DOI:
10.1039/C5RA06024A
(Paper)
RSC Adv., 2015,
5, 51662-51671
A novel electrochemical nicotine sensor based on cerium nanoparticles with anionic surfactant
Received
4th April 2015
, Accepted 21st May 2015
First published on 26th May 2015
Abstract
A novel promising electrochemical nicotine (NIC) sensor was prepared by electrodeposition of Ce-nanoparticles on a carbon paste electrode (CPE). Electrochemical techniques including cyclic voltammetry (CV), electrochemical impedance spectroscopy (EIS), scanning electron microscope (SEM) and Energy Dispersive X-ray Analysis (EDX) techniques, in both aqueous and micellar media were used. NIC measurements were investigated in Britton–Robinson (B–R) buffer solutions with a pH range (2.0–8.0) containing (1.0 mM) sodium dodecyl sulfate (SDS). The linear response range of the sensor was between 4 × 10−6 M and 5 × 10−4 M with a detection limit of 9.43 × 10−8 M. Good results were achieved for the detection of NIC in real samples and with different brands of commercial cigarettes.
1. Introduction
Nicotine, 3-(1-methyl-2-pyrrolidinyl)pyridine, is the main alkaloid in tobacco leaves (Nicotiana tabacum L., Solanaceae), the latter being best known for their use in cigarettes rather than for their therapeutical applications.1,2 Nicotine (Scheme 1) is one of the most heavily used addictive substances ever available. Pharmacologically, nicotine is a compound which acts on the central nervous system in the form of mood elevations, a sense of euphoria and revitalizing energy. However, it has some very potential health hazards, primarily cardiovascular and respiratory disorders including lung cancer. Apart from its harmful effects, studies have also been carried out to understand its medicinal value with the diseases like ulcerative colitis, Alzheimer's, and Parkinson's diseases.3 Moreover, nicotine determination is an important analysis for the tobacco industry, since the quality of the product can be determined by its nicotine content, which varies with tobacco type; but the normal nicotine level range is from 1% to 3%.1,4 Thus, its determination is important in medicine, toxicology, and the tobacco industry. In the past, there has been considerable interest in development of methods for nicotine determination including spectrophotometry,5 chromatography,6 reverse HPLC,7 and potentiometry.8,9 Some of these methods require preliminary extraction and purification of nicotine from the sample matrix plus skilled personnel manipulating sophisticated instrumentation;3 this may lead to considerable losses of the analyte. From electrochemical considerations, not many detection studies have been made on this compound. This is mainly because nicotine oxidation/reduction processes occur at extremely positive/negative potentials, which are out of the potential window of conventional electrodes.1 Suffredini et al. made progress in the area of nicotine peak separation using a boron-doped diamond electrode in basic media.10 Hannisdal et al. investigated the electroreduction of nicotine with a dropping mercury electrode to perform its quantification in chewing gum, patches, and tablets.11 Amperometric assays based on nicotine inhibition biosensors were applied and provided a promising approach to the determination of nicotine in tobacco.12 Amperometric detection of nicotine also was studied using a titanium dioxide/poly(3,4-ethylenedioxythiophene) modified electrode by a molecular imprinting technique.13 Lin et al. described a flow injection-electrochemiluminescent method based on a tri(2,2-bipyridyl)ruthenium(II) complex system for nicotine quantification.14 Sims et al. also studied the effects of thin-layer diffusion in the electrochemical detection of nicotine on basal plane pyrolytic graphite (BPPG) electrodes modified with layers of multi-walled carbon nanotubes (MWCNT-BPPG).15 Tar Wu et al. analyzed for nicotine using molecularly imprinted TiO2-modified electrodes.16 Recently, a sensitive determination of nicotine in tobacco products and anti-smoking pharmaceuticals used a boron-doped diamond electrochemical sensor.17 In this work, we introduce a sensitive electrochemical procedure for NIC detection based on cerium nanoparticles with a modified carbon paste electrode (CNMCPE) that can overcome obstacles facing conventional electrodes. Due to its low cost, high sensitivity, low background current, ease of fabrication, and renewable surface, carbon paste electrodes have been widely used for the electroanalytical applications.18 Nanoparticles research is advancing at a fast place due to their unique properties, such as having increased electrical conductivity, toughness and ductility, increased hardness and strength of metals and alloys. Electrodeposition of Ce-nanoparticles on a CPE surface was an important method for detection of NIC. Ce nanoparticles were shown to be very important in the area of electrochemical sensors, biosensors19 and batteries.20
 |
| | Scheme 1 Chemical structure of nicotine. | |
Surfactants have been widely used in chemistry and have proven to be effective in electroanalysis of biological compounds and drugs. The use of surfactant solutions as modifiers improves the sensitivity and selectivity of voltammetric measurements.1
To our knowledge, there is no work had been made on CP-Ce nanoparticles sensor for the determination of nicotine using an anionic surfactant. Thus, the aim of this work is to construct a novel and sensitive electrochemical sensor based on Ce nanoparticles, graphite, and SDS to be used for the determination of nicotine in urine samples and cigarettes.
2. Experimental
2.1. Reagent and chemicals
Nicotine standard samples (99%) were provided by the Egyptian Eastern Company for Smoking and were used as received without prior purification. Nicotine stock solutions of about 1.62 g L−1 were freshly prepared in water and then stored in a dark container since the compound is sensitive to light. Supporting electrolyte, namely, Britton–Robinson (B–R) (4.0 × 10−2 M) buffer solutions of pH 2.0–8.0 (CH3COOH + H3BO3 + H3PO4) were used for preparing the standard solutions of nicotine. pH values were adjusted using 0.2 M NaOH. Sodium dodecyl sulfate (SDS) was purchased from Sigma-Aldrich. All solutions were prepared from analytical grade chemicals and triply distilled water. Cigarette samples were purchased from local supermarkets. Two products of different international cigarette brands: L&M® and Marlboro® were chosen. All experiments were carried out at the room temperature of the laboratory for 2–3 times and gave reproducible results.
2.2. Preparation of cerium nanoparticles modified CP electrode (CNMCPE)
A carbon paste electrode (CPE) with a diameter of 3 mm was prepared using a few drops of paraffin oil mixed with graphite powder (0.5 g) and the mixture was blended thoroughly to obtain a homogeneous paste.21 The resulting mixture was used to fill into a Teflon tube and pressed tightly. The CPE was polished with ultra fine emery paper until a smooth surface was achieved. The CPE surface was pre-treated by applying a potential of +1.3 V for 30 s in the blank supporting electrolyte without stirring, in order to increase the hydrophilic properties of the electrode surface through introduction of oxygenated functionalities, accomplished with an oxidative cleaning. A CPE modified with Ce nanoparticles was formed by dipping the clean CPE into 0.1 mM ammonium ceric nitrate solution, with about 2 mL lactic acid, and the solution pH was adjusted using (33%) ammonium hydroxide solution followed by scanning the electrode in the potential range from −2.0 to +2.0 V at 25 mV s−1 for 5 cycles at 25 °C.22
2.3. Electrochemical measurements
All electrochemical measurements were investigated using an electrochemical IM6e ZAHNER-elektrik GmbH (Kronach, Germany) workstation. A typical three-electrode instrument fitted with a large platinum sheet of size 15 × 20 × 2 mm as a counter electrode (CE), saturated calomel electrode (SCE) as a reference electrode (RE), and CNMCPE as the working electrode (WE) were used. A HANNA 213 pH meter with a glass combination electrode was used for pH measurements.
Scanning electron microscopy (SEM) measurements were carried out using a SEM Model Quanta 250 FEG (Field Emission Gun) attached with EDX Unit (Energy Dispersive X-ray Analyses), with accelerating voltage 30 kV, magnification 14× up to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000, and resolution for the Gun of 1n (FEI company, Netherlands).
000000, and resolution for the Gun of 1n (FEI company, Netherlands).
All the electrochemical experiments were carried out and repeated for 2–3 times at a temperature of 25 °C and gave reproducible results.
2.4. Impedance spectroscopy measurements
Electrochemical impedance spectroscopy (EIS) was carried out using the electrochemical workstation IM6e ZAHNER-elektrik GmbH (Kronach, Germany). All impedance experiments were recorded between 0.1 Hz and 100 kHz at 10 mV amplitude. Analyses of the experimental spectra were made using Thales software provided with the workstation.
2.5. Analysis of urine
Standard NIC provided by the Egyptian Eastern Company for Smoking was dissolved in urine (diluted 400 times using 100 mL of B–R buffer, pH 2) to make a stock solution. Standard additions of nicotine were carried out from nicotine standard samples. Standard additions of urine were carried out from a solution containing NIC in 23 mL of B–R buffer (pH 2) and 1.0 mM sodium dodecyl sulfate in a voltammetric cell and analyzed under the same conditions as used to obtain a calibration graph.
2.6. Analysis of cigarettes samples
Cigarettes were taken out of their rolling paper and dried for 30 minutes at 40 °C in an oven before weighing. Then 0.1 g from a cigarette tobacco mixture of ten cigarettes taken from two packs of the same brand was placed in a 50 mL glass vial with a cap. We added 10 mL of water using a pipette, and then the contents of the vial were sonicated for 3 h in an ultrasonic water bath and filtered. Appropriate volumes (100 μL) of the clear filtrate were mixed with the B–R buffer (pH 2.0) containing 1.0 mM sodium dodecyl sulfate in the voltammetric cell and analyzed under the same conditions as used to obtain the calibration graph.1
3. Results and discussion
3.1. Morphologies of the different electrodes
SEM images of the CPE and CNMCPE are shown in Fig. 1A and B, respectively and shows their physical morphology. The SEM image of CPE (Fig. 1A) shows a microstructure with a discontinuous grain growth and also shows the graphite particles are covered by a very thin film of paraffin oil. The SEM image of CNMCPE (Fig. 1B) shows that metallic nanoparticles are located at different elevations over the CPE surface. Moreover, a random distribution and interstices among the nanoparticles were observed in the SEM image of the CNMCPE exhibiting a large random surface area.
 |
| | Fig. 1 SEM images for (A) CPE and (B) CNMCPE, and (C) EDX spectra of CeO2 nanoparticles. | |
EDX data of the CeO2 nanoparticles shown in Fig. 1C indicates the presence of Ce and O peaks, and confirms that CeO2 nanoparticles were indeed coated on the electrode surface. These results suggest that CeO2 nanoparticles were successfully prepared by this simple electrochemical method.
3.2. Electrochemistry of NIC using SDS
Fig. 2 compares cyclic voltammograms of 1.0 × 10−3 M NIC in B–R buffer (pH 2.0) at a scan rate of 25 mV s−1 recorded at three different electrodes i.e. (1) bare CPE, (2) CNMCPE, and (3) CNMCPE/1.0 mM SDS. With the bare CPE there is no oxidation peak observed, while the electrodeposition of Ce-nanoparticles onto the surface of the CPE (CNMCPE) was an effective strategy to enhance the detection of NIC. Thus, CPE represents the best electrode type for Ce-nanoparticles electrodeposition because of no adsorption of Ce3+ or Ce4+ or formation of oxides on the surfaces of graphite electrodes.19 Cerium dioxide (CeO2) is the most stable oxide of cerium. This compound is also called ceria or ceric oxide.22
 |
| | Fig. 2 CVs of bare CPE, CNMCPE, and CNMCPE/SDS in B–R buffer pH 2.0 containing 1.0 × 10−3 M NIC at 25 mV s−1. | |
The surfactant has two important roles: first it can stabilize radical ions and other reaction intermediates which effect the mechanism of the electrode reaction. Second, it modifies the double layer structure.1 The sensitivity of CNMCPE/SDS for NIC determination was clearly increased from indications that the rate of electron transfer was increased. The surfactant-modified electrode sensitivity was about 2.45 times higher than CNMCPE. Nicotine exists as a positively charged protonated form under the studied pH condition (pH 2.0).23 On the other hand, SDS adsorption onto electrode surface forms a negatively charged hydrophilic film oriented towards the water bulk phase. Based on this fact, positive charged nicotine has a tendency to accumulate in the negatively charged crown of anionic SDS micelles, which may increase the electron transfer rate.24
3.3. Effect of pH
The effect of pH was evaluated for CNMCPE/SDS using B–R buffers within the pH range of 2.0–8.0 (Fig. 3). Anodic peak potentials shifted negatively with an increase in the pH, indicating that electrocatalytic oxidation of NIC at the CNMCPE/SDS is a pH-dependent reaction and showing that protons have taken part in their electrode reaction processes. Even though the exact oxidation mechanism of nicotine has not yet been determined, according to a hypothesis proposed by Suffredini et al.10 the oxidation mechanism in alkaline solutions with a boron-doped diamond electrode involves formation of methanol and substitution of the CH3 to OH on the tertiary nitrogen of a pyrrolidine ring with two electron transitions.
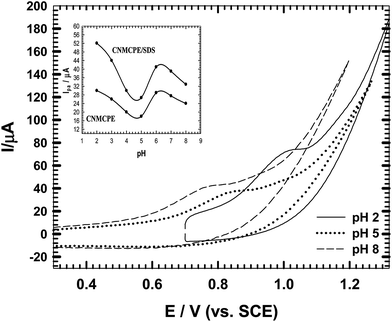 |
| | Fig. 3 CVs of CNMCPE/SDS in B–R buffer of different pH values containing 1.0 × 10−3 M NIC. Inset: variation of anodic peak current (Ipa) for CNMCPE and CNMCPE/SDS values with pH values. | |
The highest oxidation peak current was obtained at pH 2 (inset), then the peak current decreased from pH 2.0 to pH 5.0 and increased again up to pH 8.0. Nicotine is a weak diacidic base having two pKa values, pKa1 = 8.02 and pKa2 = 3.12 which correspond to the protonation of pyrrolidine nitrogen (monoprotonated form) and pyridine nitrogen (diprotonated form) present in a nicotine molecule, respectively.23 These results strongly prove that the oxidation step of nicotine is located on the pyrrolidine ring and attributed to the oxidation of the tertiary nitrogen.
3.4. Effect of scan rate
Cyclic voltammetry experiments were carried out as a function of scan rate to give information related to the adsorptive properties of the reaction studied. A plot of Ipa vs. v1/2 (v ranging from 10 to 100 mV s−1) for 1.0 × 10−3 M NIC using CNMCPE/SDS in B–R buffer (pH 2.0) gave a straight line relationship. The linearity of the relationship indicated a diffusion controlled mechanism and that the adsorption of aggregates at the electrode surface was also possible. Typical CV curves of NIC at different scan rates were shown in Fig. 4. The oxidation peak currents increased linearly (inset) with the regression coefficient r2 = 0.9997 & γ = 5 respectively, suggesting that the reaction is a diffusion-controlled electrode reaction.
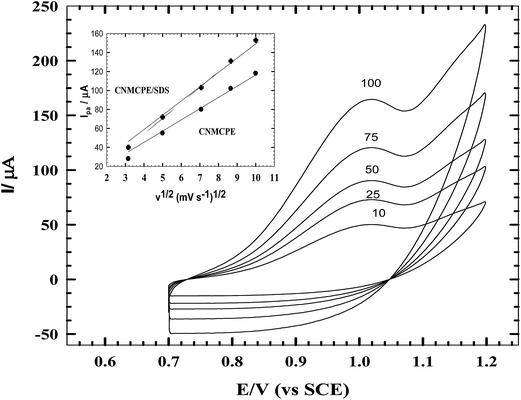 |
| | Fig. 4 CVs of CNMCPE/SDS in B–R buffer of pH 2.0 containing 1.0 × 10−3 M NIC at different scan rates. Inset: variation of anodic peak current (Ipa) with the square root of the scan rate (v1/2) for CNMCPE and CNMCPE/SDS. | |
The relation between the anodic peak current and scan rate has been used to evaluate the “apparent” diffusion coefficient, Dapp, for studied compounds. Dapp values were calculated from the Randles Sevcik equation.25
| | |
Ipa = 0.4463 nFAC0 (nFvD/RT) 1/2
| (1) |
In this equation: Ipa is the maximum current (A), n is the number of electrons transferred in the redox event, C0 is the analyte concentration (1 × 10−6 mol cm−3), A is the electrode area in cm2, D is the electroactive species diffusion coefficient (cm2 s−1), ν is the scan rate in V s−1, F is Faraday Constant in C mol−1, T is the temperature in K, and R is the Gas constant in VC K−1mol−1.
The apparent diffusion coefficients, Dapp, of NIC using CNMCPE and CNMCPE/SDS in B–R buffer (pH 2.0) were calculated from cyclic voltammetry (CV) experiments and were found to be 4.195 × 10−5 cm2 s−1 and 7.189 × 10−5 cm2 s−1, respectively; this indicates that the redox reaction of the analyte species takes place at the surface of the electrode under the control of the diffusion of the molecules from solution to the electrode surface.
3.5. Effect of accumulation time
In order to investigate the response of CNMCPE/SDS, the CV for 1.0 × 10−3 M NIC in B–R buffer (pH 2.0) solution were recorded every three minutes (Fig. 5). It was found that the anodic peak currents increased with increasing accumulation time. The highest current response for CNMCPE was reached after 21 minutes at 1.0 V vs. SCE, so the optimum time for electrode stability is 21 minutes. Repetitive measurements indicated that this electrode has good reproducibility and stability during the voltammetric measurements.
 |
| | Fig. 5 Variation of anodic peak current (Ipa) with different accumulation times: (1–40 minutes) of CNMCPE in B–R buffer pH 2.0 containing 1.0 × 10−3 M NIC. Inset: the CVs of B–R buffer (pH 2.0) solution containing 1.0 × 10−3 M NIC recorded every three minutes. | |
3.6. Electrochemical impedance spectroscopy (EIS) measurements
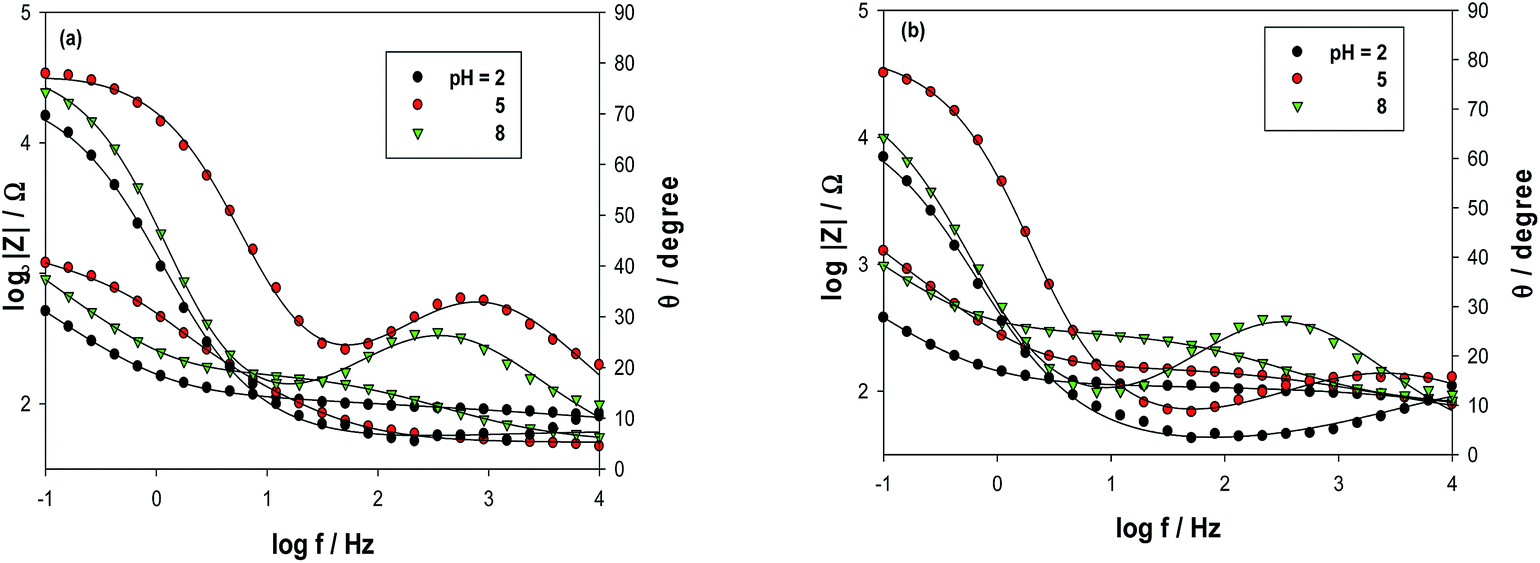
Electrochemical impedance spectroscopy (EIS) is an effective tool for studying the interfacial properties of surface-modified electrodes.26 The impedance plots are shown as both Bode (Fig. 6a and b) and Nyquist plots (Fig. 7a and b), respectively. The Bode and Nyquist plots are provided for CNMCPE at 1 × 10−3 M NIC solution only (Fig. 6a and 7a) and CNMCPE/SDS at 1 × 10−3M NIC solution (Fig. 6b and 7b), respectively. The Nyquist plots include a small semicircle corresponding to charge transfer resistance and a linear part corresponding to diffusion process. The impedance data were thus simulated to the appropriate equivalent circuit for cases exhibiting two time constants (Scheme 2). This simulation gave a reasonable fit with 1% average error. The appropriate equivalent model used to fit the high and low frequency data consists of two circuits in series from ZwCPE1 and RCTCPE2 in parallel combination and both are in series with Rs. CPE1 is related to the constant phase element (CPE) of the capacitance of the inner layer and CPE2 to the outer layer, while RCT is the charge transfer resistance of the outer layer related to the small semicircle at high frequency.27,28 Zw is a Warburg component related to the linear region at the lower frequencies in the Nyquist plot which is related to diffusion phenomena.29,30 The experimental data were fitted using Thales software provided with the workstation where the dispersion formula suitable to each model was used.27,30 In this complex formula an empirical exponent (α = 0 to 1) is introduced to account for the deviation from capacitive ideality behavior due to surface roughness.31,32 An ideal capacitor corresponds to α = 1 while α = 0.5 becomes the CPE in a Warburg component.29 In all cases, good conformity between theoretical and experimental results was obtained with an average error of 1%.
 |
| | Fig. 6 Bode plots of NIC for (a) CNMCPE, (b) CNMCPE/SDS at different pH values. | |
 |
| | Fig. 7 Nyquist plots of NIC for (a) CNMCPE, (b) CNMCPE/SDS at different pH values. | |
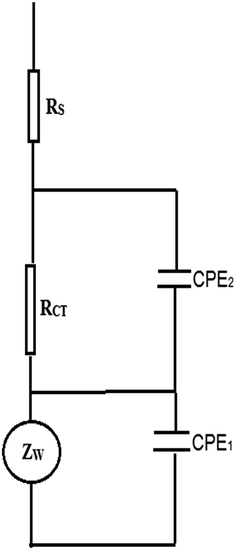 |
| | Scheme 2 The equivalent circuit used in the fitting experiments of the EIS. | |
Table 1 lists the best fitting values calculated from the equivalent circuit for the impedance data in the two different cases for the CNMCPE. The value of solution resistance, Rs, for each pH value was almost constant within the limits of the experimental errors. CNMCPE/SDS shows relatively higher values for the capacitance or lower values for the impedance compared to CNMCPE, indicating a more conducting behavior due to electrode surface ionic adsorption by SDS. Also, the same is observed for ZW values, where higher values were obtained for CNMCPE/SDS than for CNMCPE. Also, the lowest RCT or highest CPE values obtained at pH = 2 for both tested electrodes indicated higher conductivity and confirm that the highest oxidation peak current was obtained from CV's results.
Table 1 Electrochemical impedance fitting parametersa
| Electrode |
pH |
Rs/Ωcm2 |
Zw/kΩ cm2 s−1/2 |
CPE1/μF cm−2 |
α1 |
RCT/kΩ cm2 |
CPE2/μF cm−2 |
α2 |
| Rs: the solution resistance, Zw: Warburg impedance, CPE: Constant phase element, α: correlation coefficients, RCT: charge transfer resistance. |
| CNMCPE |
2.0 |
163 |
153.2 |
0.34 |
0.55 |
1.27 |
30.85 |
0.86 |
| 5.0 |
142 |
49.81 |
0.12 |
0.64 |
1.88 |
19.44 |
0.87 |
| 8.0 |
170 |
70.99 |
0.31 |
0.60 |
1.49 |
22.40 |
0.94 |
| CNMCPE/SDS |
2.0 |
148 |
562.8 |
0.49 |
0.50 |
0.87 |
62.34 |
0.78 |
| 5.0 |
126 |
201.1 |
0.13 |
0.60 |
1.45 |
37.09 |
0.91 |
| 8.0 |
112 |
552.2 |
0.43 |
0.54 |
1.13 |
57.48 |
0.91 |
ZW is characterized by a very low frequency slope of −0.5 and it intercepts on the logZ axis at f = 1 Hz of σπ−1/2, where σ is the Warburg impedance coefficient (ohm cm2 s−1/2).33,34
| |
 | (2) |
The value of σ can be obtained using eqn (2) from logZ values at f = 1, which are 2.208 and 2.176 for both CNMCPE and CNMCPE/SDS at pH = 2, respectively. The σ values are evaluated as 286.25 for CNMCPE and 266.17 for CNMPE/SDS; this means that diffusion processes take place on the electrode. The diffusion coefficient at pH = 2 can be calculated using the following equation:35
| |
 | (3) |
where
D is the diffusion coefficient (cm
2 s
−1),
A is the area of the electrode (cm
2),
σ is the Warburg coefficient (ohm cm
2 s
−1/2),
C is the concentration of nicotine (mol cm
−3),
R is the gas constant, J K
−1 mol
−1,
T is the temperature (K), and
F is Faraday constant (C mol
−1).
D is found to be equal to 8.65 × 10
−5 for CNMCPE and 10.01 × 10
−5 for CNMCPE/SDS at pH = 2. Thus, diffusion coefficient values calculated from EIS measurements are comparable to those obtained from CV's measurements. This confirms CV's data indicating that the redox reaction of the analyte species takes place under a diffusion control process.
3.7. Calibration curve
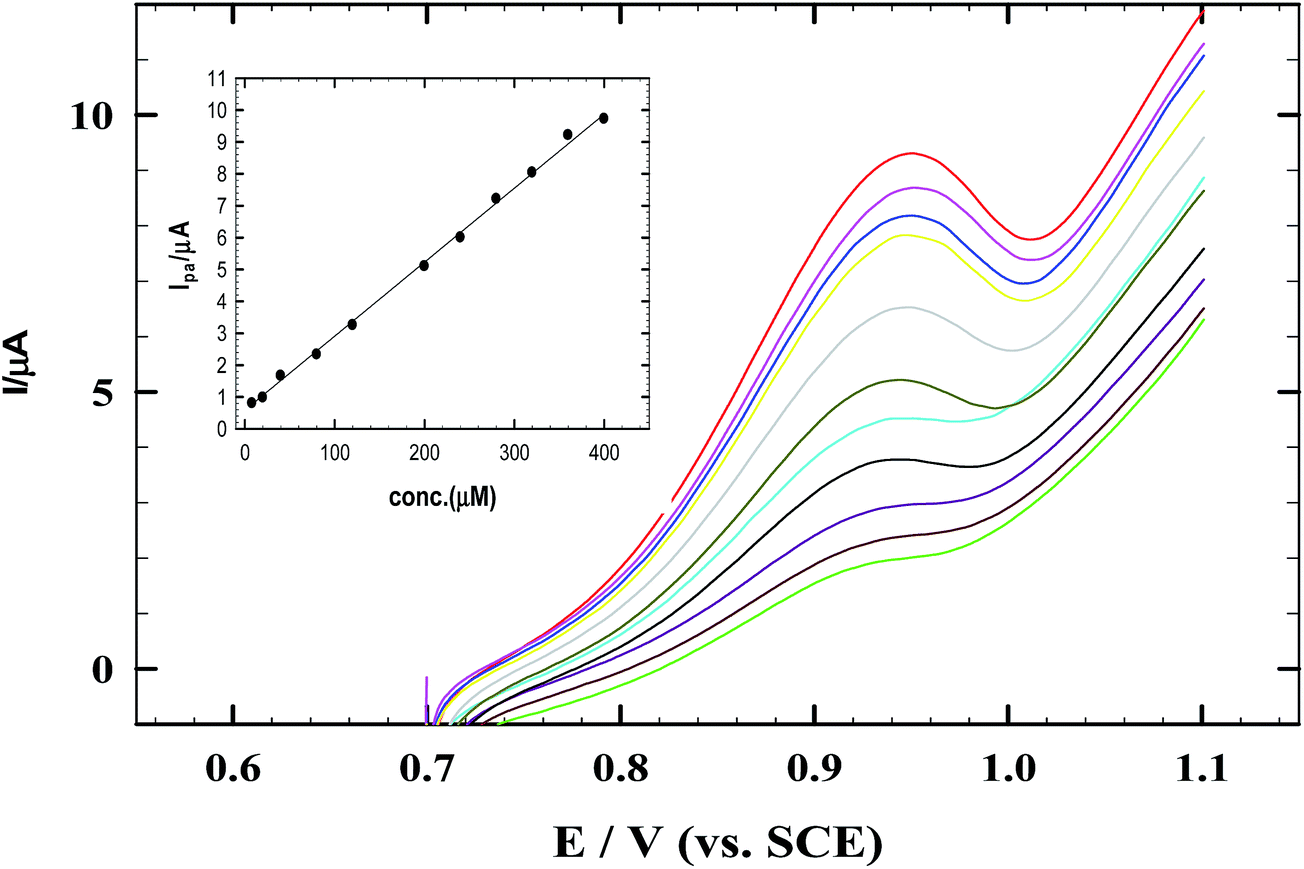
Fig. 8 represents CV data for calibration experiments processed on CNMCPE for the determination of nicotine using successive additions of 1 × 10−3 M NIC in 0.04 M B–R buffer at pH 2.0 containing 1 mM SDS solution (optimized conditions) and scan rate of 10 mV s−1.
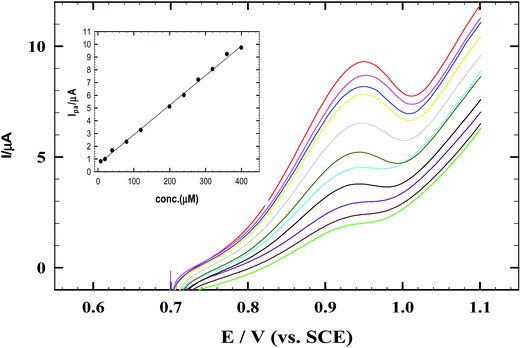 |
| | Fig. 8 The CVs of successive addition of NIC and 1.0 mM SDS in B–R buffer pH 2.0 at scan rate 10 mV s−1 using CNMCPE/SDS. Inset shows the corresponding calibration curve of NIC. | |
This CNMCPE method shows specific and well defined concentration dependence as CNMCPE showed a very fast and sensitive response to changes of NIC concentration. Fig. 8 inset represents the calibration curve obtained by successive additions of nicotine over 4.0 × 10−6 to 5.0 × 10−4 M concentration ranges at which the electrode provided a linear response toward NIC with a correlation coefficient of 0.999. The concentration of NIC was calculated from the linear regression equation of the standard calibration curve: Ipa (μA) = 0.023C (M) + 0.5886.
The sensitivity of the proposed method was evaluated using both the limit of detection (LOD) and limit of quantification (LOQ) values. The LOD and LOQ were calculated using the following equations:
where
s is the standard deviation of the oxidation peak current (three runs) and
b is the slope (μA/M) of the related calibration curves; they were found to be 9.43 × 10
−8 M and 3.14 × 10
−7 M, respectively.
3.8. Comparison of several methods for the determination of NIC
Comparisons of the data obtained for NIC determination by various methods and electrochemical techniques are shown in Table 236–39 and Table 3,1,10,12,16,40 respectively. Compared with other current techniques for the determination of NIC, the CNMCP sensor has some advantages. The sensor has the advantage of using non-poisonous and lower cost reagents giving good stability although it has nearly the same specific selectivity. Also, HPLC, GC, SPE and spectrometry instruments are more sophisticated and expensive than that of the CNMCP sensor. Furthermore, the procedures and pre-treatment for these methods are complicated while that used for CNMCP sensor are very easy and simple.
Table 2 Comparison of the CNMCP sensor with other methods for the determination of NIC
| Method |
Calibration range (M) |
Detection limit (M) |
Reference |
| HPLC |
6.8 × 10−6 to 3.4 × 10−5 |
6.2 × 10−7 |
36 |
| CE |
8.1 × 10−6 to 8.1 × 10−5 |
3.8 × 10−7 |
37 |
| Spectrometry |
Up to 7.4 × 10−5 |
|
38 |
| Flow injection |
0 to 5.8 × 10−2 |
6.2 × 10−7 |
39 |
| CNMCP sensor |
4.0 × 10−6 to 5.0 × 10−4 |
9.4 × 10−8 |
This work |
Table 3 Comparison of the CNMCP sensor with different modified electrodes for the determination of NIC
| Method |
Calibration range (M) |
Detection limit (M) |
Reference |
| Pencil graphite electrode |
7.0 × 10−6 to 1.07 × 10−4 |
2.0 × 10−6 |
1 |
| BDD |
5.0 × 10−6 to 5.0 × 10−4 |
3.1 × 10−6 |
10 |
| Cho enzyme biosensor |
9.2 × 10−5 to 2.0 × 10−4 |
1.0 × 10−5 |
12 |
| TiO2/MI-PEDOT |
0 to 5.0 × 10−3 |
4.9 × 10−6 |
16 |
| p-(AHNSA/GCE) |
1.0 × 10−6 to 2.0 × 10−4 |
0.87 × 10−6 |
40 |
| CNMCP sensor |
4.0 × 10−6 to 5.0 × 10−4 |
9.4 × 10−8 |
This work |
3.9. Application of CNMCP sensor in urine
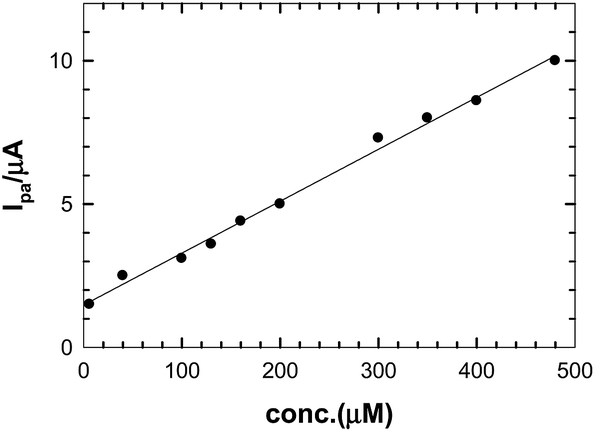
Fig. 9 shows the calibration curve for using the CNMCP sensor with real samples, such as as urine, which gave a straight line in a concentration range of 6 × 10−6 M–4.8 × 10−4 M. From the equation of the calibration curve (Ipa (μA) = 0.0181C (M) + 1.4796), the concentration of NIC in urine samples was calculated. The correlation coefficient was r2 = 0.997, the LOD was 2.06 × 10−7 M, and the LOQ was 6.89 × 10−7 M. To ensure the validation of the proposed method in urine samples, Table 4 shows evaluations of precision and accuracy of the proposed method for NIC detection for four different concentrations on the calibration curve; each was repeated five times. The recovery, standard deviation, standard error, and the confidence level were calculated as well.
 |
| | Fig. 9 Calibration curve of NIC in urine. | |
Table 4 Precision and accuracy of the CNMCP sensor for NIC detection in a urine sample
| [NIC] added (M) × 10−6 |
[NIC] founda (M) × 10−6 |
Recovery (%) |
SD × 10−6 |
S.Eb × 10−6 |
C.L.c × 10−6 |
| Mean for five determinations. Standard error = SD/n1/2. Confidence at 95% confidence level. |
| 12.00 |
12.03 |
100.2 |
0.05 |
0.02 |
0.08 |
| 60.00 |
60.20 |
100.3 |
3.61 |
0.18 |
0.58 |
| 120.0 |
119.77 |
99.80 |
0.22 |
0.11 |
0.35 |
| 300.0 |
299.25 |
99.75 |
1.70 |
0.85 |
2.77 |
3.10. Analysis of real cigarette brands samples
For a practical application, two products of different cigarette brands were analyzed (L&M and Marlboro) to ensure the validation of the proposed method with real samples. An ultrasonication method with the tobacco samples, which was used in recent studies, is considered as an alternative technique; otherwise the most commonly used methods for nicotine analysis in tobacco samples require expensive and environmentally harmful organic solvents with complex extraction procedures.1 Sample extraction efficiency can be greatly increased when bubbles created by the sonication of solutions are collapsed because that result in the generation of high local energy and a high contact between solvent and solute. The method used for preparation of samples in this study was a slight modification of the method described by Suffredini et al.10 In our study, an ultrasonic extraction time of 3 h was chosen instead of 1 h in order to achieve a quantitative transfer of nicotine into an aqueous solution. The precision and accuracy of the method can be evaluated from the results obtained in the analysis of different cigarette samples as shown in Table 5. In practice, it is necessary to know that the nicotine reported in cigarettes corresponds to the amount of nicotine which could be absorbed by a smoker when a cigarette is smoked under standard conditions.
Table 5 The recovery analysis of nicotine in cigarette tobacco
| Cigarette brand |
[Nic.] taken × 10−6 M |
[Standard] added × 10−6 M |
Found × 10−6 M |
Recovery (%) |
| L&M |
60.00 |
60.00 |
120.26 |
100.22 |
| 120.00 |
— |
179.83 |
99.91 |
| 180.00 |
— |
239.69 |
99.87 |
| 240.00 |
— |
300.13 |
100.04 |
| Marlboro |
60.00 |
— |
120.30 |
100.25 |
| 120.00 |
— |
180.06 |
100.03 |
| 180.00 |
— |
239.98 |
99.99 |
| 240.00 |
— |
300.19 |
100.06 |
In our study we made spike/recovery experiments in order to test the validation of the proposed method. Recovery studies were performed by adding the appropriate volume of a standard nicotine solution prepared in supporting electrolyte to the nicotine content previously determined in a tobacco sample. Comparing the concentration obtained from the spiked mixtures with those of the pure nicotine, we were able to calculate nicotine recovery. The recovery data ensure the accuracy of the voltammetric detection of nicotine in tobacco as it was found that nicotine amounts can be quantitatively recovered by the proposed method.
Because of the low concentrations of other minor alkaloids (0.2–0.5% of total alkaloids), they can't affect the accuracy of nicotine detection at the sensitivity level of voltammetric measurements; thus, the interference of alkaloids which may be present in tobacco was not performed in this study.4
4. Conclusions
In the present study we introduced a sensor based on a CP electrode modified with cerium nanoparticles and used it for electrochemical determination of NIC. The advantage of the cerium nanoparticles was that they significantly enhanced the sensitivity of the CP electrode. Experimental conditions such as pH, scan rate, accumulation time, and the deposition time were studied to find the highest sensitivity for determination of NIC. Under optimum conditions, calibration plots for NIC were linear in the ranges of 4.0 × 10−6 to 5.0 × 10−4 M with correlation coefficients of 0.999 and detection limits of 9.43 × 10−8 M. The results showed that the method was very easy and simple with enough sensitivity and selectivity for NIC detection in cigarettes and urine samples with satisfactory results.
References
- A. Levent, Y. Yardim and Z. Senturk, Electrochim. Acta, 2009, 55, 190–195 CrossRef CAS PubMed.
- D. J. Doolittle, R. Winegar, J. K. Lee, W. S. Caldwell, A. Wallace, J. Hayes and J. D. deBethizy, Mutat. Res., 1995, 344, 95–102 CAS.
- M. Chena, M. Alegrea, A. Durgavanshib, D. Bosec and J. Romero, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2010, 878, 2397–2404 CrossRef PubMed.
- J. M. Garrigues, A. Perez-Ponce, S. Garrigues and M. de la Guardia, Chim. Acta, 1998, 373, 63–71 CrossRef CAS.
- H. Omara and S. Attaf, World J. Pharm. Pharm. Sci., 2014, 3, 1327–1340 CAS.
- R. C. Gupta and G. D. Lundberg, Clin. Toxicol., 1977, 11, 437–442 CrossRef CAS PubMed.
- Å. Zander, P. Findlay, T. Renner and B. Sellergren, Anal. Chem., 1998, 70, 3304–3314 CrossRef.
- C. E. Efstathion, E. P. Diamandis and T. P. Hadjiioannon, Anal. Chim. Acta, 1981, 127, 173–180 CrossRef.
- S. M. Saad and E. M. Elnemma, Analyst, 1980, 114, 1033–1037 Search PubMed.
- H. B. Suffredini, M. C. Santos, D. De Souza, L. Codognoto, P. Homem-de-Mello, K. M. Honorio, A. B. F. Da Silva, S. A. S. Machado and L. A. Avaca, Anal. Lett., 2005, 38, 1587–1599 CrossRef CAS.
- A. Hannisdal, Q. Mikkelsen and K. H. Schroder, Collect. Czech. Chem. Commun., 2007, 72, 1207–1213 CrossRef CAS.
- Y. Yang, M. Yang, H. Wang, L. Tang, G. Shen and R. Yu, Anal. Chim. Acta, 2004, 509, 151–157 CrossRef CAS PubMed.
- C.-T. Wu, P.-Y. Chen, J.-G. Chen, V. Suryanarayanan and K.-C. Ho, Anal. Chim. Acta, 2009, 633, 119–126 CrossRef CAS PubMed.
- M. S. Lin, J. S. Wang and C. H. Lai, Electrochim. Acta, 2008, 53, 7775–7780 CrossRef CAS PubMed.
- M. J. Sims, N. V. Rees, E. J. F. Dickinson and R. G. Compton, Sens. Actuators, B, 2010, 144, 153–158 CrossRef CAS PubMed.
- C. T. Wu, P. Y. Chen, J. G. Chen, V. Suryanarayanan and K. C. Ho, Anal. Chim. Acta, 2009, 633, 119–126 CrossRef CAS PubMed.
- Ľ. Švorc, D. M. Stanković and K. Kalcher, Diamond Relat. Mater., 2014, 42, 1–7 CrossRef PubMed.
- H. Liu, et al., Electrochem. Commun., 2005, 7, 1357–1363 CrossRef CAS PubMed.
- Z. Xie, Q. Liua, Z. Changa and X. Zhang, Electrochim. Acta, 2013, 90, 695–704 CrossRef CAS PubMed.
- S. Liu, J. Li, S. Zhang and J. Zhao, Appl. Surf. Sci., 2005, 252, 2078–2084 CrossRef CAS PubMed.
- N. K. Chaki and K. Vijayamohanan, Biosens. Bioelectron., 2002, 17, 1–12 CrossRef CAS.
- Q. Wang, Anodic electrochemical synthesis and characterization of nanocrystalline cerium oxide and cerium oxide/montmorillonite nanocomposites, Dissertation Prepared for the Ph. D., university of north Texas, 2008.
- S. S. Yang and I. Smetana, Chromatographia, 1995, 40, 375–378 CAS.
- X. L. Wen, Y. H. Jia and Z. L. Liu, Talanta, 1999, 50, 1027–1033 CrossRef CAS.
- N. Yang, Q. Wan and J. Yu, Sens. Actuators, 2005, 110, 246–251 CrossRef CAS PubMed.
- W. Zhang, G. Xie, S. Li, L. Lu and B. Liu, Appl. Surf. Sci., 2012, 258, 8222–8227 CrossRef CAS PubMed.
- A. M. Fekry, Int. J. Hydrogen Energy, 2010, 35, 12945–12951 CrossRef CAS PubMed.
- M. A. Ameer and A. M. Fekry, Prog. Org. Coat., 2011, 71, 343–349 CrossRef CAS PubMed.
- F. El-Taib Heakal and A. M. Fekry, J. Electrochem. Soc., 2008, 155, C534–C542 CrossRef PubMed.
- F. El-Taib Heakal, A. M. Fekry and M. Z. Fatayerji, J. Appl. Electrochem., 2009, 39, 1633–1642 CrossRef.
- U. Retter, A. Widmann, K. Siegler and H. Kahlert, J. Electroanal. Chem., 2003, 546, 87–96 CrossRef CAS.
- D. D. Mackdonald, Transient Techniques in Electrochemistry, Plenum Press, New York, Ch. 6, 1977 Search PubMed.
- G. W. Walter, Corros. Sci., 1986, 26, 681–703 CrossRef CAS.
- M. A. Ameer, Mater. Corros., 2000, 51, 242–246 CrossRef CAS.
- R. Vedalakshmi, V. Saraswathy, Ha-W. Song and N. Palaniswamy, Corros. Sci., 2009, 51, 1299–1307 CrossRef CAS PubMed.
- A.-Z. C. Gudet and P. Buri, Pharm. Sci., 1998, 8, 139–144 CAS.
- G. H. Lu and S. Ralapati, Electrophoresis, 1997, 19, 19–26 CrossRef PubMed.
- M. T. Xu, R. F. Qi and Q. M. Gao, Photometric determination of nicotine in tobacco with bromocresol green, Lihua Jianyan, Huaxue Fence, 1998, 34, 78 CAS.
- J. M. Garrigues, A. Perez-Ponce, S. Garrigues and M. de la Guardia, Analyst, 1999, 124, 783–786 RSC.
- A. Geto, M. Amare, M. Tessema and S. Admassie, Electroanalysis , 24, 659–665 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.