
Abstract
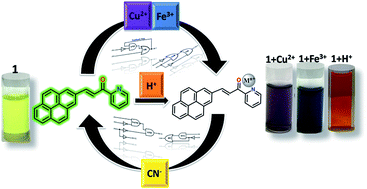
A pyrene-based optical probe for the real-time and regenerative detection of Cu2+ and Fe3+ at parts-per-million (ppm) levels is demonstrated. Moreover, the quantifiable changes in the fluorescence signal induced by chemical inputs viz. Cu2+, Fe3+, H+ and CN− have been exploited to assemble sequential and “four-input” combinatorial molecular logic circuits. A unique “two-way” security lock has also been devised for enhanced information protection at the molecular level.
 Please wait while we load your content...
Please wait while we load your content...

