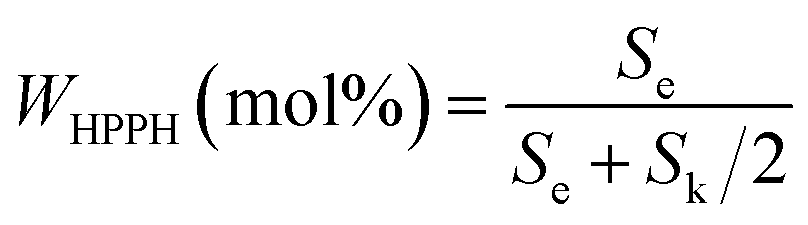DOI:
10.1039/C5RA18472J
(Paper)
RSC Adv., 2015,
5, 105038-105046
Preparation of self-promoted hydroxy-containing phthalonitrile resins by an in situ reaction
Received
9th September 2015
, Accepted 25th November 2015
First published on 26th November 2015
Abstract
The in situ reaction of a hydroxy group with a phthalonitrile system was carried out by the simple nucleophilic displacement of a nitro-substituent from 4-nitrophthalonitrile in a dipolar aprotic solvent, in a one-pot reaction. The hydroxy-containing phthalonitrile system (HPBD) was prepared by mixing 4-hydroxyphenoxy phthalonitrile (HPPH) and 1,3-bis(3,4-dicyanophenoxy)benzene (BDB), followed by heating. The curing behavior was studied using differential scanning calorimetry and dynamic rheological analysis, and the results indicated that the HPBD exhibits a large processing window (∼75 °C) and low complex viscosity (0.1–1 Pa s) at moderate temperatures. Fourier transform infrared spectroscopy (FT-IR) showed that polytriazine, polyindoline and phthalocyanine structures were formed during polymerization, and that the introduction of HPPH facilitated the curing reaction. Additionally, the prepared HPBD polymers showed outstanding thermal stability, a high modulus and a high glass transition temperature (Tg). After curing at 300 °C, the Tg of HPBD resin was raised to 410 °C. Postcuring effects on the thermal and dynamic mechanical properties were evaluated using thermogravimetric analysis (TGA) and dynamic mechanical analysis (DMA).
1. Introduction
Phthalonitrile (PN)-based polymers1–20 have emerged as ideal materials in the last 30 years. They are easily processed into a variety of shaped structures using cost effective manufacturing techniques and offer a unique combination of physical properties for advanced composites. These properties include high thermal and oxidative stability, low water absorption, and the absence of a Tg before the thermal decomposition temperature. Many potential uses, including adhesive,1 electronic,2–4 and structural applications,5–7 can be envisioned for a polymeric material such as a PN resin, compared to metals, due to its significant strength-to-weight8 and performance advantages.
The polymerization of the neat resins is extremely sluggish and requires several days at elevated temperatures before a vitrified product is obtained.9 The polymerization reaction of phthalonitrile can be initiated with many different types of curing additives such as organic amines,9 strong organic acids,10 organic acid/amine salts,11 metallic salts,12 and metals.13 During the past few decades, systematic studies have been conducted at Naval Research Laboratory (NRL) on phthalonitrile resins in terms of curing behavior, processability and the effects of various linkages between the terminal phthalonitrile units on the properties of the cured resins.14–22 They mainly focused on commercially available aromatic diamine curing additives, such as 1,3-bis(3-aminophenoxy)benzene (m-APB). The formulation of the resins at the NRL is achieved typically by physically mixing a bisphthalonitrile monomer and a curing additive below the melting temperature (Tm) of the bisphthalonitrile monomer or adding the additive to the melting monomer under vigorous stirring.23 Until now, phthalonitrile polymers, with an excellent combination of properties and good processability, and phthalonitrile-based composites have been successfully developed by NRL.5,6,24 However the volatilization of the diamine23 slows down the curing especially at high temperatures. To overcome this problem, we need stable and less reactive curing reagents to get the anticipated product. Thus, finding curing agents that do not volatilize at elevated curing reaction temperatures and that were less reactive relative to m-APB is key to promoting the processability of phthalonitrile-based composites.23
On the other hand, there are still some problems using phthalonitrile materials. The high temperature of the melting transition is a common shortcoming in phthalonitrile monomers (usually >200 °C).14,16–18 For example, the melting point of the 4,4-bis(3,4-dicyanophenoxy)biphenyl phthalonitrile monomer is 230 °C,23 and the processing window (the gap between the melting point and the polymerization temperature) is only 20 °C when curing with amine catalyzers, which brings difficulties in the processing. More importantly, the processing mode of the phthalonitrile resins described previously is mainly based on bisphthalonitrile monomer/curing additive composition systems referred to as binary composition systems. In these systems, to ensure an uniform cure rate within the phthalonitrile resins, an even molecular dispersion of a trace percent of curing additive into the bisphthalonitrile monomers is required and can be achieved merely under extreme conditions (e.g. vigorous stirring).24 Therefore, the development of a self-catalyzed phthalonitrile monomer with a low melting temperature would be interesting in the view of both scientific and industrial applications.
In our laboratory, previous investigations have demonstrated that amino or hydroxy-containing phthalonitrile derivatives (APN or HPN) showed self-promoted curing behavior even in the absence of curing additives which are required for conventional binary composition systems.25,26 Therefore, the curing of phthalonitrile derivatives containing amino or hydroxy groups offers a new route to the fabrication of phthalonitrile-based polymers or resins possessing excellent mechanical and thermal properties. Liu et al.27 synthesized phthalonitrile oligomers containing biphenyl ether nitrile cured with 4-aminophenoxy phthalonitrile. Zhou et al.28,29 prepared amino-containing (in the ortho, meta, and para positions) self-catalyzed phthalonitrile resins.
In this paper, self-promoted hydroxy-containing phthalonitrile systems were prepared using an in situ reaction. And a hydroxy group was introduced into the self-promoted phthalonitrile-based system by this method. Therefore, the blending problem of phthalonitrile monomers/curing additives under extreme conditions (e.g. vigorous stirring) was solved. Meanwhile, the phthalonitrile system, which was a blend of 1,3-bis(3,4-dicyanophenoxy)benzene (BDB) and self-promoted phthalonitrile, 4-hydroxyphenoxy phthalonitrile (HPPH), exhibited good processability, and thermal and dynamic mechanical properties. In addition, the polymerization mechanism and postcuring effects were also investigated.
2. Experimental
2.1. Materials
Resorcinol (99.5%) and dimethylsulfoxide (DMSO, AR) were supplied by Chengdu Kelong Chemical Reagent Co., Ltd, Chengdu, China. 4-Nitrophthalonitrile (99%) was purchased from Hong Kong’s Ming Tai Prosperity Chemical Co., Ltd. Potassium carbonate (99.0%) was obtained from Chengdu Kelong Chemical Reagent Co., Ltd. All solvents were used without further purification.
2.2. Synthesis of self-promoted hydroxy-containing phthalonitrile systems
To a 500 mL three-necked flask was added resorcinol, 4-nitrophthalonitrile and DMSO. During the course of the reaction, pulverized potassium carbonate was added in three portions. While stirring, the resulting mixture was heated at 40 °C for 10 h under an inert atmosphere. After cooling, the product mixture was poured into 2400 mL of water. The off-white solid was collected using suction filtration and washed with water until the filtrate was neutral, and dried in a vacuum oven (−0.09 MPa) at 80 °C for 16 h. Using this procedure, different compositions of HPPH and BDB, HPBD-18 and HPBD-28, were synthesized by controlling the feed ratio. The detailed synthesis data are listed in Table 1.
Table 1 The specific synthetic feed parameters and yield
| |
4-Nitrophthalonitrile |
Resorcinol |
DMSO |
K2CO3 |
Yield |
| HPBD-18 |
58.87 g |
22.02 g |
330 mL |
83.00 g |
87.54% |
| HPBD-28 |
51.95 g |
22.02 g |
300 mL |
83.00 g |
87.64% |
HPBD-24 was obtained through blending HPBD-18 and HPBD-28 with equal weight ratios.
2.3. Preparation of polymers
The HPBD was melted in a 100 mL reaction kettle equipped with a stirrer and was degassed for 10 min under vacuum conditions (−0.09 MPa) at 180 °C. The prepolymer was poured into a mold with dimensions of 35 mm × 15 mm × 8 mm and then cured in a nitrogen-circulating oven with a curing procedure of curing at 180 °C for 2 h, 200 °C for 5 h, 250 °C for 10 h, and 300 °C for 6 h. Postcured samples were prepared in a muffle furnace at elevated temperatures: 325 °C for 5 h; 350 °C for 5 h; and 375 °C for 5 h. The cured polymers were machined into rectangular specimens (30 mm × 12.5 mm × 3.2 mm) for the DMA experiments.
2.4. Characterizations
FT-IR spectra of HPBD and the phthalonitrile resins were recorded on a Nicolet-460 FT-IR spectrometer in KBr pellets between 4000 cm−1and 400 cm−1 in air. 1H NMR spectra were obtained using a Bruker AV II300 nuclear magnetic resonance (NMR) spectrometer at a proton frequency of 300 MHz and the corresponding proton frequency using DMSO-d6 as solvent. A differential scanning calorimetric (DSC) study was performed on a TA Instruments Q200 DSC from 50 °C to 300 °C at a heating rate of 10 °C min−1 with a nitrogen flow rate of 50 mL min−1. The rheological behaviors of the blends were investigated using a TA Instruments Rheometer AR-2000 with a frequency of 1 Hz and a heating rate of 3 °C min−1. Thermogravimetric analysis (TGA) was performed using a TA Instruments Q500 thermogravimetric analyzer at a heating rate of 10 °C min−1 under a nitrogen and air atmosphere respectively with a flow rate of 60 mL min−1. Dynamic mechanical analysis (DMA) was performed using a TA Instruments Q800 dynamic mechanical analyzer at a frequency of 1 Hz from 50 °C to 450 °C with a heating rate of 5 °C min−1. The mechanical properties of the HPBD resins were investigated using a JEOL JSM-7500F field-emission scanning electron microscope (SEM). The specimen was coated with Au prior to observation.
3. Results and discussion
3.1. Preparation
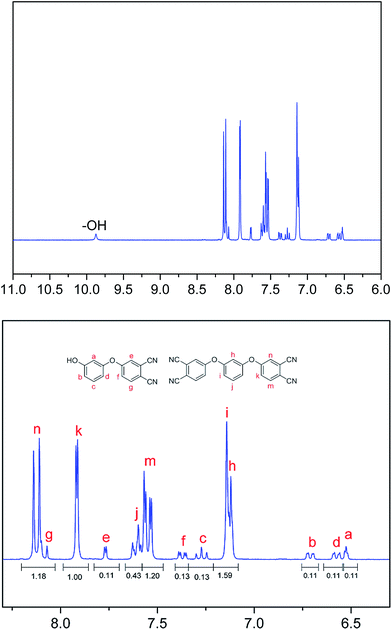
HPBD systems with a varying content of HPPH ingredient were synthesized by a nucleophilic displacement of a labile nitro substituent from 4-nitrophthalonitrile as illustrated in Scheme 1. The reaction took place in DMSO solution in the presence of an alkaline substance, K2CO3, as catalyst. The 1H NMR spectrum of HPBD-18 is given in Fig. 1 proving the success of the synthesis of HPBD and the content of HPPH in the HPBD systems. The content of HPPH was calculated using the 1HNMR integral area of HPBD.| |
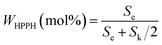 | (I) |
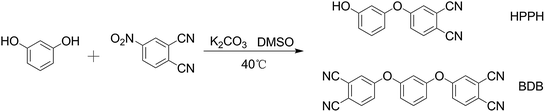 |
| | Scheme 1 General synthetic scheme for HPBD. | |
 |
| | Fig. 1 1HNMR of HPBD-18. | |
The content of HPPH was calculated according to formula (I) (Se and Sk represent the integral areas e and k in the 1HNMR spectrum, respectively). The detailed data are listed in Table 2.
Table 2 The molar ratio of HPPH in the HPBD systems
| |
Se |
Sk/2 |
HPPH content (mol%) |
| HPBD-18 |
0.11 |
0.5 |
18 |
| HPBD-24 |
0.16 |
0.5 |
24 |
| HPBD-28 |
0.20 |
0.5 |
28 |
3.2. Curing behaviors of HPBD
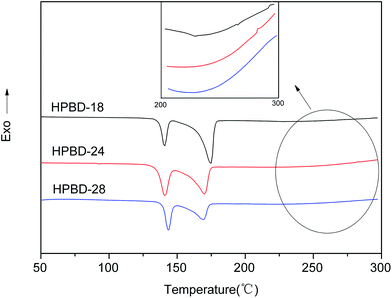
The curing behaviors of the HPBD systems were studied using DSC. As is shown in Fig. 2, all HPBD systems showed a larger processing window, and the processing window (the gap between the melting point and the polymerization temperature) of the HPBD-18 blend was up to 75 °C. Obviously, a larger processing window is favorable for processing phthalonitrile monomers into void-free resins. The DSC results also indicated that BDB can be cured into polymers with HPPH.30 In other words, the HPBD system can self-promote through heat treatment. Compared to the curing agents of aromatic amines,23,24,36 HPPH exhibited a slower reactivity. However, an advantage of the slower reactivity is that a larger processing window is available.24 Moreover, an extremely sluggish curing behavior is favorable for fabricating thick composite components using resin infusion methods (RIM) or resin transfer molding (RTM).30
 |
| | Fig. 2 DSC thermograms of the HPBD systems. | |
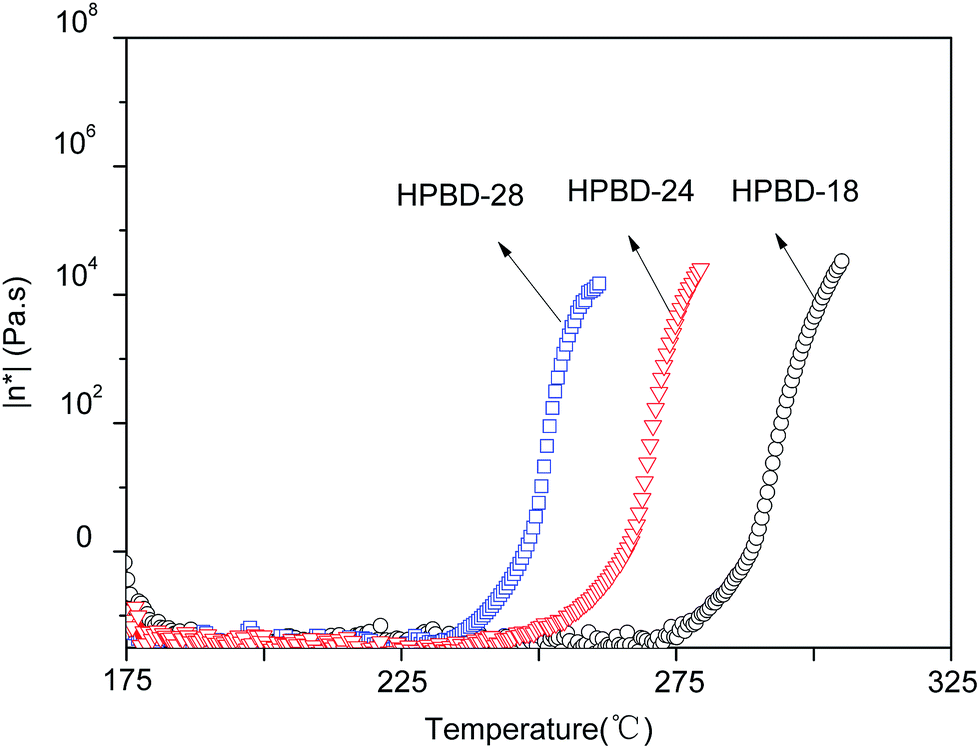
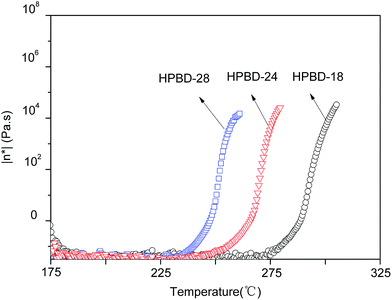
The rheological behavior plays an important part in determining the processability of materials.31 Therefore, the variation in the complex viscosity of the HPBD was determined as a function of temperature from 175 °C to 325 °C. The results, presented in Fig. 3, revealed that the complex viscosity of the blends was affected by the HPPH content. HPBD-28, HPBD-24 and HPBD-18 exhibit an abrupt complex viscosity increase at 240 °C, 260 °C, and 280 °C, respectively. The data showed that the higher the HPPH content, the faster the viscosity increase. This result was expected since the blends richer in HPPH have a greater tendency to react or cure at a faster rate. A minimum melt viscosity of around 0.1 Pa s was observed for all the blends at temperatures above 175 °C. The low melt viscosity of the blends at higher temperatures is evidence of their good melt stability. Based on the above results, the HPBD displays good processability with low complex viscosity at moderate temperatures.
 |
| | Fig. 3 Complex viscosity (η*) as a function of temperature for the various HPBD systems. | |
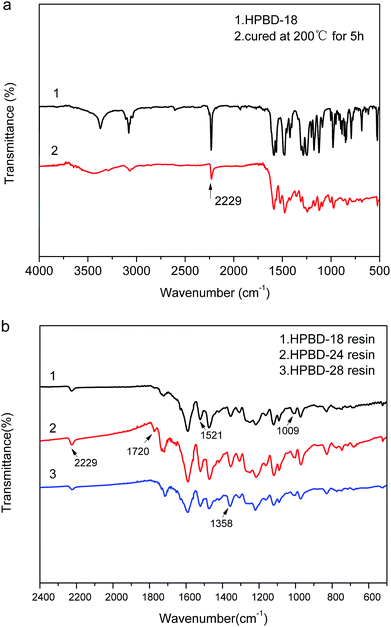
We further investigated the curing behavior through the analysis of the FTIR spectra of the HPBD resins. Fig. 4a shows the FTIR spectra of the HPBD-18 resin obtained after being heated at 200 °C for 5 h. It was obvious that the characteristic absorption band at 2229 cm−1 (Fig. 4a) was evidently weakened. Meanwhile, in Fig. 4b, the peaks at around 1009 cm−1 and 1521 cm−1 belong to the characteristic absorptions of the NH stretching and bending vibration in the phthalocyanine ring32 and the stretching vibration in the triazine ring,33 respectively. Moreover, the peak at around 1720 cm−1 was attributed to the characteristic absorption of the isoindole ring.34 Additionally, there was still a weak absorption peak attributed to the –CN groups (Fig. 4b) at around 2229 cm−1, and it became weaker gradually with an increase in the HPPH content. Thus, it can be concluded that the curing reaction happened between BDB and HPPH. However, the nitrile groups did not participate in the curing reaction completely, which can be ascribed to the formation of a triazine ring and phthalocyanine during polymerization. Only half of the nitrile groups contributed to the triazine ring structure formation because of the high steric hindrance. Based on these results, it could be concluded that HPPH could facilitate the polymerization of phthalonitrile systems and the possible polymerization mechanism of HPBD is shown in Scheme 2.
 |
| | Fig. 4 FT-IR spectra of HPBD resins, (a) HPBD resin cured at 200 °C, (b) HPBD resins. | |
 |
| | Scheme 2 Possible reaction mechanism of the HPBD systems. | |
3.3. Thermal stability
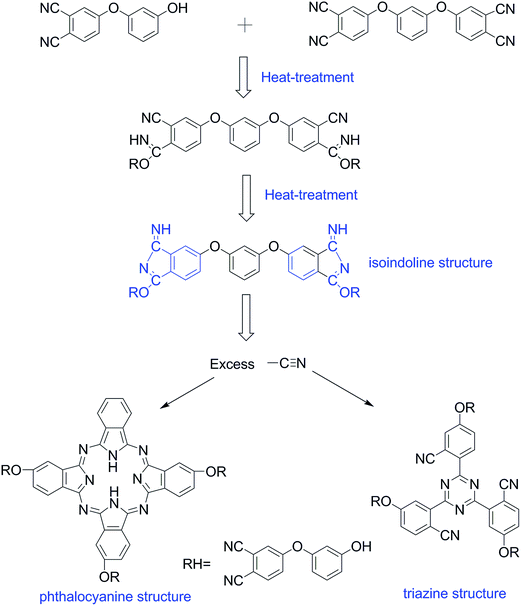
The thermal and thermo-oxidative properties of the HPBD resins cured with HPPH were evaluated using TGA as shown in Fig. 5, and the results are summarized in Table 3. Td5% and Td10% are defined, respectively, as the temperature at which 5% and 10% weight loss occurs. Whether under a nitrogen atmosphere or under an air atmosphere, as seen from Table 3, the 5 wt% and 10 wt% weight loss temperatures decreased obviously with the increased ratio of HPPH. These data showed the dependence of the thermal and thermo-oxidative stability on the HPPH content in the blends. The decrease in blend stability with an increase in the HPPH content was attributed to the weak bonds (–OH) of HPPH, which could be destroyed in the heating process. With the HPPH content increasing, the weak bonds (–OH) become more intense, leading to the possible degradation of the HPBD resins. Basically, the various HPBD resins all possessed excellent thermostability, with the temperatures corresponding to the weight loss of 5 wt% all exceeding 475 °C, and the char yields all above 72% when heated to 800 °C under a nitrogen atmosphere. These results should also be attributed to the crosslinking reaction between the BDB and HPPH. The TGA plots of the HPBD resins involve several stages which implies the oxidative degradation of different molecular segments under an air atmosphere.35 Small solid samples of the HPBD resins heated in air showed a weight retention of 95% at around 480 °C for all samples with catastrophic decomposition occurring between 500 °C and 800 °C. Based on the above results, it could be concluded that the HPBD polymers cured with HPPH show outstanding thermal stabilities and the thermal decomposition temperature was reduced regularly with an increase in the HPPH content.
 |
| | Fig. 5 TGA curves of HPBD resins cured with different ratios of HPPH: under nitrogen (a) and under air (b). | |
Table 3 Thermal properties of HPBD reins cured with HPPH
| |
HPBD |
Td5% (°C) |
Td10% (°C) |
Char yield (%) at 800 °C |
| In N2 |
HPBD-18 |
493.0 |
530.0 |
74.54 |
| HPBD-24 |
477.0 |
525.0 |
72.17 |
| HPBD-28 |
475.0 |
524.5 |
72.00 |
| In air |
HPBD-18 |
489.0 |
533.0 |
— |
| HPBD-24 |
485.5 |
535.0 |
— |
| HPBD-28 |
481.5 |
527.0 |
— |
3.4. Dynamic mechanical properties
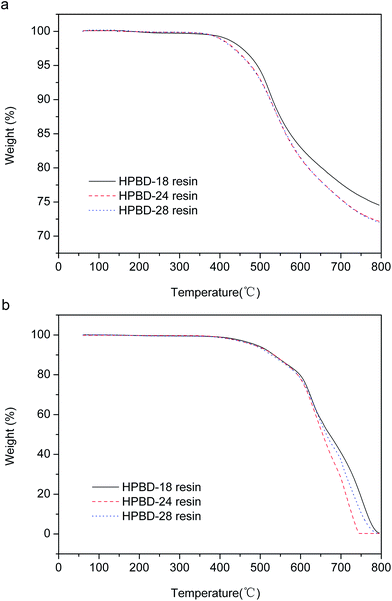
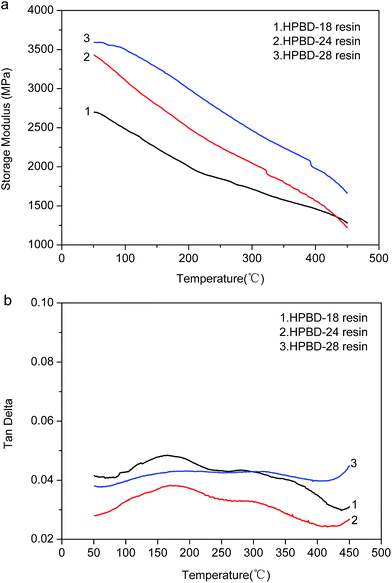
The dynamic mechanical properties of the cured HPBD polymers were evaluated to ascertain the modulus and the loss factor (Tg). Plots of the storage modulus (G′) and damping factor (tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ) are presented in Fig. 6a and (b), respectively. After curing to a maximum temperature of 300 °C, as seen from Fig. 6a, all the HPBD polymers exhibited a high storage modulus at 50 °C, which decreased gradually along with increasing temperature. The high modulus should be ascribed to the aromatic and crosslinked microstructure. The decrease of the modulus with increasing temperature was due to the stress relaxation of the polymer network. Moreover, it could be shown that the storage modulus of the HPBD-24 resin was up to 3.1 GPa at 50 °C, which is superior to the modulus of 5 wt% ODA cured BDB resin (2.9 GPa)36 and amino-containing self-catalyzed phthalonitrile resins (4-aminophenoxy-phthalonitrile, ∼1.7 GPa).37
δ) are presented in Fig. 6a and (b), respectively. After curing to a maximum temperature of 300 °C, as seen from Fig. 6a, all the HPBD polymers exhibited a high storage modulus at 50 °C, which decreased gradually along with increasing temperature. The high modulus should be ascribed to the aromatic and crosslinked microstructure. The decrease of the modulus with increasing temperature was due to the stress relaxation of the polymer network. Moreover, it could be shown that the storage modulus of the HPBD-24 resin was up to 3.1 GPa at 50 °C, which is superior to the modulus of 5 wt% ODA cured BDB resin (2.9 GPa)36 and amino-containing self-catalyzed phthalonitrile resins (4-aminophenoxy-phthalonitrile, ∼1.7 GPa).37
 |
| | Fig. 6 Dynamic mechanical properties of HPBD resins, (a) storage modulus versus temperature and (b) tanδ versus temperature. | |
The maximum of the tanδ curve represents Tg which is a particularly important parameter for polymer materials, because it generally determines the upper limit of the application temperature. The tanδ versus temperature plots for the HPPH cured resins are presented in Fig. 6b. As seen, Tg increased gradually as the HPPH content increased from 18 mol% to 28 mol%. It could be seen that the Tg of the HPBD-28 resin was up to 410 °C which was superior to the Tg of 5 wt% ODA cured BDB resin (320 °C) and 5 wt% and 10 wt% APPH cured BDB resins (320 °C and 347 °C, respectively).35 For the state-of-the-art high temperature resin, PMR-15, the maximum postcure temperature is 316 °C, and the corresponding Tg is 340 °C. Clearly, the low softening point of the PMR-15 resin would restrict its usage in structural applications that require stability at temperatures in excess of 340 °C. It has been reported that the shift in the maxima of the moduli as well as the tanδ to higher temperatures can also be explained by a higher degree of crosslinking of polymers.38,39 The greater Tg of the HPBD polymers should be also attributed to the more crosslinked structure when BDB is cured with HPPH, and a higher HPPH content leads to a higher Tg.
3.5. Postcuring effects
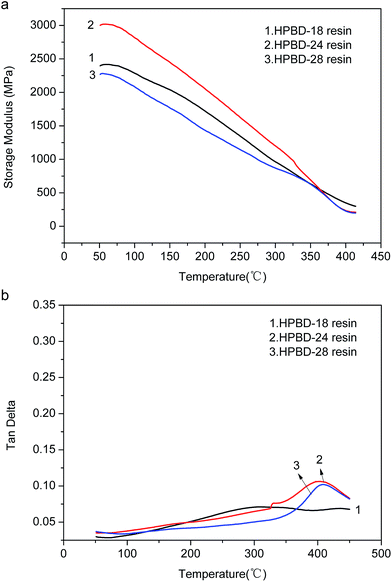
As mentioned above, the samples (HPBD resin cured with HPPH) were prepared with the curing procedure of curing at 180 °C for 2 h, 200 °C for 5 h, 250 °C for 10 h, and 300 °C for 6 h. In order to explore postcuring effects, especially the effect of different postcuring temperatures on the properties, the samples were gradually postcured at 325 °C for 5 h, 350 °C for 5 h, and 375 °C for 5 h. After postcuring, the thermal properties were evaluated, and the TGA curves are displayed in Fig. 7, and the data is collected in Table 4. As is shown in Fig. 7, all of the HPBD resins exhibited a higher char yield after postcuring under a nitrogen atmosphere. In addition, whether under a nitrogen atmosphere or under an air atmosphere, the resins exhibited much higher thermal decomposition temperatures (Td5% and Td10%) which indicated that the thermal stabilities could be further improved via postcuring using an elevated temperature process. This behavior was attributed to the corresponding increase in the curing degree and crosslinking density.
 |
| | Fig. 7 TGA thermograms of HPBD polymers after postcuring at 375 °C: under nitrogen (a) and under air (b). | |
Table 4 Thermal properties of HPBD resins after curing at 375 °C
| |
HPBD |
Td5% (°C) |
Td10% (°C) |
Char yield (%) at 800 °C |
| In N2 |
HPBD-18 |
514.5 |
549 |
74.59 |
| HPBD-24 |
504.5 |
538 |
74.65 |
| HPBD-28 |
503 |
538.5 |
75.06 |
| In air |
HPBD-18 |
505.0 |
536.0 |
— |
| HPBD-24 |
508.5 |
542.0 |
— |
| HPBD-28 |
501.5 |
536.0 |
— |
Dynamic mechanical analysis (DMA) was performed to explore the postcuring effects on the mechanical properties. Fig. 8 shows the DMA curves of the postcured HPBD resins. After curing to a maximum temperature of 375 °C, it can be found that the storage moduli of the HPBD resins with varying HPPH content were 2.7 GPa, 3.4 GPa, and 3.5 GPa at 50 °C, and they decreased with the increase in temperature gradually due to the stress relaxation of the polymer network. However, the moduli of the resins still remained at 1.3 GPa, 1.2 GPa and 1.6 GPa at 450 °C. Moreover, the viscoelastic transition characteristic Tg was not observed in the tanδ plot of the HPBD resins, indicating that a stable crosslinked polymer network had been formed through the curing process and that the polymer remained in the glassy state up to at least 450 °C. These data also showed that a higher cure temperature was needed to complete the curing of the phthalonitrile resins.
 |
| | Fig. 8 DMA curves of HPBD resins, (a) modulus versus temperature and (b) tanδ versus temperature. | |
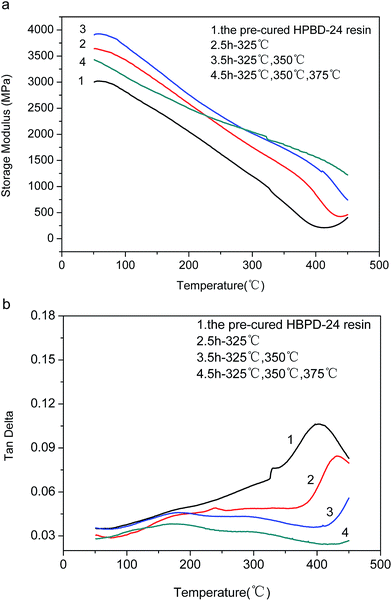
We further investigated the postcuring effect through the analysis of the DMA curves of the HPDB-24 resin at different postcured temperatures (Fig. 9a and b). As the curing temperature increased, the storage modulus of the HPBD-24 resin elevated steadily. After curing at a maximum temperature of 375 °C, the HPBD-24 resin exhibited a Tg in excess of 450 °C and maintained 35% of its initial modulus up to 450 °C. Thus, the results revealed that postcuring at elevated temperatures is favorable for improving the cross-linking degree and thermal stability of phthalonitrile resins.20,21,23
 |
| | Fig. 9 DMA plots for HPBD-24 samples postcured at elevated temperatures (a) modulus versus temperature and (b) tanδ versus temperature. | |
To sum up, the phthalonitrile resins show higher thermal and mechanical properties after postcuring, as proved by TGA and DMA, and the results also demonstrate that a higher curing temperature is necessary to improve the curing degree of phthalonitrile resin.
In addition, SEM was used to evaluate the microstructure of the cured resin. The morphology of a HPBD-28 polymer section under different magnifications is shown in Fig. 10. No voids can be observed in the HPBD-28 polymer under a magnification of 2000 times (Fig. 10c) which proves the void-free structure of the HPBD-28 polymer and also guarantees the excellent thermal and mechanical properties. Moreover, the addition polymerization mechanism of the monomer is one of the most important factors in preparing void-free components.
 |
| | Fig. 10 SEM images of the HPBD-28 resin after postcuring at 375 °C. | |
4. Conclusion
The in situ reaction of a hydroxy group with a phthalonitrile system was successfully carried out by the simple nucleophilic displacement of a nitro-substituent from 4-nitrophthalonitrile in a dipolar aprotic solvent, through a one-pot reaction. Based on DSC investigations and the rheometric studies, HPBD exhibited large processing windows (∼75 °C) and good processability with a low complex viscosity (0.01 Pa s) at moderate temperatures. FTIR spectra indicated that HPBD forms a phthalocyanine ring, isoindole ring, and triazine ring in the process of polymerization. Dynamic mechanical analysis revealed that HPPH cured resins exhibited excellent mechanical properties and glass transition temperatures. More HPPH can lead to a higher modulus and Tg, and better thermal stability was achieved though postcuring at elevated temperatures. Moreover, HPBD resins were proved to form void-free structures which guaranteed the excellent thermal and mechanical properties. Thus, they are good candidates for application as structural components, high temperature adhesives and packing materials in the aerospace and microelectronic industries.
Acknowledgements
The authors sincerely thank the National Natural Science Foundation of China (No. 51203098) for the financial support.
References and notes
- T. M. Keller and C. M. Roland, US Pat., 5, 242, 755, 1993, p. 1.
- T. M. Keller, SAMPE Symp. Proc., 1986, 31, 528 CAS.
- T. M. Keller, CHEMTECH, 1988, 18, 635 CAS.
- J. F. Giuliani and T. M. Keller, Sens. Mater., 1989, 1, 2247 Search PubMed.
- S. B. Sastri, J. P. Armistead and T. M. Keller, Polym. Compos., 1996, 17, 816 CrossRef CAS.
- S. B. Sastri, J. P. Armistead and T. M. Keller, Polym. Compos., 1997, 18, 48 CrossRef CAS.
- G. Fowler, Int. SAMPE Symp. Exhib., 2002, 1123 Search PubMed.
- A. Badshah, M. R. Kessler, Z. Heng, J. H. Zaidi, S. Hameed and A. Hasan, Polym. Chem., 2013, 4, 3617 RSC.
- T. M. Keller and T. R. Price, J. Macromol. Sci., Chem., 1982, 18, 931 CrossRef.
- T. M. Keller, Polym. Prepr., 1992, 33, 422 CAS.
- P. J. Burchill, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 1 CrossRef CAS.
- T. M. Keller and J. R. Griffith, Am. Chem. Soc. Symp. Ser., 1980, 132, 25 CrossRef CAS.
- T. R. Walton, J. R. Griffith and J. G. O’Rear, J. Adhes. Sci. Technol., 1975, 9, 665 Search PubMed.
- T. M. Keller, Chem. Mater., 1994, 6, 302 CrossRef CAS.
- T. M. Keller and R. F. Gratz, Polym. Commun., 1987, 28, 334 CAS.
- T. M. Keller, Polymer, 1993, 34, 952 CrossRef CAS.
- T. M. Keller and T. R. Price, Polym. Commun., 1984, 25, 42 CAS.
- T. M. Keller and D. D. Dominguez, Polymer, 2005, 46, 4614 CrossRef CAS.
- M. Laskoski, D. D. Dominguez and T. M. Keller, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4136 CrossRef CAS.
- D. D. Dominguez and T. M. Keller, High Perform. Polym., 2006, 18, 283 CrossRef CAS.
- D. D. Dominguez and T. M. Keller, Polymer, 2007, 48, 91 CrossRef CAS.
- M. Laskoski, D. D. Dominguez and T. M. Keller, Polymer, 2007, 48, 6234 CrossRef CAS.
- S. B. Sastri and T. M. Keller, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 1885 CrossRef CAS.
- D. D. Dominguez, H. N. Jones and T. M. Keller, Polym. Compos., 2004, 25, 544 Search PubMed.
- K. Zeng, D. M. Wu, Y. Han, G. Yang, et al., National conference on composite materials-14, 2006, p. 291 Search PubMed.
- K. Zeng, K. Zhou, W. R. Tang and G. Yang, et al., Chin. Chem. Lett., 2007, 18, 523 CrossRef CAS.
- X. L. Yang, J. D. Zhang, Y. J. Lei, J. C. Zhong and X. B. Liu, J. Appl. Polym. Sci., 2011, 121, 2331 CrossRef CAS.
- A. Badshah, H. Zhou, F. Liu and H. Aurangzeb, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5916 CrossRef.
- H. Zhou, A. Badashah, Z. H. Luo, F. Liu and T. Zhao, Polym. Adv. Technol., 2011, 22, 1459 CrossRef CAS.
- D. Wu, Y. Zhao, K. Zeng and G. Yang, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4977 CrossRef CAS.
- D. H. Zhang, L. Ye, S. Q. Deng, J. N. Zhang, Y. H. Tang and Y. F. Chen, Compos. Sci. Technol., 2012, 72, 412 CrossRef CAS.
- R. Seoudi, G. S. El-Bahy and Z. A. El Sayed, J. Mol. Struct., 2005, 753, 119 CrossRef CAS.
- A. W. Snow and J. R. Griffith, Macromolecules, 1984, 17, 1614 CrossRef CAS.
- R. K. Gujadhur and D. Venkataraman, Synth. Commun., 2001, 31, 2865 CrossRef CAS.
- X. Y. Yu, K. Naito, C. Kang, X. W. Qu and Q. X. Zhang, Macromol. Chem. Phys., 2013, 214, 361 CrossRef CAS.
- H. T. Sheng, X. G. Peng, H. Guo, X. Y. Yu, X. W. Qu and Q. X. Zhang, Thermochim. Acta, 2014, 577, 17 CrossRef CAS.
- S. B. Sastri and T. M. Keller, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 2105 CrossRef CAS.
- Z. Brunovska, R. Lyon and H. Ishida, Thermochim. Acta, 2000, 195, 357 Search PubMed.
- Y. Liu, J. Zhang, C. Liao, J. Zheng, S. Zhao and M. Run, Thermochim. Acta, 2013, 573, 138 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.