
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOperational strategies of pulsed electrolysis to enhance multi-carbon product formation in electrocatalytic CO2 reduction†
Takashi
Ito
a,
Jithu
Raj
 a,
Tianyu
Zhang
a,
Soumyabrata
Roy
b and
Jingjie
Wu
*a
a,
Tianyu
Zhang
a,
Soumyabrata
Roy
b and
Jingjie
Wu
*a
aDepartment of Chemical and Environmental Engineering, University of Cincinnati, Cincinnati, OH 45221, USA. E-mail: jingjie.wu@uc.edu
bDepartment of Materials Science and NanoEngineering, Rice University, Houston, TX 77005, USA
First published on 16th May 2024
Abstract
The electrocatalytic reduction of CO2 offers a promising avenue for converting anthropogenic CO2 into valuable chemical and fuel feedstocks. Copper (Cu) catalysts have shown potential in this regard, yet challenges persist in achieving high selectivity for multi-carbon (C2+) products. Pulsed electrolysis, employing alternating anodic and cathodic potentials (Ea/Ec) or two different cathodic potentials (Ec1/Ec2), presents a promising approach to modulate activity and selectivity. In this study, we investigate the influence of catalyst morphology and operational strategies on C2+ product formation using Cu nanoparticles (NPs) and CuO nanowires (NWs) in flow cells. In Ea/Ec mode, commercial Cu NPs show negligible promotion of C2+ selectivity while CuO NWs demonstrate enhanced C2+ selectivity attributed to facile oxidation/redox cycling and grain boundary formation. In contrast, Ec1/Ec2 pulsed electrolysis promotes C2+ yield across various catalyst morphologies by enhancing CO2 accumulation, pH effect, and supplemental CO utilization. We further extend our investigation to membrane electrode assembly cells, highlighting the potential for scalability and commercialization. Our findings underscore the importance of catalyst morphology and operational strategies in optimizing C2+ product formation pulsed electrolysis, laying the groundwork for future advancements in CO2 electroreduction technologies.
Broader contextThe electrocatalytic reduction of CO2 offers a potential platform for synthesizing valuable and energy-rich multicarbon products (C2+) such as ethylene, ethanol, acetic acid, and propanol. Cu remains the only metal capable of converting CO2 to C2+ compounds. Several strategies based on modifying the Cu composition and morphology as well as electrode and electrolyzer design are reported to enhance the C2+ selectivity. However, compared to the aforementioned procedures which are much more involved and complicated, pulsed electrolysis provides a relatively easier and reproducible strategy for the production of high-energy-density hydrocarbons and oxygenates. In the present work, we have deciphered the factors that affect C2+ selectivity in pulsed electrolysis in high current density operational conditions. We find that the increase in C2+ selectivity with pulsed electrolysis involving two different cathodic potentials (Ec1/Ec2) is invariant of catalyst morphologies and depends on enhanced CO2 accumulation, pH effect, and supplemental CO utilization. Since the work is entirely carried out in gas-diffusion electrode-based flow cells and membrane-electrode assemblies, it can be relatively easily translated to commercial electrolyzers. |
Introduction
Electrocatalytic CO2 reduction reaction (eCO2RR) has recently emerged as a key technology to convert anthropogenic CO2 into high-value chemical and fuel feedstocks. Electroproduction of HCOO− and CO from eCO2RR has reached the standard requirements for profitable commercial-scale operation.1,2 However, achieving similar industrial-scale operational efficiency in the case of more reduced eCO2RR products remains a challenge. Copper (Cu) is a well-known catalyst so far capable of reducing CO2 to the much sought-after energy-rich hydrocarbons and oxygenates at an appreciable rate with reasonable selectivity. However, the competing reaction pathways, particularly, the deep hydrogenation and the C–C coupling lower the selectivity towards the high-value multi-carbon (C2+) products, such as C2H4, C2H5OH, and 1-C3H7OH. Over thirteen products have been recorded over polycrystalline Cu, illustrating the intrinsic difficulty in obtaining a high selectivity of C2+ products.3 Systematic studies to improve the selectivity and current density of C2+ compounds in Cu have been rapidly progressing over the past couple of years. Major focuses have included modified copper-based catalysts, engineered gas-diffusion and catalyst layer structures, and refined electrolyzer designs. Efforts in these directions have yielded significant enhancements in terms of faradaic efficiency (FE) and operational current density (jtotal) for C2+ products.The method of electrolysis has emerged as an effective and simple tool to regulate the C2+ selectivity compared to the aforementioned strategies (Fig. 1). The three main modes of electrolysis are static electrolysis, pulsed electrolysis with alternating anodic and cathodic potentials (Ea/Ec), and pulsed electrolysis with two different cathodic potentials (Ec1/Ec2). Conventional static electrolysis often leads to issues such as chemical, mechanical, or thermal degradation due to continuous reduction of the catalyst, causing changes in structure, morphology, and active sites.4 Instead, pulsed electrolysis with two or more potentials can achieve goals similar to catalyst and microenvironment modification, which requires complex syntheses or pre-treatments via restructuring and roughening catalysts, improving mass transport, and controlling interfacial pH.5,6 Generally, one of the potentials considered is a cathodic potential (Ec) and the other one is anodic (Ea) or less negative compared to the first potential. The application of an anodic potential aims to tune the surface structure and oxidation state of copper catalysts. The nature of the copper catalyst during pulsed electrolysis with Ea/Ec was investigated using vacuum-transfer Auger electron spectroscopy.7 This analysis revealed the existence of Cu+/Cu0 motifs, which correlated with an enhancement in C2H5OH selectivity. Similarly, operando time-resolved XANES showed that stable Cu0–Cu1+ motifs persist during Ea/Ec pulse with CuOx catalysts while Cu1+ fraction substantially diminishes within 10 minutes of static electrolysis.8 Further, DFT calculations suggested that OH groups at the Cu0–Cu1+ boundary stabilize the carbonyl group of C2 intermediate via electrostatic interaction accounting for the enhanced C2H5OH selectivity in Ea/Ec pulse mode.8Ea/Ec pulse electrolysis at an intermediate anodic potential (0.9 V versus RHE) on Cu2O nanocubes also demonstrated increased C2+ and C2H5OH selectivity compared to static electrolysis and was attributed to highly defective interfaces and grain boundaries.6 The beforementioned results from pulsed electrolysis with Ea/Ec suggest that catalysts with pre-existing high concentration of defects (e.g., grain boundaries, GBs) can further enhance the C2+ selectivity since they have higher *CO binding energy. In previous studies, the oxide-derived copper shows enhanced performance toward C2+ products due to defects like GBs and vacancies in addition to predominated surface facets.9 These defects exhibit stronger *CO binding energies and stabilization of *COCO intermediate, leading to enhanced formation of C2+ products due to faster C–C coupling kinetics.10
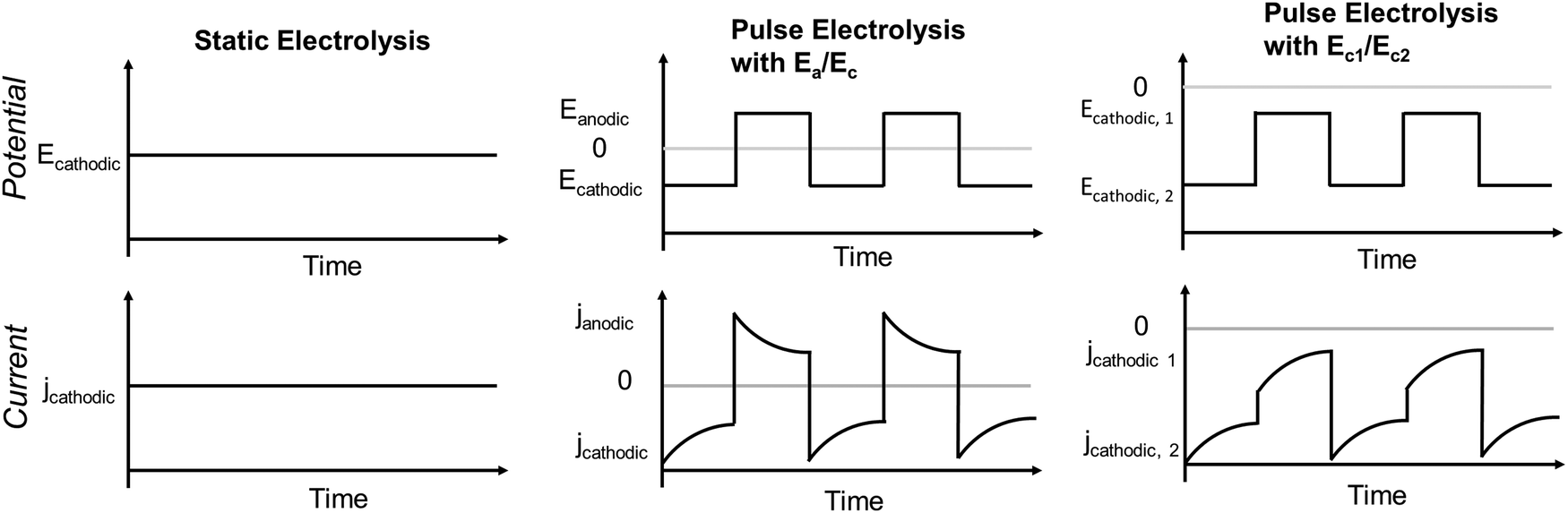 | ||
| Fig. 1 Three different modes of electrolysis in eCO2RR. | ||
The low energy efficiency associated with Ea/Ec pulsed electrolysis, because the application of periodic anodic pulse inherently consumes higher electrical input, which is not directly translated to reaction products. An alternative approach involving a sequence of cathodic potentials (Ec1/Ec2) has been proposed. Previous studies have indicated that CO2 accumulation and enhanced pH effect may influence the formation of C2+ products under the pulsed electrolysis with Ec1/Ec2 in an H-cell.11 The simulated model of transient profiles for CO2 concentration and pH have demonstrated that the pulsed electrolysis with Ec1/Ec2 results in high CO2 accumulation and high local pH in the local environment when the potential transforms to more negative one, facilitating the attainment of a higher C2+ FE.11 The suggested mechanism, based on the theoretical and experimental results for the pulsed electrolysis with Ec1/Ec2, indicates that higher CO2 concentration produces more CO, and higher CO concentration gives higher *CO surface coverage at a more cathodic potential.11 Since the mechanism of the C2+ product formation requires *CO as a key intermediate, higher coverage of *CO favors C2+ products by accelerating the C–C coupling rate.12–14
In this work, we compared two configurations of pulsed electrolysis on the improvement of C2+ products selectivity and found that the sequential Ec1/Ec2 pulsed method is more universal across various Cu catalysts. Through systematic studies employing catalysts with versatile morphologies, we identified the factors that control C2+ product enhancement in both Ea/Ec and Ec1/Ec2 pulsed electrolysis. Pulsed electrolysis experiments were conducted in the flow cell and membrane electrode assembly (MEA) cell under high current densities, simulating industrially relevant operational conditions. The first approach of pulsed electrolysis with a cycle of Ea/Ec aims to induce defects (e.g., GB) on Cu surfaces via reconstruction, thereby enhancing *CO binding energy. However, the efficacy of this configuration strongly depends on the morphology of the Cu catalysts. For example, negligible enhancement in the FE of C2+ products was observed for commercial Cu nanoparticles (NPs) using pulsed electrolysis compared to static potential electrolysis. In contrast, Cu nanowires (NWs), which are more susceptible to restructuring, demonstrated improved performance with this method. The second approach of pulsed electrolysis, involving Ec1/Ec2 pulse, aims at promoting the *CO surface coverage. By applying a less-cathodic potential (Ec1) that is selective for CO2 to CO reduction, supplemental CO was generated for subsequent reduction at a more-cathodic potential (Ec2), leading to a higher C2+ yield. Importantly, the enhancement of C2+ yield observed for the second approach of pulsed electrolysis is morphology independent. Furthermore, the energy efficiency in Ec1/Ec2 pulsed electrolysis is higher compared to Ea/Ec mode, as lower cathodic potentials were capable of CO2 reduction compared to anodic potentials.
Results and discussion
Effect of morphology on pulsed electrolysis with alternating cathodic/anodic potentials
Our experiment commenced with investigations involving Cu NPs and CuO NWs to determine the significance of catalyst morphology in pulsed electrolysis with Ea/Ec within a flow cell. Cu NPs have a diameter range of 50–100 nm, while CuO NWs are in the form of wires with approximately 20 nm diameter (Fig. S1, ESI†). To investigate the effect of oxidation reactions at Ea on pulsed electrolysis, the duration for each potential and the magnitude of Ec need to be defined. The duration for each potential (ta for anodic potential and tc for cathodic potential) was set at 1.0 second, a selection based on previous studies indicating that performance remains unchanged beyond this duration compared to 1.0 second,15 and that the oxidation was observed at this timeframe.6,15 The Ec remained constant to evaluate the influence of oxidation reactions at various anodic potentials on Cu catalysts. The Ec was determined by the outcomes of static electrolysis. Specifically, we chose an Ec of −1.5 V (vs. RHE, thereafter), without iR compensation, because the FE and current density of C2+ products, including C2H4 and C2H5OH, were maximized at this potential (Fig. S2, ESI†).To examine the trends in FE and current density for each product across varying anodic potentials, we selected anodic potentials ranging from 0.5 to 1.3 V. This range was determined based on insights gleaned from cyclic voltammograms and X-ray absorption spectroscopy (XAS) data in prior research, indicating that the oxidation of Cu catalysts typically occurs at potentials exceeding 0.6 V.4,6,16Fig. 2 illustrates the FEs and current densities of the eCO2RR products over Cu NP gas diffusion electrodes (GDEs) under Ea/Ec pulsed electrolysis, alongside benchmark static results for performance comparison. Under static conditions, the FE toward C2+ products reached 85.7% and a total current density of 293.3 mA cm−2 at a potential of −1.5 V, consistent with previous findings.12 However, upon implementing pulsed electrolysis on Cu NPs, both the selectivity and partial current density for C2+ products decreased across all anodic potentials ranging from 0.5 to 1.5 V compared to static electrolysis. For pulsed electrolysis, the highest FE for C2+ products was only 71.5%, achieved at Ea = 0.7 V, with a corresponding total current density of 180.3 mA cm−2 (Fig. 2(a) and (b)). Moreover, the partial current densities of C2+ products, C2+ liquid products, and main C2 gas product (C2H4) exhibited poorer performance during pulsed electrolysis than static electrolysis on Cu NPs (Fig. 2(c)–(e)). At anodic potentials of 1.3 V and beyond, the CH4 formation predominates over C2+ products on Cu NP. At Ea = 1.3 V, the FE of CH4 was 20.9% at a partial current density of 42.7 mA cm−2 (Fig. 2(a) and (f)). As a comparison, the FE of CH4 was only ∼1% and partial current density was 3.0 mA cm−2 under static electrolysis. The activity and selectivity to CH4 were significantly enhanced compared to static electrolysis. The mechanism underlying this phenomenon involves the reaction of OH− species with Cu to form CuxO at anodic potentials. The OH− species is quickly consumed near the catalyst surface upon cycling to the more anodic potential (e.g., ≥1.3 V), leading to a pronounced shift in local pH to lower values.6 This weak acidic condition near the catalyst surface prefers the formation of CH4 rather than C2+ products.17–19 Thus, the formation of CH4 is enhanced at anodic potentials of 1.3 V and higher.
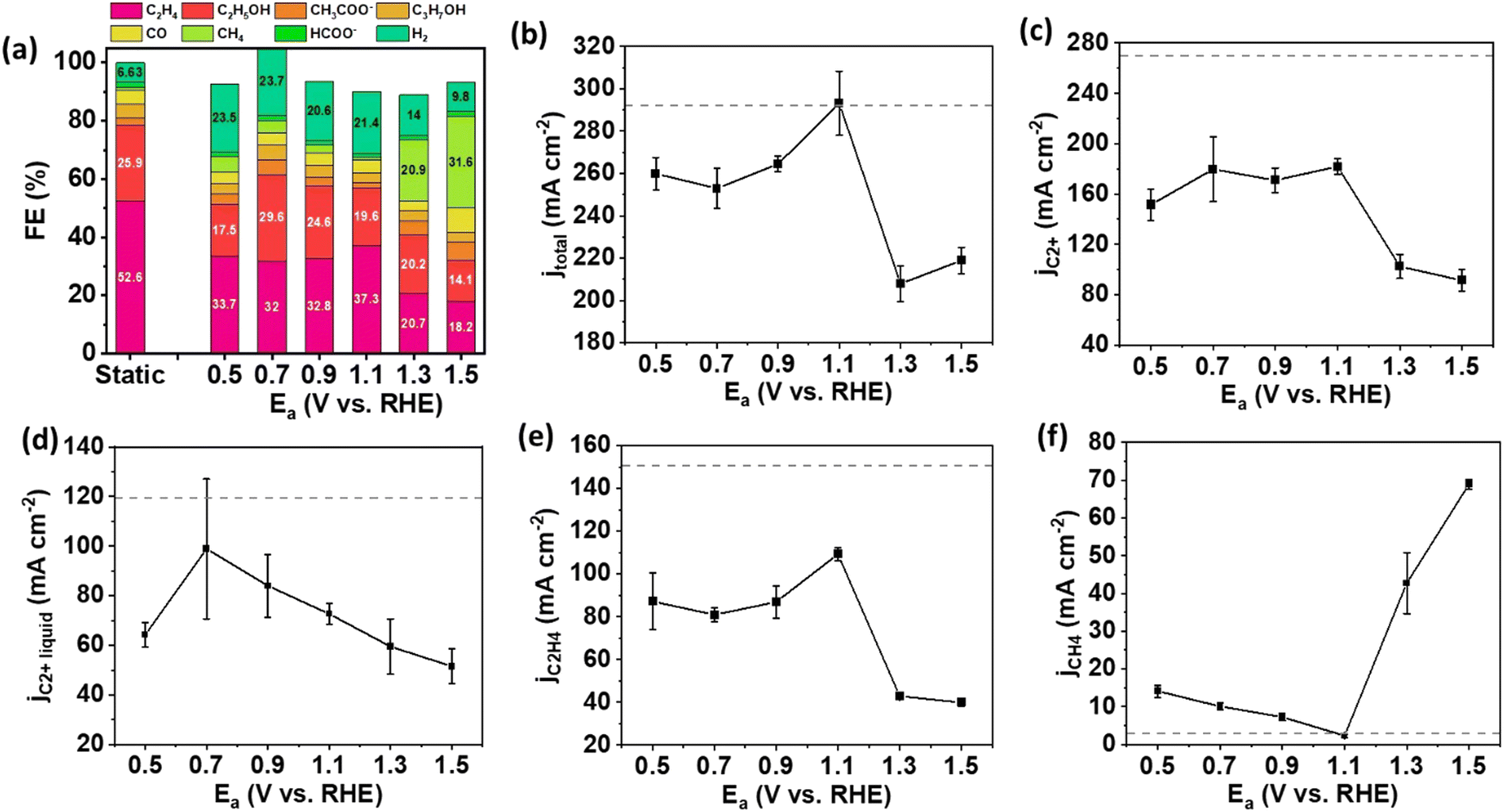 | ||
| Fig. 2 Performance of Cu NP GDEs in the flow cell with pulsed electrolysis of Ec = −1.5 V/Ea = 0.5–1.5 V and duration ta = tc = 1.0 second. (a) Product distribution of pulsed electrolysis at different anodic potentials with a comparison to static electrolysis at E = −1.5 V, (b) total current density, (c) partial current density for C2+ products, (d) partial current density for liquid C2+ products (C2H5OH, C3H7OH, and CH3COO−), (e) partial current density for C2H4, (f) partial current density for CH4, as a function of anodic potential. The gray dot lines show the static electrolysis results at E = −1.5 V. The error bar represents the standard deviation of performance for at least three independent electrodes. | ||
To further explore the impact of pulsed electrolysis using various anodic potentials, we employed CuO NWs, which represent a distinct catalyst morphology compared to Cu NPs, for eCO2RR following the same experimental protocol. The Ec for CuO NW was still set at −1.5 V as the static electrolysis revealed that the highest FEs and partial current densities for C2+ and C2H4 were achieved at this potential (Fig. S3, ESI†). Under static electrolysis conditions, CuO NW achieved an FE of 63.7% for C2+ products, with a corresponding total current density of 320 mA cm−2 (Fig. 3(a) and (b)). The selectivity to C2+ products for pulsed electrolysis was improved compared to static electrolysis until Ea increased to 1.1 V. The major contribution to the enhancement of C2+ product selectivity comes from the increase of FE of C2+ liquid products, in which C2H5OH predominates (Fig. 3(a)). In contrast, the selectivity to the major C2+ hydrocarbon, C2H4, declined monotonically as the Ea increased. The maximum activity and selectivity to C2+ products were observed at Ea = 0.7 V under the pulsed electrolysis. At Ea = 0.7 V, the partial current density of C2+ products was 264.7 mA cm−2 comparable to that of static electrolysis, while the FE of C2+ products increased from 63.7% to 83.5%. Similar to the results observed with Cu NPs, compared to static electrolysis, a significant increase in FE and partial current density for CH4 was detected on CuO NWs under pulsed electrolysis with Ea = 1.3 V and higher (Fig. 3(a) and (f)), attributed to the shift in pH towards a weak acidic environment.
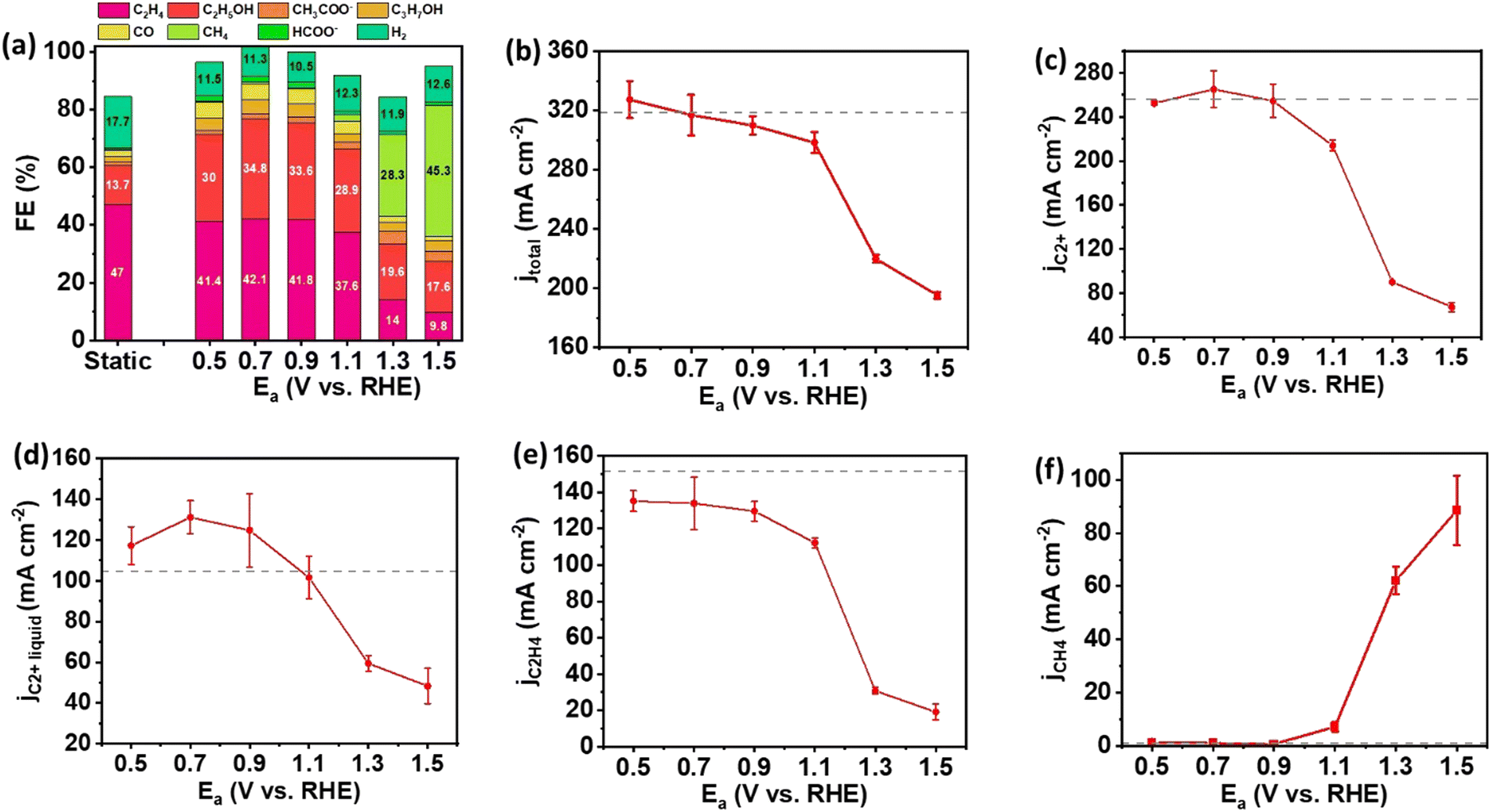 | ||
| Fig. 3 Performance of CuO NW GDEs in the flow cell with pulsed electrolysis of Ec = −1.5 V/Ea = 0.5–1.5 V, and duration ta = tc = 1.0 second. (a) Product distribution of pulsed electrolysis at different anodic potentials with a comparison to static electrolysis, (b) total current density, (c) partial current density for C2+ products, (d) partial current density for liquid C2+ products (C2H5OH, C3H7OH, and CH3COO−), (e) partial current density for C2H4, (f) partial current density for CH4, as a function of anodic potential. The gray dot lines show the static electrolysis results at E = −1.5 V. The error bar represents the standard deviation of performance for at least three independent electrodes. | ||
Compared to Cu NPs, reduced CuO NWs exhibit a higher propensity for reconstruction during pulsed electrolysis with alternating Ea/Ec.20 This cyclic process involves the oxidation of Cu to CuxO followed by rapid reduction back to Cu, facilitating the formation of GBs. TEM imaging of CuO NWs shows a significant increase in GBs after reaction (Fig. S4, ESI†). The presence of low coordinated sites across GB form Cu0/Cu1+ interface,21 leading to the enhanced selectivity towards C2H5OH during pulsed electrolysis, as anticipated based on previous research.15,22–24 The contrast of the C2+ performance between Cu NPs and CuO NWs underscores the critical role of catalyst morphology and structure in governing product selectivity during pulsed electrolysis with Ea/Ec.
Enhancing *CO surface coverage by pulsed electrolysis with alternating Ec1/Ec2
Next, we aim to investigate pulsed electrolysis with two different cathodic potentials in the flow cell. As opposed to alternating Ea/Ec which is catalyst dependent and necessitates GB rich Cu surfaces, successive Ec1/Ec1 benefits from changes in local pH and CO2 concentration.11 Besides the magnitude for each cathodic potential, the duration of each potential affects the selectivity and current density of each product.11,25,26 The evaluation of duration has been done with H-cell or similar cell configurations experimentally and theoretically. However, the pulsed electrolysis with alternating Ec1/Ec2 was not performed in the flow cell. In addition to the influence of the local microenvironment, the flow cell takes another advantage by supplemental CO utilization. The combination of CO-generation potential and C2+-generation potential results in improved CO concentration and enhanced *CO surface coverage on the catalyst. It's worth noting that different parameters need to be considered for different cell configurations. The minimum duration is determined by the time constant of double layer charging. The non-faradaic electrochemical process due to double layer charging occurs during the switch of potentials. The reported RC time constant for the double layer charging is approximately 6–30 milliseconds.25–27 The RC time constant for our flow cell was measured and calculated based on its capacitance and resistance, and it was up to 9 milliseconds (Note S1 in ESI†). Thus, only non-faradaic processes occur if the duration is too short (less than 9 milliseconds). In other words, the duration must be longer than 9 milliseconds to observe the reduction reactions. On the other hand, the maximum duration is determined by the CO2 residence time in the flow cell. The H-cell utilizes dissolved CO2 in the aqueous solution, and the concentration of CO2 in bulk solution does not change during the process due to the continuous CO2 supply. Since the performance is mainly based on the concentration of CO2 in the aqueous solution and gas-phase CO2 does not participate in the reactions, there is no upper duration limit for the H-cell. However, the flow cell demands utilizing the CO2 and the derived intermediates (e.g., CO) on-line. In the flow cell, the residence time of CO2 can be calculated according to the flow channel volume and flow rate of CO2. The residence time of CO2 to pass through the flow channel is within 3 seconds according to our flow cell configuration and CO2 flow rate (Note S1 in ESI†). Therefore, the shortest duration is 9 milliseconds due to double layer charging and the longest duration is 3 seconds due to the CO2 residence time. A longer duration of the more cathodic potential (Ec2) was reported to provide slightly higher current density.11 However, the focus for pulsed electrolysis is better performance with less energy consumption. Thus, the equal duration was selected for two cathodic potentials to determine clear trends for overall performance in this study. The duration was selected between 0.15 seconds and 1 second.To determine the optimal duration for the flow cell, the suitable potentials for less negative cathodic (Ec1) and more negative cathodic (Ec2) potentials were selected. The Ec1 was selected based on the formation of CO. CO utilization in the flow cell has unique advantages compared to the H-cell, when CO is produced upstream and carried through the flow channel. Thus, local CO concentration is increased to enhance C–C coupling kinetics downstream of the electrode. The highest CO formation rate was observed at −1.2 V under the static electrolysis over Cu NPs GDEs (Fig. S2, ESI†). Differently, the Ec2 was selected based on the selectivity of C2+ products. The trend of static electrolysis shows that the highest FE of C2+ occurred at −1.5 V (Fig. S2, ESI†). Therefore, Ec1 was −1.2 V for the highest CO formation rate, while Ec2 was −1.5 V for the highest FE of C2+. The best duration among the selected conditions was determined as 0.30 seconds/0.30 seconds for Ec1/Ec2 because the FE of C2+ products reached the highest (Fig. S5, ESI†). Although the current density of C2+ was slightly higher at the duration of 0.15 seconds, our EnergyLab XM potentiostat system reported errors frequently with a shorter duration than 0.3 seconds. Since the result with a duration of 0.15 seconds was similar to the result with 0.3 seconds, the duration of 0.3 seconds for each potential was selected to obtain the valid result and ensure the system was safe during operations.
The effect of potential pair (Ec1/Ec2) on C2+ yield was investigated with the 0.30 seconds duration. The result of pulsed electrolysis was compared to the static electrolysis at a time-average potential to assess performance based on the identical voltage efficiency (defined as the standard reduction potential divided by the applied cathodic potential) (Fig. 4). The FE of C2+ products was slightly higher in pulsed electrolysis than in static electrolysis. At Ec1/Ec2 = −1.2/−1.5 V, a FE of 87.2% was achieved for C2+ products, compared to 84.2% for static electrolysis (Fig. 4(a)). This enhancement in C2+ selectivity primarily resulted from an increase in the FE of C2H4. However, pulsed electrolysis led to an increase in the FE of C2H4 at the expense of the FE of C2H5OH. This is in accordance with previous findings that increased CO coverage promotes C2H4 selectivity up to a limit before shifting to oxygenates at a much higher CO concentration.28 The dominance of C2H4 selectivity with increased *CO coverage is also observed in tandem electrodes by our group.29 In contrast to the minor increase in FE of C2+ products, total current density significantly increased, resulting from a significant enhancement of partial current density of C2+ products and C2H4 (Fig. 4(b)–(d)). For example, the partial current density of C2+ products increased from 206 mA cm−2 during static electrolysis at −1.35 V to 297 mA cm−2 during pulsed electrolysis with Ec1/Ec2 of −1.2 V/−1.5 V, and further to 356 mA cm−2 with Ec1/Ec2 of −1.0 V/−1.7 V (Fig. 4(c)).
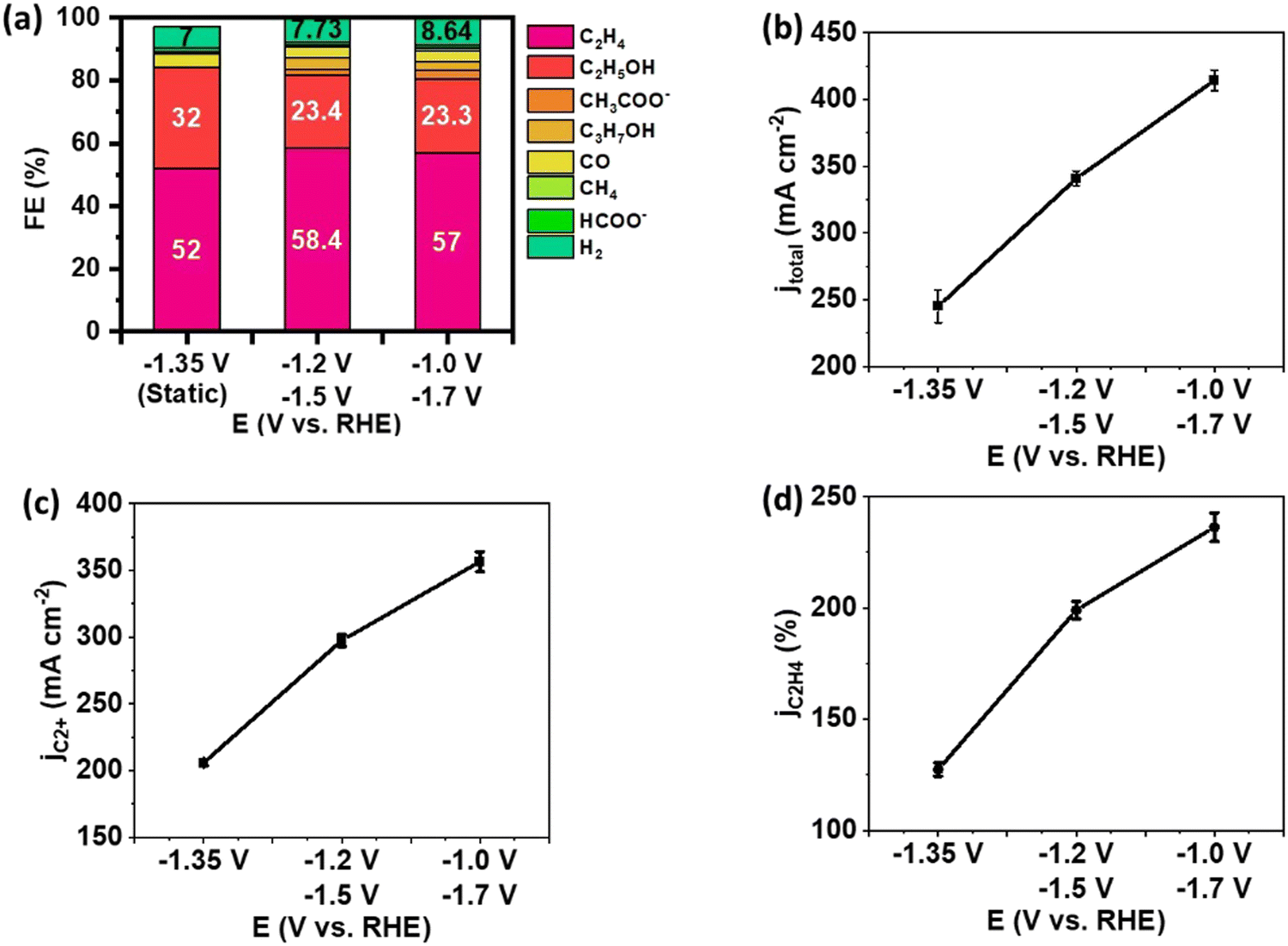 | ||
| Fig. 4 Performance of Cu NP GDEs in the flow cell under pulsed electrolysis with Ec1/Ec2 of −1.2 V/−1.5 V and −1.0 V/−1.7 V with a comparison to static electrolysis at an average potential of −1.35 V. (a) Product distribution, (b) total current density for all products, (c) partial current density for C2+ products, (d) partial current density for C2H4. The error bar represents the standard deviation of performance for at least three independent electrodes. | ||
The CO2 accumulation and pH effect resulted from pulsed electrolysis are transferable from an H-cell configuration to a flow cell.5 The pH effect and CO2 accumulation are strongly related to each other. The Ec1 of pulsed electrolysis has less current density, and the OH− concentration is lower and the CO2 utilization is less than the one at Ec2. Thus, the CO2 accumulation occurs because of the difference in CO2 utilization between each cathodic potential. The increase of CO2 concentration at Ec2 leads to a higher rate of CO2 reduction reactions, and it leads to the increase of *CO formation correspondingly. On the other hand, the CO formed at Ec1 also accumulates at Ec2 upon potential switching. Therefore, the adsorbed *CO surface coverage on the catalyst surface increases, favoring C–C coupling toward the formation of C2+ products. Due to the enhanced concentration of adsorbed *CO on the Cu surface, pulsed electrolysis exhibited a significant increase of partial current density of C2+ products compared to static electrolysis at the same average potential. CO utilization is an advantage of using a flow cell configuration. The lower FE of CO was observed during pulsed electrolysis compared to static electrolysis (Fig. S6, ESI†). This result indicates that the consumption rate of CO under the pulsed electrolysis is higher than under the static electrolysis with a time-average potential. Thus, this outcome suggests the utilization of supplementary CO from Ec1 can facilitate C–C coupling rate at the subsequent Ec2 in the flow cell, a similar mechanism to that in the tandem electrode design.29–31
The facilitated C–C coupling rate was also observed on CuO NW following the same pulsed electrolysis procedure with alternating Ec1/Ec2 (Fig. 5). The increase in FE of C2+ products was trivial. However, the total current density increased monotonically as Ec2 became more negative. Likewise, the partial current density of C2+ products was promoted from 203 mA cm−2 during static electrolysis at −1.35 V to 299.9 mA cm−2 during pulsed electrolysis at −1.0 V/−1.7 V. Pulsed electrolysis with Ec1/Ec2 can universally apply to all morphologies of Cu-based catalysts, leading to promoted C2+ yield at the same voltage efficiency.
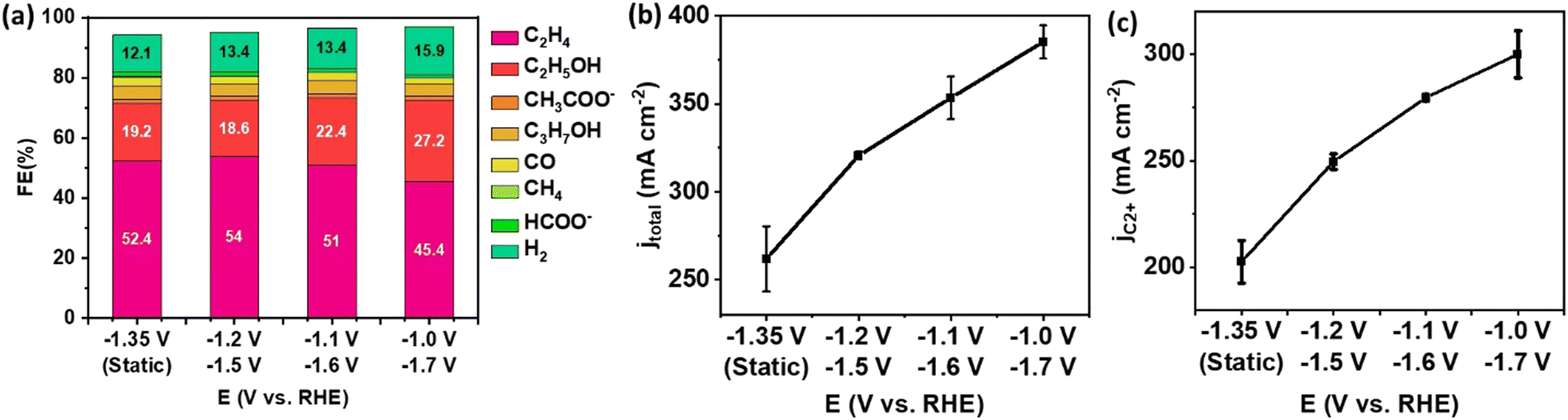 | ||
| Fig. 5 Performance of CuO NW GDEs in the flow cell under pulsed electrolysis with the potential setup of −1.2 V/−1.5 V, −1.1 V/−1.6 V and −1.0 V/−1.7 V and comparison to static electrolysis at an average potential of −1.35 V. (a) Product distribution, (b) total current density for all products, (c) partial current density for C2+ products. The error bar represents the standard deviation of performance for at least three independent electrodes. | ||
Implementation of pulsed electrolysis with Ec1/Ec2 in the MEA cell
To improve energy efficiency via lowering the applied cell voltage, the pulsed electrolysis with Ec1/Ec2 extends to an MEA cell. Similar to the flow cell configuration, the enhancement of CO formation and *CO surface coverage is the strategy to achieve in an MEA cell with pulsed electrolysis. The duration of 0.3 seconds for each potential was optimal to achieve the highest FE and partial current density of C2+ products and C2H4 over Cu NP GDEs (Fig. S8, ESI†). Since the flow channel volume and the flow rate of CO2 are the same as the flow cell configuration, the duration of 0.3 seconds is acceptable for the residence time to utilize CO. The pulsed electrolysis was performed at various Ec2 near by 2.5 V since the highest FE for C2+ products was observed at a cell voltage of 2.5 V during static electrolysis (Fig. S7, ESI†).All three setups of pulsed electrolysis (Ec1/Ec2 = 2.3 V/2.5 V, 2.2 V/2.6 V and 2.1 V/2.7 V) showed improvement in FE and partial current density of C2+ compared to static electrolysis at the time-average cell voltage of 2.4 V (Fig. 6). The FE (76.9%) and partial current density (124.4 mA cm−2) of C2+ product achieved the highest at cell voltages Ec1/Ec2 = 2.1 V/2.7 V among three setups. As a control, the FE and current density of C2+ products were 61.5% and 81.5 mA cm−2 at the time-average cell voltage of 2.4 V. The time-dependent voltage efficiency of pulsed electrolysis is the same as static electrolysis. However, the FE for C2+ products was enhanced by 20%, and the partial current density for C2+ products was increased by 56% during pulsed electrolysis compared to static electrolysis. The combined factors of enhanced pH, CO2 accumulation, and increased CO utilization contribute to enhanced C–C coupling rate (Fig. S9, ESI†).
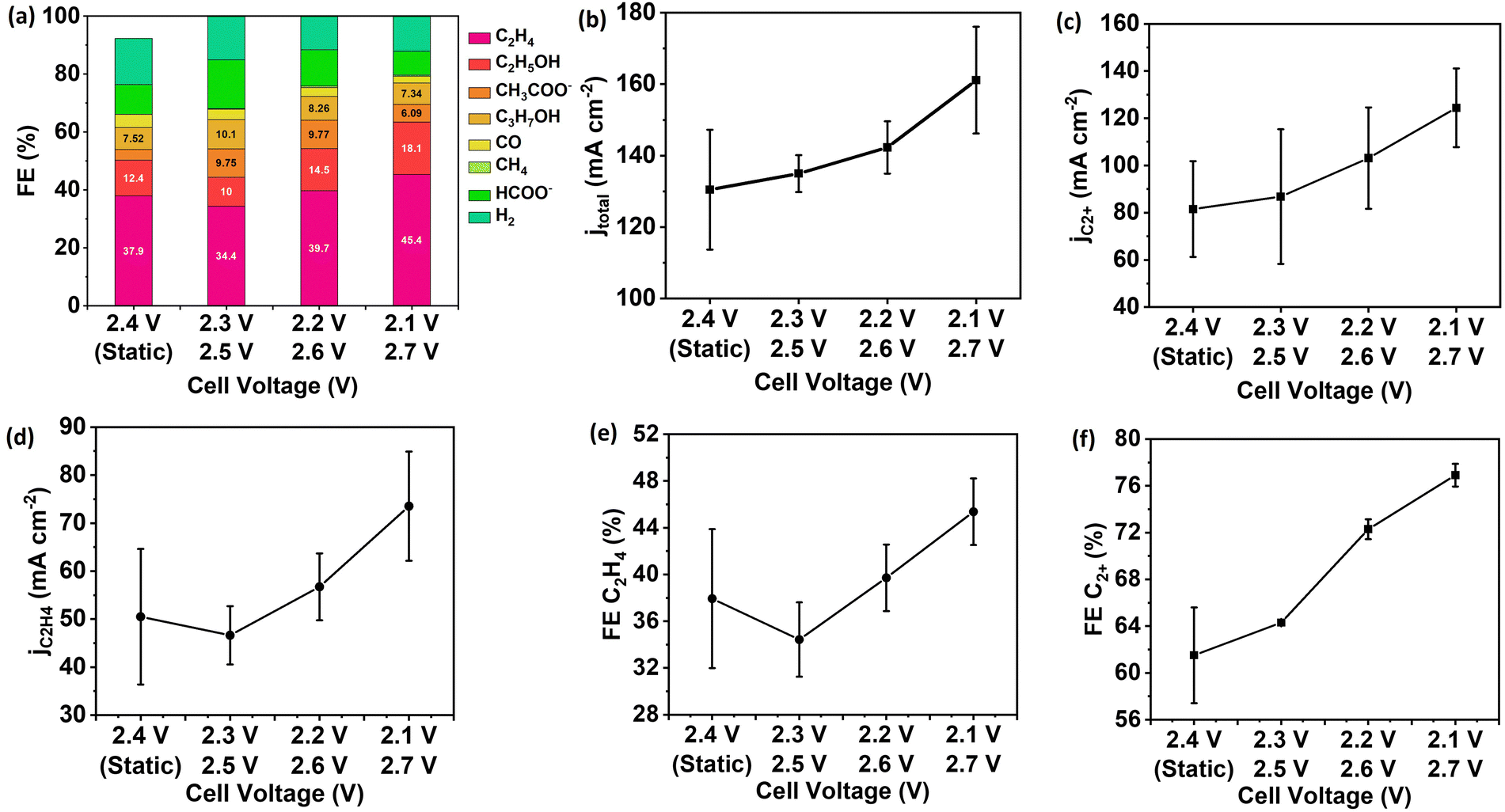 | ||
| Fig. 6 Performance of pulsed electrolysis for Cu NP GDEs in the MEA cell with alternating cell voltage of 2.3 V/2.5 V, 2.2 V/2.6 V and 2.1 V/2.7 V and tc1 = tc2 = 0.3 seconds, and comparison to static electrolysis at an time-average cell potential of 2.4 V. (a) Product distribution, (b) total current density for all products, (c) partial current density for C2+ products, (d) partial current density of C2H4, (e) FE of C2+ products, (f) FE of C2H4. The error bar represents the standard deviation of performance for at least three independent electrodes. | ||
Finally, pulsed electrolysis in Ec1/Ec2 mode was also carried out with CuO NW in an MEA cell to conclude its universality. Static electrolysis showed HER is significant at voltages >2.4 V due to defects in NW (Fig. S10, ESI†). Hence low voltages (2.2 V and 2.3 V) were chosen as the base for pulse electrolysis in Ec1/Ec2 mode. Pulse electrolysis at Ec1/Ec2 = 2.1 V/2.3 V showed a moderate C2H4 selectivity of 31.3%, which was a reasonable increment compared to static electrolysis at 2.2 V considering operation at lower current density compared to Cu NP. However, jC2H4 increases by almost 1.5 times (Fig. S11, ESI†). Larger Ec2 resulted in a decrease of FE of C2H4 due to increased HER. A similar trend was obtained in experiments with 2.3 V as the base where Ec1/Ec2 = 2.2 V/2.4 V showed the best FE of C2H4 and the most increment of jC2H4 (Fig. S12, ESI†).
Conclusion
In conclusion, we have determined that the causes of enhancement of C2+ activity and selectivity in pulsed electrolysis are sensitive to the mode of operation under high current density operation conditions. In Ea/Ec mode, commercial Cu NPs show no apparent promotion of C2+ selectivity which is contravening to the results obtained from H-cell operations. However, CuO NWs showed increased C2+ selectivity in the same mode with enhancement in FE of C2H5OH compared to static electrolysis. That is attributed to the fact that CuO NWs easily generate GBs while undergoing facile oxidation/redox cycling during Ea/Ec as opposed to Cu NPs. GB rich surface provides ample defects and Cu0/Cu1 interfaces which possess enhanced CO binding energy and faster C–C coupling kinetics which can account for higher C2+ and C2H5OH selectivity. Hence, multicarbon product formation in Ea/Ec method is strongly dependent on catalyst morphology. In contrast, the Ec1/Ec2 method was found to enhance C2+ yield in both Cu NPs and CuO NWs and hence is invariant to catalyst morphology. The Ec1 is not sufficient to cause catalyst surface oxidation even in CuO NWs. Ec1/Ec2 pulsed electrolysis provides enhancement in C2+ and C2H4 selectivity due to CO2 accumulation, enhanced pH effect, and supplemental CO utilization in the flow and MEA cells. In addition to the local microenvironmental changes such as CO2 concentration and pH, the enhanced *CO surface coverage by CO selective formation at Ec1 is an advantage of using a flow cell and an MEA cell. The intermediate increment in *CO coverage generated by Ec1/Ec2 cycling selectively facilitates C2H4 formation in Cu NPs at the expense of C2H5OH.Note that while the current MEA cell comprises a 1 cm2 reaction area, the impact of pulsed electrolysis on larger reaction areas remains uncertain. To advance towards commercialization, further upscale experiments are imperative. Pulse electrolysis warrants further investigation using large-area electrodes to assess its effectiveness, considering the heterogeneous distribution of current density and selectivity across larger surfaces. Such experiments will be instrumental in optimizing the scalability and applicability of pulsed electrolysis systems for industrial implementation.
Experimental method
CO2 reduction in the flow cell
The selectivity and productivity of gaseous and liquid products were first tested in a customized flow cell. All potential mentioned in the text are referenced versus a reversible hydrogen electrode (RHE) unless stated otherwise. The cell system consists of a GDE cathode, a Sustainion anion-exchange membrane, and Ni foam as an anode. 1 M KOH was supplied as the catholyte and anolyte through the electrolyte buffer layers between membrane and cathode/anode at a rate of 0.8 mL min−1 controlled by a peristaltic pump (Harvard Apparatus P70-7000). The dry CO2 feedstock was supplied to the cathode at a rate of 20 standard cubic centimeters per minute (sccm) controlled by a mass flow controller (Alicat Scientific MC-100SCCM-D). The applied potential for a flow cell was controlled by a potentiostatic/galvanostatic station (EnergyLab XM, Solatron Analytical). In the case of Ea/Ec mode,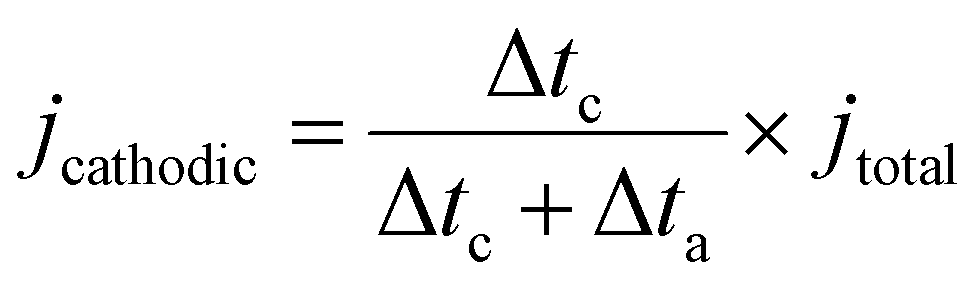 where Δta represents the duration of the oxidation period and Δtc the duration of the cathodic period. The term
where Δta represents the duration of the oxidation period and Δtc the duration of the cathodic period. The term 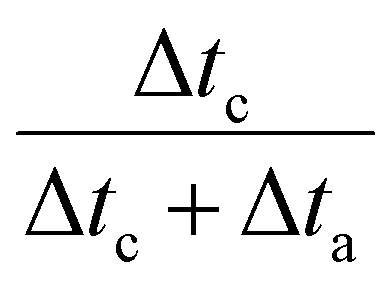 accounts for the effective cathodic part of jtotal while pulse mode is on. In Ec1/Ec2, the time-averaged value of the current is taken since both are cathodic currents. The representative potential versus time and current versus time plots are given in Fig. S13 and S14 (ESI†). The solution from the catholyte buffer layer was collected to analyze liquid products. The gas products were quantified by gas chromatography (GC, Agilent 7890B), and the liquid products were measured by 1H NMR spectroscopy (Bruker AV500). For the correct quantification of outlet CO2 and gas products, a constant stream of Ar gas (10 sccm) was used as an internal reference and evenly mixed with the cell outlet gas stream before it was injected into the GC column. The injection of gas products for GC is set at 200 seconds after the electrolysis started to keep consistency. The solution containing trisodium phosphate (TSP) and D2O was utilized as the internal reference for NMR spectroscopy.
accounts for the effective cathodic part of jtotal while pulse mode is on. In Ec1/Ec2, the time-averaged value of the current is taken since both are cathodic currents. The representative potential versus time and current versus time plots are given in Fig. S13 and S14 (ESI†). The solution from the catholyte buffer layer was collected to analyze liquid products. The gas products were quantified by gas chromatography (GC, Agilent 7890B), and the liquid products were measured by 1H NMR spectroscopy (Bruker AV500). For the correct quantification of outlet CO2 and gas products, a constant stream of Ar gas (10 sccm) was used as an internal reference and evenly mixed with the cell outlet gas stream before it was injected into the GC column. The injection of gas products for GC is set at 200 seconds after the electrolysis started to keep consistency. The solution containing trisodium phosphate (TSP) and D2O was utilized as the internal reference for NMR spectroscopy.
The detailed preparation of CuO nanowires was demonstrated in the previous research.20 For the preparation of Cu NP and CuO NW electrodes, 10 mg of Cu NPs (Sigma) or CuO NW was dispersed in 10 mL IPA (isopropyl alcohol). The suspension was then sonicated for 1 hour to form catalyst ink. The electrodes were prepared by air spraying the ink onto the carbon paper with a microporous carbon gas diffusion layer (Sigracet 39BB) followed by drying at 130 °C. The Cu loading was kept constant at approximately 1.0 mg cm−2 by measuring the weight of electrodes before and after the spraying.
CO2 reduction in the MEA cell
The pulsed electrolysis with combinations of different cathodic potentials was tested in a customized MEA cell. The MEA cell consists of a sandwiched structure of a GDE cathode, Sustainion anion-exchange membrane, and a Ni foam anode, which are mechanically pressed together. For the MEA cell, only 1 M KOH anolyte was supplied at a rate of 2.5 mL min−1 controlled by a peristaltic pump (Gilson Minipuls 3 Pump) since no catholyte compartment was assembled. The dry CO2 feedstock was supplied to the cathode at a rate of 20 standard cubic centimeters per minute (sccm) controlled by a mass flow controller (Alicat Scientific MC-100SCCM-D). The applied cell voltage for an MEA cell was controlled by a potentiostatic/galvanostatic station (EnergyLab XM; Solatron Analytical). The product analysis followed the same procedure as that of the flow cell.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially financially supported by NSF CBET-2033343.References
- J. Li, et al., Selective CO2 electrolysis to CO using isolated antimony alloyed copper, Nat. Commun., 2023, 14, 340, DOI:10.1038/s41467-023-35960-z.
- T. Zheng, et al., Copper-catalysed exclusive CO2 to pure formic acid conversion via single-atom alloying, Nat. Nanotechnol., 2021, 16, 1386–1393, DOI:10.1038/s41565-021-00974-5.
- T. Hatsukade, K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Insights into the electrocatalytic reduction of CO2 on metallic silver surfaces, Phys. Chem. Chem. Phys., 2014, 16, 13814–13819, 10.1039/C4CP00692E.
- R. Casebolt, K. Levine, J. Suntivich and T. Hanrath, Pulse check: Potential opportunities in pulsed electrochemical CO2 reduction, Joule, 2021, 5, 1987–2026, DOI:10.1016/j.joule.2021.05.014.
- J. C. Bui, et al., Engineering Catalyst–Electrolyte Microenvironments to Optimize the Activity and Selectivity for the Electrochemical Reduction of CO2 on Cu and Ag, Acc. Chem. Res., 2022, 55, 484–494, DOI:10.1021/acs.accounts.1c00650.
- H. S. Jeon, et al., Selectivity Control of Cu Nanocrystals in a Gas-Fed Flow Cell through CO2 Pulsed Electroreduction, J. Am. Chem. Soc., 2021, 143, 7578–7587, DOI:10.1021/jacs.1c03443.
- R. M. Arán-Ais, F. Scholten, S. Kunze, R. Rizo and B. Roldan Cuenya, The role of in situ generated morphological motifs and Cu(I) species in C2+ product selectivity during CO2 pulsed electroreduction, Nat. Energy, 2020, 5, 317–325, DOI:10.1038/s41560-020-0594-9.
- S.-C. Lin, et al., Operando time-resolved X-ray absorption spectroscopy reveals the chemical nature enabling highly selective CO2 reduction, Nat. Commun., 2020, 11, 3525, DOI:10.1038/s41467-020-17231-3.
- Y. Lum, B. Yue, P. Lobaccaro, A. T. Bell and J. W. Ager, Optimizing C–C Coupling on Oxide-Derived Copper Catalysts for Electrochemical CO2 Reduction, J. Phys. Chem. C, 2017, 121, 14191–14203, DOI:10.1021/acs.jpcc.7b03673.
- Z. Chen, et al., Grain-Boundary-Rich Copper for Efficient Solar-Driven Electrochemical CO2 Reduction to Ethylene and Ethanol, J. Am. Chem. Soc., 2020, 142, 6878–6883, DOI:10.1021/jacs.0c00971.
- J. C. Bui, C. Kim, A. Z. Weber and A. T. Bell, Dynamic Boundary Layer Simulation of Pulsed CO2 Electrolysis on a Copper Catalyst, ACS Energy Lett., 2021, 6, 1181–1188, DOI:10.1021/acsenergylett.1c00364.
- S. Nitopi, et al., Progress and Perspectives of Electrochemical CO2 Reduction on Copper in Aqueous Electrolyte, Chem. Rev., 2019, 119, 7610–7672, DOI:10.1021/acs.chemrev.8b00705.
- Y. Hori, C. Vayenas, R. White and M. Gamboa-Aldeco, Electrochemical CO2 Reduction on Metal Electrodes, Modern Aspects of Electrochemistry, Springer, New York, 2008, pp. 89–189 Search PubMed.
- X. Kong, et al., Understanding the Effect of *CO Coverage on C–C Coupling toward CO2 Electroreduction, Nano Lett., 2022, 22, 3801–3808, DOI:10.1021/acs.nanolett.2c00945.
- J. Timoshenko, et al., Steering the structure and selectivity of CO2 electroreduction catalysts by potential pulses, Nat. Catal., 2022, 5, 259–267, DOI:10.1038/s41929-022-00760-z.
- G. Nogami, H. Itagaki and R. Shiratsuchi, Pulsed Electroreduction of CO2 on Copper Electrodes-II, J. Electrochem. Soc., 1994, 141, 1138 CrossRef CAS.
- A. S. Varela, M. Kroschel, T. Reier and P. Strasser, Controlling the selectivity of CO2 electroreduction on copper: The effect of the electrolyte concentration and the importance of the local pH, Catal. Today, 2016, 260, 8–13 CrossRef CAS.
- S. Ringe, et al., Understanding cation effects in electrochemical CO2 reduction, Energy Environ. Sci., 2019, 12, 3001–3014, 10.1039/C9EE01341E.
- E. R. Cofell, U. O. Nwabara, S. S. Bhargava, D. E. Henckel and P. J. A. Kenis, Investigation of Electrolyte-Dependent Carbonate Formation on Gas Diffusion Electrodes for CO2 Electrolysis, ACS Appl. Mater. Interfaces, 2021, 13, 15132–15142, DOI:10.1021/acsami.0c21997.
- J. Zhang, et al., Reconstructing two-dimensional defects in CuO nanowires for efficient CO2 electroreduction to ethylene, Chem. Commun., 2021, 57, 8276–8279, 10.1039/D1CC03171F.
- J. Zhang, et al., Switching CO2 Electroreduction Selectivity Between C1 and C2 Hydrocarbons on Cu Gas-Diffusion Electrodes, Energy Environ. Mater., 2023, 6, e12307, DOI:10.1002/eem2.12307.
- C. W. Li, J. Ciston and M. W. Kanan, Electroreduction of carbon monoxide to liquid fuel on oxide-derived nanocrystalline copper, Nature, 2014, 508, 504–507 CrossRef CAS PubMed.
- D. Ren, et al., Selective electrochemical reduction of carbon dioxide to ethylene and ethanol on copper(I) oxide catalysts, ACS Catal., 2015, 5, 2814–2821 CrossRef CAS.
- Y. Lum and J. W. Ager, Evidence for product-specific active sites on oxide-derived Cu catalysts for electrochemical CO2 reduction, Nat. Catal., 2019, 2, 86–93, DOI:10.1038/s41929-018-0201-7.
- C. Kim, L.-C. Weng and A. T. Bell, Impact of pulsed electrochemical reduction of CO2 on the formation of C2+ products over Cu, ACS Catal., 2020, 10, 12403–12413 CrossRef CAS.
- R. C. DiDomenico and T. Hanrath, Pulse Symmetry Impacts the C2 Product Selectivity in Pulsed Electrochemical CO2 Reduction, ACS Energy Lett., 2022, 7, 292–299, DOI:10.1021/acsenergylett.1c02166.
- S. Ha and K. Doblhofer, The electrochemical interface between copper (111) and aqueous electrolytes, J. Electroanal. Chem., 1995, 380, 185–191 CrossRef.
- J. Li, et al., Constraining CO coverage on copper promotes high-efficiency ethylene electroproduction, Nat. Catal., 2019, 2, 1124–1131, DOI:10.1038/s41929-019-0380-x.
- T. Zhang, et al., Highly selective and productive reduction of carbon dioxide to multicarbon products via in situ CO management using segmented tandem electrodes, Nat. Catal., 2022, 5, 202–211, DOI:10.1038/s41929-022-00751-0.
- C. G. Morales-Guio, et al., Improved CO2 reduction activity towards C2+ alcohols on a tandem gold on copper electrocatalyst, Nat. Catal., 2018, 1, 764–771, DOI:10.1038/s41929-018-0139-9.
- F. Li, et al., Cooperative CO2-to-ethanol conversion via enriched intermediates at molecule–metal catalyst interfaces, Nat. Catal., 2020, 3, 75–82, DOI:10.1038/s41929-019-0383-7.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ey00039k |
| This journal is © The Royal Society of Chemistry 2024 |