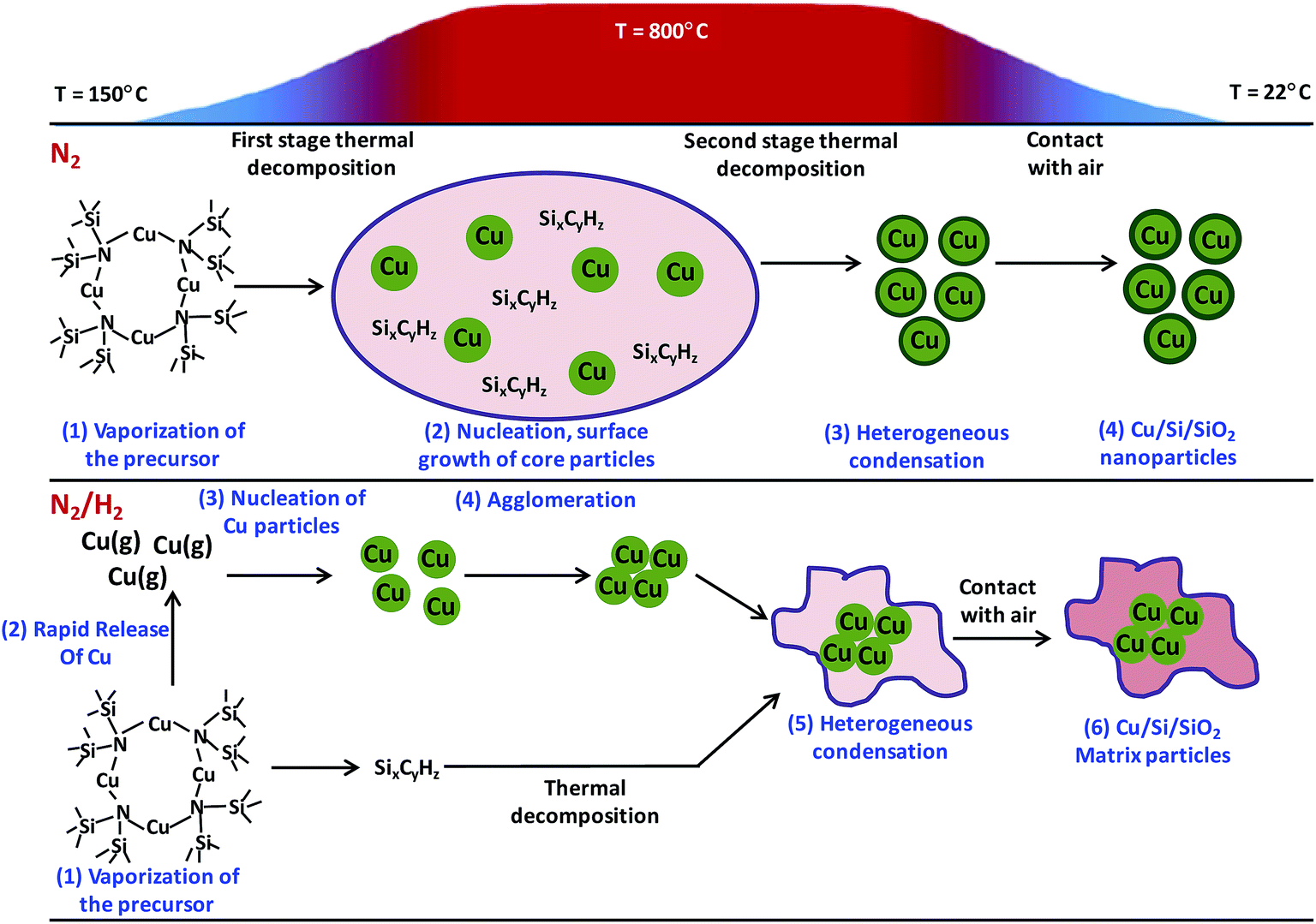 | ||
| Fig. 11 Schematic representation of formation of Cu–Si/SiO2 NPs by APCVS process. Reproduced with permission from ref. 233. | ||
Further Lee et al. reported the fabrication of fullerene-like WS2 NPs using CVS process.234 The W(CO)6 was used as precursor for tungsten (W), which was decomposed in the temperature between 420 and 1000 °C. The evaporation temperature was varied from 80 to 110 °C at flowing He gas with varied flow rate from 2000 cm3 min−1 up to 4000 cm3 min−1. In order to form WS2 NPs, the decomposition of tungsten hexacarbonyl W(CO)6 was performed in the presence of sulphur vapour with their partial pressure varied from 0.02 to 0.04 atm. Finally, at optimized conditions, the CVC process produces fullerene-like WS2 NPs having mean particle size in the range from 20 to 70 nm.
CVS process was also used widely for the synthesis of iron oxide NPs of mean particle size from 80–90 nm and various other metal oxide NPs.235,236
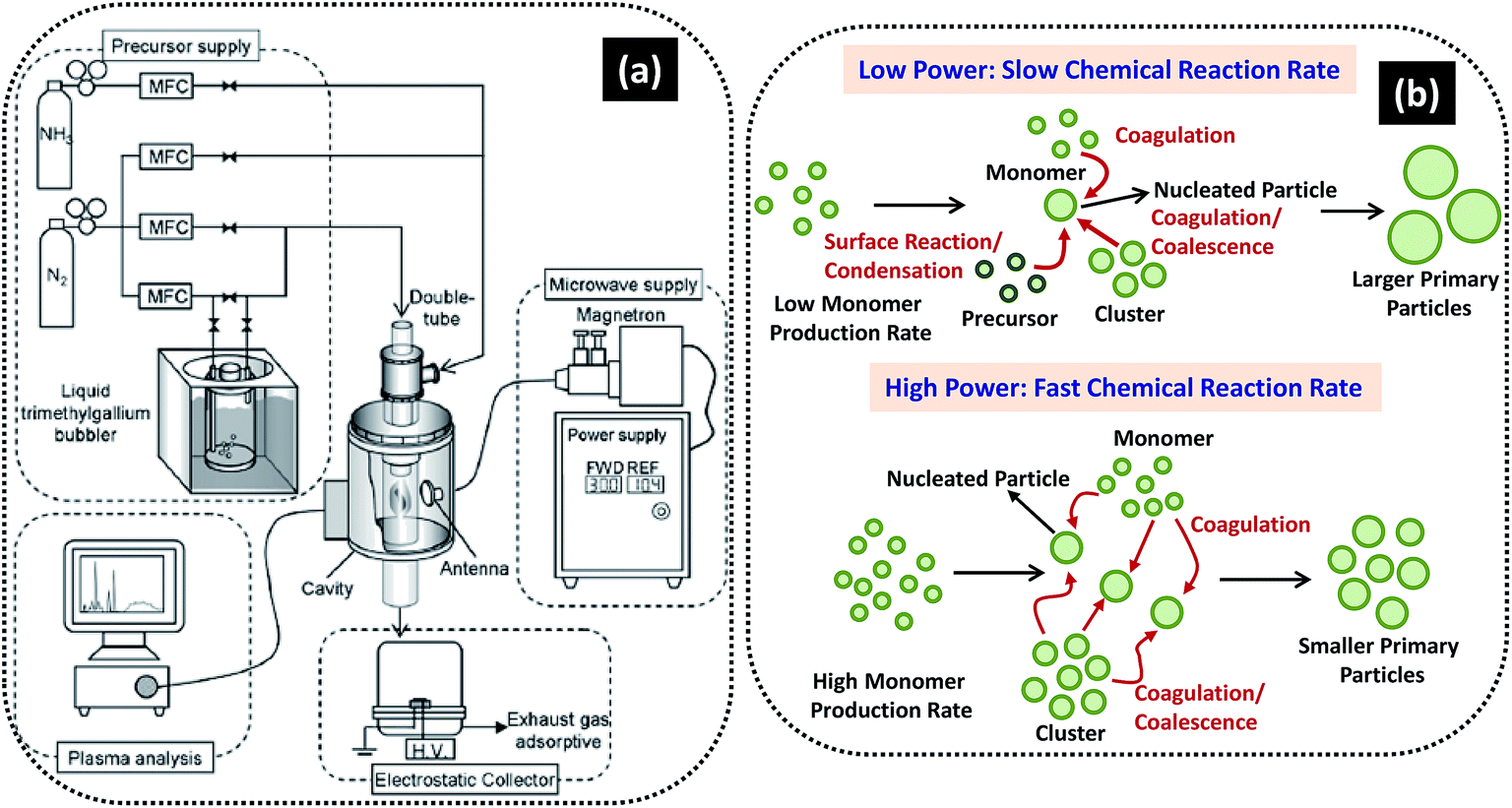 | ||
| Fig. 12 (a) Schematic of MPECVD system and (b) possible mechanism of growth of NPs at low and high powers. Reproduced with permission from ref. 237. | ||
The mechanism involved in the synthesis of NPs of varied sizes is depicted in Fig. 12b. At low input power, the plasma reactivity remains low causing slow chemical reaction rate and hence, results in the formation of monomers. Because of slow reaction rate, relatively small number of nuclei are formed and at the same time significant amount of unreacted precursor molecules exit. When large numbers of unreacted species condense and react with relative small number of nuclei, the size of the resulting particles becomes larger. However, at higher power the plasma reactivity and chemical reaction got enhanced, leading to formation of more nuclei and disappearance of unreacted species. Under this condition the coagulational growth can be prevented and formation of small size NPs would be possible. Thus, the size of GaN NPs can be efficiently controlled by the input power. Dwivedi et al. used PECVD method to synthesize hard carbon nanoparticle based films by tuning the self-bias, introducing different doping strategies and using metal/carbon bi- and multilayer designs; where nitrogen was used as foreign atoms while Ti, Cu and Ag were used as metal layer in metal/carbon bi- and multilayer structures.238–244 Designed carbon nanoparticles films showed excellent electrical, field emission, optical and mechanical properties with potential applications in the field of emission devices, protective coatings, solar cells, etc.
The synthesis of carbon NPs and hydrogenated Si NPs was also reported by MPECVD and radio frequency PECVD (RF-PECVD) techniques, respectively.245,246 Yun et al. demonstrated the synthesis of silica NP array on flexible polymer substrate using PECVD process involving following two steps:247
(i) Pre-plasma treatment of polymer surface followed by
(ii) Coating of silica NPs array
To perform plasma pre-treatment, the polymer substrate (polyethylene terephthalate; PET) was placed on electrode housed in vacuum system (base pressure of 6.7 Pa). The plasma pre-treatment was performed on PET at constant RF power of 200 W (1.1 W cm−2), working pressure of 22.7 Pa and Ar flow rate of 50 sccm, while the plasma treatment time was varied. The Ar plasma pre-treatment of the PET surfaces led to the formation of polymer protrusions due to morphological modifications of polymer chains as shown in Fig. 13. Height of protrusions (h) and the distance between neighbouring protrusions (d) was found to increase continuously with increasing pre-treatment time from 1 min to 7 min (Fig. 13). Subsequently silica NPs are deposited on pre-treated polymer surfaces by plasma polymerization of hexamethyldisiloxane (HMDSO). First in evaporator, the vapors of HMDSO are formed by thermal evaporation of precursor liquid HMDSO at a heating temperature of 130 °C and then they are mixed with O2 and Ar gases at a flow rate ratio of HMDSO:O2:Ar = 1.8:10:100 before entering into the synthesis chamber. Finally silica NPs are grown having O:Si atomic ratio of 1.9–2.0. Fig. 14 shows the growth of silica NPs on pre-treated polymer surfaces.
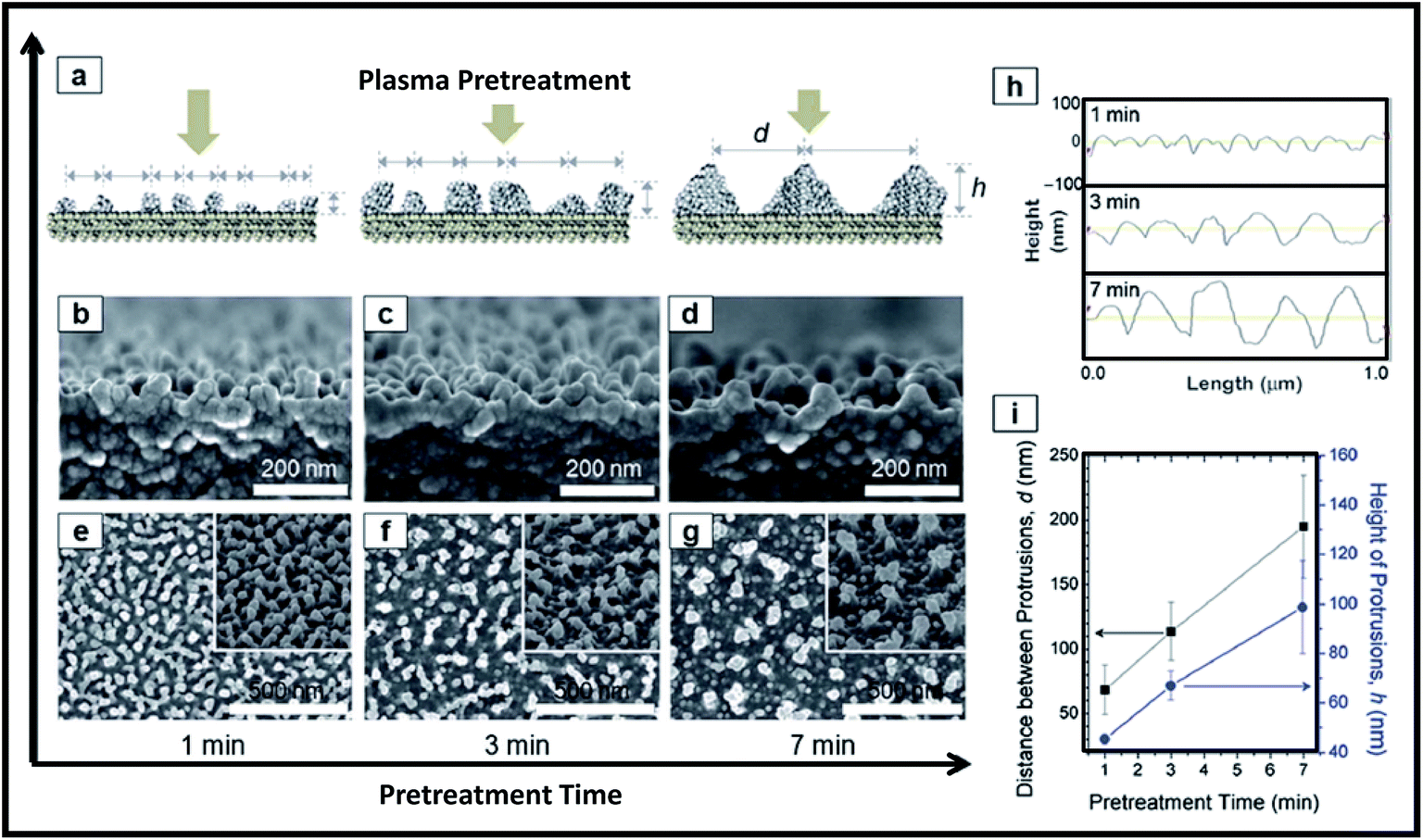 | ||
| Fig. 13 Plasma pre-treatment for creation of polymer protrusions. (a) Schematic diagram and (b–d) cross section as well as (e–g) plane view FE-SEM images showing the effect of treatment time on aspect ratio of formed polymer protrusions. (h) AFM line profiles and (i) the corresponding quantitative data of the protrusions (h and d) with plasma treatment times. Reproduced with permission from ref. 247. | ||
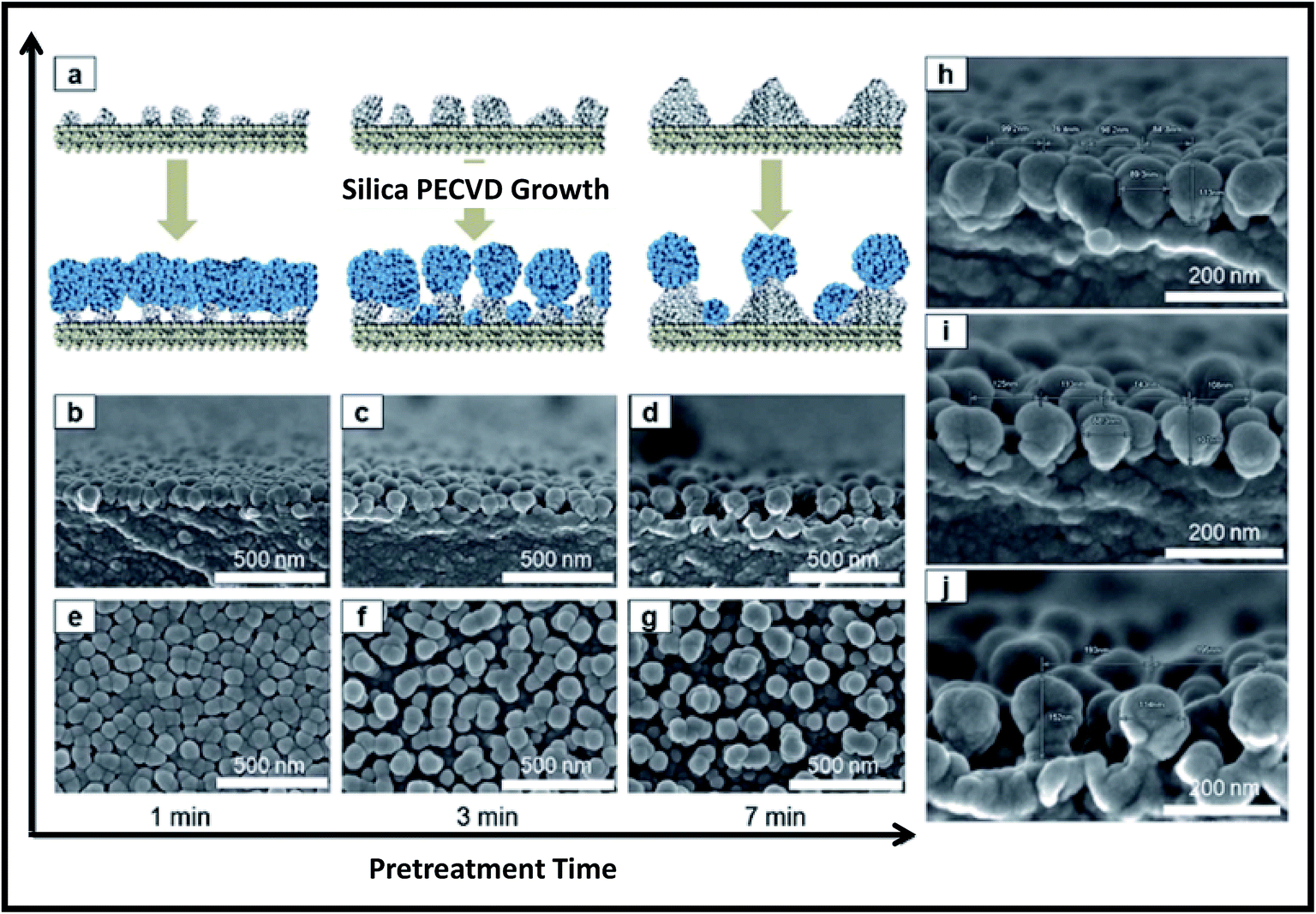 | ||
| Fig. 14 (a) Schematic showing the role of pre-treated polymer surfaces on the synthesis of silica NPs. (b–d) Cross-sectional and (e–g) plane view FE-SEM images of silica NPs formed on pre-treated polymer surfaces. (h–j) High magnification cross-sectional FESEM images of silica NPs formed on pre-treated polymer surfaces for different treatment times of (h) 1 min, (i) 3 min, and (j) 7 min. Reproduced with permission from ref. 247. | ||
Of note, plasma pre-treatment time of polymer surfaces significantly affects the growth of NPs. As the polymer protrusions reduce with increasing plasma pre-treatment time, the density of the formed silica NPs also reduces with increasing plasma-pre-treatment time. The globular silica NPs synthesized by this method showed excellent antireflective property.
2.3 Bio-assisted methods for the synthesis of nanoparticles
Bio-assisted methods, biosynthesis or green synthesis provides an environmentally benign, low-toxic, cost-effective and efficient protocol to synthesize and fabricate NPs. These methods employ biological systems like bacteria, fungi, viruses, yeast, actinomycetes, plant extracts, etc. for the synthesis of metal and metal oxide NPs. Bio-assisted methods can be broadly divided into three categories:(i) Biogenic synthesis using microorganisms
(ii) Biogenic synthesis using biomolecules as the templates
(iii) Biogenic synthesis using plant extracts
Fungal-mediated green chemistry approach for the synthesis of NPs has many advantages like higher bioaccumulation, economic viability, easily scaled up synthesis method due to simple downstream processing and biomass handling. In this context, Mukherjee et al. demonstrated a fungal-assisted biological method to synthesize silver NPs using fungus Verticillium.261 Exposure of the fungal biomass to the aqueous Ag+ ions leads to their intracellular reduction to silver NPs of dimension 25 ± 12 nm. Microscopic investigation reveal mycelia surface as the predominant site for the NPs synthesis which is mediated by the enzymes available on the mycelia cell wall. Biosynthesis of Ag NPs was also reported using Aspergillus terreus fungus and herein the speculated mechanism is reported to be NADH dependant enzyme catalyzed extracellular reaction process.262 There are number of other reports available on the biosynthesis of Ag NPs using versatile fungal species including Pleurotus ostreatus, Aspergillus flavus, Bryophilous Rhizoctoni, etc.263–265 Chauhan and co-workers synthesized Au NPs (20–40 nm) employing Candida albicans mediated bioreduction strategy.266 The mechanism of fungal biosynthesis of Au NPs is very well explained by Kitching et al.267 Bansal et al. demonstrated the biotransformation of amorphous silica in cheap agro-based rice husk to nanocrystalline silica (2–6 nm).268 There are several reports on the NPs biosynthesis of magnatite, TiO2 and ZrO2 using various fungal species.269–271
Yeast and actinomycetes also established their applicability for the bio-inspired synthesis of NPs. Yeast is an eukaryotic microorganisms belonging to the fungi kingdom. Kowshik et al. reported bulk extracellular synthesis of silver NPs (2–5 nm) using silver tolerant yeast strain MKY3.272 Kandasamy et al. shown the applicability of Hanensula anomala species to reduce gold salt generating AuNPs.273 Yarrowia lipolytica was also reported as efficient reducing agent for AuCl4 to produce gold NPs.274 Carboxyl, hydroxyl and amide groups on the cell surface are anticipated to be responsible for AuNPs synthesis. Yeast was also reported to be useful in the synthesis of Cd and PbS NPs using Candida glabrata and Rhodosporidium diobovatum species, respectively.275,276 Enriched with various enzymes, actinomycetes have been for the reduction of AuCl4− to generate Au NPs (8 nm) by Thermomonospora sp.277 Torres-Chavolla et al. reported the synthesis of comparatively larger monodispersed Au NPs (30–60 nm) in the presence of Thermomonospora curvata, Thermomonospora fusca and Thermomonospora chromogena species.278 Table 4 lists various NPs synthesized using a variety of microorganisms.
| Microorganism | Type of nanoparticles | Size of nanoparticles | Mode of synthesis | Reference | |
|---|---|---|---|---|---|
| a Abbreviations: NPs – nanoparticles, QDs – quantum dots. | |||||
| Bacteria | Staphylococcus aureus | Ag NPs | 160–180 nm | Extracellular | 255 |
| Bacillus cereus | Ag NPs | 4–5 nm | Extracellular | 258 | |
| Marinobacter pelagius | Au NPs | 2–10 nm | Extracellular | 260 | |
| Actinobacter sp. | Au NPs | 50–500 nm | Extracellular | 279 | |
| Enterobacter sp. | Hg NPs | 2–5 nm | Intracellular | 280 | |
| Lactobacillus | TiO2 NPs | 40–60 nm | Extracellular | 257 | |
| Escherichia coli | CdS QDs | 2–5 nm | Intracellular | 259 | |
| Escherichia coli | CdTe QDs | 2–3.2 nm | Extracellular | 281 | |
| Fungi | Verticillium | Ag NPs | 25 ± 12 nm | Intracellular | 261 |
| Aspergillus terreus | Ag NPs | 1–20 nm | Extracellular | 262 | |
| Bryophilous Rhizoctoni | Ag NPs | 20–50 nm | Extracellular | 263 | |
| Aspergillus flavus | Ag NPs | 8.92 ± 1.61 nm | Extracellular | 264 | |
| Pleurotus ostreatus | Ag NPs | 8–50 nm | Extracellular | 265 | |
| Fusarium oxysporum | Au NPs | 8–40 nm | Extracellular | 282 | |
| Fusarium oxysporum | SiO2 NPs | 2–4 nm | Extracellular | 268 | |
| Fusarium oxysporum | Magnetite NPs | 20–50 nm | Extracellular | 269 | |
| Fusarium oxysporum | TiO2 NPs | 6–13 nm | Extracellular | 270 | |
| Fusarium oxysporum | ZrO2 NPs | 7–8 nm | Extracellular | 271 | |
| Yeast & actinomycetes | MKY3 | Ag NPs | 2–5 nm | Extracellular | 272 |
| Candida albicans | Au NPs | 20–40 nm | Extracellular | 266 | |
| Hansenula anomala | Au NPs | 14 nm | Extracellular | 273 | |
| Yarrowia lipolytica NCIM 3589 | Au NPs | 9–23 nm | Extracellular | 274 | |
| Candida glabrata | CdS NPs | 2 nm | Intracellular | 275 | |
| Rhodosporidium diobovatum | PbS NPs | 2–5 nm | Intracellular | 276 | |
| Thermomonospora sp. | Au NPs | 8–40 nm | Extracellular | 277 | |
| Rhodococcus sp. | AuNPs | 5–15 nm | Intracellular | 283 | |
| Self assembled nanocluster of | Shape of nanocluster | Dimensions | Applications | References |
|---|---|---|---|---|
| a Abbreviations: AgNPs – silver nanoparticles, NPs – nanoparticles, ϕP – particle diameter, ϕC – chain diameter, ϕW – wire diameter, L – chain length, SERS – surface enhanced Raman spectroscopy. | ||||
| Ag NPs | Wire like | ϕP = 17 ± 3 nm, IPD = 1.7 ± 0.2 nm | As ultrasensitive SERS substrate | 285 |
| Os NPs | Wire like | ϕP ∼2 ± 0.5 nm, ϕW ∼290 ± 20 nm | Catalysis and SERS | 298 |
| Honeycomb like | ϕP = 1.5 ± 0.2 nm, ϕW = 400 nm | |||
| Os NPs (organosol) | Wire like | ϕP = 2.6 ± 0.2 nm, L = 0.54 ± 0.03 μm | Catalysis and SERS | 289 |
| Aggregated wires | ϕP = 1.2 ± 0.2 nm, L = 8–10 micron | |||
| ZnO NPs | Wire-like | ϕP = 150 ± 15 nm, L = 1–2 μm | Catalysis and dye-sensitized solar cells | 288 |
| Flower-like | ϕP = 350 ± 50 | |||
| Flake-like | ϕP = 80 ± 10 nm | |||
| β-MnO2 NPs | Wire-like | L = 1.9 ± 0.2 mm, ϕP = 35 ± 5 nm | Catalysis and supercapacitor | 299 |
| Flake-like | L = 275 ± 25 nm, ϕP = 25 ± 5 nm | |||
| TiO2 NPs | Wire like cluster (large) | ϕP = 15 ± 5 nm, ϕC = 180 ± 20 nm | Supercapacitor and dye sensitized solar cells | 300 |
| Wire like cluster (small) | ϕP = 10 ± 2 nm, ϕC = 40 ± 5 nm | |||
| NiWO4 NPs | Chain like (small) | L = 2 ± 0.2 μm, ϕC = 175 ± 15 nm, ϕP = 20 ± 5 nm | Catalysis and supercapacitor | 291 |
| Chain like (large) | L = 3.4 ± 0.2 μm, ϕC = 245 ± 15 nm, ϕP = 26 ± 4 nm | |||
| ZnWO4 NPs | Aggregated, chain like | ϕP = 75 ± 5 nm, ϕC = ∼75 ± 15 nm, L = ∼3 μm | High performance supercapacitor and catalysis | 292 |
| MnWO4 NPs | Wire-like | ϕP = 75 ± 15 nm, L ≥ 700 nm | Magnetic, catalysis and supercapacitor studies | 293 |
| Flake-like | L = 90–180 nm | |||
| Rice-like | L = 90 ± 10 nm, ϕP 25 ± 5 nm | |||
Likewise, owing to the presence of ultra-fine pores in their structure, biological membranes were also utilized as templates to design NPs. Rubber membrane made from the Hevea brasiliensis trees was used as a preservative in the production of Au NPs synthesized at the surface of membranes through the reduction of Au(III) in a solution at 80 °C for various reaction times.301 Alternatively, a uniform size and morphology of NPs could be obtained by the use of viruses as they contain hollow spaces in the center that can be used as the template for production of particles.302 Diatoms such as Amphora-46 was also reported to synthesize polycrystalline Ag NPs of size ranging 20 to 25 nm under the exposure to light. This process is done by the utilization of its extract and silver nitrate (AgNO3), in which the presence of a pigment known as fucoxanthin reduces the Ag ion that leads to further production of Ag NPs.303
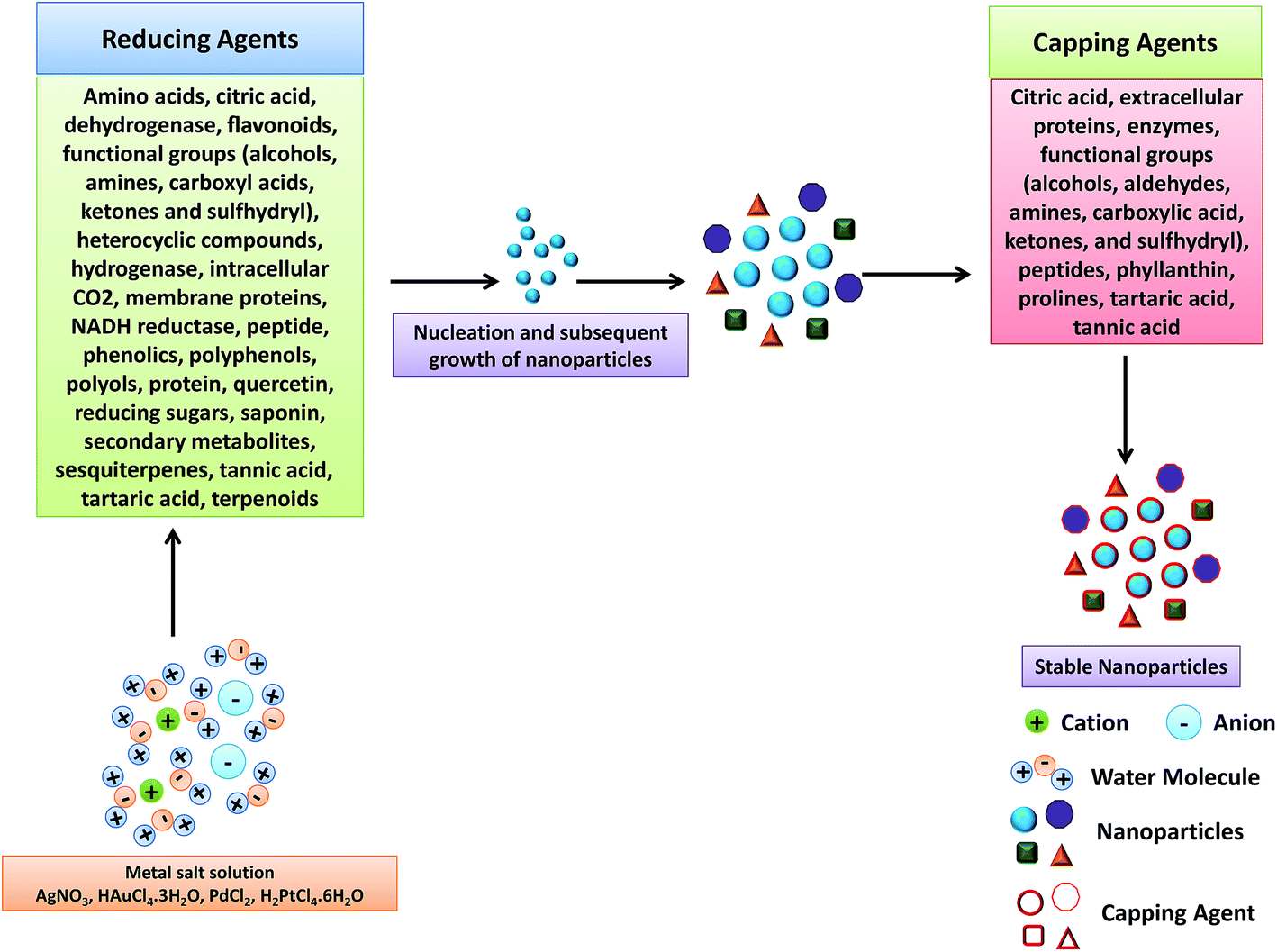 | ||
| Fig. 15 Mechanism for the biogenic synthesis of metal NPs using plant extract. Reproduced with permission from ref. 305. | ||
Kinetics of phytosynthesis of NPs is comparatively much higher to that of other biosynthesis methods and sometime equivalent to the rate of chemical routes. Shankar et al. reported the preparation of gold nanotriangles by treating the lemongrass leaf extract with the aqueous AuCl− ions.306 Similarly the leaf extract of different plants like Tamarindus indica, Aloe vera, Emblica officinalis, etc. were reported to be useful for designing Au NPs.307–309 Few-nanometer sized Pd NPs and Pt NPs were prepared using the extract taken out from various parts of different plants.310,311 Shankar et al. also reported highly concentrated Ag NPs obtained from the leaf extract of Azadirachta indica and from the fruit extract of Emblica officinalis.309,312 Li et al. and Leela et al. used the leaf extract of Aloe vera, Capsicum annuum and Helianthus annuus for the efficient synthesis of Ag NPs.313,314 Extracellular production of Cu NPs was carried out using stem latex of Euphorbia nivulia (medicinal plant) which were found to be stabilized by the peptides and terpenoids present within the latex.315 In a very interesting report, Seraphin et al. demonstrated the synthesis of In2O3 NPs (5–50 nm) using plant extract from Aloe vera.316 Furthermore, Hou et al., synthesized wurtzite ZnO NPs from Zn-hyper-accumulator plant Sedum alfredii with mean size of 53.7 nm.317 Ascencio et al. reported the bio-assisted synthesis of iron-oxide NPs by using the biomass of Medicago sativa (alfalfa) plant.318 Glutathione, an antioxidant tripeptide in plants, animals, fungi and archaea, was reported to design well-ordered aggregates of Au NPs by favouring the inter-particle interactions.319 Kundu and Nithiyanantham also demonstrated the in situ fabrication of shape-selective silver nanostructures using curcumin as stabilizing and reducing agent.320
2.4 Key applications of different nanoparticles
NPs have wide range of applications from electronic, optoelectronic, magnetic, data storage, energy and energy storage, nanomedicines, bioimaging, etc. We compiled the key applications of commonly used NPs in Table 6.| Nanoparticles | Possible key applications |
|---|---|
| Ag | Nanomedicines: antibacterial agent (healthcare, food technology, textile coating, environmental cleaning, water disinfection),321 wound healing,322,323 cancer therapeutics324 |
| Diagnosis: designing highly sensitive biosensor electrodes/chips,325 biological tagging for quantitative detection326 | |
| Solar cells,327 electroluminescent displays,328 optical sensors, surface enhanced Raman spectroscopy99,285,329 | |
| To generate eco-friendly electrical contacts for electrical devices330 | |
| Supercapacitors93 | |
| Catalysis331,286 | |
| Au | Nanomedicines: drug and gene delivery,332 hyperthermia-assisted cancer phototherapy,333,334 photodynamic therapy335 |
| Biosensors: highly sensitive optical,336 electrochemical337 and plasmon resonance biosensors338 | |
| Sensors90 | |
| Ex vivo and in vivo bio-imaging:339 two photon luminescence imaging, magnetic resonance imaging, photoacoustic tomography, Raman imaging, etc. | |
| Antibacterial agents340 | |
| Solar cells,341 energy storage devices90 | |
| Fe | Biomedical applications: biosensors,342 MRI contrast enhancement,343 hyperthermia344 |
| Magnetic and electrical applications: magnetic recording media, soft magnetic materials, motors | |
| Catalytic applications | |
| Pt | Biomedical applications: affinity probe for the detection of small biomolecules,345 catalytic nanomedicines,346 biosensors347 |
| Catalysis348 | |
| Solar cells349 | |
| Pd | Surface enhanced Raman scattering and electrocatalysis350 |
| Catalysis351 | |
| Antibacterial activity352 | |
| Fuel cells353 | |
| Biosensors354 | |
| Ge | Electronic/optoelectronic82 |
| CeO2 | Potential regenerative antioxidant355 |
| Therapeutic applications for reactive oxygen species (ROS)-related diseases e.g. cancer, diabetes, arthritis, infertility, macular degeneration356,357 | |
| Catalytic convertor for removing toxic gases355 | |
| Solid oxide fuel cells355 | |
| Biosensors358 | |
| Photocatalysis125 | |
| Antibacterial activity359 | |
| TiO2 | Drug delivery321,360 |
| For cell delivery361 | |
| Antimicrobial coatings | |
| As UV filters in sunscreens,362 toothpaste, cosmetics, etc. | |
| Biosensors363 | |
| ZnO | Solar cells364 |
| Photocatalytic and hygienic coatings365 | |
| Biosensors366 | |
| Important additive for different cosmetics, ointments and food products | |
| Cure activator for rubbers of different kinds367 | |
| Antibacterial65 | |
| Fe2O3 | Due to its excellent magnetic properties, they are being used in magnetic seals and inks, magnetic recording media, catalysts, and ferrofluids368 |
| Development of immunoassays, magnetic resonance imaging contrast agents, and targeted drug delivery vehicles, as well as in magnetic hyperthermia369 | |
| CdS | As fluorescent probes370 |
| Optical, electrochemical and photoelectrochemical biosensors371,372 | |
| Solar cells373 | |
| Photoluminescence devices374 | |
| CdSe | Solar cells338,375 |
| Light emitting diodes339,376 | |
| Magnetic nanoparticles | Biomedical applications: gene delivery,377 magnetic resonance imaging,378 magnetic carriers for bio-separation379 |
| Catalyst supports380 | |
| Upconversion (lanthanides) nanoparticles | In vivo cell imaging381 |
| Near-infrared photodynamic therapy382 | |
| Light nanotransducers for photoactivation applications383 | |
| Solar cells384 | |
| Carbon nanoparticles | Cancer diagnostic and therapy385,386 |
| Antibacterial387 | |
| Lithium sulfur batteries388 | |
| Designing of fluorescent imaging probes389 | |
| Catalysis62 | |
| Solar cells, protective coatings and emission devices238–244 | |
| Graphene oxide nanoparticles | For cell delivery248,251,252 |
| In drug delivery for cancer therapeutics390 | |
| Non-bleaching optical probe for two-photon luminescence imaging253 | |
| Cell therapy253 | |
| Biosensors391 | |
| Polymeric NPs | Nanomedicines and drug delivery392–394 |
| Fluorescent polymeric NPs for cell imaging395 | |
| Photoacoustic imaging396 | |
| Biosensors200 |
3 Conclusions and future prospects
Since the inception of NPs over half a century ago, scientists are continuously exploring advanced novel methods of synthesizing NPs with the optimal size and morphology that would be beneficial for various disciplines. To engineer and fabricate an ideal size and morphology of NPs, depending upon their application/use, diverse range of physical, chemical or biological methods are already available. In this review, we provided a comprehensive overview of the commonly employed/explored strategies for the synthesis of NPs in term of their working principle, NPs designing stratagem and relevant literature. It is anticipated that this review will act as a guide article for the researchers to selectively choose particular synthesis strategy for designing the desired NPs with particular material type, size, surface properties, targeted application, etc.Owing to its enormous technological potential, nanoparticles research is at the forefront. Being a leading nanomaterial in targeted drug delivery and nanomedicines, precise size with specified surface characteristics are the key desirables during nanoparticle synthesis. Cell toxicity, potential target, drug release profile, cost-effectiveness, etc. are the key concerns which are needed to be taken care of by the optimization of size and surface properties of nanocarriers by selecting appropriate synthesis methods. Electrospraying is growing enormously as a dry technique to design polymeric nanoparticles without using any hazardous chemicals. It provides prospects to develop nanoformulations incorporating drugs/growth factors/biomolecules with versatile structural designs like core–shell NPs, hybrid NPs, composite NPs, etc., that will be useful in drug delivery, tissue engineering, biosensors, etc. Furthermore, electrospraying is still almost unexplored for the production of metal and ceramic nanoparticles. Plasmonics, optoelectronics, information and data storage, etc. are also the key areas where the synthesis strategies for the scalable production of nanoparticles needs to be further explored and optimized which can help to develop economically viable technologies.
Acknowledgements
We acknowledge the financial support received from Singapore National Research Foundation under its Translational and Clinical Research Flagship Programme (NMRC/TCR/008-SERI/2013) and administered by the Singapore Ministry of Health's National Medical Research Council.” RL thanks the SingHealth Foundation Transition Project (SHF/FG508P/2012). CD thanks the funding support from SingHealth Foundation Research Grant (SHF/FG637S/2014). NKV acknowledges Nanyang Technological University and Ministry of Education, Singapore for funding support (Grant # L0412130.010, L0412290.010 and L0421050.010).References
- R. P. Feynman, Eng. Sci., 1960, 23, 22–36 Search PubMed
.
- J. Ramsden, Essentials of Nanotechnology, Ventus Publishing ApS, Denmark, 2009 Search PubMed
- J. Lee, S. Mahendra and P. J. J. Alvarez, ACS Nano, 2010, 4, 3580–3590 CrossRef CAS PubMed
- D. M. Smith, J. K. Simon and J. R. Baker Jr, Nat. Rev. Immunol., 2013, 13, 592–606 CrossRef CAS PubMed
- S. Nie, Y. Xing, G. J. Kim and J. W. Simons, Annu. Rev. Biomed. Eng., 2007, 9, 257–288 CrossRef CAS PubMed
- Y. Sun, B. T. Mayers and Y. Xia, Nano Lett., 2002, 2, 481–485 CrossRef CAS
- S. E. Skrabalak, J. Chen, Y. Sun, X. Lu, L. Au, C. M. Cobley and Y. Xia, Acc. Chem. Res., 2008, 41, 1587–1595 CrossRef CAS PubMed
- Y. Xia, W. Li, C. M. Cobley, J. Chen, X. Xia, Q. Zhang, M. Yang, E. C. Cho and P. K. Brown, Acc. Chem. Res., 2011, 44, 914–924 CrossRef CAS PubMed
- C. Suryanarayana and C. C. Koch, Hyperfine Interact., 2000, 130, 5–44 CrossRef CAS
- C. B. Murray, S. Sun, W. Gaschler, H. Doyle, T. A. Betley and C. R. Kagan, IBM J. Res. Dev., 2001, 45, 47–56 CrossRef CAS
- X. Wang, J. Zhuang, Q. Peng and Y. Li, Nature, 2005, 437, 121–124 CrossRef CAS PubMed
- J. Park, J. Joo, S. G. Kwon, Y. Jang and T. Hyeon, Angew. Chem., Int. Ed., 2007, 46, 4630–4660 CrossRef CAS PubMed
- T. Zhu, S. G. Cloutier, I. Ivanov, K. L. Knappenberger Jr, I. Robel and F. Zhang, J. Nanomater., 2012, 2012, 1–2 Search PubMed
- M. G. Panthani and B. A. Korgel, Annu. Rev. Chem. Biomol. Eng., 2012, 3, 287–311 CrossRef CAS PubMed
- Z. W. Pan, Z. R. Dai and Z. L. Wang, Science, 2001, 291, 1946–1949 CrossRef
- C. Hu, H. Liu, W. Dong, Y. Zhang, G. Bao, C. Lao and Z. L. Wang, Adv. Mater., 2007, 19, 470–474 CrossRef CAS
- X. Wang, J. Song and Z. L. Wang, J. Mater. Chem., 2007, 17, 711–720 RSC
- D. Li and Y. Xia, Adv. Mater., 2004, 16, 1151–1170 CrossRef CAS
- W. E. Teo and S. Ramakrishna, Nanotechnology, 2006, 17, R89 CrossRef CAS
- A. S. Tayi, E. T. Pashuck, C. J. Newcomb, M. T. McClendon and S. I. Stupp, Biomacromolecules, 2014, 15, 1323–1327 CrossRef CAS PubMed
- W. E. Teo and S. Ramakrishna, Nanotechnology, 2006, 17, 89–106 CrossRef
- R. Vasita and D. S. Katti, Int. J. Nanomed., 2006, 1, 15–30 CrossRef CAS PubMed
- C. J. Ellison, A. Phatak, D. W. Giles, C. W. Macosko and F. S. Bates, Polym. J., 2007, 48, 3306–3316 CAS
- V. Leung and F. Ko, Polym. Adv. Technol., 2011, 22, 350–365 CrossRef CAS
- V. Beachley and X. Wen, Mater. Sci. Eng., C, 2009, 29, 663–668 CrossRef CAS PubMed
- Q. Wei, Functional Nanofibers and their Applications, Woodhead Publishing Limited, Cambridge, 2012 CrossRef
- X. Luo, A. Morrin, A. J. Killard and M. R. Smyth, Electroanalysis, 2006, 4, 319–326 CrossRef
- I. J. Joye and D. J. McClements, Curr. Opin. Colloid Interface Sci., 2014, 19, 1–11 CrossRef
- R. H. Baughman, A. A. Zakhidov and W. A. de Heer, Science, 2002, 297, 787–792 CrossRef CAS PubMed
- C. Zhang, Y. Yan, Y. S. Zhao and J. Yao, Annu. Rep. Prog. Chem., Sect. C: Phys. Chem., 2013, 109, 211–239 RSC
- H. Dai, Acc. Chem. Res., 2002, 35, 1035–1044 CrossRef CAS PubMed
- C. Dhand, P. R. Solanki, K. N. Sood, M. Datta and B. D. Malhotra, Electrochem. Commun., 2009, 11, 1482–1486 CrossRef CAS
- M. F. L. de Volder, S. H. Tawfick, R. H. Baughman and A. John Hart, Science, 2013, 339, 535–539 CrossRef CAS PubMed
- H. He, L. A. Pham-Huy, P. Dramou, D. Xiao, P. Zuo and C. Pham-Huy, Int. J. Biomed. Res., 2013, 2013, 1–12 Search PubMed
- V. Schmidt, J. V. Wittemann, S. Senz and U. Gosele, Adv. Mater., 2009, 21, 2681–2702 CrossRef CAS
- S. Kundu and H. Liang, Adv. Mater., 2008, 20, 826–831 CrossRef CAS
- Y. Li, F. Qian, J. Xiang and C. M. Lieber, Mater. Today, 2006, 9, 18–27 CrossRef CAS
- J. K. Hyun, S. Zhang and L. J. Lauhon, Annu. Rev. Mater. Res., 2013, 43, 451–479 CrossRef CAS
- M. K. Sheela Modani and M. Nijhawan, Int. J. Curr. Pharm. Res., 2013, 5, 55–59 Search PubMed
- S. M. Farkhani and A. Valizadeh, IET Nanobiotechnol., 2014, 8, 59–76 CrossRef CAS PubMed
- N. Z. Frederik Hetsch, S. V. Kershaw and A. L. Rogach, Mater. Today, 2013, 16, 312–325 CrossRef
- T. Jamiesona, R. Bakhshi, D. Petrova, M. Imani and A. M. Seifalian, Biomaterials, 2007, 28, 4717–4732 CrossRef PubMed
- H. Dhyani, C. Dhand, B. D. Malhotra and P. Sen, J. Biosens. Bioelectron., 2011, 3, 112 Search PubMed
- H. Fischer, Mater. Sci. Eng., C, 2003, 23, 763–772 CrossRef
- C. Dhand, S. K. Arya, S. P. Singh, B. P. Singh, M. Datta and B. D. Malhotra, Carbon, 2008, 46, 1727–1735 CrossRef CAS
- P. H. C. Camargo, K. G. Satyanarayana and F. Wypych, Mater. Res., 2009, 12, 1–39 CrossRef CAS
- P. Thoniyot, M. J. Tan, A. A. Karim, D. J. Young and X. J. Loh, Adv. Sci., 2015, 2, 1400010 Search PubMed
- E. Ye and X. J. Loh, Aust. J. Chem., 2013, 66, 997–1007 CrossRef CAS
- W. H. de Jong and P. J. Borm, Int. J. Nanomed., 2008, 3, 133–149 CrossRef CAS
- M. L. Hans and A. M. Lowman, Curr. Opin. Solid State Mater. Sci., 2002, M6, 319–327 CrossRef
- S. Zeng, K. Yong, I. Roy, X. Dinh, X. Yu and F. Luan, Plasmonics, 2011, 6, 491–506 CrossRef CAS
- A. M. Coto-García, E. Sotelo-González, M. T. Fernández-Argüelles, R. Pereiro, J. M. Costa-Fernández and A. Sanz-Medel, Anal. Bioanal. Chem., 2011, 399, 29–42 CrossRef PubMed
- O. V. Salata, J. Nanobiotechnol., 2004, 2, 1–6 CrossRef PubMed
- M. Das, N. Saxena and P. D. Dwivedi, Nanotoxicology, 2009, 3, 10–18 CrossRef CAS
- A. Becheri, M. Durr, P. L. Nostro and P. Baglioni, J. Nanopart. Res., 2008, 10, 679–689 CrossRef CAS
- S. Taheri, A. Cavallaro, S. N. Christo, L. E. Smith, P. Majewski, M. Barton, J. D. Hayball and K. Vasilev, Biomaterials, 2014, 35, 4601–4609 CrossRef CAS
- O. N. Gadomskii and Y. Y. Kharitonov, Quantum Electron., 2004, 34, 249–254 CrossRef CAS
- G. E. Rachkovskaya, G. B. Zakharevich, K. V. Yumashev, A. M. Malyarevich and M. S. Gaponenko, Glass Ceram., 2004, 61, 9–10 CrossRef CAS
- B. V. N. Nagavarma, H. K. S. Yadav, A. Ayaz, L. S. Vasudha and H. G. Shivakumar, Asian J. Pharm. Clin. Res., 2012, 5, 16–23 CAS
- M. Abhilash, Int. J. Pharma Bio Sci., 2010, 1–12 Search PubMed
- Synthesis and Patterning Methods for Nanostructures Useful for Biological Applications, in Nanotechnology for Biology and Medicine, ed. C. Daraio, S. Jin, G. A. Silva and V. Parpura, Springer, New York, 2012, pp. 27–44 Search PubMed
- T. Xing, J. Sunarso, W. Yang, Y. Yin, A. M. Glushenkov, L. H. Li, P. C. Howlett and Y. Chen, Nanoscale, 2013, 5, 7970–7976 RSC
- J. F. de Carvalho, S. N. de Medeiros, M. A. Morales, A. L. Dantas and A. S. Carrico, Appl. Surf. Sci., 2013, 275, 84–87 CrossRef CAS
- S. Kar, S. Logad, O. P. Choudhary, C. Debnath, S. Verma and K. S. Bartwal, Univers. J. Mater. Sci., 2013, 1, 18–24 Search PubMed
- N. Salah, S. S. Habib, Z. H. Khan, A. Memic, A. Azam, E. Alarfaj, N. Zahed and S. Al-Hamedi, Int. J. Nanomed., 2011, 6, 863–869 CrossRef CAS PubMed
- D. Chen, X. Yi, Z. Chen, Y. Zhang and B. Chen, Int. J. Appl. Ceram. Technol., 2014, 11, 954–959 CrossRef CAS
- M. Ullah, M. E. Ali and S. B. A. Hamid, Rev. Adv. Mater. Sci., 2014, 37, 1–14 Search PubMed
- L. Zheng, B. Cui, L. Zhao, W. Li and G. C. Hadjipanayis, J. Alloys Compd., 2012, 539, 69–73 CrossRef CAS
- K. N. Islam, A. B. Z. Zuki, M. E. Ali, M. Z. B. Hussein, M. M. Noordin, M. Y. Loqman, H. Wahid, M. A. Hakim and S. B. A. Hamid, J. Nanomater., 2012, 2012, 1–5 CrossRef
- C. Chen, Y. Chen and W. J. Tseng, J. Mater. Process. Technol., 2007, 190, 61–64 CrossRef CAS
- M. B. Ward, R. Brydson and R. F. Cochrane, J. Phys.: Conf. Ser., 2006, 26, 296–299 CrossRef
- E. Perez-Tijerina, S. Mejia-Rosales, H. Inada and M. Jose-Yacaman, J. Phys. Chem. C, 2010, 114, 6999–7003 CAS
- M. Maicu, R. Schmittgens, D. Hecker, D. Glob, P. Frach and G. Gerlach, J. Vac. Sci. Technol., A, 2014, 32, 02B113 Search PubMed
- M. Raffi, A. K. Rumaiz and M. M. Hasan, J. Mater. Res., 2007, 22, 3378–3384 CrossRef CAS
- M. G. Pinilla, E. Martinez, G. S. Vidaurri and E. Perez-Tijerina, Nanoscale Res. Lett., 2010, 5, 180–188 CrossRef PubMed
- M. Benelmekki, J. Vernieres, J. Kim and R. Diaz, Mater. Chem. Phys., 2015, 151, 275–281 CrossRef CAS
- K. Okuyama and I. W. Lenggoro, Chem. Eng. Sci., 2003, 58, 537–547 CrossRef CAS
- Y. Hatakeyama, K. Onishi and K. Nishikawa, RSC Adv., 2011, 1, 1815–1821 RSC
- G. M. Veith, A. R. Lupini, S. J. Pennycook, A. Villa, L. Prati and N. J. Dudney, Catal. Today, 2007, 122, 248–253 CrossRef CAS
- V. Bouchat, N. Moreau, J. F. Colomer and S. Lucas, J. Surf. Eng. Mater. Adv. Technol., 2013, 3, 184–189 Search PubMed
- P. Asanithi, S. Chaiyakun and P. Limsuwan, J. Nanomater., 2012, 2012, 963609 Search PubMed
- D. Ichida, G. Uchida, H. Seo, K. Kamataki, N. Itagaki, K. Koga and M. Shiratani, J. Phys.: Conf. Ser., 2014, 518, 012002 CrossRef
- P. K. Ghosh, S. F. Ahmed, S. Jana and K. K. Chattopadhyay, Opt. Mater., 2007, 29, 1584–1590 CrossRef CAS
- G. M. Veith, A. R. Lupini, S. J. Pennycook, G. W. Ownby and N. J. Dudney, J. Catal., 2005, 231, 151–158 CrossRef CAS
- B. Ramalingam, S. Mukherjee, C. J. Mathai, K. Gangopadhyay and S. Gangopadhyay, Nanotechnology, 2013, 24, 205602 CrossRef PubMed
- Y. Zhang, J. Wan, V. Skumryev, S. Stoyanov, Y. Huang, G. C. Hadjipanayis and D. Weller, Appl. Phys. Lett., 2004, 85, 5343–5345 CrossRef CAS
- Y. K. Takahashi, T. Koyama, M. Ohnuma, T. Ohkubo and K. Hono, J. Appl. Phys., 2004, 95, 2690–2696 CrossRef CAS
- O. Dmitrieva, M. Acet, G. Dumpich, J. Kastner, C. Antoniak, M. Farle and K. Fauth, J. Phys. D: Appl. Phys., 2006, 39, 4741–4745 CrossRef CAS
- F. Stellacci, C. A. Bauer, T. Meyer-Friedrichsen, W. Wenseleers, V. Alain, S. M. Kuebler, S. J. K. Pond, Y. Zhang, S. R. Marder and J. W. Perry, Adv. Mater., 2002, 14, 175–198 CrossRef
- T. Hsieh, C. Chuang, Y. Chou and C. Shu, Mater. Des., 2010, 31, 1684–1687 CrossRef CAS
- S. Uhm, D. Song, J. Kwon, S. Lee, J. Han, K. Kim and K. Kim, Surf. Coat. Technol., 2013, 228, 360–366 CrossRef
- L. Castaldi, K. Giannakopoulosa, A. Travlosa, D. Niarchosa, S. Boukarib and E. Beaurepaire, J. Magn. Magn. Mater., 2005, 290–291, 544–546 CrossRef CAS
- A. Bello, M. Fabiane, D. Dodoo-Arhin, K. I. Ozoemena and N. Manyala, J. Phys. Chem. Solids, 2014, 75, 109–114 CrossRef CAS
- Y. Chen and C. Yeh, Colloids Surf., A, 2002, 197, 133–139 CrossRef CAS
- K. Naessens, P. van Daele and R. Baets, SPIE-Int. Soc. Opt. Eng., Proc., 2001, 4426, 124–127 Search PubMed
- S. C. Singh and R. Gopal, Bull. Mater. Sci., 2007, 30, 291–293 CrossRef CAS
- Happy, R. S. Rawat, R. V. Ramanujan, P. Lee, A. Patran, T. L. Tan, S. V. Springham and S. Lee, 31st EPS Conference on Plasma Phys., London, 2004, vol. 28G, pp. 1–4 Search PubMed
- P. L. Ong, S. Mahmood, T. Zhang, J. J. Lin, R. V. Ramanujan, P. Lee and R. S. Rawat, Appl. Surf. Sci., 2008, 254, 1909–1914 CrossRef CAS
- C. D. Andrea, F. Neri, P. M. Ossi, N. Santo and S. Trusso, Nanotechnology, 2009, 20, 245606 CrossRef PubMed
- Y. Jing, H. Wang, X. Chen, X. Wang, H. Wei and Z. Guo, Appl. Surf. Sci., 2014, 316, 66–71 CrossRef CAS
- D. Kumar, S. Yarmolenko, J. Sankar, J. Narayan, H. Zhou and A. Tiwari, Composites, Part B, 2004, 35, 149–155 CrossRef
- M. Quintana, E. Haro-Poniatowski, J. Morales and N. Batina, Appl. Surf. Sci., 2002, 195, 175–186 CrossRef CAS
- J. J. Lin, L. S. Loh, P. Lee, T. L. Tan, S. V. Springham and R. S. Rawat, Appl. Surf. Sci., 2009, 255, 4372–4377 CrossRef CAS
- S. Amoruso, G. Ausanio, C. de Lisio, V. Iannotti, M. Vitiello, X. Wang and L. Lanotte, Appl. Surf. Sci., 2005, 247, 71–75 CrossRef CAS
- M. S. Dhlamini, J. J. Terblans, O. M. Ntwaeaborwa, J. M. Ngaruiya, K. T. Hillie, J. R. Botha and H. C. Swart, J. Lumin., 2008, 128, 1997–2003 CrossRef CAS
- M. K. Akbari, R. Derakhshan and O. Mirzaee, Chem. Eng. J., 2015, 259, 918–926 CrossRef
- J. P. Lei, X. L. Dong, X. G. Zhu, M. K. Lei, H. Huang, X. F. Zhang, B. Lu, W. J. Park and H. S. Chung, Intermetallics, 2007, 15, 1589–1594 CrossRef CAS
- G. A. J. Amaratunga, M. Chhowalla, C. J. Kiely, I. Alexandrou, R. Aharonov and R. M. Devenish, Nature, 1996, 383, 321–323 CrossRef CAS
- M. Chhowalla and G. A. J. Amaratunga, Nature, 2000, 407, 164–167 CrossRef CAS PubMed
- R. D. Amato, M. Falconieri, S. Gagliardi, E. Popovici, E. Serra, G. Terranova and E. Borsella, J. Anal. Appl. Pyrolysis, 2013, 104, 461–469 CrossRef
- M. Scarisoreanu, R. Alexandrescu, I. Morjan, R. Birjega, C. Luculescu, E. Popovici, E. Dutu, E. Vasile, V. Danciu and N. Herlin-Boime, Appl. Surf. Sci., 2013, 278, 295–300 CrossRef CAS
- E. Marino, B. Bouchet-Fabre, D. Porterat and C. Reynaud, Diamond Relat. Mater., 2005, 14, 1120–1125 CrossRef CAS
- I. Llamas-Jansa, C. Jager, H. Mutschke and T. Henning, Carbon, 2007, 45, 1542–1557 CrossRef CAS
- M. T. Swihart, Curr. Opin. Colloid Interface Sci., 2003, 8, 127–133 CrossRef CAS
- S. Veintemillas-Verdaguer, Y. Leconte, R. Costo, O. Bomati-Miguel, B. Bouchet-Fabre, M. P. Morales, P. Bonville, S. Perez-Rial, I. Rodriguez and N. Herlin-Boime, J. Magn. Magn. Mater., 2007, 311, 120–124 CrossRef CAS
- O. Bomatí-Miguel, P. Tartaj, M. P. Morales, P. Bonville, U. Golla-Schindler, X. Q. Zhao and S. Veintemillas-Verdaguer, Small, 2006, 2, 1476–1483 CrossRef PubMed
- F. Erogbogbo, K. Yong, I. Roy, G. X. Xu, P. N. Prasad and M. T. Swihart, ACS Nano, 2008, 2, 873–878 CrossRef CAS PubMed
- W. Y. Teoh, R. Amal and L. Madler, Nanoscale, 2010, 2, 1324–1347 RSC
- M. Sokolowski, A. Sokolowska, A. Michalski and B. Gokieli, J. Aerosol Sci., 1977, 8, 219–230 CrossRef CAS
- T. R. Hinklin, S. C. Rand and R. M. Laine, Adv. Mater., 2008, 20, 1270–1273 CrossRef CAS
- T. Sahma, L. Madler, A. Gurlo, N. Barsan, S. E. Pratsinis and U. Weimar, Sens. Actuators, B, 2004, 98, 148–153 CrossRef
- H. Schulz, L. Mädler, S. E. Pratsinis, P. Burtscher and N. Moszner, Adv. Funct. Mater., 2005, 15, 830–837 CrossRef CAS
- R. N. Grass, E. K. Athanassiou and W. J. Stark, Angew. Chem., Int. Ed., 2007, 46, 4909–4912 CrossRef CAS PubMed
- O. Arutanti, A. B. D. Nandiyanto, T. Ogi, F. Iskandar, T. O. Kim and K. Okuyama, J. Alloys Compd., 2014, 591, 121–126 CrossRef CAS
- D. Channei, B. Inceesungvorn, N. Wetchakun, S. Phanichphant, A. Nakaruk, P. Koshy and C. C. Sorrell, Ceram. Int., 2013, 39, 3129–3134 CrossRef CAS
- C. Siriwong, N. Tamaekong and S. Phanichphant, Mater. Lett., 2012, 68, 97–100 CrossRef CAS
- J. A. Bhushani and C. Anandharamakrishnan, Trends Food Sci. Technol., 2014, 38, 21–33 CrossRef
- R. Sridhar, R. Lakshminarayanan, K. Madhaiyan, V. A. Barathi, K. H. C. Lim and S. Ramakrishna, Chem. Soc. Rev., 2015, 44, 790–814 RSC
- P. H. M. Bottger, Z. Bi, D. Adolph, K. A. Dick, L. S. Karlsson, M. N. A. Karlsson, B. A. Wacaser and K. Deppert, Nanotechnology, 2007, 18, 1–6 CrossRef
- R. Sridhar and S. Ramakrishna, Biomatter, 2013, 3, e24281 CrossRef PubMed
- M. Zamani, M. P. Prabhakaran, E. S. Thian and S. Ramakrishna, Int.
J. Pharm., 2014, 473, 134–143 CrossRef CAS PubMed
- G. Pyrgiotakis, J. McDevitt, A. Bordini, E. Diaz, R. Molina, C. Watson, G. Deloid, S. Lenard, N. Fix, Y. Mizuyama, T. Yamauchi, J. Brain and P. Demokritou, Environ. Sci.: Nano, 2014, 1, 15–26 RSC
- A. Jaworek, A. T. Sobczyk, A. Krupa, M. Lackowski and T. Czech, Bull. Pol. Acad. Sci.: Tech. Sci., 2009, 57, 63–70 Search PubMed
- W. Cai, T. Gao, H. Hong and J. Sun, Nanotechnol., Sci. Appl., 2008, 1, 17–32 CAS
- M. Valvo, U. Lafont and D. Munao, J. Power Sources, 2009, 189, 297–302 CrossRef CAS
- S. Prabhu and E. K. Poulose, Int. Nano Lett., 2012, 2, 1–10 CrossRef
- T. Krishnakumar, R. Jayaprakash, V. N. Singh, B. R. Mehta and A. R. Phani, J. Nano Res., 2008, 4, 91–101 CrossRef CAS
- S. S. Chee and J. H. Lee, Trans. Nonferrous Met. Soc. China, 2012, 22, 707–711 CrossRef
- A. Jaworek and A. T. Sobczyk, J. Electrost., 2008, 66, 197–219 CrossRef CAS
- L. Tang and J. Cheng, Nano Today, 2013, 8, 290–312 CrossRef CAS PubMed
- S. M. Haidary, E. P. Corcoles and N. K. Ali, J. Nanomater., 2012, 2012, 15 Search PubMed
- S. Witharana, C. Hodges, D. Xu, X. Lai and Y. Ding, J. Nanopart. Res., 2012, 14, 1–11 CrossRef
- F. Alonso, P. Riente, J. A. Sirvent and M. Yus, Appl. Catal., A, 2010, 378, 42–51 CrossRef CAS
- H. Shi, R. Magaye, V. Castranova and J. Zhao, Part. Fibre Toxicol., 2013, 10, 1–33 CrossRef CAS PubMed
- M. Eshed, S. Pol, A. Gedanken and M. Balasubramanian, Beilstein J. Nanotechnol., 2011, 2, 198–203 CrossRef CAS PubMed
- S. A. Jamal, Chem. Sci. J., 2013, 2013, 1–10 Search PubMed
- J. V. Erven, R. Moerman and J. C. M. Marijnissen, Aerosol Sci. Technol., 2005, 39, 941–946 CrossRef
- S. S. Nkosi, B. W. Mwakikunga, E. Sideras-Haddad and A. Forbes, Nanotechnol., Sci. Appl., 2012, 5, 27–36 CAS
- M. R. Diaz and P. E. Vivas-Mejia, Pharmaceuticals, 2013, 6, 1361–1380 CrossRef PubMed
- L. Y. Yeo, Z. Gagnon and H. C. Chang, Biomaterials, 2005, 26, 6122–6128 CrossRef CAS PubMed
- L. Cismaru and M. Popa, Rev. Roum. Chim., 2010, 55, 433–442 CAS
- C. J. Luo, T. Okubo, M. Nangrejob and M. Edirisinghe, Polym. Int., 2015, 64, 183–187 CrossRef CAS
- K. Matsukawa, M. Watanabe, T. Hamada, T. Nagase and H. Naito, Int. J. Polym. Sci., 2012, 2012, 10 Search PubMed
- E. C. Cho, Y. K. Hwang and U. Jeong, Bull. Korean Chem. Soc., 2014, 35, 1784–1788 CrossRef CAS
- L. Buruaga and J. A. Pomposo, Polymers, 2011, 3, 1673–1683 CrossRef CAS
- Y. Wu and R. L. Clark, J. Biomater. Sci., Polym. Ed., 2008, 9, 573–601 CrossRef PubMed
- N. Dubey, R. Varshney, J. Shukla, A. Ganeshpurkar, P. P. Hazari, G. P. Bandopadhaya, A. K. Mishra and P. Trivedi, Drug Delivery, 2012, 19, 132–142 CrossRef CAS PubMed
- Y. Ma, Y. Zheng, X. Zeng, L. Jiang, H. Chen, R. Liu, L. Huang and L. Mei, Int. J. Nanomed., 2011, 6, 2679–2688 CAS
- U. Nagarajan, K. Kawakami, S. Zhang, B. Chandrasekaran and B. U. Nair, Chem. Pharm. Bull., 2014, 62, 422–428 CrossRef CAS PubMed
- M. Papi, V. Palmieri, G. Maulucci, G. Arcovito, E. Greco, G. Quintiliani, M. Fraziano and M. de Spirito, J. Nanopart. Res., 2011, 13, 6141–6147 CrossRef CAS
- L. Peltonen, H. Valo, R. Kolakovic, T. Laaksonen and J. Hirvonen, Expert Opin. Drug Delivery, 2010, 7, 705–719 CrossRef CAS PubMed
- S. K. Nitta and K. Numata, Int. J. Mol. Sci., 2013, 14, 1629–1654 CrossRef CAS PubMed
- N. Karak, Fundamentals Of Polymers: Raw Materials To Finish Products, Prentice-hall Of India Pvt Ltd, New Delhi, 1st edn, 2009 Search PubMed
- B. Lin, U. Sundararaj and P. Potschke, Macromol. Mater. Eng., 2006, 291, 227–238 CrossRef CAS
- B. Sevil and K. Zuhal, Macromol. Symp., 2010, 295, 59–64 CrossRef CAS
- H. S. Lee, L. Zhu and R. A. Weiss, Polymer, 2005, 46, 10841–10853 CrossRef CAS
- Sol-Gel Science: The Physics and Chemistry of Sol-Gel Processing, ed. C. J. Brinker and G. W. Scherer, Academic Press, USA, 1990, e-book, http://depts.washington.edu/solgel/documents/class_docs/MSE502/SolGel_Science_The_physics_and_chemistry_of_sol-gel_processing_-_Brinker_1990.pdf Search PubMed
- M. A. Behnajady, H. Eskandarloo, N. Modirshahla and M. Shokri, Desalination, 2011, 278, 10–17 CrossRef CAS
- M. Aziz, S. S. Abbas and W. R. W. Baharom, Mater. Lett., 2013, 91, 31–34 CrossRef CAS
- L. F. F. F. Goncalves, F. K. Kanodarwala, J. A. Stride, C. J. R. Silva, M. R. Pereira and M. J. M. Gomes, J. Photochem. Photobiol., A, 2014, 285, 21–29 CrossRef CAS
- N. Bayal and P. Jeevanandam, J. Alloys Compd., 2012, 516, 27–32 CrossRef CAS
- P. Nautiyal, M. M. Seikh, O. I. Lebedev and A. K. Kundu, J. Magn. Magn. Mater., 2015, 377, 402–405 CrossRef CAS
- S. M. Reda, Mater. Sci. Semicond. Process., 2010, 13, 417–425 CrossRef CAS
- Y. Zhang, R. Li, Y. Jiang, B. Zhao, H. Duan, J. Li and Z. Feng, J. Solid State Chem., 2011, 184, 2047–2052 CrossRef CAS
- N. Chumha, S. Kittiwachana, T. Thongtem, S. Thongtem and S. Kaowphong, Mater. Lett., 2014, 136, 18–21 CrossRef CAS
- T. Sreethawong, S. Ngamsinlapasathian and S. Yoshikawa, J. Mol. Catal. A: Chem., 2013, 374–375, 94–101 CrossRef CAS
- J. N. Solanki and Z. V. P. Murthy, Ind. Eng. Chem. Res., 2011, 50, 12311–12323 CrossRef CAS
- M. A. Malik, M. Y. Wani and M. A. Hashim, Arabian J. Chem., 2012, 5, 397–417 CrossRef CAS
- M. Boutonnet, J. Kizling and P. Stenius, Colloids Surf., 1982, 5, 209–225 CrossRef CAS
- R. A. Martınez-Rodriguez, F. J. Vidal-Iglesias, J. Solla-Gullon, C. R. Cabrera and J. M. Feliu, J. Am. Chem. Soc., 2014, 136, 1280–1283 CrossRef PubMed
- W. Zhang, X. Qia, J. Chen and H. Wang, J. Colloid Interface Sci., 2006, 302, 370–373 CrossRef CAS
- W. Zhang, X. Qia and J. Chen, J. Chem. Phys., 2006, 330, 495–500 CAS
- M. P. Pileni and I. Lisiecki, Colloids Surf., A, 1993, 80, 63–68 CrossRef CAS
- C. Petit, P. Lixon and M. P. Pileni, J. Phys. Chem., 1990, 94, 1598–1603 CrossRef CAS
- D. H. Chen and S. H. Wu, Chem. Mater., 2000, 12, 1354–1360 CrossRef CAS
- S. Kundu, J. Mater. Chem. C, 2013, 1, 831–842 RSC
- S. Kundu, K. Wang and H. Liang, J. Phys. Chem. C, 2009, 113, 18570–18577 CAS
- S. Praharaj, S. K. Ghosh, S. Nath, S. Kundu, S. Panigrahi, S. Basu and T. Pal, J. Phys. Chem. B, 2005, 109, 13166–13174 CrossRef CAS PubMed
- S. Y. Chang, L. Liu and S. A. Asher, J. Am. Chem. Soc., 1994, 116, 6739–6744 CrossRef CAS
- D. H. Chen and C. J. Chen, J. Mater. Chem., 2002, 12, 1557–1562 RSC
- S. Vaucher, J. Fielden, M. Li, E. Dujardin and S. Mann, Nano Lett., 2002, 2, 225–229 CrossRef CAS
- M. Mandal, S. Kundu, S. K. Ghosh and T. Pal, J. Photochem. Photobiol., A, 2004, 167, 17–22 CrossRef CAS
- S. S. Atik and J. K. Thomas, J. Am. Chem. Soc., 1981, 103, 4279–4280 CrossRef CAS
- M. P. Pileni, C. R. Chim., 2003, 6, 965–978 CrossRef CAS
- S. Holdcroft and J. E. Guillet, J. Polym. Sci., Part A: Polym. Chem., 1990, 28, 1823–1829 CrossRef CAS
- W. R. P. Raj, M. Sasthav and H. M. Cheung, Langmuir, 1991, 7, 2586–2591 CrossRef CAS
- C. Destree and J. B. Nagy, Adv. Colloid Interface Sci., 2006, 123–126, 353–367 CrossRef CAS PubMed
- C. Yang, Z. Junhua and W. Guanghui, J. Wuhan Univ. Technol., Mater. Sci. Ed., 2013, 28, 787–792 CrossRef
- Z. Wang, Y. Wang, D. Xu, E. S. W. Kong and Y. Zhang, Synth. Met., 2010, 160, 921–926 CrossRef CAS
- C. Dhand, M. Das, G. Sumana, A. K. Srivastava, M. K. Pandey, C. G. Kim, M. Datta and B. D. Malhotra, Nanoscale, 2010, 2, 747–754 RSC
- H. Hayashi and Y. Hakuta, Materials, 2010, 3, 3794–3817 CrossRef CAS
- A. Abedini, A. R. Daud, M. A. A. Hamid, N. K. Othman and E. Saion, Nanoscale Res. Lett., 2013, 8, 1–10 CrossRef PubMed
- J. J. Du, C. Chen, Y. L. Gan, R. H. Zhang, C. Y. Yang and X. W. Zhou, Int. J. Hydrogen Energy, 2014, 39, 17634–17637 CrossRef CAS
- Y. Ma, M. Chen and M. Li, Mater. Lett., 2015, 139, 22–25 CrossRef CAS
- X. D. Liu, H. Chen, S. S. Liu, L. Q. Ye and Y. P. Li, Mater. Res. Bull., 2015, 62, 217–221 CrossRef CAS
- M. Tadic, M. Panjan, V. Damnjanovic and I. Milosevic, Appl. Surf. Sci., 2014, 320, 183–187 CrossRef CAS
- K. Sue, S. Kawasaki, M. Suzuki, Y. Hakuta, H. Hayashi, K. Arai, Y. Takebayashi, S. Yoda and T. Furuya, Chem. Eng. J., 2011, 166, 947–953 CrossRef CAS
- D. Zhao, X. Wu, H. Guan and E. Han, J. Supercrit. Fluids, 2007, 42, 226–233 CrossRef CAS
- J. Yang and J. Pan, Acta Mater., 2012, 60, 4753–4758 CrossRef CAS
- J. Guo, X. Zhou, Y. Lu, X. Zhang, S. Kuang and W. Hou, J. Solid State Chem., 2012, 196, 550–556 CrossRef CAS
- B. H. Choi, S. Park, B. K. Park, H. H. Chun and Y. Kim, Mater. Res. Bull., 2013, 48, 3651–3656 CrossRef CAS
- Y. Cao, P. Hu and D. Jia, Appl. Surf. Sci., 2013, 265, 771–777 CrossRef CAS
- A. Behbahani, S. Rowshanzamir and A. Esmaeilifar, Procedia Eng., 2012, 42, 908–917 CrossRef
- E. Maryanti, D. Damayanti, I. Gustian and S. Y. Salprima, Mater. Lett., 2014, 118, 96–98 CrossRef CAS
- P. Rahman and M. Green, Nanoscale, 2009, 1, 214–224 RSC
- T. Herricks, J. Chen and Y. Xia, Nano Lett., 2004, 2, 2367–2371 CrossRef
- B. K. Park, S. Jeong, D. Kim, J. Moon, S. Lim and J. S. Kim, J. Colloid Interface Sci., 2007, 311, 417–424 CrossRef CAS PubMed
- D. Kim, S. Jeong and J. Moon, Nanotechnology, 2006, 17, 4019–4024 CrossRef CAS PubMed
- M. H. Kim, B. Lim, E. P. Lee and Y. Xia, J. Mater. Chem., 2008, 18, 4069–4073 RSC
- R. J. Joseyphus and B. Jeyadevan, J. Phys. Chem. Solids, 2011, 72, 1212–1217 CrossRef CAS
- S. Lee, S. Jeong, D. Kim, S. Hwang, M. Jeon and J. Moon, Superlattices Microstruct., 2008, 43, 330–339 CrossRef CAS
- M. Ahrén, L. Selegard, F. Soderlind, M. Linares, J. Kauczor, P. Norman, P. Kall and K. Uvdal, J. Nanopart. Res., 2012, 14, 1–17 CrossRef
- C. Quievryn, S. Bernard and P. Miele, Nanomater. Nanotechnol., 2014, 4, 1–8 CrossRef
- N. Songvorawit, K. Tuitemwong and P. Tuitemwong, ISRN Nanotechnol., 2011, 2011, 483129 Search PubMed
- C. Cheng, F. Xu and H. Gu, New J. Chem., 2011, 35, 1072–1079 RSC
- K. Isogai and T. Itoh, Proceedings of International Symposium on EcoTopia Science, ISETS07, 2007, pp. 155–157 Search PubMed
- M. Zamanpour, Y. Chen, B. Hu, K. Carroll, Z. J. Huba, E. E. Carpenter, L. H. Lewis and V. G. Harris, J. Appl. Phys., 2012, 111, 07B528 CrossRef
- M. T. Swihart, Curr. Opin. Colloid Interface Sci., 2003, 8, 127–133 CrossRef CAS
- S. Polarz, A. Roy, M. Merz, S. Halm, D. Schroder, L. Schneider, G. Bacher, F. E. Kruis and M. Driess, Small, 2005, 1, 540–552 CrossRef CAS PubMed
- S. Hartner, M. Ali, C. Schulz, M. Winterer and H. Wiggers, Nanotechnology, 2009, 20, 445701 CrossRef PubMed
- W. Jin, I. Lee, A. Kompch, U. Dorfler and M. Winterer, J. Eur. Ceram. Soc., 2007, 27, 4333–4337 CrossRef CAS
- J. Suffner, P. Agoston, J. Kling and H. Hahn, J. Nanopart. Res., 2010, 12, 2579–2588 CrossRef CAS
- A. Lahde, N. Kokkonen, A. J. Karttunen, S. Jaaskelainen, U. Tapper, T. A. Pakkanen and J. Jokiniemi, J. Nanopart. Res., 2011, 13, 3591–3598 CrossRef CAS
- D. Lee, O. V. Tolochko, F. R. Turaev, D. Kim and B. Kim, J. Nanosci. Nanotechnol., 2009, 9, 1–6 CrossRef
- J. Ruusunen, M. Ihalainen, T. Koponen, T. Torvela, M. Tenho, J. Salonen, O. Sippula, J. Joutsensaari, J. Jokiniemi and A. Lahde, J. Nanopart. Res., 2014, 16, 1–11 CrossRef
- J. Lee, C. Lee and K. Lee, IOP Conf. Ser.: Mater. Sci. Eng., 2011, 18, 1–6 Search PubMed
- M. Shimada, W. Wang and K. Okuyama, Chem. Vap. Deposition, 2010, 16, 151–156 CrossRef CAS
- N. Dwivedi, S. Kumar and H. K. Malik, ACS Appl. Mater. Interfaces, 2011, 3, 4268–4278 CAS
- N. Dwivedi, S. Kumar, Ishpal, S. Dayal, Govind, C. M. S. Rauthan and O. S. Panwar, J. Alloys Compd., 2011, 509, 1285–1293 CrossRef CAS
- N. Dwivedi, S. Kumar, R. K. Tripathy, J. D. Carey, H. K. Malik and M. K. Dalai, ACS Appl. Mater. Interfaces, 2012, 4, 5309–5316 CAS
- N. Dwivedi, S. Kumar, J. D. Carey, R. K. Tripathy, H. K. Malik and M. K. Dalai, ACS Appl. Mater. Interfaces, 2013, 5, 2725–2732 CAS
- N. Dwivedi, S. Kumar, H. K. Malik, C. M. S. Rauthan and O. S. Panwar, Mater. Chem. Phys., 2011, 130, 775–785 CrossRef CAS
- N. Dwivedi, S. Kumar and H. K. Malik, J. Appl. Phys., 2012, 112, 023518 CrossRef
- N. Dwivedi, S. Kumar, R. K. Tripathy, H. K. Malik and O. S. Panwar, Appl. Phys. A, 2011, 105, 417–425 CrossRef CAS
- X. Wang, Z. Hu, X. Chen and Y. Chen, Scr. Mater., 2001, 44, 1567–1570 CrossRef CAS
- H. Vach, Q. Brulin, N. Chaabane, T. Novikova, P. R. Cabarrocas, B. Kalache, K. Hassouni, S. Botti and L. Reining, Comput. Mater. Sci., 2006, 35, 216–222 CrossRef CAS
- J. Yun, T. Bae, J. Kwon, S. Lee and G. Lee, Nanoscale, 2012, 4, 7221 RSC
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS
- X. Zhao, L. Liu, X. Li, J. Zeng, X. Jia and P. Liu, Langmuir, 2014, 30, 10419–10429 CrossRef CAS PubMed
- B. Jana, A. Biswas, S. Mohapatra, A. Saha and S. Ghosh, Chem. Commun., 2014, 50, 11595–11598 RSC
- B. Jana, G. Mondal, A. Biswas, I. Chakraborty, A. Saha, P. Kurkute and S. Ghosh, Macromol. Biosci., 2013, 13, 1478–1484 CrossRef CAS PubMed
- J. L. Li, H. C. Bao, X. L. Hou, L. Sun, X. G. Wang and M. Gu, Angew. Chem., Int. Ed., 2012, 51, 1830–1834 CrossRef CAS
- X. Zhang, S. Yan, R. D. Tyagi and R. Y. Surampalli, Chemosphere, 2011, 82, 489–494 CrossRef CAS PubMed
- A. Nanda and M. Saravanan, Nanomedicine, 2009, 5, 452–456 CrossRef CAS PubMed
- L. Sintubin, W. D. Windt, J. Dick, J. Mast, D. V. Ha, W. Verstraete and N. Boon, Appl. Microbiol. Biotechnol., 2009, 84, 741–749 CrossRef CAS PubMed
- K. Prasad, A. K. Jha and A. R. Kulkarni, Nanoscale Res. Lett., 2007, 2, 248–250 CrossRef CAS
- M. M. G. Babu and P. Gunasekaran, Colloids Surf., B, 2009, 74, 191–195 CrossRef CAS PubMed
- R. Y. Sweeney, C. Mao, X. Gao, J. L. Burt, A. M. Belcher, G. Georgiou and B. L. Iverson, Chem. Biol., 2004, 11, 1553–1559 CrossRef CAS PubMed
- N. Sharma, A. K. Pinnaka, M. Raje, A. Fnu, M. S. Bhattacharyya and A. R. Choudhury, Microb. Cell Fact., 2012, 11, 1–6 CrossRef PubMed
- P. Mukherjee, A. Ahmad, D. Mandal, S. Senapati, S. R. Sainkar, M. I. Khan, R. Parishcha, P. V. Ajaykumar, M. Alam, R. Kumar and M. Sastry, Nano Lett., 2001, 1, 515–519 CrossRef CAS
- G. Li, D. He, Y. Qian, B. Guan, S. Gao, Y. Cui, K. i. Yokoyama and L. Wang, Int. J. Mol. Sci., 2012, 13, 466–476 CrossRef CAS PubMed
- D. B. Raudabaugh, M. B. Tzolov, J. P. Calabrese and B. E. Overton, Nanomater. Nanotechnol., 2013, 3, 1–6 Search PubMed
- N. Vigneshwaran, N. M. Ashtaputre, P. V. Varadarajan, R. P. Nachane, K. M. Paralikar and R. H. Balasubramanya, Mater. Lett., 2007, 61, 1413–1418 CrossRef CAS
- R. Devika, S. Elumalai, E. Manikandan and D. Eswaramoorthy, Sci. Rep., 2012, 1, 1–5 Search PubMed
- A. Chauhan, S. Zubair, S. Tufail, A. Sherwani, M. Sajid, S. C. Raman, A. Azam and M. Owais, Int. J. Nanomed., 2011, 6, 2305–2319 CAS
- M. Kitching, M. Ramani and E. Marsili, Microb. Biotechnol., 2014, 1–14, DOI:10.1111/1751-7915.12151
- V. Bansal, A. Ahmad and M. Sastry, J. Am. Chem. Soc., 2006, 128, 14059–14066 CrossRef CAS PubMed
- A. Bharde, D. Rautaray, V. Bansal, A. Ahmad, I. Sarkar, S. M. Yusuf, M. Sanyal and M. Sastry, Small, 2006, 2, 135–141 CrossRef CAS PubMed
- V. Bansal, D. Rautaray, A. Bharde, K. Ahire, A. Sanyal, A. Ahmad and M. Sastry, J. Mater. Chem., 2005, 15, 2583–2589 RSC
- V. Bansal, D. Rautaray, A. Ahmad and M. Sastry, J. Mater. Chem., 2004, 14, 3303–3305 RSC
- M. Kowshik, S. Ashtaputre, S. Kharrazi, W. Vogel, J. Urban, S. K. Kulkarn and K. M. Paknikar, Nanotechnology, 2003, 14, 95–100 CrossRef CAS
- K. K. Sathish, R. Amutha, P. Arumugam and S. Berchmans, ACS Appl. Mater. Interfaces, 2011, 3, 1418–1425 CAS
- P. S. Pimprikar, S. S. Joshi, A. R. Kumar, S. S. Zinjarde and S. K. Kulkarni, Colloids Surf., B, 2009, 74, 309–316 CrossRef CAS PubMed
- C. T. Dameron, R. N. Reese, R. K. Mehra, A. R. Kortan, P. J. Carroll, M. L. Steigerwald, L. E. Brus and D. R. Winge, Nature, 1989, 338, 596–597 CrossRef CAS
- S. Seshadri, K. Saranya and M. Kowshik, Biotechnol. Prog., 2011, 27, 1464–1469 CrossRef CAS PubMed
- A. Ahmad, S. Senapati, M. I. Khan, R. Kumar and M. Sastry, Langmuir, 2003, 19, 3550–3553 CrossRef CAS
- E. Torres-Chavolla, R. J. Ranasinghe and E. C. Alocilja, IEEE Trans. Nanotechnol., 2010, 9, 533–538 CrossRef
- A. Bharde, A. Kulkarni, M. Rao, A. Prabhune and M. Sastry, J. Nanosci. Nanotechnol., 2007, 7, 4369–4377 CrossRef CAS PubMed
- A. Sinha and S. K. Khare, Bioresour. Technol., 2011, 102, 4281–4284 CrossRef CAS PubMed
- H. Bao, Z. Lu, X. Cui, Y. Qiao, J. Guo, J. M. Anderson and C. M. Li, Acta Biomater., 2010, 6, 3534–3541 CrossRef CAS PubMed
- P. Mukherjee, S. Senapati, D. Mandal, A. Ahmad, M. I. Khan, R. Kumar and M. Sastry, ChemBioChem, 2002, 3, 461–463 CrossRef CAS PubMed
- A. Ahmad, S. Senapati, M. I. Khan, R. Kumar, R. Ramani, V. Srinivas and M. Sastry, Nanotechnology, 2003, 14, 824–828 CrossRef CAS
- A. Zinchenko, Y. Miwa, L. I. Lopatina, V. G. Sergeyev and S. Murata, ACS Appl. Mater. Interfaces, 2014, 6, 3226–3232 CAS
- D. Majumdar, A. Singha, P. K. Mondal and S. Kundu, ACS Appl. Mater. Interfaces, 2013, 5, 7798–7807 CAS
- S. Kundu, Phys. Chem. Chem. Phys., 2013, 15, 14107–14119 RSC
- S. Kundu, V. Maheshwari and R. F. Saraf, Langmuir, 2008, 24, 551–555 CrossRef CAS PubMed
- S. Kundu and U. Nithiyanantham, Ind. Eng. Chem. Res., 2014, 53, 13667–13679 CrossRef CAS
- S. Anantharaj, U. Nithiyanantham, S. R. Ede and S. Kundu, Ind. Eng. Chem. Res., 2014, 53, 19228–19238 CrossRef CAS
- S. R. Ede, S. Anantharaj, U. Nithiyanantham and S. Kundu, Phys. Chem. Chem. Phys., 2015, 17, 5474–5484 RSC
- U. Nithiyanantham, S. R. Ede, S. Anantharaj and S. Kundu, Cryst. Growth Des., 2015, 15, 673–686 CAS
- S. R. Ede, A. Ramadoss, U. Nithiyanantham, S. Anantharaj and S. Kundu, Inorg. Chem., 2015, 54, 3851–3863 CrossRef CAS PubMed
- U. Nithiyanantham, S. R. Ede, T. Kesavan, P. Ragupathy, M. D. Mukadam, S. M. Yusuf and S. Kundu, RSC Adv., 2014, 4, 38169–38181 RSC
- S. Kundu, K. Wang, D. Huitink and H. Liang, Langmuir, 2009, 25, 10146–10152 CrossRef CAS PubMed
- S. Kundu, H. Lee and H. Liang, Inorg. Chem., 2009, 48, 121–127 CrossRef CAS PubMed
- S. Kundu and H. Liang, Langmuir, 2008, 24, 9668–9674 CrossRef CAS PubMed
- S. Kundu and M. Jayachandran, RSC Adv., 2013, 3, 16486–16498 RSC
- U. Nithiyanantham, S. R. Ede and S. Kundu, J. Mater. Chem. C, 2014, 2, 3782–3794 RSC
- S. R. Ede, A. Ramadoss, S. Anantharaj, U. Nithiyanantham and S. Kundu, Phys. Chem. Chem. Phys., 2014, 16, 21846–21859 RSC
- U. Nithiyanantham, A. Ramadoss, S. R. Ede and S. Kundu, Nanoscale, 2014, 6, 8010–8023 RSC
- F. C. Cabrera, H. Mohan, R. J. D. Santos, D. L. S. Agostini, R. F. Aroca, M. A. Rodríguez-Pérez and A. E. Job, J. Nanomater., 2013, 2013, 1–10 CrossRef
- J. K. Pokorski and N. F. Steinmetz, Mol. Pharm., 2011, 8, 29–43 CrossRef CAS PubMed
- J. Jena, N. Pradhan, B. P. Dash, P. K. Panda and B. K. Mishra, J. Saudi Chem. Soc., 2014, 1–6 Search PubMed
- S. Iravani, Green Chem., 2011, 13, 2638–2650 RSC
- M. S. Akhtar, J. Panwar and Y. Yun, ACS Sustainable Chem. Eng., 2013, 1, 591–602 CrossRef CAS
- S. S. Shankar, A. Rai, B. Ankamwar, A. Singh, A. Ahmad and M. Sastry, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2014, 3, 482–488 Search PubMed
- S. P. Chandran, M. Chaudhary, R. Pasricha, A. Ahmad and M. Sastry, Biotechnol. Prog., 2006, 22, 577–583 CrossRef CAS PubMed
- B. Ankamwar, M. Chaudhary and M. Sastry, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2005, 35, 19–26 CrossRef CAS
- B. Ankamwar, C. Damle, A. Ahmad and M. Sastry, J. Nanosci. Nanotechnol., 2005, 5, 1665–1671 CrossRef CAS PubMed
- F. Coccia, L. Tonucci, D. Bosco, M. Bressand and N. Alessandro, Green Chem., 2012, 14, 1073–1078 RSC
- M. Sathishkumar, K. Sneha, I. S. Kwak, J. Mao, S. J. Tripathy and Y. S. Yun, J. Hazard. Mater., 2009, 171, 400–404 CrossRef CAS PubMed
- S. S. Shankar, A. Rai, A. Ahmad and M. Sastry, J. Colloid Interface Sci., 2004, 275, 496–502 CrossRef CAS PubMed
- A. Leela and M. Vivekanandan, Afr. J. Biotechnol., 2008, 7, 3162–3165 Search PubMed
- S. Li, Y. Shen, A. Xie, X. Yu, L. Qiu, L. Zhang and Q. Zhang, Green Chem., 2007, 9, 852–858 RSC
- V. Vellora, T. Padil and M. Cernik, Int. J. Nanomed., 2013, 8, 889–898 Search PubMed
- S. Maensiri, P. Laokul, J. Klinkaewnarong, S. Phokha, V. Promarak and S. Seraphin, J. Optoelectron. Adv. Mater., 2008, 10, 161–165 Search PubMed
- J. Qu, C. Luo and J. Hou, Micro Nano Lett., 2011, 6, 174–176 CAS
- R. Herrera-Becerra, C. Zorrilla, J. L. Rius and J. A. Ascencio, Appl. Phys. A, 2008, 91, 241–246 CrossRef CAS
- S. Basu, S. K. Ghosh, S. Kundu, S. Panigrahi, S. Praharaj, S. Pande, S. Jana and T. Pal, J. Colloid Interface Sci., 2007, 313, 724–734 CrossRef CAS PubMed
- S. Kundu and U. Nithiyanantham, RSC Adv., 2013, 3, 25278–25290 RSC
- M. M. Kholoud, A. El-Nour, A. Eftaiha, A. Al-Warthan and R. A. A. Ammar, Arabian J. Chem., 2010, 3, 135–140 CrossRef
- C. Rigo, L. Ferroni, I. Tocco, M. Roman, I. Munivrana, C. Gardin, W. R. L. Cairns, V. Vindigni, B. Azzena, C. Barbante and B. Zavan, Int. J. Mol. Sci., 2013, 14, 4817–4840 CrossRef CAS
- V. Ambrogi, A. Donnadio, D. Pietrella, L. Latterini, F. A. Proietti, F. Marmottini, G. Padeletti, S. Kaciulis, S. Glovagnoli and M. Ricci, J. Mater. Chem. B, 2014, 2, 6054–6063 RSC
- M. Jeyaraj, G. Sathishkumar, G. Sivanandhan, D. MubarakAli, M. Rajesh, R. Arun, G. Kapildev, M. Manickavasagam, N. Thajuddin, K. Premkumar and A. Ganapathi, Colloids Surf., B, 2013, 106, 86–92 CrossRef CAS PubMed
- M. Fan, M. Thompson, M. L. Andrade and A. G. Brolo, Anal. Chem., 2010, 82, 6350–6352 CrossRef CAS PubMed
- S. Mukherjee, D. Chowdhury, R. Kotcherlakota, S. Patra, B. Vinothkumar, M. P. Bhadra, B. Sreedhar and C. R. Patra, Theranostics, 2014, 4, 316–335 CrossRef PubMed
- B. Wu, X. Wu, C. Guan, K. F. Tai, E. K. L. Yeow, H. J. Fan, N. Mathews and T. C. Sum, Nat. Commun., 2004, 4, 1–7 Search PubMed
- J. Park, M. H. Ullah, S. S. Park and C. Ha, J. Mater. Sci.: Mater. Electron., 2007, 18, 393–397 CrossRef
- K. G. Stamplecoskie and J. C. Scaiano, J. Phys. Chem. C, 2011, 115, 1403–1409 CAS
- M. Lungu, S. Gavriliu, E. Enescu, M. Lucaci, V. Tsakiris and G. Rimbu, Metall. Mater. Trans. A, 2012, 1–6 Search PubMed
- K. O. Santos, W. C. Elias, A. M. Signori, F. C. Giacomelli, H. Yang and J. B. Domingos, J. Phys. Chem. C, 2012, 116, 4594–4604 CAS
- A. K. Khan, R. Rashid, G. Murtaza and A. Zahra, Trop. J. Pharm. Res., 2014, 13, 1169–1177 CAS
- D. K. Chatterjee, P. Diagaradjane and S. Krishnan, Ther. Delivery, 2011, 2, 1001–1014 CrossRef CAS
- E. Ye, M. D. Regulacio, S. Y. Zhang, X. J. Loh and M. Y. Han, Chem. Soc. Rev., 2015, 44, 6001–6017 RSC
- M. K. K. Oo, X. Yang, H. Du and H. Wang, Nanomedicine, 2008, 3, 777–786 CrossRef CAS PubMed
- P. Pandey, S. P. Singh, S. K. Arya, V. Gupta, M. Datta, S. Singh and B. D. Malhotra, Langmuir, 2007, 23, 3333–3337 CrossRef CAS PubMed
- J. M. Pingarro, P. Y. Seden and A. G. Cortes, Electrochim. Acta, 2008, 53, 5848–5866 CrossRef
- Y. Du, L. Shi, M. Hong, H. Li, D. Li and M. Liu, Opt. Commun., 2013, 298–299, 232–236 CrossRef CAS
- W. Cai, T. Gao, H. Hong and J. Sun, Nanotechnol., Sci. Appl., 2008, 1, 17–32 CAS
- E. Lima, R. Guerra, V. Lara and A. Guzman, Chem. Cent. J., 2013, 7, 1–7 CrossRef PubMed
- C. C. D. Wang, W. C. H. Choy, C. Duan, D. D. S. Fung, W. E. I. Sha, F. X. Xie, F. Huang and Y. Cao, J. Mater. Chem., 2012, 22, 1206–1211 RSC
- R. Sharma, V. V. Agrawal, A. K. Srivastava, Govind, L. Nain, M. Imran, S. R. Kabi, R. K. Sinha and B. D. Malhotra, J. Mater. Chem. B, 2013, 1, 464–474 RSC
- N. Lee and T. Hyeon, Chem. Soc. Rev., 2012, 41, 2575–2589 RSC
- D. L. Huber, Small, 2015, 1, 482–501 CrossRef PubMed
- K. Shrivas, K. Agrawal and H. F. Wu, Analyst, 2011, 136, 2852–2857 RSC
- T. Lopez, F. Figueras, J. Manjarrez, J. Bustos, M. Alvarez, J. Silvestre Albero, F. Rodriguez Reinoso, A. Martinez Ferre and E. Martinez, Eur. J. Med. Chem., 2010, 45, 1982–1990 CrossRef CAS PubMed
- J. C. Claussen, A. Kumar, D. B. Jaroch, M. H. Khawaja, A. B. Hibbard, D. M. Porterfield and T. S. Fisher, Adv. Funct. Mater., 2012, 22, 3399–3405 CrossRef CAS
- S. Mostafa, F. Behafarid, J. R. Croy, L. K. Ono, L. Li, J. C. Yang, A. I. Frenkel and B. R. Cuenya, J. Am. Chem. Soc., 2010, 132, 15714–15719 CrossRef CAS PubMed
- K. Q. Peng, X. Wang, X. L. Wu and S. T. Lee, Nano Lett., 2009, 9, 3704–3709 CrossRef CAS PubMed
- H. Chen, G. Wei, A. Ispas, S. G. Hickey and A. Eychmuller, J. Phys. Chem. C, 2010, 114, 21976–21981 CAS
- L. Xu, X. C. Wu and J. J. Zhu, Nanotechnology, 2008, 19, 305603 CrossRef PubMed
- C. P. Adams, K. A. Walker, S. O. Obare and K. M. Docherty, PLoS One, 2014, 9, e85981 Search PubMed
- I. F. Ramos, B. C. Montes, M. M. García-Maldonado, C. L. Menendez, A. R. Mayol, L. M. Díaz-Vazquez and C. R. Cabrera, J. Chem. Educ., 2015, 92, 360–363 CrossRef PubMed
- S. H. Lim, J. Wei, J. Lin, Q. Li and J. K. You, Biosens. Bioelectron., 2005, 20, 2341–2346 CrossRef CAS PubMed
- S. Das, J. M. Dowding, K. E. Klump, J. F. McGinnis, W. Self and S. Seal, Nanomedicine, 2013, 8, 1483–1508 CrossRef CAS PubMed
- Y. Gao, K. Chen, J. Ma and F. Gao, OncoTargets Ther., 2014, 7, 835–840 CrossRef CAS PubMed
- M. S. Wason and J. Zhao, Am. J. Transl. Res., 2013, 5, 126–131 CAS
- P. R. Solanki, C. Dhand, A. Kaushik, A. A. Ansari, K. N. Sood and B. D. Malhotra, Sens. Actuators, B, 2009, 141, 551–556 CrossRef CAS
- V. Shah, S. Shah, H. Shah, F. J. Rispoli and K. T. McDonnell, Nanoparticles, 2012, 7, 1–13 Search PubMed
- G. D. Venkatasubbu, S. Ramasamy, V. Ramakrishnan and J. Kum, Adv. Powder Technol., 2013, 24, 947–954 CrossRef
- B. Jana, G. Mondal, A. Biswas, I. Chakraborty and S. Ghosh, RSC Adv., 2013, 3, 8215–8219 RSC
- A. Besinis, T. D. Peralta and R. D. Handy, Nanotoxicology, 2014, 8, 1–16 CrossRef CAS PubMed
- E. Casero, C. Alonso, M. D. Petit-Domínguez, L. Vazquez, A. M. Parra-Alfambra, P. Merino, S. Alvarez-Garcia, A. de Andres, E. Suarez, F. Pariente and E. Lorenzo, Microchim. Acta, 2014, 181, 79–87 CrossRef CAS
- M. A. Ibrahem, H. Y. Wei, M. H. Tsai, K. C. Ho, J. J. Shyue and C. W. Chu, Sol. Energy Mater. Sol. Cells, 2013, 108, 156–163 CrossRef CAS
- V. Jaskova, L. Hochmannova and J. Vytrasova, Int. J. Photoenergy, 2013, 2013, 795060 CrossRef
- X. Ren, D. Chen, X. Meng, F. Tang, X. Hou, D. Han and L. Zhang, J. Colloid Interface Sci., 2009, 334, 183–187 CrossRef CAS PubMed
- S. Sahoo, M. Maiti, A. Ganguly, J. J. George and A. K. Bhowmick, J. Appl. Polym. Sci., 2007, 105, 2407–2415 CrossRef CAS
- L. M. Rossi, N. J. S. Costa, F. P. Silva and R. Wojcieszak, Green Chem., 2014, 16, 2906–2933 RSC
- A. S. Teja and P. Y. Koh, Prog. Cryst. Growth Charact. Mater., 2009, 55, 22–45 CrossRef CAS
- L. Y. Wang, L. Wang, F. Gao, Z. Y. Yu and Z. M. Wu, Analyst, 2002, 127, 977–980 RSC
- J. Sun, Y. Zhu, X. Yang and C. Li, Particuology, 2009, 7, 347–352 CrossRef CAS
- H. Dhyani, C. Dhand, B. D. Malhotra and P. Sen, Biosens. Bioelectron., 2011, 3, 1–7 Search PubMed
- H. Cortinaa, C. M. Alonsoa, M. Castillo-Ortegab and H. Hu, Mater. Sci. Eng., B, 2012, 177, 1491–1496 CrossRef
- A. M. Suhail, M. J. Khalifa, N. M. Saeed and O. A. Ibrahim, Eur. Phys. J.: Appl. Phys., 2010, 49, 1–5 CrossRef
- I. Lokteva, N. Radychev, F. Witt, H. Borchert, J. Parisi and J. Kolny-Olesiak, J. Phys. Chem. C, 2010, 114, 12784–12791 CAS
- M. A. Schreuder, K. Xiao, I. N. Ivanov, S. M. Weiss and S. J. Rosenthal, Nano Lett., 2010, 10, 573–576 CrossRef CAS PubMed
- X. J. Loh, T. C. Lee, Q. Dou and G. R. Deen, Biomater. Sci., 2016 10.1039/c5bm00277j
- J. M. Perez, L. Josephson, T. O'Loughlin, D. Hogemann and R. Weissleder, Nat. Biotechnol., 2002, 20, 816–820 CrossRef CAS PubMed
- P. S. Doyle, J. Bibette, A. Bancaud and J. L. Viovy, Science, 2002, 295, 2237 CrossRef CAS PubMed
- T. J. Yoon, W. Lee, Y. S. Oh and J. K. Lee, New J. Chem., 2003, 27, 227–229 RSC
- H. T. Wong, M. K. Tsang, C. F. Chan, K. L. Wong, B. Feic and J. Hao, Nanoscale, 2013, 5, 3465–3473 RSC
- Q. Q. Dou, C. P. Teng, E. Ye and X. J. Loh, Int. J. Nanomed., 2015, 10, 419–432 CrossRef CAS PubMed
- N. M. Idris, M. K. G. Jayakumar, A. Bansal and Y. Zhang, Chem. Soc. Rev., 2015, 44, 1449–1478 RSC
- P. Ramasamy, P. Manivasakan and J. Kim, RSC Adv., 2014, 4, 34873–34895 RSC
- V. Kumar, G. Toffoli and F. Rizzolio, ACS Med. Chem. Lett., 2013, 4, 1012–1013 CrossRef CAS PubMed
- X. Tu, Y. Ma, Y. Cao and J. Huang, J. Mater. Chem. B, 2014, 2, 2184–2192 RSC
- Q. Dou, X. Fang, S. Jiang, P. L. Chee, T. C. Lee and X. J. Loh, RSC Adv., 2015, 5, 46817–46822 RSC
- J. Schuster, G. He, B. Mandlmeier, T. Yim, K. T. Lee, T. Bein and L. F. Nazar, Angew. Chem., Int. Ed., 2012, 51, 3591–3595 CrossRef CAS PubMed
- S. K. Bhunia, A. Saha, A. R. Maity, S. C. Ray and N. R. Jana, Sci. Rep., 2012, 3, 1–7 Search PubMed
- X. Zhao, L. Yang, X. Li, X. Jia, L. Liu, J. Zeng, J. Guo and P. Liu, Bioconjugate Chem., 2015, 26, 128–136 CrossRef CAS PubMed
- M. Das, C. Dhand, N. Dwivedi, B. P. Singh, G. Sumana, V. V. Agarwal, J. S. Tawale and B. D. Malhotra, Sens. Actuators, B, 2015, 210, 281–289 CrossRef
- C. I. C. Crucho, ChemMedChem, 2015, 10, 24–38 CrossRef CAS PubMed
- C. Dhand, M. P. Prabhakaran, R. W. Beuerman, R. Lakshminarayanan, N. Dwivedi and S. Ramakrishna, RSC Adv., 2014, 4, 32673–32689 RSC
- M. Elsabahy and K. L. Wooley, Chem. Soc. Rev., 2012, 41, 2545–2561 RSC
- K. Wang, X. Zhang, X. Zhang, B. Yang, Z. Li, Q. Zhang, Z. Huang and Y. Wei, J. Mater. Chem. C, 2015, 3, 1854–1860 RSC
- K. Lia and B. Liu, Chem. Soc. Rev., 2014, 43, 6570–6597 RSC
| This journal is © The Royal Society of Chemistry 2015 |