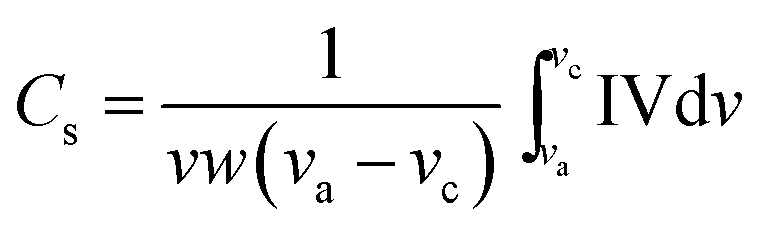DOI:
10.1039/C5RA20763K
(Paper)
RSC Adv., 2015,
5, 104991-105002
Rapid and large-scale synthesis of Co3O4 octahedron particles with very high catalytic activity, good supercapacitance and unique magnetic properties†
Received
7th October 2015
, Accepted 30th November 2015
First published on 1st December 2015
Abstract
The scarcity of rapid and large scale synthesis of functional materials hinders the progress from the laboratory scale to commercial applications. In this study, we report a rapid and large scale synthesis of micron size (1.3 μm) Co3O4 octahedron particles enclosed by (111) facets. The octahedron particles were composed of ±25 nm rectangular/cube shaped particles as seen from the TEM images. We have characterized and evaluated the catalytic, supercapacitance and magnetic properties of the as prepared material. The Co3O4 octahedron particles were highly active in heterogeneous PMS activation reaction. Formation of Co–OH bonding due to water molecule dissociation on the (111) surface of the particles was evident from the ELNEFS analysis. The as prepared octahedron materials showed >4 times higher pseudocapacitance properties (182 F g−1) with good capacity retention ability (up to the 1000 cycles studied) compared to commercial microcrystalline Co3O4 powder (43 F g−1). The material showed interesting magnetic properties at low temperature. The coexistence of superparamagnetic single domain and linear/quadratic behaviours was observed at low temperature for the as prepared Co3O4 octahedron particles.
1. Introduction
Advanced oxidation technologies (AOTs) for environmental remediation purposes are gathering attention these days. Widespread interest is based on their high theoretical conversion of toxic compounds to CO2 and H2O. The efficiency of the AOTs relies on the generation of strong oxidising species, e.g. hydroxyl radicals (˙OH, HRs), with standard reduction potential in the range of 1.8–2.7 V.1 Recently, sulphate radicals (SR) have demonstrated high reduction potentials of 2.5–3.1 V at neutral pH.1 Sulphate radicals are usually generated by both homogeneous and heterogeneous activation of peroxymonosulphate (PMS). Homogeneous catalysis is more efficient compared to heterogeneous catalysis due to the fact that every single catalytic entity can act as a unified active site.2 Transition metals such as Co(II), Ru(III), Fe(II), Ce(III), Mn(II), Fe(III), Ni(II) etc. have been used to activate PMS to generate SR in a homogeneous system, with Co(II) reported as the best activator for PMS.3 Pristine cobalt oxide (Co3O4), and Co3O4 supported on other metal oxide supports has been studied as potential heterogeneous catalyst for PMS activations for degradation of organic pollutants.4–11 However, absence of large scale synthesis of highly active Co3O4 catalysts restricts the application of Co3O4 in AOT applications.
The discovery of supercapacitance/supercapacitors in 1950s have sparked off a growing research interest in their design and testing. This is because supercapacitors are invaluable energy storage units in domestic and military applications. Supercapacitors are categorized into three major classes depending on charge storage mode, i.e. electric double layer based (EDLC), faradic pseudocapacitor and hybrid capacitor. Pseudocapacitors rely on reversible surface redox reactions to store charge.12 Supercapacitors based on faradic pseudocapacitance materials are particularly important due to their higher energy density and specific capacitance compared to carbonaceous materials. Co3O4 is considered to be one of the most attractive candidates among many other pseudocapacitive materials owing to its high theoretical specific capacitance values (up to 3560 F g−1), good capacity retention and high redox reactivity.13,14 However, slow kinetics of ion and electron transport in electrodes and at the electrode/electrolyte interface forces a compromise between the power performance and reversibility in pseudocapacitive materials.13 Extensive research has been conducted to synthesize materials with interesting architecture to improve redox kinetics in pseudocapacitive materials. Recently Co3O4 material with; hollow sphere (346 F g−1),15 nanowire array (754 F g−1),13 ultralayered highly porous (548 F g−1),16 nanotubes (574 F g−1),17 hollow boxes (278 F g−1),18 nanoflake array (351 F g−1)19 etc. have been used as pseudocapacitive materials. Spinel Co3O4 octahedron material enclosed by (111) facets has shown excellent electrochemical properties.20–23
Antiferromagnetic (AFM) materials are very interesting materials to study various quantum phenomena. Cobalt oxide, Co3O4 has been reported to be an ideal candidate for studying quantum phenomenon.24 Recently Co3O4 has been used in recording technology and medical science because of their interesting magnetic properties.25,26 Many recent reports have also discussed the application of Co3O4 nanomaterials as electrochemical and gas sensors.27–30
Various synthesis routes have been reported to produce Co3O4 nanoparticles of different shape and size to enhance catalytic, supercapacitance, and magnetic properties of the materials. Methods such as; hydrothermal, sol–gel, thermal decomposition, solvothermal decomposition have been reported.31,32 Process that favours shape control; i.e. hydro and solvothermal routes and liquid phase precipitation are considered the most powerful synthesis strategies.33 However, longer synthesis time and smaller product yield are the two main factors that increase the manufacturing cost of nanoparticles, which in turn restricts Co3O4 application as a heterogeneous catalyst, supercapacitance and/or magnetic material. Continuous hydrothermal flow synthesis (CHFS) allows rapid and large scale synthesis of nanoparticles. Synthesis of cubic shaped Co3O4 nanoparticles in CHFS system have been reported before.32 However, report on the synthesis of octahedron Co3O4 particles enclosed by (111) facets by CHFS technique is absent in literature to the very best of our knowledge. Large scale synthesis of octahedron particles enclosed by (111) facets is vital owing to their excellent catalytic and electrochemical activity. Moreover, report on the supercapacitance and catalytic properties of Co3O4 particles of any morphology synthesized by CHFS technique are scarce despite the dependence of nanomaterial properties on factors, such as size, shape and preparative route. Hence, evaluation of physical and chemical properties of Co3O4 octahedron particles synthesized by CHFS technique is important as it can allow the commercial scale production of Co3O4 materials and spark practical applications, e.g. heterogeneous catalysis; pseudocapacitive, magnetic material etc.
In this contribution, we synthesized Co3O4 octahedron particles enclosed by (111) facets in gram scale and evaluated its catalytic, supercapacitance as well as magnetic properties. CHFS technique was used to precipitate cobalt hydroxide octahedron material and subsequently annealed to transform into Co3O4 octahedron particles. The as prepared material exhibited very high PMS activation ability, good supercapacitance (∼4 times higher than commercial Co3O4 nanoparticles) and capacity retention ability and unique magnetic properties.
2. Experimental
2.1 Materials and methods for Co3O4 synthesis
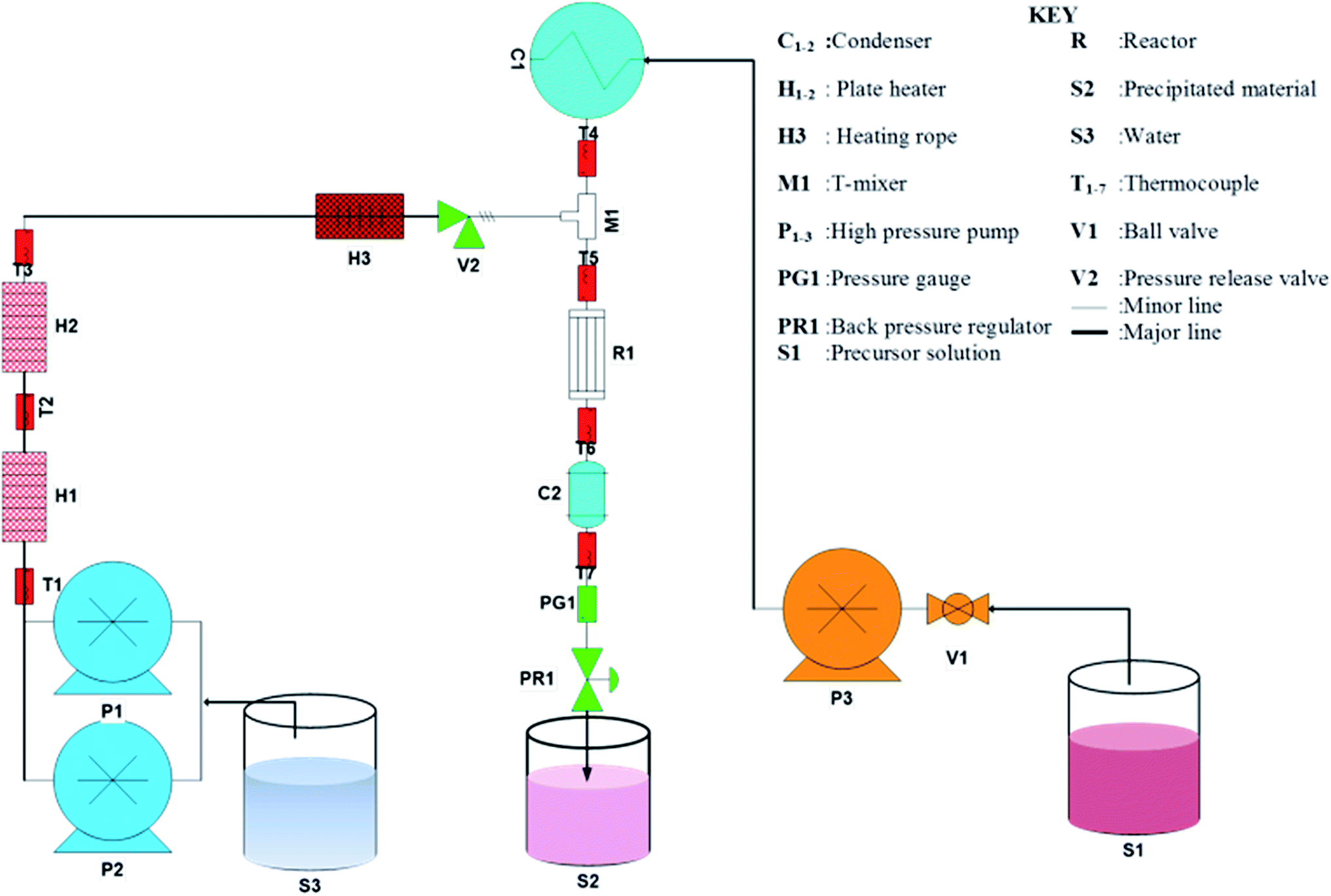
Analytical grade CoCl2 and urea were purchased from Sigma Aldrich South Africa and used without further purification. A schematic diagram of the CHFS process used to synthesis Co3O4 octahedron nanoparticles are presented in Fig. 1. A description of the CHFS system is reported in our previous publication elsewhere.34 Briefly, two Isco Teledyne 260D syringe pumps in series supplied deionised water (18 MΩ cm) through the two plate heater (1 kW) to the reactor. The reactor temperature was 280 °C. To prepare the precursor solution 15 g cobalt(II) chloride salt was dissolved in 250 mL of water. In a separate beaker, 0.75 g urea was dissolved in 250 mL water. The urea solution was introduced to the cobalt solution drop wise. Precursor volume can be changed according to the length of the run or the amount of product needed. The precursor solution at ambient temperature was pumped to the reactor by a Milton Roy metallic diaphragm dosing pump where it was mixed with the heated water. This resulted in rapid precipitation of pink, materials. The precipitated pink powders were collected at the exit of the back pressure regulator. The overall system pressure was controlled at 20 MPa. All the stainless steel 316 tubing and fittings used was provided by Swagelok. A typical sample of 1 litre nanoparticle laden slurry was collected after each run. The slurry was centrifuged, washed and dried in an oven at 60 °C and subsequently calcined at 300 °C. After calcining the pink powder transformed into black powder. This black powder was used only for characterization and evaluation of supercapacitance, magnetic and catalytic properties. Addition of urea was essential for the precipitation of the materials. In the absence of urea no precipitation occurred. The role of urea was to release OH− ions at elevated temperature and pressure in the reactor, which reacts with Co2+ to precipitate Co(OH)2. Decomposition of urea affords NH3 and CO2 followed by the release of OH−.35 For a typical run Co(OH)2 at a rate of 18 g h−1 on average could be achieved in this study.
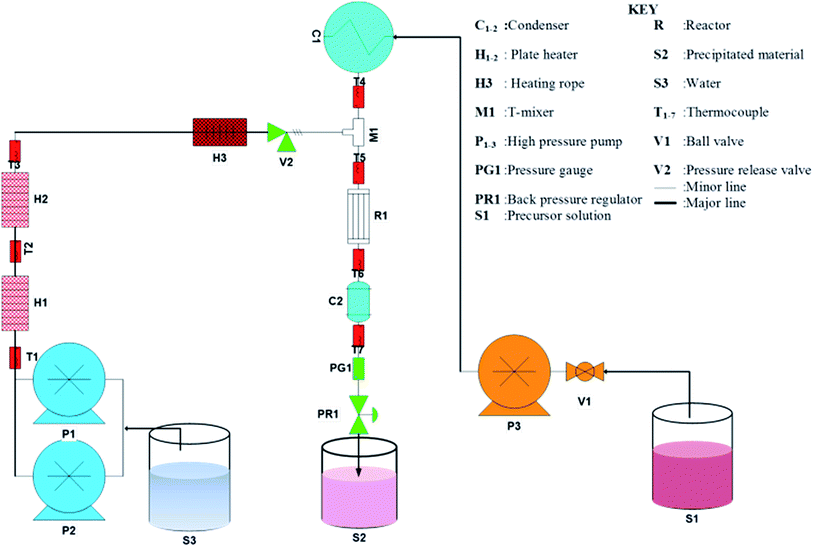 |
| | Fig. 1 Schematic of the CHFS system for the synthesis of Co3O4 material. | |
2.2 Electrode fabrications and supercapacitance testing
Electrochemical measurements were performed in a three-electrode cell configuration utilizing Co3O4 nanoparticles with carbon black and polyvinylidene difluoride (PVdF) coated on nickel foam current collector as the working electrode. Prior to use, the nickel foam was cleaned by heat treatment under Ar/H2 (95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 ratio) environment at a temperature of 1000 °C for about 30 minutes. A beaker-type three-electrode cell was fabricated utilizing Co3O4 nanoparticles with carbon black and polyvinylidene difluoride (PVdF) as the working electrode. Co3O4 nanoparticles (60 wt%), carbon black (30 wt%), and PVdF binder (10 wt%) were ground thoroughly and dispersed in N-methyl-2-pyrrolidinone (NMP) solvent to form a slurry. Then the slurry was spread onto a piece of nickel foam and maintained at 120 °C in a vacuum oven overnight. Platinum mesh and a Ag/AgCl were used as the counter electrode and the reference electrode, respectively. The electrochemical properties were evaluated in 2 M KOH electrolyte at room temperature by the cyclic voltammetry (CV) method. CV measurements were conducted over the voltage range from −0.25 to 0.55 V at various scan rates (5, 10, and 20 mV s−1). Galvanostatic charge–discharge cycling tests were performed in the same voltage range, with a current density of 0.5 A g−1 for 1000 cycles.
:5 ratio) environment at a temperature of 1000 °C for about 30 minutes. A beaker-type three-electrode cell was fabricated utilizing Co3O4 nanoparticles with carbon black and polyvinylidene difluoride (PVdF) as the working electrode. Co3O4 nanoparticles (60 wt%), carbon black (30 wt%), and PVdF binder (10 wt%) were ground thoroughly and dispersed in N-methyl-2-pyrrolidinone (NMP) solvent to form a slurry. Then the slurry was spread onto a piece of nickel foam and maintained at 120 °C in a vacuum oven overnight. Platinum mesh and a Ag/AgCl were used as the counter electrode and the reference electrode, respectively. The electrochemical properties were evaluated in 2 M KOH electrolyte at room temperature by the cyclic voltammetry (CV) method. CV measurements were conducted over the voltage range from −0.25 to 0.55 V at various scan rates (5, 10, and 20 mV s−1). Galvanostatic charge–discharge cycling tests were performed in the same voltage range, with a current density of 0.5 A g−1 for 1000 cycles.
2.3 Magnetic properties measurement
Magnetic properties were measured using two vibrating sample magnetometers: a MagLab from Oxford Instruments and an MPMS3 from Quantum Design. The first one operates using conventional electric pick-up coils, it has a sensitivity of 10 μemu and was used to measure hysteresis loops up to 9 T from 2.5 to 100 K. The second uses a superconducting quantum interference device (SQUID) to detect the magnetic signal, and it was also used to measure hysteresis loops up to 5 T, AC/DC susceptibility and magnetisation at temperatures from 2 to 350 K with a sensitivity better than 10−8 emu. Both instruments showed the same results, including a non-linear susceptibility term at low temperatures as described in the later section.
2.4 Evaluation of catalytic activity
Catalytic activity of the octahedral Co3O4 particles was conducted in a 200 mL glass vessel. Methyl orange (MO) was used as an organic probe to evaluate the catalytic activity of the material. In a typical reaction 0.03 g L−1 of the octahedron Co3O4 particles were dispersed in 40 mg L−1 of MO. The solution was stirred for 30 min to reach adsorption equilibrium. Commercial oxone can be used to generate peroxymonosulfate, PMS. Small amount of 0.9 mM of oxone was added to the solution. Sample was withdrawn at regular interval using a membrane syringe filter. Adsorption of dye on the membrane filter was negligible as was measured via control experiments. The MO concentration was measured using UV/vis technique at a wavelength of 464 nm. The solution pH was not controlled to minimise the number of processing parameters involved in the catalytic reaction. Intermediate products during catalytic degradation of MO were identified using Liquid chromatography mass spectroscopy (LCMS). An Agilent 6530 LCMS-TOF (Time of flight) running on mass hunter software, using a Symmetry C8 150 mm column preceded by a 2 mm guard column was used for intermediate identification. Elution was based on the gradient; 2 mM ammonium acetate (A): methanol (B); A 85% (t = 0 min): A 0% (t = 30 minutes) at a flow of 0.3 mL min−1. Ionization was by electron spray (ESI source temperature 350 °C) and ions were acquired in negative mode. Filtered samples (GHP Acrodisc filters) were analyzed without prior treatment.
2.5 Characterisations
X-ray diffraction patterns of the synthesized products were determined using a Phillips PW 3830/40 Generator with Cu-Kα radiation. The surface morphology and the electron energy loss spectroscopy (EELS) of the sample was studied using a FEI Tecnai F20 field emission gun TEM operated at 200 kV, equipped with a Gatan GIF-2001 energy filter. EELS was utilized to study the electron energy loss near edge fine structure (ELNEFS) signals of Co and Oxygen present within the specimen. Each spectrum was collected in normal parallel beam, bright-field TEM mode for 10 seconds. Infrared spectra were collected with a Perkin Elmer 1000 series FTIR spectrometer in the range of 4000–200 cm−1 in a KBr matrix. FTIR spectra were recorded using a Perkin Elmer spectrum 1000. Photoluminescence was measured using a Perkin Elmer LS55 luminescence spectrometer. Electrochemical properties were measured using a Biologic VMP3 analyser. Micromeritics Gemini VII 2390 surface area analyser was used to measure the BET surface area.
3. Results and discussion
3.1 Characterisation of the Co3O4 particles
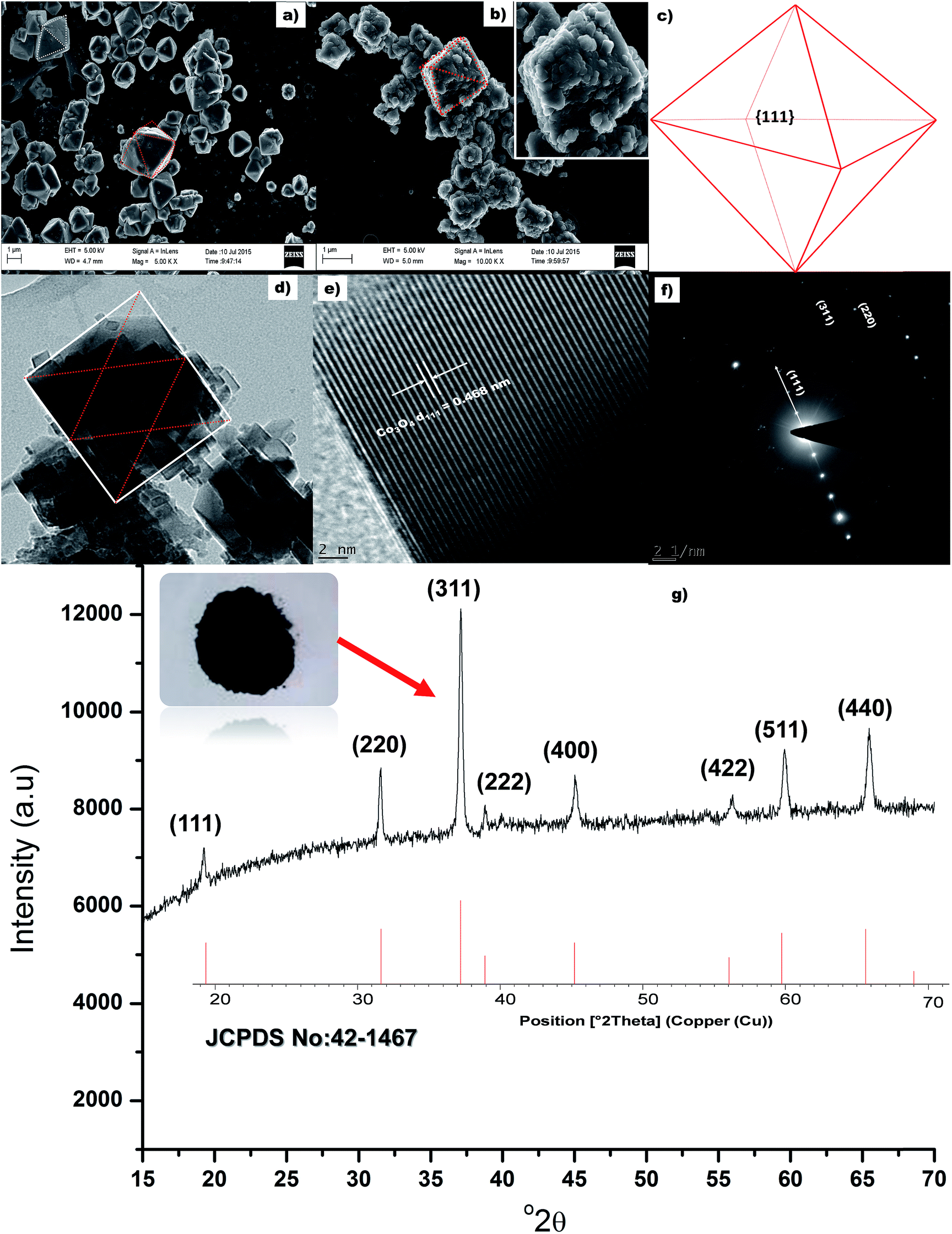
Fig. 2a presents the SEM micrograph of the uncalcined material. It is clearly visible from the SEM image that the uncalcined material possesses octahedron morphology. Fig. 2b presents the SEM image of the calcined (300 °C) sample. It can be seen from Fig. 2b that the morphology of the calcined, Co3O4, and uncalcined sample are similar (inset Fig. 2b), i.e. octahedron. The size of the calcined materials was found to be ±1.3 μm, similar size of a bacterium. All eight faces of the Co3O4 octahedron particles are enclosed with (111) faces as shown in Fig. 2c. The colour of the calcined material changed from pink (Co(OH)2) to black (Co3O4) indicating oxidation of Co(II) to Co(III) in the presence of atmospheric oxygen. TEM image of a single Co3O4 particle bounded by face normal projection of an octahedron (the rectangular red lines) is shown in Fig. 2d. Average size of the rectangular particles which acted as a building block (ESI, Fig. S-1†) for Co3O4 particles was measured to be ±25 nm. The d-spacing (Fig. 2e) was measured and found to be 0.468 nm corresponding to the (111) plane of Co3O4 material. The d-spacing of hexagonal (001) lattice plane of (Co(OH)2) is also 0.46 nm (JCPDS card no.: 30-0443). This closed orientation relationship between Co(OH)2 and Co3O4 has been determined previously as (the hexagonal (001) lattice plane parallel to the cubic (111) plane) the direct result of phase transformation of CO(OH)2 to Co3O4.36,37 Selected area electron diffraction (SAED) pattern of the cubic/rectangular shaped Co3O4 sample is shown in Fig. 2f. The SAED spot pattern represents a rectangular dot pattern which compliments the TEM results and is also indicative of the monocrystalline character of the material, with a Co3O4 d111 = 0.469 nm. X-ray diffraction pattern of the calcined material is presented in Fig. 2g. XRD pattern of the calcined material matches the diffraction pattern of pure spinel Co3O4 structure (JCPDS card no.: 42-1467). No impurity peaks or Co(OH)2 peaks were observed in the diffraction pattern. This indicates complete transformation of Co(OH)2 to Co3O4. The growth rate ratio, R, along the (001) and (111) directions are responsible for controlling the shape of a face centred crystal.38 Xiao and co-authors reported that enhanced growth along the (001) direction due to the presence of high NaOH was responsible for Co3O4 octahedron particle formation.21 Hence, in our case the release of OH− from urea successfully enhanced the growth rate along the (001) direction for the formation of octahedron particles. The BET surface area of the octahedron particles was measured to be 12 m2 g−1.
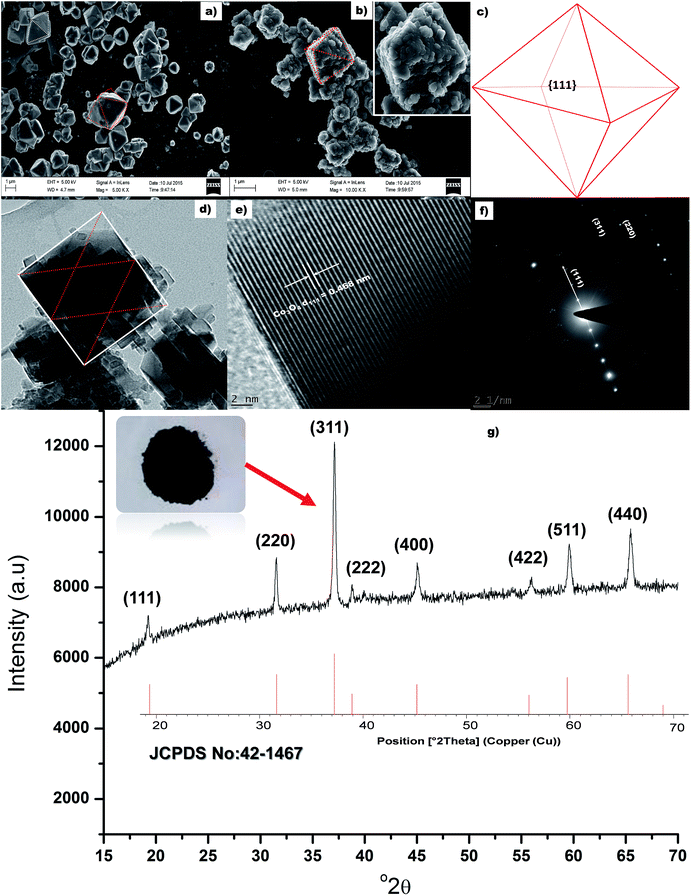 |
| | Fig. 2 (a & b) SEM micrograph of the uncalcined and calcined sample respectively, (c) schematic of the octahedral Co3O4 particles, (d) TEM image of a single Co3O4 particle, (e) lattice fringe of the rectangular particle, (f) SAED of the Co3O4 particles and (g) XRD of the Co3O4 octahedron particles. | |
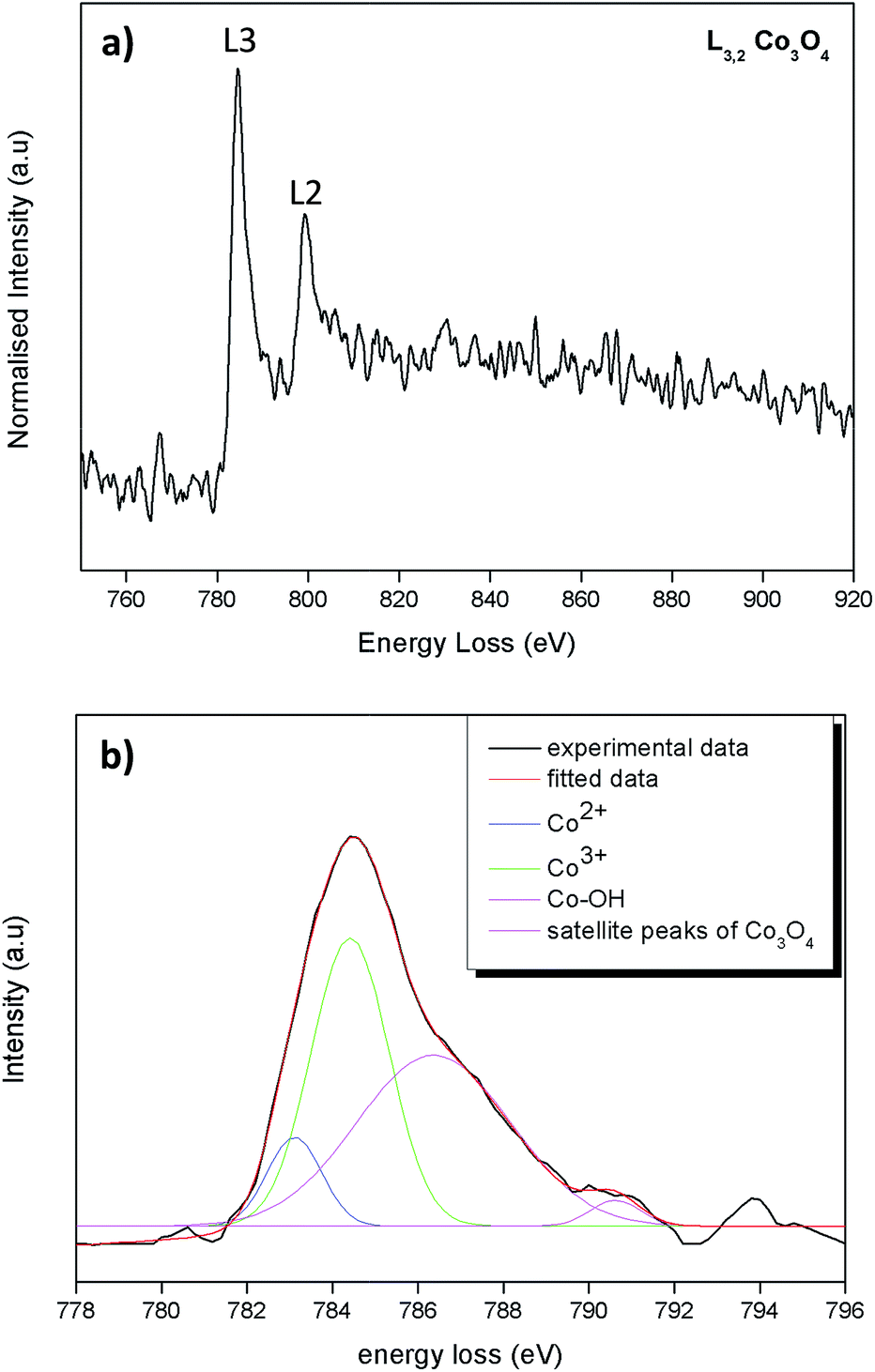
The ratio between Co2+ and Co3+ on the predominantly exposed planes of Co3O4 plays a role on its catalytic property.39 For catalytic oxidation reactions Co3+ is the more active species than the Co2+.40 The Co valence states of the as synthesized octahedral particles were determined by energy loss near edge fine structures (ELNEFS) analysis. ELNEFS of the cobalt L2,3-edge at 779 eV is sensitive to the chemical environment of the respective elements, thus offers a valuable means for selective identification of Co valence states. From Fig. 3a an average L3/L2 ratio of 2.7 was calculated for the synthesized Co3O4 octahedron particles. The L3/L2 ratio of 2.7 for the Co3O4 particles is indicative of a 0.33Co2+ + 0.67Co3+ configuration present within the structure.41 To further investigate the L2,3 line-shapes of the Co3O4 samples, the L3 peak of the sample was deconvoluted and is presented in Fig. 3b. The line-shape was filtered with a Savitzky–Golay algorithm and the background was subtracted using a simple least squares model to get accurate quantitative results from the deconvolution procedure. It can be observed from Fig. 3b that the L3 line-shape comprises of peaks of Co2+ and Co3+ also of the surface hydroxyl, Co–OH bond. The hydroxylated Co–OH peaks forms because hydroxyl groups are ubiquitous in Co3O4 because of air42,43 and chemisorbed water. From the deconvoluted data of Fig. 3b, a ratio 1:2.03 for Co2+:Co3+ was obtained from the integrated areas under peaks. This is in good agreement with the L3/L2 line-shape ratios of Co3O4 material.
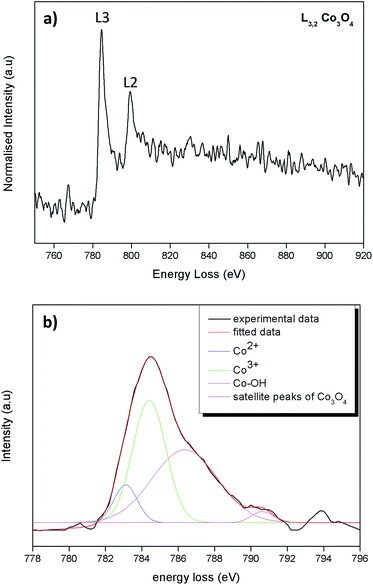 |
| | Fig. 3 (a) ELNEFS spectra comparing the cobalt – L2,3 edges of the Co3O4 octahedral particles & (b) deconvolution of the L3 line-shape of the Co3O4 octahedral particles. | |
3.2 Evaluation of catalytic properties
Catalytic activity of the Co3O4 octahedron particles are presented in Fig. 4. Control experiments were conducted to distinguish between catalytic activity, adsorption and PMS self-oxidation. It can be seen from Fig. 4a that the degradation of MO due to adsorption is negligible. Only 2% MO was removed due to adsorption process. After 33 minute of PMS self-oxidation, 81% of MO was degraded, implying that absence of catalyst was not effective to produce enough reactive species for significant MO degradation. In the presence of the synthesized Co3O4 octahedron particles and 0.9 mM oxone, 100% of MO was degraded just after 2 minutes of reaction. This fast degradation of MO highlights the very good catalytic PMS activation property of the synthesized Co3O4 octahedron particles. Catalyst reproducibility and catalytic activity repeatability is an issue in catalyst science. In this study the Co3O4 octahedron materials were produced five times in different batches and tested for their catalytic activity. It can be seen from Fig. 4a that the catalysts show excellent reproducibility and catalytic activity repeatability (data for three repeated experiments are presented in Fig. 4a). The MO degradation followed a first order reaction kinetics (R2 = 0.93). The MO degradation rate constant for catalysis is significantly higher than the PMS self-oxidation (Fig. 4b). From the environmental point of view, conversion of any organic pollutant, as MO, is only the first step in the ultimate objective, i.e. to attain the mineralization of the corresponding solutions. It can be seen from Fig. 4c that 68% of COD was removed after 60 minutes of catalytic reaction compared to 32% of PMS self-oxidation. This highlights the potential of the as prepared material in practical waste water treatment application. The role of PMS concentration on the Co3O4 catalytic activity was evaluated in the concentration range of 0.5–2.3 mM (Fig. 4d). It can be seen from Fig. 4d that the reaction rate constant increases with increasing PMS concentration from 0.5 to 1.14 mM. A catalyst should be active with minimum external input. Hence 0.9 mM oxone concentration was used for all experiments in this study. The reaction rate constant decreases with further increase of PMS concentration to 2.3 mM. It is possible that a higher oxone concentration has a radical quenching effect. Table 1 presents a comparison of catalytic activity between the as prepared material and the literature. The as prepared pristine material exhibits a very high reaction rate constant for 0.12 mM MO degradation at very low [PMS] and catalyst load compared to the literature.
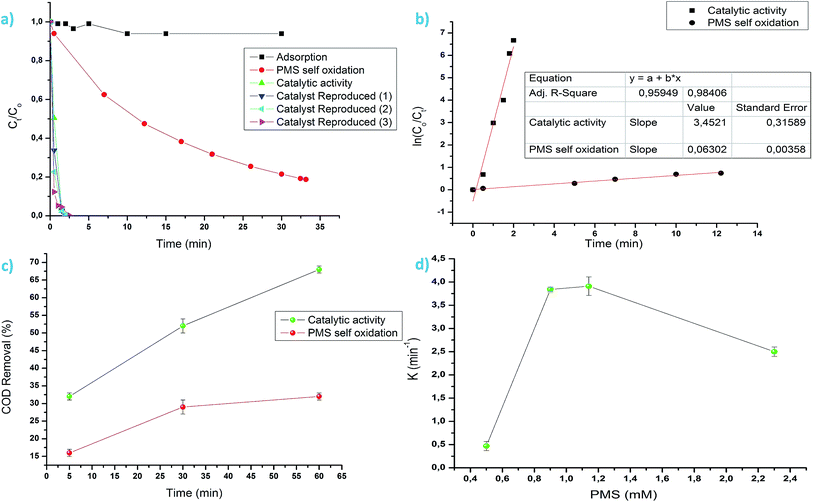 |
| | Fig. 4 (a) MO degradation via various route, (b) MO degradation kinetics, (c) COD removal via catalysis and PMS self-oxidation and (d) role of [PMS] on reaction rate constants. | |
Table 1 Comparison of reaction rate constant between literature and this studya
| Ref. |
Catalyst |
Organic compound |
PMS [mM] |
Catalyst load (g L−1) |
Reaction rate constant (K, min−1) |
| In all cases, it was reported that the composite material showed higher catalytic activity and lower Co leaching compared to the pristine material. |
| 5 |
Co3O4 |
Acid orange 7 (0.2 mM) |
2 |
0.5 |
0.19 |
| 11 |
Co3O4/MgO |
Methyl blue (0.05 mM) |
0.5 |
0.05 |
1.08 |
| 43 |
Co3O4/GO |
Acid orange 7 (0.6 mM) |
6 |
0.05 |
0.97 |
| 44 |
Co3O4 |
Phenol (0.2 mM) |
6.5 |
0.2 |
0.039 |
| 45 |
CoCl2·6H2O |
2,4-Dichlorophenol (0.307 mM) |
6 |
0.07 |
0.07 |
| 46 |
Co3O4/GO |
Phenol (0.2 mM) |
6.5 |
0.06 |
0.10 |
| 47 |
Co3O4/titanate |
Methyl orange (0.03 mM) |
0.5 |
0.5 |
0.29 |
| 48 |
Co3O4/TiO2 |
2,4-Dichlorophenol (0.307 mM) |
0.92 |
0.1 |
0.01 |
| 49 |
Commercial Co3O4 |
2,4-Dichlorophenol (0.12 mM) |
2.67 |
0.01 |
0.4 |
| This study |
Co3O4 |
Methyl orange (0.12 mM) |
0.9 |
0.15 |
3.45 |
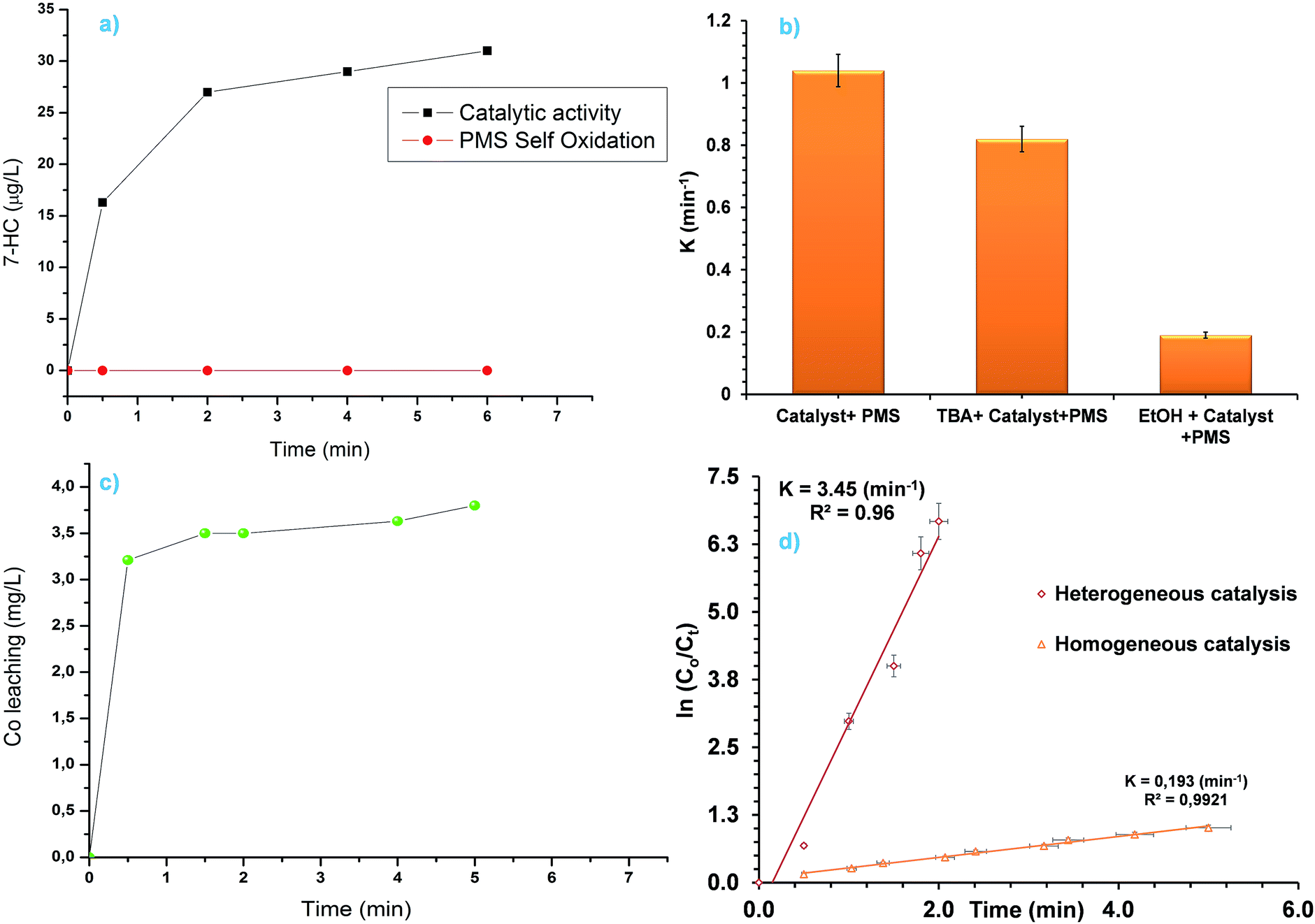
Generation of sulphate radicals (SR) by catalytic decomposition of PMS in homogeneous and/or heterogeneous system in the presence of Co3O4 was considered to be the primary reactive species for oxidation of organic pollutant. Wang and co-worker44 used electron paramagnetic resonance (EPR) technique to show that the generation of OH˙ also takes place in Co3O4 mediated catalytic activation of PMS. However, measurement of OH˙ using EPR technique is costly. In this study, we used a fluorescence based technique to confirm and show that photoluminescence (PL) based technique can also be used to verify the generation of OH˙ in catalytic PMS activation. Coumarin (COU) was used (10 mM) as a fluorescence probe. COU reacts with OH˙ to convert into highly luminescent 7-hydroxycoumarin (7-HC). The concentration of 7-HC is proportional to the OH˙ concentration. Though this method has been used previously to measure OH˙ in photocatalysis50,51 and catalytic ozonation,52 it has never been used in catalytic decomposition of PMS where SR and OH˙ are present simultaneously. It can be seen from Fig. 5a that in the absence of Co3O4 no 7-HC was detected highlighting that PMS self-oxidation doesn't generate OH˙. In the case of Co3O4 catalysed PMS activation reaction significant amount of 7-HC was detected. Hence, the Co3O4 octahedral particle catalysed PMS activation reaction not only produce SR but also OH˙. Moreover, COU based fluorescence method can also be used to measure OH˙ in catalytic PMS activation reaction. Equation showing formation of OH˙ from Co3O4 catalysed PMS activation reaction is given below:44
| | |
Co(II) + HSO5− → Co(III) + SO42− + ˙OH
| (1) |
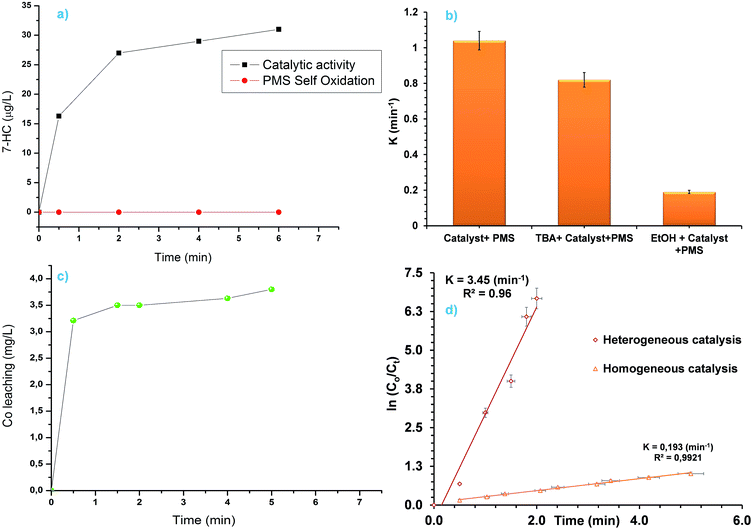 |
| | Fig. 5 (a) Generation of 7-HC as a reaction between COU and OH˙, (b) effect of quenching reagents on the MO degradation reaction rate constants, (c) Co leaching during PMS activation reaction and (d) comparison between homogeneous and heterogeneous PMS activation. | |
Ethanol (EtOH) and tert-butanol (TBA) was added as a SR and OH˙ scavenger respectively. It can be seen from Fig. 5b that the addition of TBA did not decrease the reaction rate constant significantly. However, addition of EtOH decreased the rate constant considerably due to the quenching of generated SR. So, in Co3O4 catalysed PMS activation reaction, SR is the dominant reactive species produced. Leaching of Co from unsupported pristine Co3O4 is a common phenomenon in Co3O4 catalysed PMS activation reaction. Any leached Co from the solution can be extracted using classical resin column or ion exchange process before discharge if required. The amount of leached cobalt in the solution was found to be 3.8 ppm (Fig. 5c) in this study. Homogeneous activation of PMS for MO degradation was studied, using concentration higher (8 mg L−1 was studied compared to 3.8 mg L−1 leached in solution) than the leached Co amount to evaluate the effect of homogeneous and heterogeneous catalysis. It can be seen from Fig. 5d that the heterogeneous catalysis is more effective than the homogeneous catalysis. This highlights the excellent catalytic properties of the as prepared octahedron material. From the TEM analysis it was shown earlier that the octahedron particles are enclosed by (111) facets. Zasada and co-workers showed previously that water molecules dissociates when adsorbed on Co3O4 (111) surfaces and forms Co–OH groups, where as molecular adsorption is favoured on 〈110〉 surfaces.53 Evidence of Co–OH bonding was found from the ELNEFS analysis of the octahedron particles as it was discussed in earlier section (Fig. 3b). Formation of surface hydroxyl groups or Co–OH bonds are critical for the catalytic activation of PMS, as the radical generation is dependent on the CoOH+ formation as shown in the eqn (2)–(4).43
| | |
Co2+ + H2O ↔ CoOH+ + H+
| (2) |
| | |
CoOH+ + HSO5− → CoO+ + SO4−˙ + H2O
| (3) |
| | |
CoO+ + 2H+ → Co3+ + H2O
| (4) |
The effect of reaction temperature was investigated to calculate the activation energy. Reactions were carried at 20, 35 and 40 °C (data not shown). Using classical Arrhenius equation the activation energy was found to be 49 kJ mol−1.
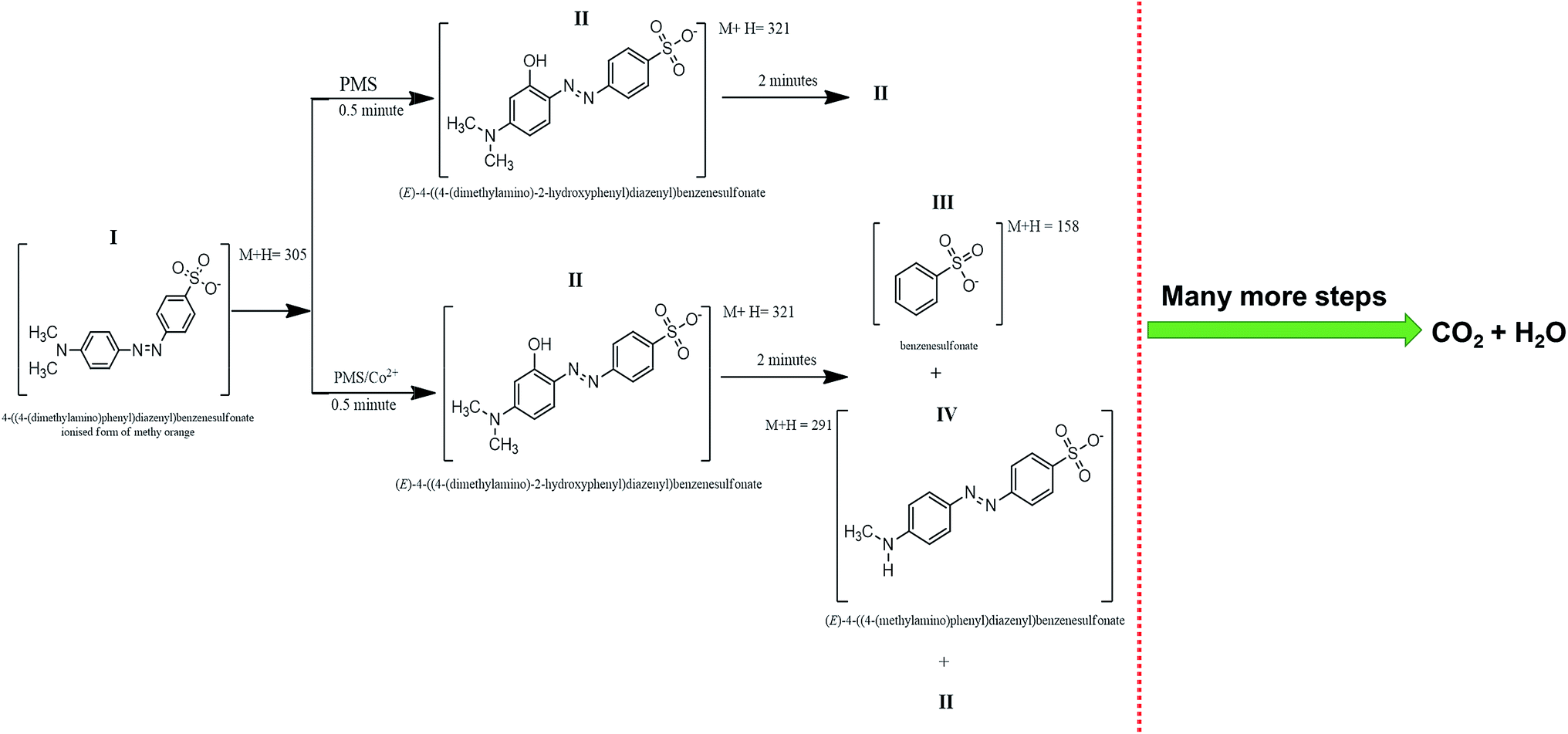
Intermediate compounds were sought based on their molecular features and masses were assigned formula based on algorithm restriction C ≤ 20, H ≤ 20, N ≤ 5, O ≤ 10 and S ≤ 2. Allowances were therefore given to accommodate intermediates formed from reaction of MO with SO4˙−, OH˙ and CH3˙. Fig. 6 represents a time based degradation scheme for MO as followed by LCMS using PMS alone and the Co3O4 catalysed breakdown of PMS respectively. At t = 0.5 minutes, intermediate identified in both systems (PMS self-oxidation and Co3O4/PMS system) had an M + H = 321 (C14H15N3O4S) and is depicted by structure (II) (structure I and II is given in Fig. 6). This is based on the compelling argument presented by Baiocchi and co-workers.54 This suggests that mono-hydroxylation of MO was the starting mechanism leading to its subsequent breakdown. It also supports our experimental results from PL study that activation of PMS also produce OH˙ (Fig. 5a). This compound has also been confirmed as catalytic breakdown product of MO degradation in earlier research.54–56 Chen et al.,56 had reported that the mono-hydroxylation could occur on any of five positions on the MO molecule. However, only one compound with M + H = 321 was observed in this study. After 2 minutes of oxidation, there is a marked difference in (ESI (Table S1†)), the intermediates identified by LCMS for both reaction systems. Two new intermediates were identified: compound III (M + H = 158) and compound IV (M + H = 291) (Fig. 6). Compound III is the product of cleavage of the azo link to the benzenesulfonate group, while compound IV is the product of cleavage of one of the methyl groups on MO. Compound III and IV were not detected in all LCMS runs for the PMS self-oxidation system and have been identified as catalytic cleavage products of MO in earlier studies.54–56 This highlights the fact that the PMS self oxidation and catalytic PMS activation might follow a separate reaction path way. The cleavage compounds III and IV may well possibly be why the cobaltous route presented better degradation rates compared to the PMS route. Further time based intermediates observed during PMS and Co3O4/PMS degradation are summarized in ESI (Table S1†). Compound III and IV were not observed after 5 minutes of reaction for both reaction systems. The remaining signals (Table S1†) M + H = 282, M + H = 332 and M + H 382 could not be simply assigned to any simultaneous introduction and/or detachment of any OH, CH2, CH3, SO2, SO3 groups and may probably be compounds formed from rearrangement reactions or reaction of specific transient intermediates. Compound M + H = 332 (m/z = 321) has also been previously identified, however, no probable structure for the compound was provided.56 The absence of assigned formula for several compounds in Table S1† highlights the point that a search based on the molecular feature of the parent compound is inadequate in identifying products of this type of reaction; a limitation based on the understanding of the reaction process itself and would require further in depth study of the reaction mechanism. Chromatogram at different time interval is given in Fig. S2.†
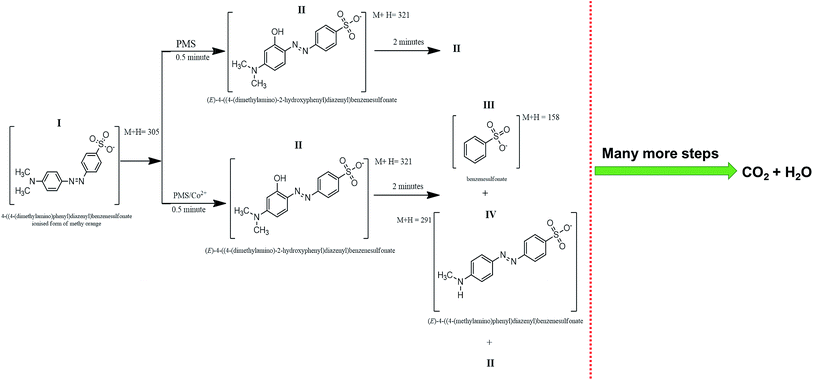 |
| | Fig. 6 Time based degradation products observed within the first 2 minutes of reaction. | |
In addition, to the obvious environmental benefits, good heterogeneous PMS activation ability of the Co3O4 octahedron particles discussed here synthesized using a reproducible rapid and large scale synthesis technique can be considered as an advancement of the catalysis field of research.
3.3 Supercapacitance properties of the material
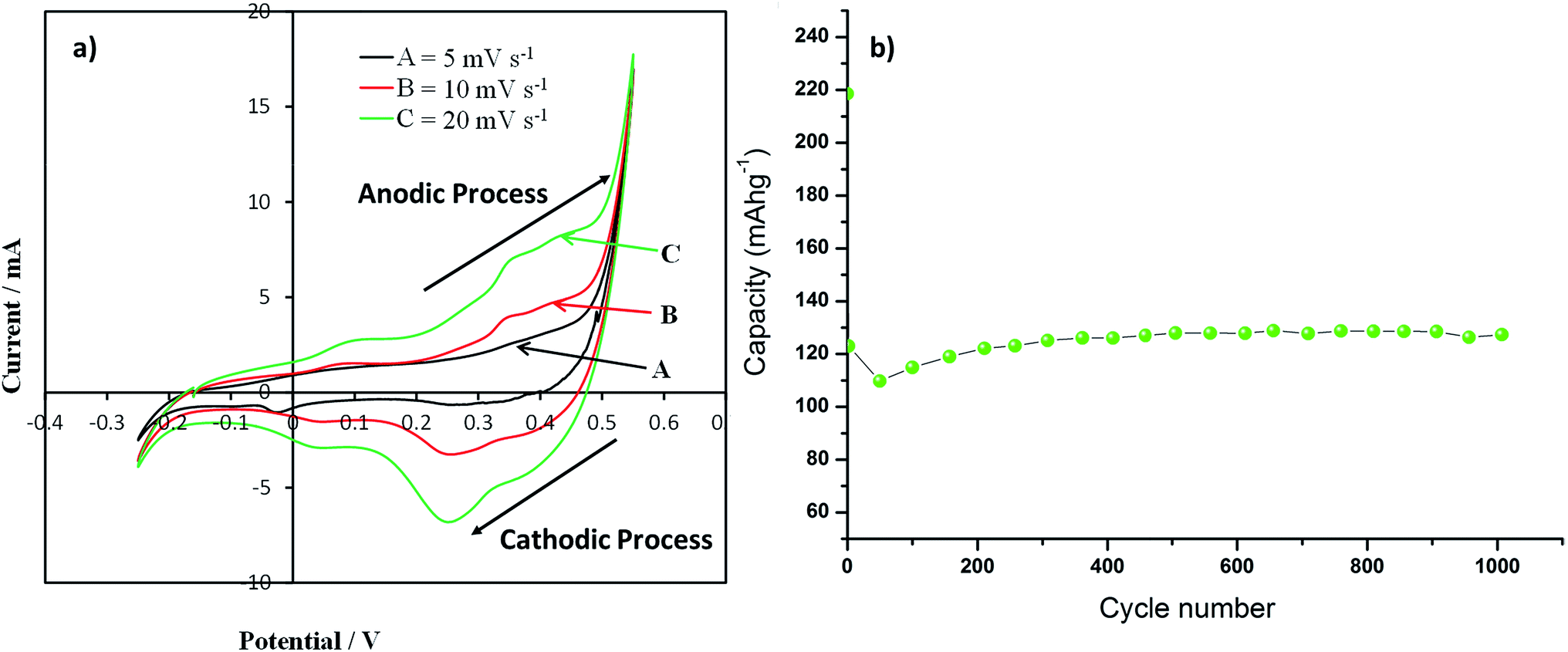
Cyclic voltammograms recorded for the Co3O4 nanoparticles supercapacitance electrode at a potential sweep rates of 5, 10 and 20 mV s−1are presented in Fig. 7a. An increase in scan rate results in increased peak current was observed in CV profile. All the CV profile exhibits asymmetry of the CV scans which essentially indicate faradic reactions and a consequence of ohmic resistance as a result of electrolyte diffusion within the electrode, which are the typical characteristics of faradaic pseudocapacitance behaviour. The anodic peak at 0.37 V and cathodic peak at 0.267 V in the CV curves are due to the oxidation and reduction processes, respectively. The redox reactions which are responsible for pseudocapacitance of Co3O4 in the alkaline solutions are given by eqn (5) and (6).| | |
CoOOH + OH− ↔ CoO2 + H2O + e−
| (5) |
| | |
Co3O4 + H2O + OH− ↔ 3CoOOH + e−
| (6) |
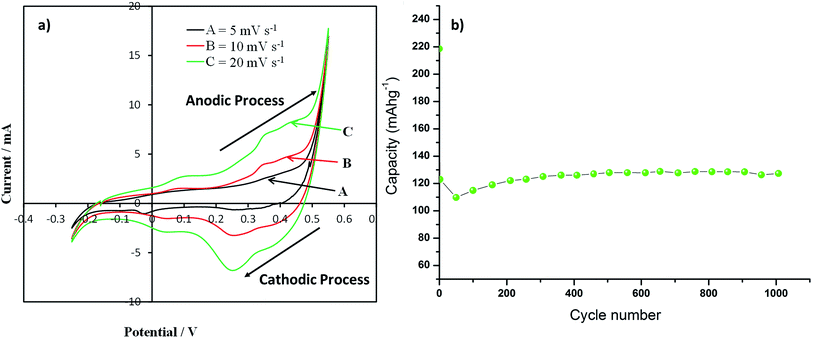 |
| | Fig. 7 (a) Cyclic voltammograms (CVs) of as-synthesized Co3O4 particle electrode films & (b) discharge capacity of as-synthesized Co3O4 particle electrode films at a current density of 0.5 A g−1 up to 1000 cycle [potentials are presented as V vs. Ag/AgCl]. | |
The specific capacitance (Cs) of Co3O4 was calculated using the CV curves shown in Fig. 7, using the equation below:
| |
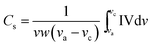 | (7) |
where,
v is sweep rate (V s
−1),
w is the mass of the Co
3O
4 and (
va −
vc) is the potential window. The maximum specific capacitance of 182 F g
−1 was obtained at a 5 mV s
−1 scan rate in 2 M KOH solution. Our result is higher as compared with reported commercial Co
3O
4 powders giving a maximum specific capacitance value of 43 F g
−1 as reported by Wang and co-workers.
31 The applicability of the supercapacitor can also be evaluated by means of the galvanostatic charge–discharge studies. Typical charge–discharge curves are presented in Fig. S3 (ESI
†). The supercapacitive performance properties of Co
3O
4 nanoparticles sample were evidenced by the galvanostatic charge–discharge experiments, as shown in
Fig. 7b. We have examined the charge–discharge cycles of the sample at 0.5 A g
−1 current density for 1000 cycles with absolute operating voltage windows in agreement with the CV data. It is noted that the specific capacitance of the Co
3O
4 nanoparticles decreases in the first 50 cycles and then starts to increase. In the first 50 cycles the capacity of the sample decreases by 50%, then the capacity of the sample starts to increase. The sample retains higher capacity after 1000 cycles (128 mA h g
−1) than its 2
nd cycle capacity (123 mA h g
−1). This highlights the long term stability and potential application of the Co
3O
4 materials as supercapacitor material. A possible reason for the decrease in capacity during the first 50 cycles may be the incomplete activation of all the reversible redox couples (Co
2+/Co
3+) prior to the second cycle after which their activation yields and increased capacitance. Pseudocapacitors rely on their reversible surface redox reactions to store charge. The sample starts to deliver a continuously increasing capacity upon repetitive cycling for remaining 1000 cycles as more redox couples were being activated. A comparison of the different Co
3O
4 synthesis technique used and subsequent supercapacitance properties reported between the literature and this study is presented in
Table 2. The advantage of rapid and large scale synthesis of the as prepared Co
3O
4 octahedron particles outweighs the loss in specific capacitance when compared to other reported Co
3O
4 material in literature. The reported synthesis technique will allow the commercial scale production of Co
3O
4 material with good specific capacitance and capacity retention ability for supercapacitor application, which is absent in the other reported method in literature. It is worthy to mention that the reported Co
3O
4 material shows ∼4 times higher specific capacitance than the commercially available Co
3O
4 powder (43 F g
−1).
31
Table 2 Comparison of synthesis technique used and reported specific capacitance of Co3O4 between the literature and this study
| Ref. |
Morphology |
Synthesis time (h) |
Technique |
Quantity produced |
Calcination temperature (°C) |
Specific capacitance (F g−1) |
Reported stability (cycle) |
| 13 |
Nanowire array |
9 h |
Hydrothermal (120 °C) |
Small |
350 (2 h) |
754 |
4000 |
| 15 |
Hollow sphere on reduced graphene oxide |
24 h |
Hydrothermal (180 °C) |
Small |
n/a |
346 |
50 |
| 16 |
Ultralayered |
>24 h |
Hydrothermal (120 °C) |
Small |
300 (not reported) |
548 |
4000 |
| 17 |
Nanotube |
5 h |
Templated growth |
Small |
500 (4 h) |
574 |
1000 |
| 18 |
Hollow boxes |
Days |
Soft chemistry |
Small |
300 (10 min) |
278 |
500 |
| 19 |
Nanoflake array |
5 h |
Electrpdeposition |
Small |
250 (2 h) |
289 |
4000 |
| 31 |
Nanorods |
6 |
Hydrothermal (105 °C) |
Small |
300 (for 3 h) |
281 |
Not reported |
| 37 |
Porous hierarchical morphologies |
1 |
Soft chemistry |
Small |
200 (for 1 h) |
185 |
>1000 |
| 57 |
Hollow octahedra |
>4 h |
Soft chemistry + hydrothermal (200 °C) |
Small |
n/a |
192 |
1200 |
| This study |
Octahedra |
Instantaneous |
Continuous hydrothermal flow (280 °C) |
Large scale production |
300 (3 h) |
181 |
1000 |
3.4 Evaluation of magnetic properties
The magnetisation versus temperature (M vs. T) of our samples is very similar to that reported previously for particles synthesized by a hydrothermal method,31 with a magnetisation peak at 35 K (Fig. 8a). At high temperatures, the samples show a large linear susceptibility and a (T + θ)−1 dependence of the magnetisation with θ ∼ 110 K. However, interestingly below 35 K, in addition to the linear susceptibility there is a remarkable quadratic dependence of the magnetisation with the applied field, M α H2. This has previously been observed in antiferromagnetic nanoparticles below the Néel temperature58 and has been associated to multiferroic systems with weak magnetisation.59,60 However, quadratic dependence of the magnetisation with the applied field is scarcely reported for pristine Co3O4 material. The quadratic component is anti-correlated to the linear susceptibility and it has a maximum at 10–15 K (Fig. 8b). If the linear and quadratic components of the susceptibility are subtracted, the particles show a typical superparamagnetic behaviour, with a sigma-shaped loop at 300 K and a ferromagnetic characteristic at low temperatures (see Fig. 8c and d). The ferromagnetic loop shows a different initial susceptibility, which could be attributed to a training effect seen in coexistent antiferromagnetic–ferromagnetic hybrids. The coexistent superparamagnetic single domain and linear/quadratic behaviours could correspond to particles of different size/composition, or to different magnetic properties in the core and the surface of the nanoparticle. It was shown from the TEM images that the octahedral particles are composed of nano size rectangular/cube shaped particles. Given the magnetic moment at 2 K after polynomial correction (150 μemu in a 3 mm3 sample), the single domain phase can be roughly estimated at just ∼0.1% of the total.
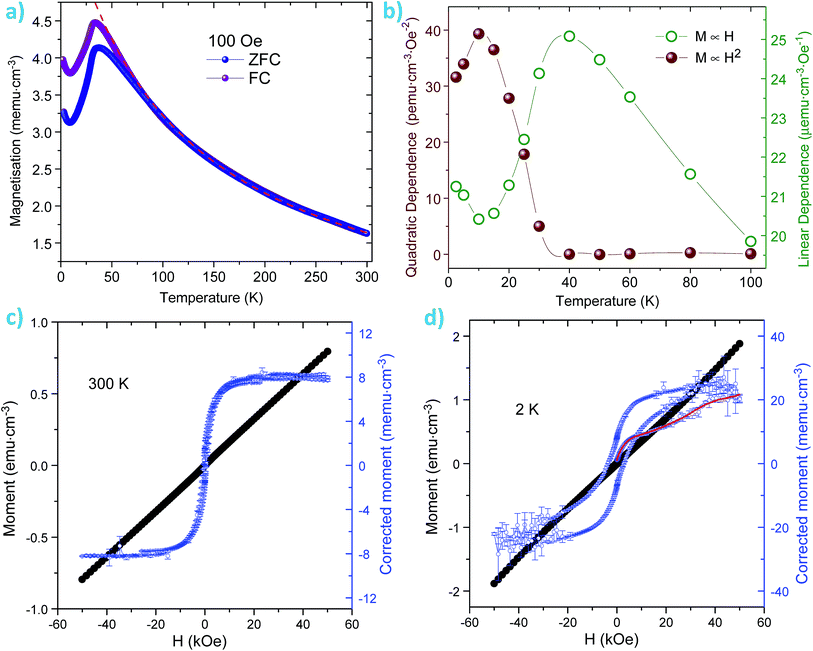 |
| | Fig. 8 (a) Zero field cooled–field cooled (ZFC–FC) magnetisation measured at 100 Oe. The dashed red line is a high temperature fit to a (T + θ)−1 dependence with θ ≈ 110 K. (b) Variation with temperature of the linear and quadratic dependence of the susceptibility with the applied field as calculated from hysteresis loops measured up to 9 T. The unusual quadratic component emerges below the Néel temperature (∼35 K), it is anti-correlated to the linear susceptibility and has a maximum at ∼10–15 K, the temperature for the magnetisation minimum. (c) Magnetisation at 300 K. The blue dots show the remanent magnetisation after the linear component has been subtracted. (d) Magnetisation at 2 K. The blue dots show the magnetisation after both the linear and quadratic components have been subtracted – the red line is the initial magnetisation in the virgin state. | |
4. Conclusions
In this study, we demonstrated a scalable large scale synthesis of octahedral Co3O4 microparticles (1.3 μm) enclosed by (111) facets using continuous hydrothermal flow process. The octahedron particles exhibited very high catalytic PMS activation properties. Dissociation of adsorbed water molecules on (111) surfaces resulting in Co–OH bonding was evident from the ELNEFS analysis. The Co–OH complexes, which efficiently activate PMS to oxidise MO, were formed on the surface via interaction of Co2+ with hydroxyl groups. This process facilitates the heterogeneous activation of PMS. Presence of both hydroxyl and sulphate radicals were found during PMS activation reaction. The prepared material showed ∼4 times higher pseudocapacitance properties when compared to commercial Co3O4 microcrystalline material. The as prepared material showed very interesting magnetic properties at low temperature. The material exhibited a coexistence of superparamagnetic single domain and linear/quadratic behaviours at low temperature.
Acknowledgements
This study was supported by the National Research Foundation of South Africa (Grant no. 88220; UID: 78697).
References
- Q. Yang, H. Choi, S. R. Al-Abed and D. D. Dionysiou, Appl. Catal., B, 2009, 88, 462–469 CrossRef CAS.
- H. Miyamura, R. Matsubara, Y. Miyazaki and S. Kobayashi, Angew. Chem., Int. Ed., 2007, 119, 4229–4232 CrossRef.
- G. P. Anipsitakis and D. D. Dionysiou, Environ. Sci. Technol., 2004, 38, 3705–3712 CrossRef CAS.
- E. Saputra, S. Muhammad, H. Sun, H.-M. Ang, M. O. Tadé and S. Wang, J. Colloid Interface Sci., 2013, 407, 467–473 CrossRef CAS PubMed.
- X. Chen, J. Chen, X. Qiao, D. Wang and X. Cai, Appl. Catal., B, 2008, 80, 116–121 CrossRef CAS.
- P. Shukla, S. Wang, K. Singh, H. M. Ang and M. O. Tadé, Appl. Catal., B, 2010, 99, 163–169 CrossRef CAS.
- S. Muhammad, E. Saputra, H. Sun, H.-M. Ang, M. O. Tadé and S. Wang, Ind. Eng. Chem. Res., 2012, 51, 15351–15359 CrossRef CAS.
- E. Saputra, S. Muhammad, H. Sun, H. M. Ang, M. O. Tadé and S. Wang, Catal. Today, 2012, 190, 68–72 CrossRef CAS.
- P. R. Shukla, S. Wang, H. Sun, H. M. Ang and M. Tadé, Appl. Catal., B, 2010, 100, 529–534 CrossRef CAS.
- H. Liang, H. Sun, A. Patel, P. Shukla, Z. H. Zhu and S. Wang, Appl. Catal., B, 2012, 127, 330–335 CrossRef CAS.
- W. Zhang, H. L. Tay, S. S. Lim, Y. Wang, Z. Zhong and R. Xu, Appl. Catal., B, 2010, 95, 93–99 CrossRef CAS.
- H. Wang, L. Zhang, X. Tan, C. M. B. Holt, B. Zahiri, B. C. Olsen and D. Mitlin, J. Phys. Chem. C, 2011, 115, 17599–17605 CAS.
- X.-H. Xia, J.-P. Tu, Y.-Q. Zhang, Y.-J. Mai, X.-L. Wang, C.-D. Gu and X.-B. Zhao, RSC Adv., 2012, 2, 1835–1841 RSC.
- X.-H. Xia, J.-P. Tu, X.-L. Wang, C.-D. Gu and X.-B. Zhao, Chem. Commun., 2011, 47, 5786–5788 RSC.
- D. Zhang and W. Zou, Curr. Appl. Phys., 2013, 13, 1796–1800 CrossRef.
- S. K. Meher and G. R. Rao, J. Phys. Chem. C, 2011, 115, 15646–15654 CAS.
- J. Xu, L. Gao, J. Cao, W. Wang and Z. Chen, Electrochim. Acta, 2010, 56, 732–736 CrossRef CAS.
- W. Du, R. Liu, Y. Jiang, Q. Lu, Y. Fan and F. Gao, J. Power Sources, 2013, 227, 101–105 CrossRef CAS.
- Y. Q. Zhang, L. Li, S. J. Shi, Q. Q. Xiong, X. Y. Zhao, X. L. Wang, C. D. Gu and J. P. Tu, J. Power Sources, 2014, 256, 200–205 CrossRef CAS.
- G.-L. Xu, J.-T. Li, L. Huang, W. Lin and S.-G. Sun, Nano Energy, 2013, 2, 394–402 CrossRef CAS.
- X. Xiao, X. Liu, H. Zhao, D. Chen, F. Liu, J. Xiang, Z. Hu and Y. Li, Adv. Mater., 2012, 24, 5762–5766 CrossRef CAS PubMed.
- X. Wang, L. Yu, X.-L. Wu, F. Yuan, Y.-G. Guo, Y. Ma and J. Yao, J. Phys. Chem. C, 2009, 113, 15553–15558 CAS.
- H. Zhou, B. Lv, D. Wu and Y. Xu, CrystEngComm, 2013, 15, 8337–8344 RSC.
- F. Moro, S. V. Yu Tang, F. Tuna and E. Lester, J. Magn. Magn. Mater., 2013, 348, 1–7 CrossRef CAS.
- M. J. Benitez, O. Petracic, E. L. Salabas, F. Radu, H. Tüysüz, F. Schüth and H. Zabel, Phys. Rev. Lett., 2008, 101, 097206 CrossRef CAS PubMed.
- L. M. Parkes, R. Hodgson, L. T. Lu, L. D. Tung, I. Robinson, D. G. Fernig and N. T. K. Thanh, Contrast Media Mol. Imaging, 2008, 3, 150–156 CrossRef CAS PubMed.
- M. M. Shahid, P. Rameshkumar, A. Pandikumar, H. N. Lim, Y. H. Ng and N. M. Huang, J. Mater. Chem. A, 2015, 3, 14458–14468 CAS.
- S. Wang, C. Xiao, P. Wang, Z. Li, B. Xiao, R. Zhao, T. Yang and M. Zhang, Mater. Lett., 2014, 137, 289–292 CrossRef CAS.
- T. Chen, X. Li, C. Qiu, W. Zhu, H. Ma, S. Chen and O. Meng, Biosens. Bioelectron., 2014, 53, 200–206 CrossRef CAS.
- J. Zhang, W. Gao, M. Dou, F. Wang, J. Liu, Z. Li and J. Ji, Analyst, 2015, 140, 1686–1692 RSC.
- G. Wang, X. Shen, J. Horvat, B. Wang, H. Liu, D. Wexler and J. Yao, J. Phys. Chem. C, 2009, 113, 4357–4361 CAS.
- E. Lester, G. Aksomaityte, J. Li, S. Gomez, J. Gonzalez-Gonzalez and M. Poliakoff, Prog. Cryst. Growth Charact. Mater., 2012, 58, 3–13 CrossRef CAS.
- X. Xie and W. Shen, Nanoscale, 2009, 1, 50–60 RSC.
- B. Kriedemann and V. Fester, Chem. Eng. J., 2015, 281, 312–321 CrossRef CAS.
- Z. H. Ibupoto, S. Elhag, M. S. AlSalhi, O. Nur and M. Willander, Electroanalysis, 2014, 26, 1773–1781 CrossRef CAS.
- L. Tian, K. Huang, Y. Liu and S. Liu, J. Solid State Chem., 2011, 184, 2961–2965 CrossRef CAS.
- J. P. Cheng, X. Chen, J.-S. Wu, F. Liu, X. B. Zhang and V. P. Dravid, CrystEngComm, 2012, 14, 6702–6709 RSC.
- Z. L. Wang, J. Phys. Chem. B, 2000, 104, 1153–1175 CrossRef CAS.
- P. Shi, X. Wang, X. Zhou, Y. Min, J. Fan and W. Yao, RSC Adv., 2015, 5, 34125–34133 RSC.
- X. Xie, Y. Li, Z.-Q. Liu, M. Haruta and W. Shen, Nature, 2009, 458, 746–749 CrossRef CAS PubMed.
- Z. L. Wang, J. S. Yin and Y. D. Jiang, Micron, 2000, 31, 571–580 CrossRef CAS PubMed.
- R. Xu and H. C. Zeng, Langmuir, 2004, 20, 9780–9790 CrossRef CAS PubMed.
- P. Shi, X. Dai, H. Zheng, D. Li, W. Yao and C. Hu, Chem. Eng. J., 2014, 240, 264–270 CrossRef CAS.
- Y. Wang, L. Zhou, X. Duan, H. Sun, E. L. Tin, W. Jin and S. Wang, Catal. Today, 2015, 258, 576–584 CrossRef CAS.
- G. P. Anipsitakis and D. D. Dionysiou, Environ. Sci. Technol., 2003, 37, 4790–4797 CrossRef CAS PubMed.
- Y. Yao, Z. Yang, H. Sun and S. Wang, Ind. Eng. Chem. Res., 2012, 51, 14958–14965 CrossRef CAS.
- Z. Chen, S. Chen, Y. Li, X. Si, J. Huang, S. Massey and G. Chen, Mater. Res. Bull., 2014, 57, 170–176 CrossRef CAS.
- Q. Yang, H. Choi and D. D. Dionysiou, Appl. Catal., B, 2007, 74, 170–178 CrossRef CAS.
- G. P. Anipsitakis, E. Stathatos and D. D. Dionysiou, J. Phys. Chem. B, 2005, 109, 13052–13055 CrossRef CAS PubMed.
- Q. Xiang, J. Yu and P. K. Wong, J. Colloid Interface Sci., 2011, 357, 163–167 CrossRef CAS PubMed.
- M. Chowdhury, M. Ntiribinyange, K. Nyamayaro and V. Fester, Mater. Res. Bull., 2015, 68, 133–141 CrossRef CAS.
- O. Oghenochuko, M. Chowdhury, N. Kudzanai, F. Cummings, V. Fester and O. S. Fatoki, RSC Adv., 2015, 5, 59513–59521 RSC.
- F. Zasada, W. Piskorz, S. Cristol, J.-F. Paul, A. Kotarba and Z. Sojka, J. Phys. Chem. C, 2010, 114, 22245–22253 CAS.
- C. Baiocchi, M. C. Brussino, E. Pramauro, A. B. Prevot, L. Palmisano and G. Marcì, Int. J. Mass Spectrom., 2002, 214, 247–256 CrossRef CAS.
- K. Dai, H. Chen, T. Peng, D. Ke and H. Yi, Chemosphere, 2007, 69, 1361–1367 CrossRef CAS PubMed.
- T. Chen, Y. Zheng, J.-M. Lin and G. Chen, J. Am. Soc. Mass Spectrom., 2008, 19, 997–1003 CrossRef CAS PubMed.
- Y. Cao, F. Yuan, M. Yao, J. H. Bang and J.-H. Lee, CrystEngComm, 2014, 16, 826–833 RSC.
- N. F. Kharchenko, R. Szymczak and M. Baran, J. Magn. Magn. Mater., 1995, 140–144(1), 161–162 CrossRef CAS.
- N. F. Kharchenko, Y. N. Kharchenko, R. Szymczak, M. Baran and H. Schmid, J. Low Temp. Phys., 2001, 27, 895–898 CrossRef CAS.
- B. B. van Aken, J.-P. Rivera, H. Schmid and M. Fiebig, Nature, 2007, 449, 702–705 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra20763k |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.