
Structure–activity relationship study for design of highly active covalent peroxidase-mimicking DNAzyme†
Anastasia V. Gribasa,
Sergey P. Korolevab,
Timofey S. Zatsepinabc,
Marina B. Gottikhb and
Ivan Yu. Sakharov*a
aDepartment of Chemistry, Lomonosov Moscow State University, Leninskie Gory, Moscow 119991, Russia. E-mail: sakharovivan@gmail.com; Fax: +7 495 9395417
bBelozersky Institute of Physical and Chemical Biology, Lomonosov Moscow State University, Leninskie Gory, Moscow 119991, Russia
cCentral Research Institute of Epidemiology, Novogireevskaya 3a, Moscow 111123, Russia
First published on 1st June 2015
Abstract
We synthesized a series of conjugates of hemin and its aptamer EAD2, named covalent peroxidase-mimicking DNAzymes (PMDNAzymes), varying the length, rigidity and 5′-/3′-position of a linker between the oligonucleotide and hemin. Systemic structure–activity relationship study of these PMDNAzymes showed that covalent PMDNAzyme with hemin bound to the 5′-end of EAD2 via T10 spacer (PMDNAzyme(T10)) demonstrated the highest activity in luminol oxidation assay. Its activity was significantly higher in comparison to the non-covalent complex of hemin and aptamer EAD2 (non-covalent PMDNAzyme). Comparison of the detection limit values for the PMDNAzyme(T10) in the reactions of oxidation of luminol and ABTS, which were equal to 0.2 and 1.6 pM, respectively, showed that the chemiluminescent method of PMDNAzyme(T10) detection is preferred over the colorimetric one. Similarity of the detection limit values for the PMDNAzyme(T10) and horseradish peroxidase, whose activity was measured in an enhanced chemiluminescence reaction (0.25 pM), opens up very promising perspectives for the development of highly sensitive PMDNAzyme(T10)-based assays and devices.
1. Introduction
Nowadays, horseradish peroxidase (HRP, EC 1.11.1.7) is extensively used in analytical practice due to its high activity, stability and commercial availablility.1–3 However, the high cost of the enzyme has stimulated a search of its alternatives. Some peroxidase mimetics such as hemin, metal-containing porphyrins and phthalocyanines, FeIII–TAML activators and nanoparticles of different chemical nature have been reported to possess peroxidase-like activity.4–7 Among them, complexes of hemin and its G-quadruplex aptamers attract great interest, since their catalytic activity is significantly higher than that of hemin itself.8,9 Such complexes named peroxidase-mimicking DNAzymes (PMDNAzymes) have been used in construction of various analytical assays and biosensors.10–14Nevertheless, the widespread application of non-covalent PMDNAzymes is restricted because of their relatively low sensitivity. Detection limit of a non-covalent PMDNAzyme formed by hemin and its EAD2 aptamer, even in the chemiluminescent assay, was only 350 pM,15 whereas the detection limit of the native HRP was reported to be 0.25 pM.16 One of the main reasons of the low sensitivity of non-covalent PMDNAzyme determination is dissociation of the PMDNAzyme molecules to the initial components (DNA and hemin) at nano- and picomolar concentrations because of a poor affinity of hemin to its aptamers (Kd was ca. 10−7 M).15,17–19 However, this drawback could be overcome by a covalent binding of hemin to its aptamer resulting in formation of a so called covalent PMDNAzyme.
The first covalent PMDNAzyme synthesis has been reported in 2010, when Thirstup and Baird prepared a conjugate of hemin and its aptamer PS2.M.20 Hemin was bound to the 3′-amino group of 5′-biotin-dA15-PS2.M-NH2. A detection limit of this PMDNAzyme estimated with 3,3′-diaminobenzidine as a substrate was 10 nM, whereas the detection limit of a noncovalent complex of hemin and PS2.M was 100 nM. The obtained covalent PMDNAzyme was applied in the immunohistochemical analysis of prostate specific antigen in prostate tissue as an alternative of HRP.20
Later Nakayama et al. have synthesized covalent PMDNAzymes by covalent coupling of hemin to aptamers with parallel and anti-parallel G-quadruplex topology.21 Hemin was attached to the 5′-ends of the aptamers or internally to the G-quadruplex loop. All obtained PMDNAzymes were active catalysts, their catalytic activity depending strongly on substrates used and experimental conditions.
Although covalent PMDNAzymes demonstrate significantly lower detection limits compared to the non-covalent PMDNAzymes, our present knowledge about the effect of the covalent PMDNAzyme structure on their catalytic activity is extremely limited. The high peroxidase activity of PMDNAzymes is considered to result from the efficient end stacking of hemin with its aptamer having G-quadruplex structure.21 In all covalent PMDNAzymes mentioned above hemin was bound to amino modified aptamers without any spacer. This manner of the hemin coupling limits its mobility and can therefore lead to serious steric hindrances for 3D organization of catalytically active covalent PMDNAzymes. Such hindrances could be removed by insertion of spacers between hemin and its aptamers.
In the present work we described first the systemic structure–activity relationship study of a covalent PMDNAzyme based on the EAD2 aptamer (CTGGG(AGGG)3A). This aptamer was chosen for the covalent PMDNAzyme construction, since its complex with hemin is one of the most active non-covalent PMDNAzymes.15,22,23 The length and rigidity of spacers between hemin and EAD2 as well as hemin position at 3′- or 5′-end of the aptamer were varied. This approach allowed producing covalent PMDNAzymes with extremely high catalytic activity. Among all the hemin–EAD2 conjugates obtained, the PMDNAzyme with hemin bound at the 5′-end of EAD2 via dT10 spacer (PMDNAzyme(T10)) was the most active catalyst. The activity of PMDNAzyme(T10) measured towards both luminol (chemiluminescent substrate) and 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) (ABTS, colorimetric substrate) turned out to be significantly higher than that of the noncovalent hemin–EAD2 complex. The detection limit value for the PMDNAzyme(T10) was similar to that one for the native HRP and significantly lower than that for the parent non-covalent PMDNAzyme.
2. Experimental
2.1. Chemicals
Luminol, hemin, NaCl, KCl, and tris(hydroxymethyl)aminomethane (Tris), dicyclohexylcarbodiimide (DCC), N-hydroxysuccinimide (NHS), ABTS and N,N-diisopropylethylamine (DIPEA) were obtained from Sigma (USA). Universal Controlled Pore Glass (CPG) (loading 40 μmol g−1), protected 2′-deoxynucleoside (A(Bz), G(iBu), C(Ac), T) phosphoramidites, C3 (1,3-propanediol) linker and 5′-amino-modifier C6 phosphoramidites were purchased from ChemGenes (USA). C7-amino CPG (loading 45 μmol g−1) was purchased from GlenResearch (USA).Hydrogen peroxide (H2O2, 30%) was obtained from ChemMed (Russia). The concentration of H2O2 was estimated by measuring absorbance using ε240 = 43.6.
Amino modified oligonucleotides (Table 1) were synthesized by standard phosphoramidite method under conditions recommended by manufacturer on an ABI 3400 DNA synthesizer (Applied Biosystems, USA) with minor modifications: detrytilation was carried out with 6% dichloroacetic acid in 1,2-dichloroethane, coupling of all modified phosphoramidites was carried out using 4,5-dicyanoimidazole as an activator for 5 minutes. The oligonucleotides obtained were deprotected using ammonium hydroxide–methylamine solution for 20 min at 65 °C.
| N | Covalent PMDNAzyme 5′ → 3′ | Chemiluminescent activity | |
|---|---|---|---|
| 106 RLU | % | ||
| a Prd – 1,3-propandiol. | |||
| 1 | Hemin-T15-EAD2 | 7.9 | 71 |
| 2 | Hemin-T10-EAD2 | 11.1 | 100 |
| 3 | Hemin-T5-EAD2 | 10.0 | 90 |
| 4 | Hemin-EAD2 | 6.7 | 60 |
| 5 | EAD2-T15-hemin | 7.5 | 68 |
| 6 | EAD2-T10-hemin | 8.6 | 77 |
| 7 | EAD2-T5-hemin | 5.5 | 50 |
| 8 | EAD2-hemin | 5.2 | 47 |
| 9 | Hemin-Prd10-EAD2a | 8.1 | 73 |
| 10 | EAD2-Prd10-hemina | 2.2 | 20 |
The oligonucleotides electroeluted from gel by Elutrap electroelution system (Whatman) were additionally purified by HPLC on an ÄKTA Purifier (GE Healthcare, USA) equipped with a Jupiter C18 column (Phenomenex, Jupiter 5 μm, 300 Å, 250 × 4.6 mm) and a UV-Vis detector. The oligonucleotides were eluted using 0.05 M ammonium acetate (pH 7) with an acetonitrile gradient (0–80%). The oligonucleotides were desalted by ethanol precipitation and redissolved in distilled water.
2.2. Synthesis of hemin–oligonucleotide conjugates
The NHS ester of hemin was prepared accordingly to ref. 21 (Fig. 1) by adding 6 equiv. each of DCC, NHS and DIPEA in 100 μL of anhydrous dimethylformamide (DMF) to 3 μmole of hemin and the reaction was carried out overnight at room temperature. Then a 10-fold volume of isopropanol was added to the reaction mixture, and the black precipitate was collected by centrifugation for 10 min at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g and washed twice with 1 mL of isopropanol. The obtained activated ester of hemin was dissolved in 400 μL of DMF and used for preparation of hemin–DNA conjugates instantly.
000g and washed twice with 1 mL of isopropanol. The obtained activated ester of hemin was dissolved in 400 μL of DMF and used for preparation of hemin–DNA conjugates instantly.
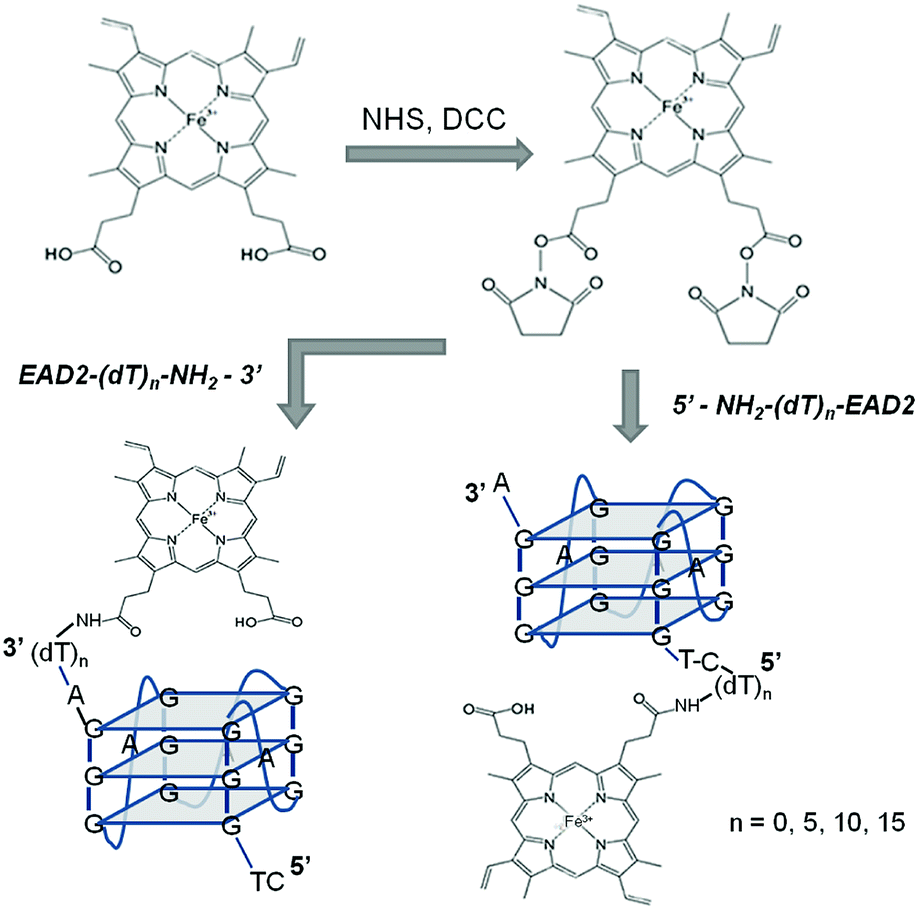 | ||
| Fig. 1 Scheme of the synthesis of covalent PMDNAzymes. | ||
Hemin–DNA conjugates were prepared by adding the activated ester of hemin (600 nmoles) to amino-functionalized oligonucleotides (10 nmoles) in 200 μL of 50 mM NaHCO3, pH 9.0 containing 50% DMF, and incubating at room temperature for 20 h (Fig. 1). The obtained conjugates were precipitated with 1 mL of 2% LiClO4 solution in acetone at room temperature for 10 min. The precipitate was collected by centrifugation for 10 min at 10000g and re-dissolved in 80% aqueous formamide. The hemin–DNA conjugates were purified by electrophoresis in a 20% polyacrylamide/7 M urea gel. The main band corresponded to a hemin–DNA conjugate was cut out, eluted from gel using aqueous 2 M LiClO4 and then precipitated adding 1 mL of acetone. Finally the precipitate obtained by centrifugation (10 min, 10000g) was re-dissolved in distilled water. The band corresponded to the initial oligonucleotide was also cut and treated as described above. The DNA amount eluted from both bands was determined by UV spectroscopy, and on the base of these data the conjugation efficiency was estimated as 80–90%. The low yield (<5%) of a conjugate with a molar ratio of hemin–EAD2 = 1:2 (see Fig. S1† as example) is explained by the use of a 60-fold molar excess of hemin in the reaction.
The absorbance spectra of the hemin–DNA conjugates were recorded on Carry 50 Bio (Varian). The extinction coefficients of DNA at 260 nm were calculated as a sum of the corresponding nucleoside extinction coefficients. The extinction coefficients of hemin at 260 nm and 405 nm were taken from the paper published by Travascio et al.8 Using these extinction coefficients, the absorbance spectra of the covalent derivatives were consistent with a 1:1 DNA–hemin complex. The concentration of the covalent PMDNAzymes prepared was determined measuring absorbance at 405 nm. The overall yields of the covalent PMDNAzyme preparations varied between 15 and 20%.
2.3. Characterization of hemin–oligonucleotide conjugates
Colorimetric determination of the covalent PMDNAzyme was carried out in wells of transparent polystyrene plates for enzyme immunoassay (Costar, USA). For this, covalent PMDNAzyme samples were mixed with aqueous solutions of ABTS and H2O2. The final concentrations of ABTS and H2O2 in the wells were 4 mM and 12 mM, respectively, unless otherwise stated. Change of adsorption at 405 nm was measured for 20 min after the initiation of ABTS oxidation at room temperature on a reader (Zenyth 3100, Austria). The determination of the non-covalent PMDNAzyme was carried out under the same conditions.
Chemiluminescent determination of the covalent PMDNAzyme was carried out in wells of black polystyrene plates for enzyme immunoassay (Costar, USA). For this, covalent PMDNAzyme samples were mixed with aqueous solutions of luminol and H2O2. The final concentrations of luminol and H2O2 in the wells were 5 μM and 2 mM, respectively, unless otherwise stated. CL intensity was measured 1 min after the initiation of luminol oxidation at room temperature on a luminometer (Spectra Max L, USA). Chemiluminescent activity of the non-covalent PMDNAzyme was measured as described previously.15 The light intensity was expressed in relative luminescence units (RLU).
3. Results and discussion
3.1. Optimization of the covalent PMDNAzyme structure
Limited knowledge about the structure–activity relationships for covalent PMDNAzymes stimulated optimization of their structure. We synthesized two sets of covalent PMDNAzymes composed of hemin and its aptamer EAD2 covalently bound through spacers of different lengths and nature. The role of the spacer within covalent PMDNAzyme structure is to covalently attach hemin to its aptamer thus providing formation of an intramolecular complex of hemin and EAD2 with high peroxidase-like activity.The conjugation was carried out by acylation of amino-modified oligonucleotides with hemin NHS ester (Fig. 1). In the first set of covalent PMDNAzymes hemin was attached to the 5′-end, whereas covalent PMDNAzymes from the second set contained hemin at the 3′-end of DNA (Table 1). All the covalent PMDNAzymes prepared were purified by electrophoresis in 20% polyacrylamide gel/7 M urea and their structure was confirmed by the electrophoresis mobility (Fig. S1†), LC-MS (Table 1S, ESI†) and absorbance spectra. The absorbance spectrum of one of the conjugates (Table 1, item 2) is presented in Fig. 2. The data obtained clearly indicated that a molar ratio of hemin and DNA in the synthesized conjugates was 1:1. Interestingly, the spectra of the covalent PMDNAzymes in the range of 350–370 nm have no shoulder that is characteristic of free hemin in aqueous solutions (Fig. 2, curve 3). The existence of such shoulder for water solution of hemin was reported earlier.8,25 This means that hemin within the covalent PMDNAzymes exists in monomeric form unlike free hemin. This is an important characteristic of any hemin–aptamer complex, because a transfer of hemin from dimeric to monomeric form increases the catalytic activity of the complex compared to free hemin.8
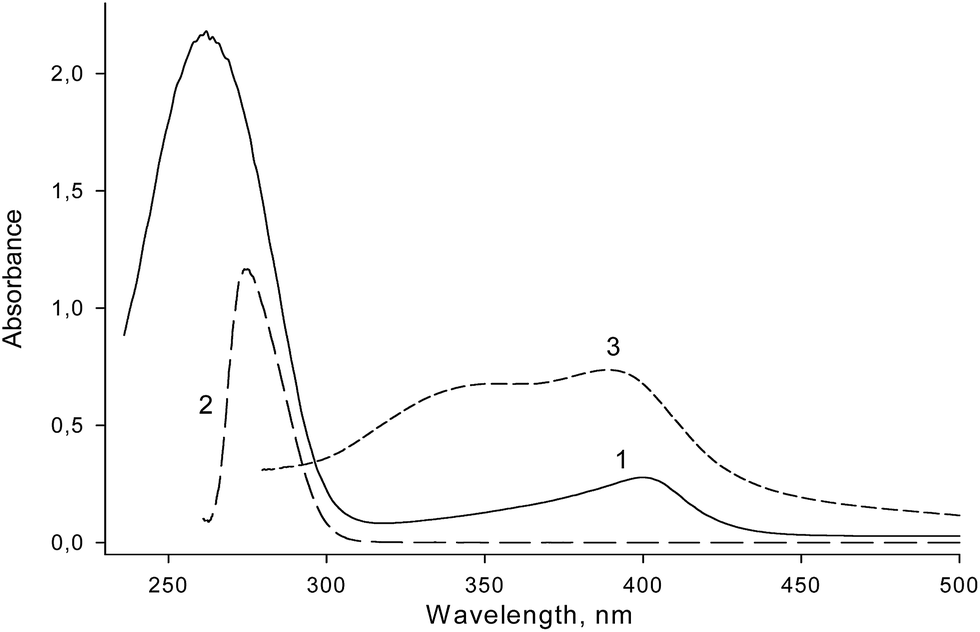 | ||
| Fig. 2 UV-vis spectra of PMDNAzyme(T10) (1), amino-containing aptamer NH2-T10-EAD2 (2) and hemin alone (3) dissolved in distilled water. | ||
We measured catalytic activity of the EAD2–hemin conjugates prepared towards luminol since it usually gives an ultrasensivity in peroxidase-based assays.26–28 All the covalent PMDNAzymes possessed HRP-like activity (Table 1). Nevertheless, we observed a clear effect of the hemin position on the covalent PMDNAzymes catalytic properties. All covalent PMDNAzymes with hemin bound at the 5′-end had higher catalytic activity than their analogs bearing hemin at the 3′-end. This effect may arise from different conformational changes in the proper G-quadruplex structure induced with its 5′- or 3′-end modifications. Further our work will focus on the detailed study of this question. It should be also noted that our data correlate with results of Zhang et al. who showed that adding dTn fragment to the 5′-end of hemin aptamer DZ (5′-GGTA(G)3C(G)3TT(G)3-3′) improved the catalytic activity of a noncovalent complexes of such modified aptamers with hemin, whereas the addition of dTn to the 3′-end decreased their activity.29
In addition, the covalent PMDNAzyme activity was found to depend on the spacer length. In both series of the conjugates (Table 1, items 1–4 and 5–8) the highest activity was observed for the covalent PMDNAzymes with T10 spacer (Table 1, items 2 and 6). It appears that this spacer is optimal to provide the intramolecular stacking interactions between hemin and G-quadruplex structure of EAD2 necessary for the organization of the catalytically active conformation of the PMDNAzyme.
To evaluate the effect of the spacer flexibility on the covalent PMDNAzyme activity, we replaced the spacer dT10 with (1,3-propanediol)10 spacer (Prd10), which had a similar length, but higher mobility. It turned out that increasing the flexibility of the spacer decreased the activity of the covalent PMDNAzyme, and this effect was observed for the compounds containing hemin at both 5′- and 3′-ends (Table 1, items 1 and 8). Taking into account the above-mentioned, the further work was focused on the study of catalytic properties of the most active covalent PMDNAzyme with hemin bound at the 5′-end via dT10 spacer (PMDNAzyme(T10)).
3.2. Study of the synthesized covalent PMDNAzymes topology
The most active G-quadruplex aptamers of hemin in the presence of K+ cations are known to have parallel or mixed-hybrid topology.21,30,31 We proceeded to investigate the topology of the synthesized covalent PMDNAzymes using circular dichroism (CD) spectroscopy and gel shift assay.CD spectra of PMDNAzyme(T10) and its parent aptamer NH2-T10-EAD2 are shown in Fig. 3. In the presence of K+ cations CD spectrum of PMDNAzyme(T10) had a positive signal at around 265 nm and a negative signal at 242 nm, which are characteristic of parallel G-quadruplexes.21 The similar spectrum was measured for the aptamer NH2-T10-EAD2 itself. Therefore, these results demonstrated that EAD2 after its covalent binding with hemin remained the parallel G-quadruplex structure found earlier for the studied aptamer.21,32 Moreover, we showed that CD spectra of PMDNAzyme(T10) in the presence and in the absence of K+ cations were practically identical, whereas CD spectra of NH2-T10-EAD2 changed (Fig. 3). The invariability of CD-spectrum of PMDNAzyme(T10) in the presence and absence of K+ cations allowed us to suppose that its G-quadruplex structure is stabilized by Na+ ions and hemin together, hemin stabilization being due to its interactions with the adjacent G-quartet.
 | ||
| Fig. 3 CD spectra of (1,3) PMDNAzyme(T10) and (2,4) NH2-T10-EAD2 aptamer in the presence (1,2) and absence (3,4) of 20 mM KCl. The samples were dissolved in 25 mM Tris buffer, pH 8.6, 200 mM NaCl. Oligonucleotides concentration was 2 μM. | ||
Capacity of the synthesized covalent PMDNAzymes to maintain quadruplex topology in the absence of potassium was confirmed by gel shift assay experiments (Fig. 2S†). The conjugate EAD2-T10-hemin and its parent aptamer EAD2-T10-NH2 were used, both 5′-end 32P-labelled to simplify the analysis. As seen in Fig. 2S,† the migration rate of EAD2-T10-hemin did not depend on the presence or absence of K+ cations, whereas the mobility of EAD2-T10-NH2 decreased in the absence of potassium because of destruction of its compact quadruplex structure.
Of note, the existence of a parallel G-quadruplex structure in the absence of potassium (but in presence of sodium) ions has been shown for a covalent PMDNAzyme prepared on the base of another hemin aptamer (5′-(G)3TA(G)3C(G)3TT(G)3T-3′).21
3.3. Optimization of conditions of PMDNAzyme(T10) catalysis
It is well known that both native enzymes and their mimetics implement their catalytic potential only under defined conditions. In order to optimize the experimental conditions for PMDNAzyme(T10), we varied concentrations of a substrate-hydrogen donor (luminol and ABTS used in chemiluminescent and colorimetric detection of the PMDNAzyme, respectively) and a substrate-oxidant (hydrogen peroxide) as well as pH value of the substrate solution (Figs. S3–S8†). The most favorable conditions for PMDNAzyme(T10) catalysis of luminol oxidation were found to be 25 mM Tris buffer, pH 8.6 containing 200 mM NaCl, 5 μM luminol and 2 mM H2O2. The ABTS oxidation by PMDNAzyme(T10) was the most efficient in 25 mM citrate-phosphate buffer, pH 6.0 containing 200 mM NaCl, 4 mM ABTS and 12 mM H2O2. It should be noted that contrary to the non-covalent PMDNAzyme, the activity of PMDNAzyme(T10) did not depend on the presence of potassium cations in the reaction solution (data not shown), and this is in good accordance with the above-mentioned CD experiments. In addition, Triton X100 in the concentration up to 0.1% did not affect the PMDNAzyme(T10) activity. This surfactant is commonly used upon preparation of non-covalent PMDNAzymes to transfer hemin from dimeric to monomeric state. Therefore, the absence of Triton X100 effect additionally points to the monomeric form of hemin within PMDNAzyme(T10) (see above).3.4. Analytical parameters of PMDNAzyme(T10) detection
The dependence of the initial rate of ABTS oxidation on the PMDNAzyme(T10) concentration studied under favorable conditions is presented in Fig. 4. The obtained results demonstrated clearly that hemin in the covalent PMDNAzyme is significantly more active than hemin within the non-covalent counterpart. The calibration curve for the PMDNAzyme(T10) allowed calculating a detection limit value (3σ) equal to 1.6 pM. Comparison of this value with the detection limit of the parent non-covalent PMDNAzyme (18 nM) showed that the sensitivity of the synthesized PMDNAzyme(T10) determination was remarkably higher than that of its non-covalent analog (Fig. 4). | ||
| Fig. 4 Dependence of the initial rate of catalytic oxidation of ABTS on the concentration of PMDNAzyme(T10) (1) and the non-covalent PMDNAzyme (2). Experimental conditions: 25 mM citrate-phosphate buffer, pH 6.0, 200 mM NaCl, 20 mM KCl and 0.1% Triton X-100, [ABTS] = 4 mM, [H2O2] = 12 mM. | ||
Although many researchers studying PMDNAzymes and applying them in analytical practice use the ABTS-based colorimetric method to estimate their catalytic activity,33–36 the chemiluminescent method based on luminol oxidation is well-known to be more sensitive. This fact is widespread used in the development of ultrasensitive analytical assays.26–28 Therefore, we studied the dependence of the chemiluminescence intensity produced upon luminol oxidation on the concentration of PMDNAzyme(T10) and its parent non-covalent counterpart (hemin–EAD2 complex) and constructed the calibration curves for both studied HRP mimetics (Fig. 5). The detection limit value of PMDNAzyme(T10) in the chemiluminescent method was 0.2 pM.
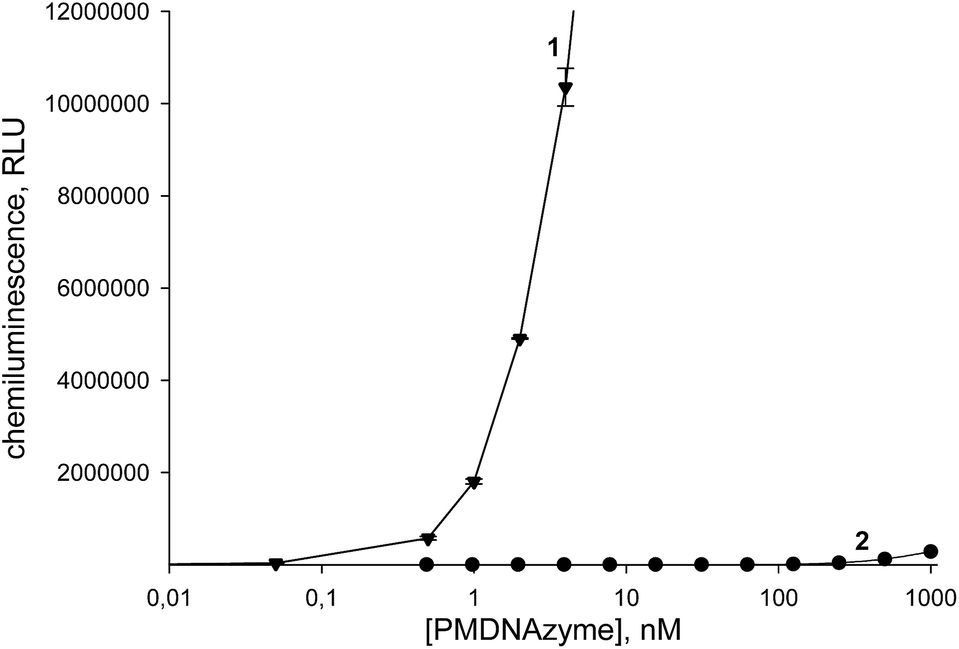 | ||
| Fig. 5 Dependence of the chemiluminescence intensity produced upon catalytic oxidation of luminol under favorable conditions on the concentration of PMDNAzyme(T10) (1) and the non-covalent PMDNAzyme (2). Experimental conditions: (1) 25 mM Tris buffer, pH 8.6 with 200 mM NaCl, [luminol] = 5 μM, [H2O2] = 2 mM; (2) 25 mM Tris buffer, pH 8.0 with 200 mM NaCl, 20 mM KCl, 0.05% Triton X-100, and 1% DMSO, [luminol] = 5 μM, [H2O2] = 1.3 mM. | ||
This value was remarkably lower than that of its non-covalent analog (350 pM). Moreover, the detection limit of the PMDNAzyme(T10) estimated in the chemiluminescent method was one order lower than that of the same catalyst in the colorimetric method. These results clearly demonstrated that upon development of highly sensitive assays it is preferable to apply PMDNAzyme(T10) instead of traditionally used non-covalent PMDNAzymes.
4. Conclusions
In this study we optimized the structure of a covalent PMDNAzyme, a conjugate of hemin with its aptamer EAD2. The influence of the hemin position at 3′- or 5′-end of the aptamer, as well as the effect of the length and nature of spacers between the oligonucleotide and hemin on the covalent PMDNAzyme catalytic activity towards luminol was studied in detail. Among all the conjugates studied, the covalent PMDNAzyme with hemin bound at the 5′-end of EAD2 via dT10 spacer possessed the highest activity. The chemiluminescent activity of the produced PMDNAzyme(T10) was significantly higher in comparison to the non-covalent hemin–EAD2 complex. Taking into account that the detection limit for PMDNAzyme(T10) is similar to that for HRP (0.25 pM), whose enzymatic activity was measured in an enhanced chemiluminescence reaction with the most active enhancers (3-(10′-phenothiazinyl)propane-1-sulfonate or 3-(10′-phenothiazinyl)propionic acid with 4-morpholinopyridine) so far,16,26 the PMDNAzyme(T10) has very promising perspectives for its wide application to develop assays with improved sensitivity. Further our work will be focused on the development of covalent PMDNAzyme-based analytical kits and assays.Acknowledgements
The authors thank K. Sukhoverkov for help with CD measurements and the Russian Foundation for Basic Research (NK-13-04-91164/14) for financial support.Notes and references
- C. Regalado, B. E. Garcia-Almendarez and M. A. Duarte-Vazquez, Phytochem. Rev., 2004, 3, 243 CrossRef CAS.
- B. J. Ryan, N. Carolan and C. O'Fagain, Trends Biotechnol., 2006, 24, 355 CrossRef CAS PubMed.
- M. Seidel and R. Niessner, Anal. Bioanal. Chem., 2014, 406, 5589 CrossRef CAS PubMed.
- S. Baj, R. Slupska and T. Krawczyk, Talanta, 2013, 103, 172 CrossRef CAS PubMed.
- M. M. Vdovenko, A. S. Demiyanova, K. E. Kopylov and I. Y. Sakharov, Talanta, 2014, 125, 361 CrossRef CAS PubMed.
- S. Xu, W. Liu, B. Hu, W. Cao and Z. Liu, J. Photochem. Photobiol., A, 2013, 227, 32 CrossRef PubMed.
- L. Gao, J. Zhuang, L. Nie, J. Zhang, Y. Zhang, N. Gu, T. Wang, J. Feng, D. Yang, S. Perrett and X. Yan, Nat. Nanotechnol., 2007, 2, 577 CrossRef CAS PubMed.
- P. Travascio, Y. Li and D. Sen, Chem. Biol., 1998, 5, 505 CrossRef CAS.
- D. Sen and L. C. H. Poon, Crit. Rev. Biochem. Mol. Biol., 2011, 46(6), 478 CAS.
- J. Kosman and B. Juskowiak, Anal. Chim. Acta, 2011, 707, 7 CrossRef CAS PubMed.
- F. Wang, C.-H. Lu and I. Willner, Chem. Rev., 2014, 114, 2881 CrossRef CAS PubMed.
- S. Nakayama and H. O. Sintim, Mol. BioSyst., 2010, 6, 95 RSC.
- M. Luo, X. Chen, G. Zhou, X. Xiang, L. Chen, X. Ji and Z. He, Chem. Commun., 2012, 48, 1126 RSC.
- L. Gong, Z. Zhao, Y.-F. Lv, S.-Y. Huan, T. Fu, X.-B. Zhang, G.-L. Shen and R.-Q. Yu, Chem. Commun., 2015, 51, 979 RSC.
- A. V. Gribas, S. Zhao and I. Y. Sakharov, Anal. Biochem., 2014, 466, 19 CrossRef CAS PubMed.
- M. M. Vdovenko, A. S. Demiyanova, T. A. Chemleva and I. Y. Sakharov, Talanta, 2012, 94, 223 CrossRef CAS PubMed.
- L. Stefan, H.-J. Xu, C. P. Gros, F. Denat and D. Monchaud, Chem.–Eur. J., 2011, 17, 10857 CrossRef CAS PubMed.
- L. Zhu, C. Li, Z. Zhu, D. Liu, Y. Zou, C. Wang, H. Fu and C. J. Yang, Anal. Chem., 2012, 84, 8383 CrossRef CAS PubMed.
- T. Li, E. Wang and S. Dong, PLoS One, 2009, 4, e5126 Search PubMed.
- D. Thirstrup and G. S. Baird, Anal. Chem., 2010, 82, 2498 CrossRef CAS PubMed.
- S. Nakayama, J. Wang and H. O. Sintim, Chem.–Eur. J., 2011, 17, 5691 CrossRef CAS PubMed.
- X. Cheng, X. Liu, T. Bing, Z. Cao and D. Shangguan, Biochemistry, 2009, 48, 7817 CrossRef CAS PubMed.
- C. Qi, N. Zhang, J. Yan, X. Liu, T. Bing, H. Mei and D. Shangguan, RSC Adv., 2014, 4, 1441 RSC.
- A. C. McGinnis, E. C. Grubb and M. G. Bartlett, Rapid Commun. Mass Spectrom., 2013, 27, 2655 CrossRef CAS PubMed.
- T. Xue, S. Jiang, Y. Q. Qu, Q. Su, R. Cheng, S. Dubin, C.-Y. Chiu, R. Kaner, Y. Huang and X. F. Duan, Angew. Chem., Int. Ed., 2012, 51, 3822 CrossRef CAS PubMed.
- I. Y. Sakharov, A. S. Demiyanova, A. V. Gribas, N. A. Uskova, E. E. Efremov and M. M. Vdovenko, Talanta, 2013, 115, 414 CrossRef CAS PubMed.
- C. A. Marquette and L. J. Blum, Bioanalysis, 2009, 1, 1259 CrossRef CAS PubMed.
- L. Zhao, L. Sun and X. Chu, Trends Anal. Chem., 2009, 28, 404 CrossRef CAS PubMed.
- M. Zhang, H. Li, M. Deng, X. Weng, H. Ma, S. Feng, Y. Zhou and X. Zhou, Chem. Biodiversity, 2012, 9, 170 CAS.
- D. M. Kong, W. Yang, J. Wu, C. X. Li and H. X. Shen, Analyst, 2010, 135, 321 RSC.
- S. Nakayama and H. O. Sintim, J. Am. Chem. Soc., 2009, 131, 10320 CrossRef CAS PubMed.
- P. Tóthová, P. Krafcíková and V. Víglasky, Biochemistry, 2014, 53, 7013 CrossRef PubMed.
- M. Deng, D. Zhang, Y. Zhou and X. Zhou, J. Am. Chem. Soc., 2008, 130, 13095 CrossRef CAS PubMed.
- X. Zhu, H. Zhang, C. Feng, Z. Ye and G. Li, RSC Adv., 2014, 4, 2421 RSC.
- L. Zhou, F. Du, Y. Zhao, A. Yameen, H. Chen and Z. Tang, Biosens. Bioelectron., 2013, 45, 141 CrossRef CAS PubMed.
- Y. Weizmann, M. K. Beissenhirtz, Z. Cheglakov, R. Nowarski, M. Kotler and I. Willner, Angew. Chem., Int. Ed., 2006, 45, 7384 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional information as noted in the text (Table S1 and Fig. S1–S6). See DOI: 10.1039/c5ra03167b |
| This journal is © The Royal Society of Chemistry 2015 |