
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural selectivity of supported Pd nanoparticles: selective ethanol ammoxidation to acetonitrile†
Khaled
Mohammed
a,
Reza
Vakili
bc,
Donato
Decarolis
de,
Shaojun
Xu
df,
Luke
Keenan
 e,
Apostolos
Kordatos
a,
Nikolay
Zhelev
a,
Chris K.
Skylaris
a,
Marina
Carravetta
a,
Emma K.
Gibson
dgh,
Haresh
Manyar
b,
Alexandre
Goguet
b and
Peter P.
Wells
*ade
e,
Apostolos
Kordatos
a,
Nikolay
Zhelev
a,
Chris K.
Skylaris
a,
Marina
Carravetta
a,
Emma K.
Gibson
dgh,
Haresh
Manyar
b,
Alexandre
Goguet
b and
Peter P.
Wells
*ade
aSchool of Chemistry, University of Southampton, University Road, Southampton, SO17 1BJ, UK. E-mail: ppwells@soton.ac.uk
bSchool of Chemistry and Chemical Engineering, Queen's University Belfast, David Keir Building, Stranmillis Rd, Belfast BT9 5AG, UK
cDepartment of Chemical Engineering, Imperial College London, London SW7 2AZ, UK
dUK Catalysis Hub, Research Complex at Harwell, Rutherford Appleton Laboratory, Harwell Oxon, Didcot, Oxfordshire OX11 0FA, UK
eDiamond Light Source, Harwell Science and Innovation Campus, Didcot OX11 0DE, Oxfordshire, UK
fDepartment of Chemistry, the University of Manchester, Oxford Road, Manchester, M13 9PL, UK
gSchool of Chemistry, University of Glasgow, Glasgow G12 8QQ, UK
hUK Catalysis Hub, Research Complex at Harwell, Rutherford Appleton Lab, Harwell, Oxfordshire OX11 0FA, UK
First published on 18th April 2024
Abstract
The need to achieve net zero requires decarbonisation across all areas of our industrialised society, including the production of chemicals. One example is the production of acetonitrile, which currently relies on fossil carbon. Recently, supported Pd nanoparticles have been shown to promote the selective transformation of bio-derived ethanol to acetonitrile. Elsewhere, current research has demonstrated the importance of interstitial structures of Pd in promoting specific transformations. In this study, we demonstrate through a spatially resolved operando energy-dispersive-EXAFS (EDE) technique that selectivity to acetonitrile (up to 99%) is concurrent with the formation of a PdNx phase. This was evidenced from the features observed in the X-ray absorption near edge structure that were validated against PdNx samples made via known synthesis methods. Above 240 °C, the Pd nanoparticles became progressively oxidised which led to the production of unwanted byproducts, primarily CO2. The spatially resolved analysis indicated that the Pd speciation was homogeneous across the catalyst profile throughout the series of studies performed. This work resolved the structural selectivity of Pd nanoparticles that directs ethanol ammoxidation towards acetonitrile, and provides important information on the performance descriptors required to advance this technology.
Broader contextDecarbonising the chemicals industry is an important part of the roadmap for industrialised societies achieving net zero. Key chemical feedstocks, such as acetonitrile, can exhibit significant market volatility – both in price and availability – where their production is linked to the petrochemicals industry. In this regard, there is a need to decouple the supply of such feedstocks from the petrochemicals industry; not only to decarbonise the process but to improve security of supply. In this study, we assessed the direct ammoxidation of ethanol (which can be delivered through biorefining) to acetonitrile, over a Pd/Al2O3 catalyst. Using advanced operando synchrotron characterisation methods, we were able to develop key structure function relationships, that provide important insights into how the process can be further optimized. |
1. Introduction
The intergovernmental panel on climate change has indicated that decarbonisation activities are required across all areas of our global industrialised activity, if we are to limit the temperature rise to 1.5 °C of pre-industrial levels. Some of the biggest gains come from decarbonising the energy sector, however, the chemicals industry is a significant blind spot that is interconnected with the exploitation of fossil-derived carbon. Acetonitrile is an important fine chemical, especially for the pharmaceutical industry which uses over 70% of the ∼80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons produced per annum.1 The conventional approach to produce acetonitrile is as a by-product of acrylonitrile production. This method makes acetonitrile supply dependent on the demand for acrylonitrile and the availability of propylene, which comes traditionally from fossil derived carbon.2 Achieving a secure route to important platform chemicals that are not dependent on fossil derived feedstocks is an important aspect of global net zero strategies.
000 tons produced per annum.1 The conventional approach to produce acetonitrile is as a by-product of acrylonitrile production. This method makes acetonitrile supply dependent on the demand for acrylonitrile and the availability of propylene, which comes traditionally from fossil derived carbon.2 Achieving a secure route to important platform chemicals that are not dependent on fossil derived feedstocks is an important aspect of global net zero strategies.
Conversely, the ammoxidation of bioethanol affords a sustainable route to acetonitrile.3 The process of direct ammoxidation of ethanol was reported initially in the 1990s, with early studies utilising SAPOs (including the addition of vanadium doping) and supported vanadium phosphorous oxide catalysts.4,5 These works established that high selectivity (up to 99%) could be achieved under certain conditions, but relatively high temperatures were required (>350 °C). Subsequently, the catalysts scope was extended to supported Co based materials, with or without additional promoters (e.g., Nickel).6–8 These materials showed enhanced durability, achieving an acetonitrile yield of 82% for 720 h of operation. Active vanadium centres have remained an avenue of research, and both vanadyl pyrophosphate (VPP) and supported vanadium oxide have been studied for the ammoxidation of ethanol; supported vanadium oxides exhibited a strong temperature dependence on selectivity, with an optimum yield achieved at ∼380 °C. The work on VPP did not achieve high selectivity to acetonitrile but reported valuable mechanistic insights into the different reaction networks that impact on acetonitrile yield.4
Recently, there have been reports of producing acetonitrile from ethanol ammoxidation at mild temperatures (150–200 °C) using Pd based catalysts.9 These studies demonstrated that although high yield (>90%) could be sustained for extended periods (>70 hours) there was a clear temperature effect on the selectivity; above 250 °C, the selectivity rapidly declined. The authors postulated that a structure sensitive reaction was occurring, although they indicated that this was not investigated as part of their work.
Elsewhere, our work has established the dynamic behaviour of Pd nanoparticles in an atmosphere of NH3.10–12 It has long been established that metallic Pd nanoparticles can form interstitial structures of hydride, carbide and boride.13–16 We recently identified that, during low temperature selective ammonia oxidation towards nitrogen, an interstitial nitride structure was formed. Moreover, the presence of this structure was crucial in maintaining a high selectivity towards nitrogen. This structural change was evidenced through operando X-ray absorption fine structure (XAFS) studies,11 as well as ex situ X-ray photoelectron spectroscopy (XPS) and magic angle spinning (MAS) solid-state nuclear magnetic resonance (NMR) spectroscopy.12
Motivated by the analogous nature of the conditions we postulated that the formation of Pd nitride could be responsible for the structural selectivity that was alluded to for ethanol ammoxidation. In this work, we demonstrate through a spatially resolved operando energy dispersive EXAFS (EDE) investigation that high selectivity to acetonitrile is only achieved when the interstitial Pd structure is present for a Pd/Al2O3 catalyst. Unwanted selectivity, e.g., towards CO2 is concurrent with the progressive oxidation of the Pd nanoparticles.
2. Experimental
Catalyst preparation
A 1.5 wt% Pd/γ-Al2O3 catalyst was synthesized via incipient wetness impregnation (IWI), followed by calcination in static air at 500 °C (5 °C min−1) for 2 hours. This catalyst has been characterised previously, with structural information reported elsewhere.10,11SEM characterisation
Images studying the morphology of the materials were collected using a Carl Zeiss Sigma 500 VP field emission scanning electron microscope (FE-SEM). Powders of the samples were dropped onto a conductive adhesive carbon tab. Any excess loose material was tapped off and blown away via nitrogen stream. The microscope was operated at 3 kV accelerating voltage in order to minimise charging effects common to studying even conductive powders, as well as provide improved surface signal sensitivity.EDE measurements and data analysis
Measurements utilising EDE were carried out at the I20 beamline at the Diamond Light Source at the Pd K-edge (24358 eV) using a Si(311) polychromator and an X-ray detector consisting of a FReLoN CCD camera.17 The X-ray beam was focused onto the sample with horizontal and vertical spot sizes of 0.5 and 0.15 mm, respectively. To avoid any potential radiation damage to the sample, the beam intensity was reduced by a factor of 5 through the opening of the wiggler gap. EDE spectra of a Pd foil were collected between measurements for data calibration. An analogous capillary filled with Al2O3 was used as I0.
Fig. 1 illustrates the experimental setup used for the operando investigations, which includes a compact reaction cell for controlling the sample temperature and atmosphere during measurements. The cell design, construction, and operation have been described in detail elsewhere.18 In brief, the reactor cell features a designated area where up to two horizontally aligned quartz reactors (3 mm ∅) can be mounted, with a 5 cm spacing between them. To ensure accurate sample heating without obstructing the X-ray beam, tungsten coils are positioned above and below each reactor tube. Temperature control, reaching up to 1273 K, is achieved using an Eurotherm PID controller. A K-type thermocouple inserted through the end of each reactor allows localized temperature measurement at the catalyst bed's position. Additionally, the cell is equipped with a motorized track that enables precise alignment of the X-ray beam across the sample in the x, y, and z coordinates.
 | ||
| Fig. 1 Reaction cell and experimental setup used at I20-EDE beamline: (a) ceramic jacket, (b) quartz capillary tube containing the catalyst bed, (c) quartz capillary containing Al2O3 support as a reference, and (d) tungsten coil for heating. | ||
In a typical EDE experiment, 400 mg of Pd/γ-Al2O3 catalyst was sieved to a pellet fraction of 250–350 μm and then loaded into the reactor. The catalyst bed length was 10 mm and was centred between the heating coils to maintain uniform heating across the bed prior to any measurements. The EDE measurements were collected at 10 different axial positions along the catalyst bed length. Prior to the EDE measurements, the catalyst was reduced at 50 °C under a flow of 5% H2/He, and then heated to 100 °C under pure Ar gas, to remove any traces of PdHx that may have formed, before switching to the reactant gas feed. The reactant gas feed consisted of 4200 ppm NH3, 525 ppm ethanol, and 6800 ppm O2 balanced in Ar, at a total flow rate of 40 cm3 min−1. EDE spectra were collected at each axial position while the temperature was increased from 100 to 300 °C in 20 °C increments (i.e., at 100, 120, 140, 160, 180, 200, 220, 240, 260, 280, and 300 °C).
The processing of the time-resolved EDE data involved the use of the DAWN software package19 to crop and calibrate the XAFS spectra. EXAFS data processing was performed using the Horae package (Athena and Artemis) with the IFEFFIT software.20,21 Athena was used to calibrate, align, and normalize the spectra with respect to the Pd foil, whereby E0 was fixed at 24358 eV.
Ex situ EXAFS measurements
Ex situ XAS spectra at the Pd K edge were acquired at the B18 beamline at Diamond Light Source, UK. Samples were measured in transmission mode, using a Si(311) monochromator and ion chamber detectors. The samples were pelletised with cellulose as a binder to optimise their absorption characteristics.Ethanol ammoxidation testing
The ethanol ammoxidation reaction was carried out within a quartz tubular reactor located inside a programmable furnace, under atmospheric pressure and with a flow rate of 100 cm3 min−1. The gas mixture was composed of 4200 ppm ammonia (NH3), 525 ppm ethanol, and 6800 ppm oxygen (O2), balanced with argon (Ar). The Pd/γ-Al2O3 catalyst was pelletized and sieved to achieve a particle size of 150–300 μm and was packed into the reactor. Prior to the reaction, the catalyst was pre-treated for 1 hour in 10 vol% H2/Ar, at a flow rate of 100 cm3 min−1 and a temperature of 120 °C. During the tests, the total flow rate (100 or 200 cm3 min−1) and catalyst loadings (300, 150 or 35 mg) were modified to achieve a GHSV between 2 and 34.3 × 104 cm3 g−1 h−1. The reactions were conducted at temperatures ranging from 100 to 300 °C, and the gas products were analysed using a mass spectrometer (Hiden HPR20). Mass-to-charge (m/z) ratios of 17, 31, 32, 36, and 41, were monitored corresponding to ammonia, ethanol, oxygen, argon, and acetonitrile, respectively. The temperature of the catalyst bed was monitored by a K-type thermocouple positioned adjacent to the bed. Ethanol conversion (X) and acetonitrile selectivity (S) were determined as shown below (eqn (1) and (2)).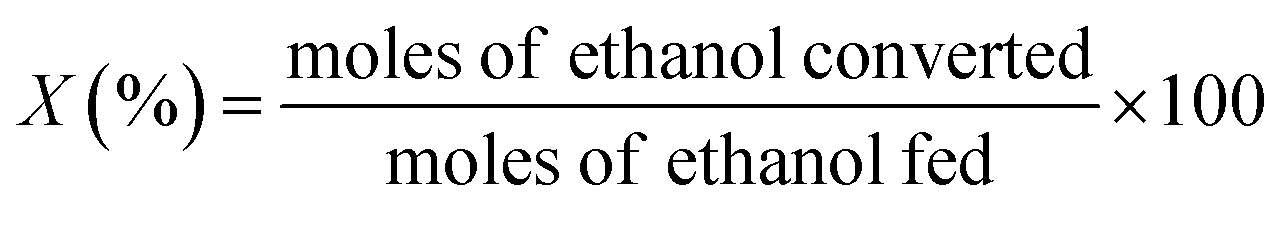 | (1) |
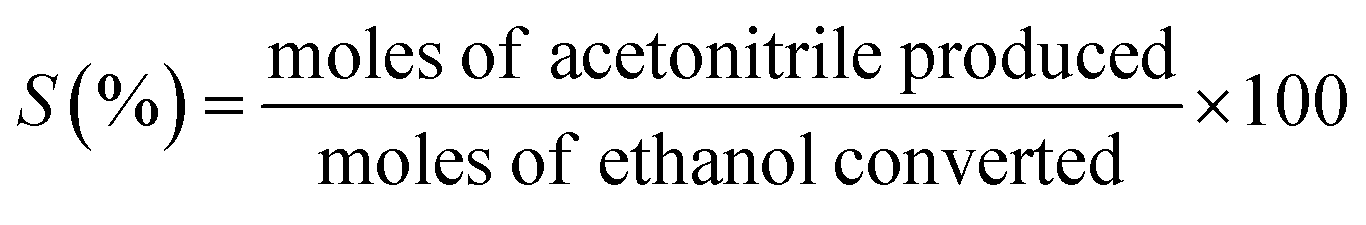 | (2) |
 | (3) |
Ethanol pulse testing
30 μl of ethanol (or acetaldehyde) was injected using a Hamilton syringe through a septum at the front of the reactor, while helium served as the carrier gas to deliver ethanol over the freshly, in situ, prepared samples. Approximately 250 mg of 1.5 wt% Pd/γ-Al2O3 was situated between glass wool nests in an 8-inch ID stainless steel tube reactor internally coated with fused silica. All samples were maintained isothermally at 150 °C for all measurements performed. The total flow rate was maintained at 40 ml min−1. Mass-to-charge ratios (m/z) of 17, 18, 28, 31, 41, and 44 were recorded with an online Hiden HPR-20 R&D mass spectrometer to assess the products evolved during ethanol injection.3. Results and discussion
The synthesis and characterization of the 1.5 wt% Pd/γ-Al2O3 catalyst used within this work, has been described extensively elsewhere; the nanoparticles are well-dispersed with an average particle size of 2 nm as determined through transmission electron microscopy.10,11 Prior to the operando EDE study, catalytic testing was performed to demonstrate the efficacy of our Pd/γ-Al2O3 for ethanol ammoxidation (Table 1 and Fig. 2). The percentage of ethanol conversion (C%) and acetonitrile selectivity (S%) were monitored as a function of temperature (100–300 °C) under steady-state conditions. Previous work on the performance of Pd/γ-Al2O3 towards the ammoxidation of ethanol,9 demonstrated a strong temperature dependence on selectivity; at high temperatures the acetonitrile selectivity progressively decreases. In the present study, the same behaviour was observed across all different GHSV values. The maximum selectivity was achieved at low residence times and low temperatures; 99% selectivity was achieved at a GHSV of 8 × 104 cm3 g−1 h−1 and a reaction temperature of 150 °C. At longer residence times (lower GHSV) there was a trend towards poorer selectivity, e.g., at GHSV values of 2 and 4 × 104 cm3 g−1 h−1, the selectivity towards acetonitrile at 150 °C was 60 and 67%, respectively. This could, in part, be ascribed to the different conversion levels and local cascade reactions, where the generated acetonitrile can further oxidise. However, if we compare the respective ethanol conversion and acetonitrile selectivity at GHSV values of 4 and 8 × 104 cm3 g−1 h−1 at temperatures of 150 °C and 200 °C, respectively, we observe an effect which cannot solely by attributed to the level of conversion; the 8 × 104 cm3 g−1 h−1 GHSV study achieved higher relative selectivity to acetonitrile, for the ethanol conversion point, e.g., 72% acetonitrile selectivity at 54% ethanol conversion.| Entry | GHSV (cm3 g−1 h−1) | T (°C) | C(ethanol) (%) | S(acetonitrile) (%) |
|---|---|---|---|---|
| Reaction conditions: gas mix = NH3 (4200 ppm), ethanol (525 ppm) and O2 (6800) balanced in Ar. Total flow and catalyst weight = 100 cm3 min−1, 300 mg (entry 1), 100 cm3 min−1, 150 mg (entry 2), 200 cm3 min−1, 150 mg (entry 3), and 200 cm3 min−1, 35 mg (entry 4). | ||||
| 1 | 2 × 104 | 100 | 12 | 60 |
| 150 | 34 | 50 | ||
| 200 | 96 | 48 | ||
| 250 | 98 | 2 | ||
| 300 | 99 | 1.7 | ||
| 2 | 4 × 104 | 100 | 6 | 67 |
| 150 | 16.3 | 60 | ||
| 200 | 92 | 52 | ||
| 250 | 97 | 1.5 | ||
| 300 | 98 | 1.2 | ||
| 3 | 8 × 104 | 150 | 8.8 | 99 |
| 200 | 54 | 72 | ||
| 250 | 97 | 1.2 | ||
| 4 | 34.3 × 104 | 250 | 49.4 | 1.4 |
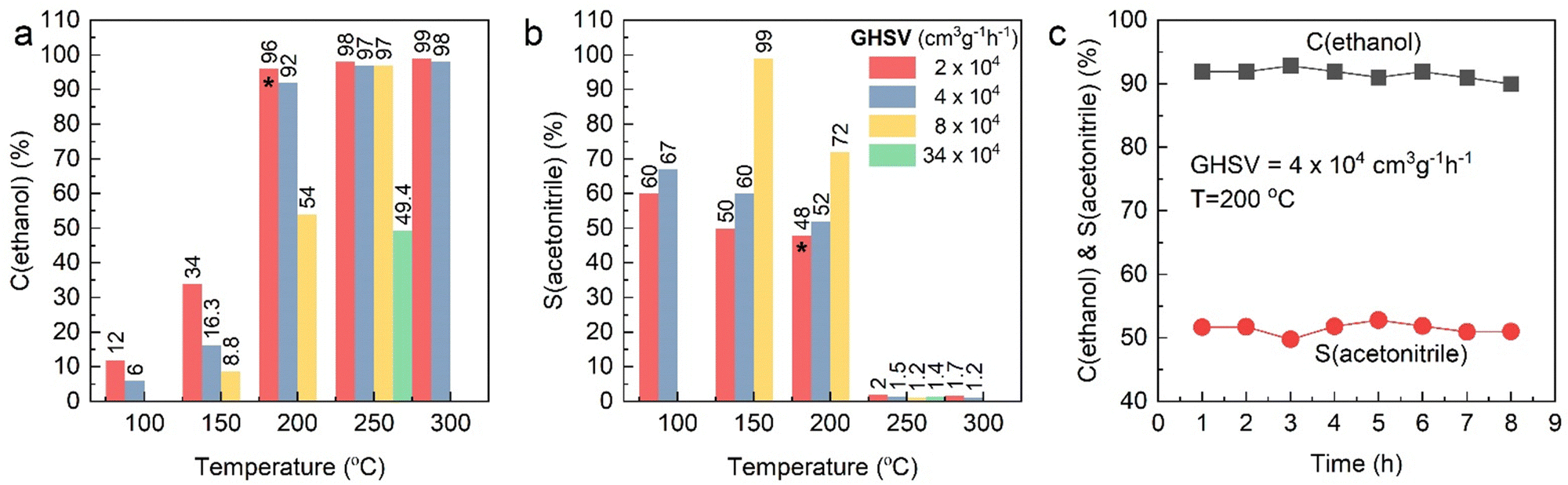 | ||
| Fig. 2 Activity testing profile over Pd/Al2O3 catalyst as a function of temperature (100–300 °C) at different GHSV, as indicated. (a) Ethanol conversion (%), (b) acetonitrile selectivity (%) and (c) stability test over 8 h on reactant feed stream at 200 °C on 150 mg of catalyst at flow of 100 cm3 min−1 (same conditions denoted with asterisks in Fig. 2a and b collected over 8 h). | ||
This variance of selectivity as a function of GHSV is indicative of a parasitic side reaction that directly consumes ethanol that can be moderated by reducing the contact time. Cavani and co-workers looked at the reaction mechanism of ethanol ammoxidation over VPP.22 In their work, they identified that the initial step in the process was oxidative dehydrogenation to yield acetaldehyde; this then can form the imine on reaction with NH3 or undergo further oxidation. They also show that the acetonitrile can react with O2 directly to yield further oxidation products. Our data suggest that parasitic side reactions are highly dependent on GHSV, which is not the case for acetonitrile formation. However, the data also showed that the selectivity drops significantly as a function of temperature. Irrespective of the conversion point or GHSV the selectivity to acetonitrile was negligible (∼2%) at and above 250 °C.
The Pd catalysed conversion of ethanol to acetaldehyde and ethyl acetate is well established.23 Indeed, ethanol is known to oxidise to CO2 over Pd based catalysts with acetaldehyde known to be a key intermediate.24 These studies demonstrated that dehydrogenation to acetaldehyde, was the primary step before rapid oxidation. The temperature of this process was 190–230 °C. However, in our study, meaningful selectivity (>70%) to acetonitrile at 200 °C was achieved when working at high GHSV. Assuming that acetaldehyde is still formed initially, what limits the rapid reaction to CO2 as demonstrated previously? Interestingly, all studies at 250 °C predominantly produced CO2, even though, in some cases, there was a negligible change in conversion, e.g., at a GHSV of 2 × 104 cm3 g−1 h−1 the acetonitrile selectivity drops from 48 to 2%, whilst the conversion remains broadly constant (96 vs. 98%). We postulated that this stark change in selectivity would be consistent with a large structural change in the catalyst at this temperature, which subsequently impacts on the yield of acetonitrile. Elsewhere, an extended time-online studies (Fig. 2, pane C) demonstrated consistent selectivity and activity over an 8-hour window at 200 °C. These results suggested that Pd based materials are highly effective and durable at high ethanol conversion and that a future need is to better understand what causes the high-temperature loss in selectivity and learn how to moderate the undesirable over-oxidation processes; the 99% selectivity achieved at one testing condition suggests that the latter of these points is achievable.
To follow the structural changes that may occur at these different temperatures, a spatially resolved operando EDE study with online mass spectrometry was performed. EDE was chosen to allow us to assess transient changes (time resolution of ms) if required. However, the spectral resolution of EDE is inherently poorer in comparison to traditional XAFS data acquisition. We initially collected in situ reference spectra of a reduced form of our catalyst and that of interstitial PdNx (Fig. 3). The metallic form (Pd0) of our supported nanoparticles was achieved by reduction at 50 °C, followed by switching to argon and heating to 100 °C to decompose the formed Pd hydride.13,25,26 The interstitial nitride (PdNx) structure was formed using our established methods reported elsewhere.10–12 The EDE XANES data shows clear differences between the metallic and nitridic forms of Pd; the main-edge transition (often referred to as the white line) broadens for the PdNx, with a subsequent peak decreasing in intensity and shifting towards lower energy. These changes are consistent with what has been reported elsewhere,11 with differences in spectral appearance attributable to decreased resolution of EDE measurements.
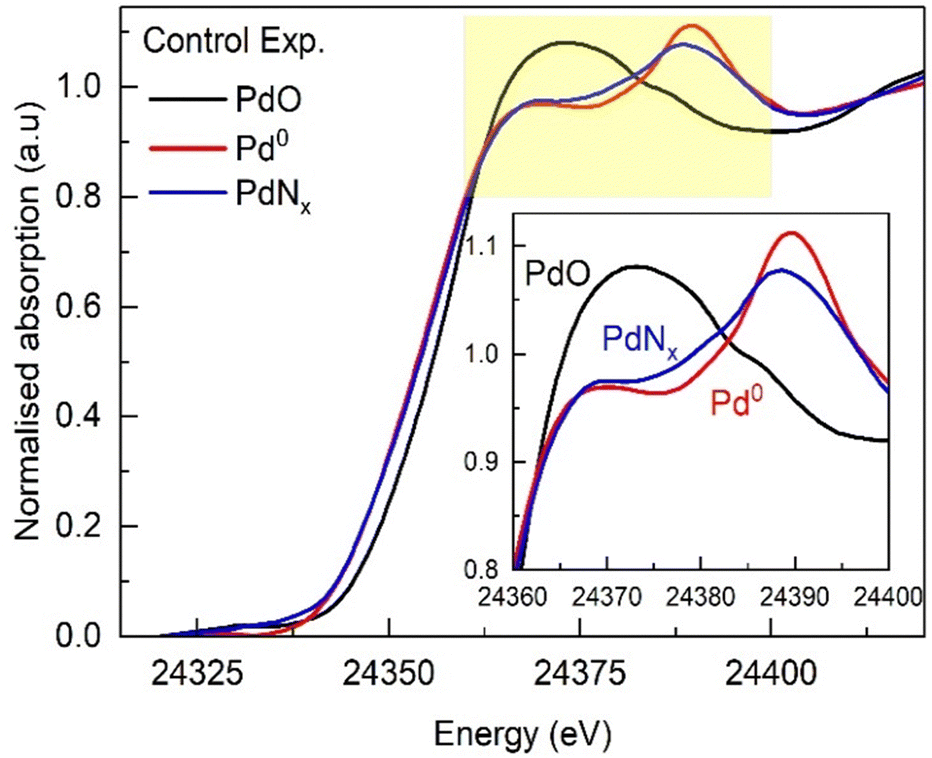 | ||
| Fig. 3 Normalized Pd K-edge XANES spectra of Pd0 (Ar-100 °C), PdNx (NH3-100 °C), and PdO (Ar-RT) forms obtained for 1.5 wt% Pd/γ-Al2O3 catalyst. | ||
To follow up on the catalytic testing studies, the Pd speciation was assessed at the same steady-state conditions, previously discussed (at temperatures of 100 to 300 °C, at discrete 20 °C intervals). Moreover, since a single-point analysis operando analysis often fails to account for the axial variation of environment throughout the catalyst bed;11 the catalyst speciation was assessed at discrete 1 mm intervals along the bed. Prior to the operando EDE study, the Pd/γ-Al2O3 was subjected to a reduction process using a 5% H2/He flow at 50 °C, and then heated to 100 °C under He, eliminating any Pd hydride. For the ethanol ammoxidation study, we introduced a balanced reactant mixture of NH3, O2, and ethanol in He into the reactor at 100 °C, which was then followed by the isothermal measurements, where EDE was performed at different axial positions and the end-pipe mass spectrometry data was recorded (Fig. 4). EDE data is subject to subtle variations in the bed homogeneity and packing density at different axial positions. However, these artefacts are consistent for each discrete axial position. To account for this, the normalised XANES data was compared at each axial position and separated into three temperature windows: 100–200 °C, 200–240 °C, and 240–300 °C (Fig. 5A–C, respectively). The full set of individual normalised XANES and additional XAFS analyses are reported in the ESI† (Fig. S1–S9 and Table S1).
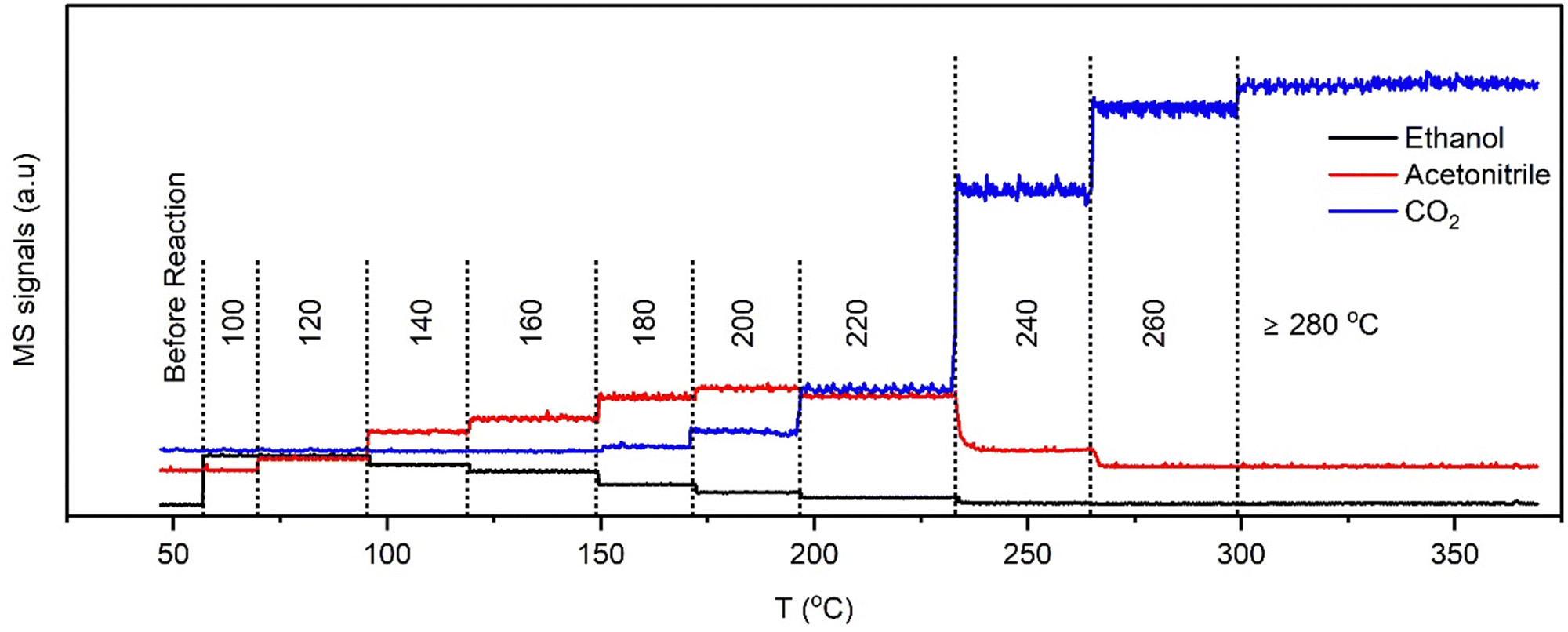 | ||
| Fig. 4 End-pipe MS signals of ethanol (m/z = 31), acetonitrile (m/z = 41), and CO2 (m/z = 44) recorded concurrently during EDE measurements. | ||
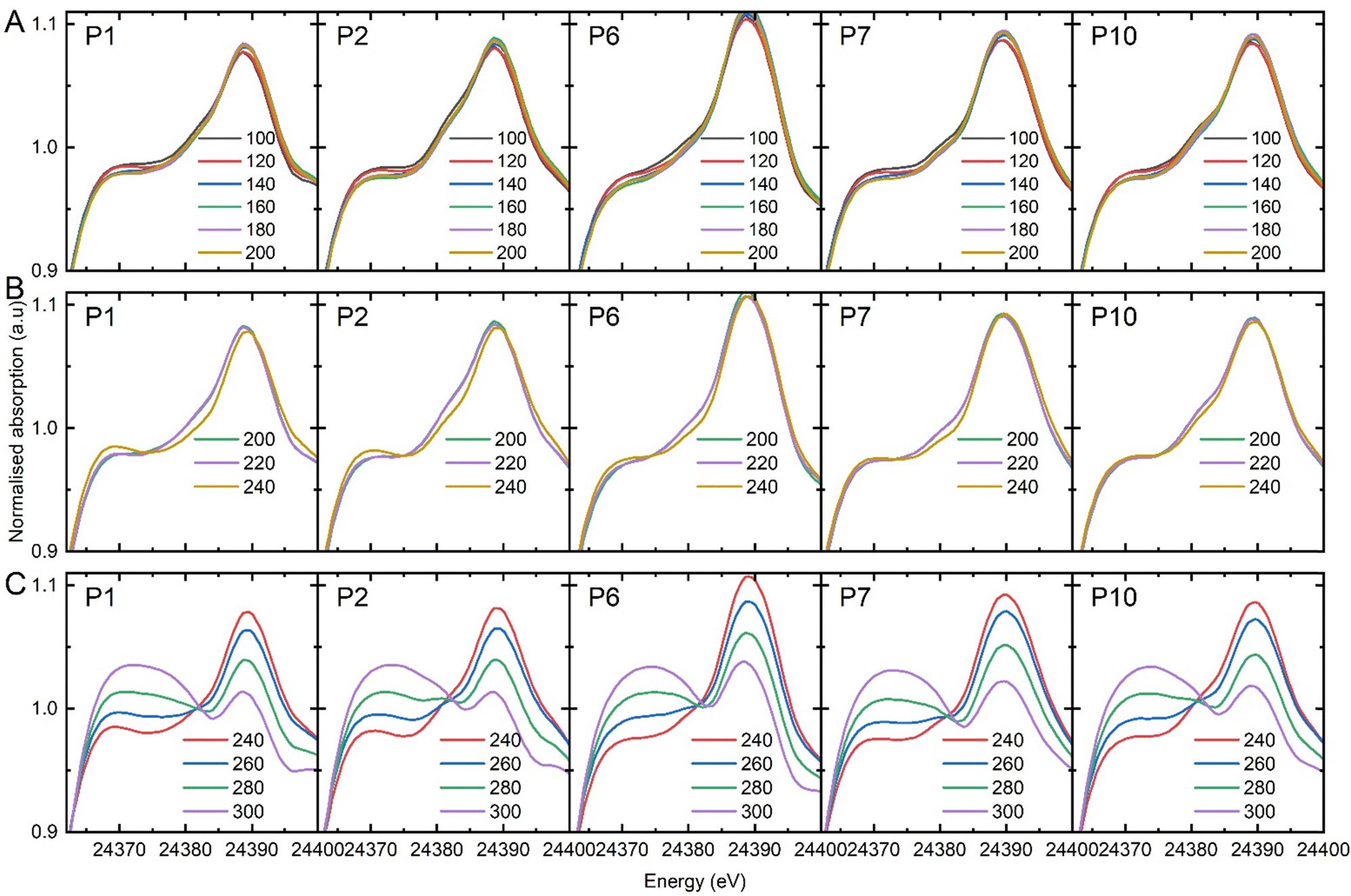 | ||
| Fig. 5 Normalized Pd K-edge XANES spectra obtained at various spatial positions from the front of the bed: 0 mm (P1), 1 mm (P2), 5 mm (P6), 6 mm (P7), and 9 mm (P10) recorded during EDE measurement. Spectra categorised into three temperature windows: (A) 100–200 °C, (B) 200–240 °C, (C) and 240–300 °C. Refer to ESI,† for the complete set of individual measurements. | ||
The end-pipe mass spectrometry data show the signals associated with ethanol (m/z = 31), acetonitrile (m/z = 41), and carbon dioxide (m/z = 44) at the different isothermal conditions. The results show a sharp decline in acetonitrile selectivity at and above 240 °C, which is consistent with the catalytic testing studies, where a drop in acetonitrile production was observed from 250 °C onwards. The palladium speciation at temperatures between 100 and 200 °C (Fig. 5A) showed strong uniformity in the spectra, irrespective of the axial position. A comparison to the metallic Pd and PdNx reference spectra (Fig. 3), indicated that heteroatom insertion in the FCC lattice of the Pd nanoparticles had occurred. Further evidence came from the absence of a rise in the main-edge transition; Pd nanoparticles tend to form a passivating surface oxide layer in net-oxidising environments, which manifests itself in the XANES as an increase in the main-edge transition.27–29 The absence of a change in this region is indicative of an interstitial structure type being present, which has been shown to inhibit the adsorption of oxygen.10,28
Conversely, when we assessed the catalyst speciation in the temperature range 200–240 °C (Fig. 5B), observable differences were observed. For the isothermal study conducted at 240 °C, changes in the XANES for the initial three positions were evident, with the inlet position experiencing the largest alteration. The data showed an increase in the main-edge transition, which is consistent with the removal of the heteroatom from the FCC lattice and the partial adsorption of surface oxygen. The subsequent sampling points within the catalyst bed, were consistent with an interstitial structure (Fig. 5C). Considering that the reaction took place under net-oxidising conditions, and that there was evidence of surface oxidation at the reaction inlet, why would the downstream speciation remain consistent with that of an interstitial structure type? As discussed previously, it has been proposed that a key intermediate in the formation of acetonitrile is acetaldehyde. If the catalyst is no longer selective towards acetonitrile what is the fate of the acetaldehyde? Bowker and co-workers studied the reaction of acetaldehyde on single crystals of Pd on different metal oxide supports. Their work demonstrated that the acetaldehyde reacts with the Pd surface, and inserts carbon into the FCC lattice, forming Pd carbide.30 At 240 °C, we propose we have reached the limit of the Pd nitride stability, which is consistent with the work we have reported elsewhere.10 At the inlet of the reactor, where there is largest concentration of oxygen, the conversion of ethanol is limited with subsequent low concentration of acetaldehyde. Consequently, the catalyst is partially oxidised. As the concentration of acetaldehyde increases moving downstream, in the absence of a Pd nitride interstitial structure, carbon is incorporated into the lattice. Our previous work has demonstrated that Pd carbide structures are more stable than Pd nitride and likely to persist for longer.12 Elsewhere, it has been shown that Pd carbide surfaces can be cleaned by pulsing oxygen at temperatures in excess of 250 °C. Above 260 °C (Fig. 5C), a more pronounced degree of oxidation of the Pd surface at all axial positions is evident. The depth of oxidation increased as a function of temperature – as evidenced through a steady rise in the main edge transition. However, at no point did complete oxidation of the Pd nanoparticles to PdO occur.
The spectral resolution of EDE was not entirely sufficient to confirm whether PdCx or PdNx was present during the point of maximum acetonitrile selectivity (i.e., at 150 °C). To assess this further, the XANES of the spent catalyst from the catalyst testing studies at 150 °C was compared to the XANES of analogous (same batch of catalyst) ex situ PdNx and PdCx prepared reference materials (Fig. 6). The fresh and spent catalysts were also assessed through SEM (Fig. S10, ESI†). The data confirmed that, at the highest selectivity towards acetonitrile, a PdNx phase was present. It is our assertion that the presence of the PdNx phase – at a minimum – limits the decomposition of acetaldehyde, by restricting the adsorption of oxygen.
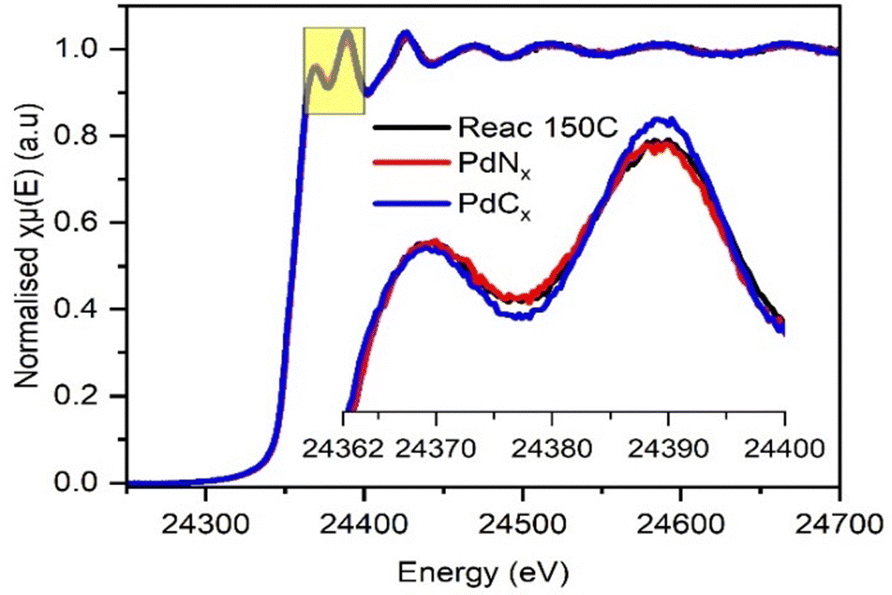 | ||
| Fig. 6 Ex situ XANES spectra at Pd K edge recorded on Pd/γ-Al2O3 catalyst after reaction at 150 °C (black), PdNx (red) and PdCx (blue) phases. | ||
To further investigate the role of PdNx in selective ammonia oxidation, we performed ethanol injection studies over a series of Pd/Al2O3 catalysts: (i) reduced Pd/γ-Al2O3, (ii) PdNx/γ-Al2O3 (produced in a NH3/O2 atmosphere), (iii) PdNx/γ-Al2O3 (produced in a NH3 atmosphere), (iv) Pd/γ-Al2O3 with adsorbed oxygen, and reduced Pd/γ-Al2O3, formed after PdNx decomposition.
For the reduced Pd/γ-Al2O3 catalyst we identified products of acetaldehyde, water, ethene, and carbon monoxide after pulsing ethanol (Fig. 7a and f). Acetaldehyde was identified through an increase in m/z 44, where the increase cannot be accounted for by the minor cracking fragment of ethanol (Fig. S11–S13, ESI†). The acetaldehyde was formed initially and was followed by an increase in water (m/z = 18), and m/z 28. The formation of water is indicative of acid catalysed dehydration of ethanol to ethene, as has previously been established over γ-Al2O3 supports.31,32 The relative fractions of m/z 28 and m/z 18 changed as a function of time, indicating a secondary process contributing towards m/z 28. Bowker et al. have previously confirmed that acetaldehyde decomposes to CH4, PdCx, Cad, and CO over Pd surfaces, which is consistent to the variation in m/z 28 identified here. It is also noteworthy that there is a time delay between the formation of acetaldehyde and the acid catalysed processes.
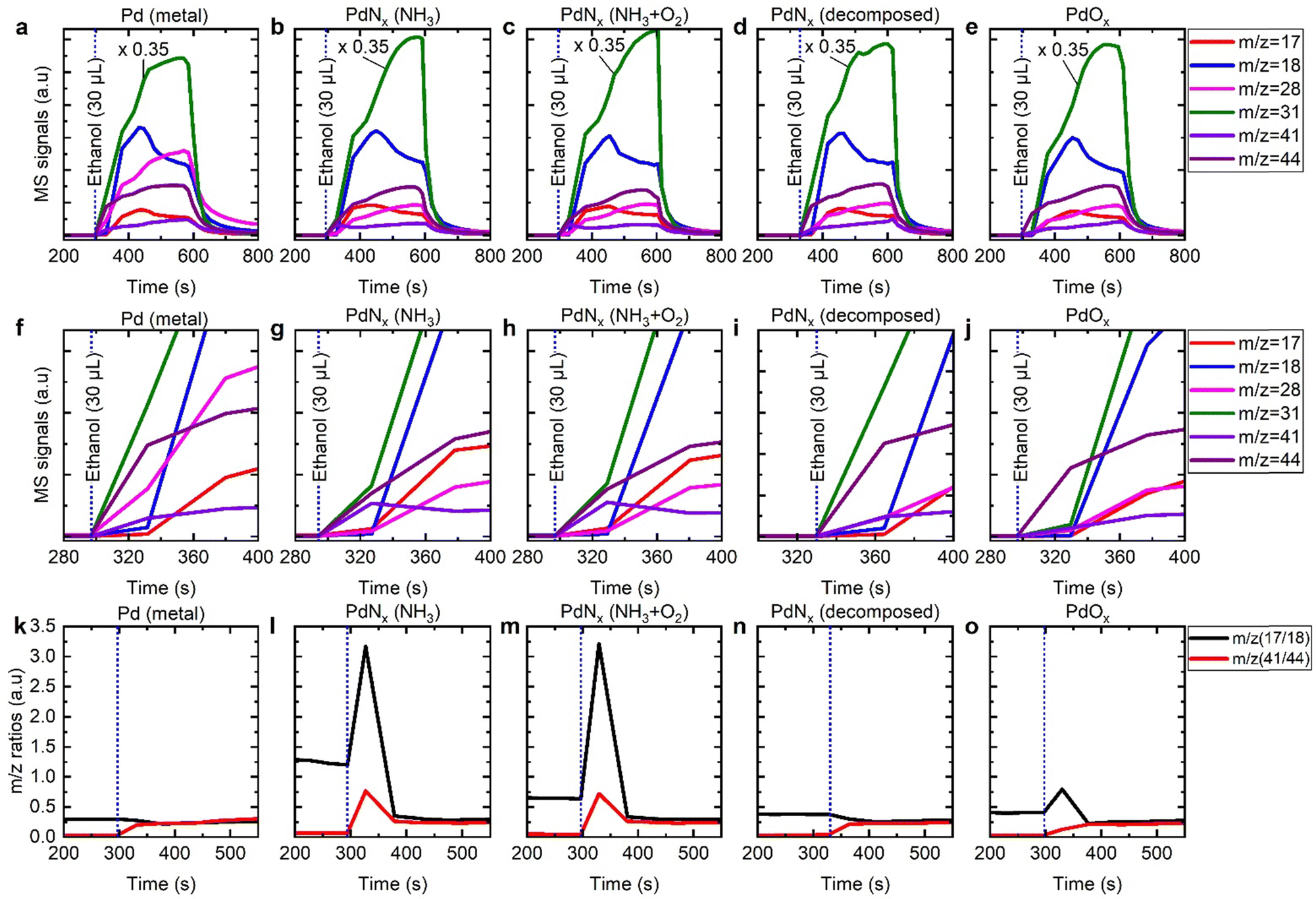 | ||
| Fig. 7 Traces of m/z = 31, 17, 18, 28, 31, 41, and 44 during the pulsing of 30 ml of ethanol over a 1.5 wt% Pd/γ-Al2O3 catalyst with different forms of supported Pd nanoparticles: (a) Pd metal, (b) PdNx (formed using 5% NH3/He only), (c) PdNx (formed using 37 ml min−1 of 5% NH3/He and 3 ml min−1 of 5% O2/He), (d) PdNx decomposed under helium at 250 °C, and PdOx formed by exposing in situ formed Pd metal to 5% O2/He prior to pulsing. Total flow of 40 ml min−1. Middle subfigures: (f)–(j), are zoomed-in areas of the corresponding subfigures (a)–(e) above, respectively. Bottom subfigures: (k)–(o), represent m/z (17/18) and m/z (41/44) ratios over the different forms of Pd presented above, respectively. | ||
All other catalysts investigated in this study were exposed to NH3 at some point during their formation. For both PdNx/γ-Al2O3 samples there were marked differences to the metallic Pd sample, when pulsed with ethanol (Fig. 7b, c, g and h); there is a significant increase in m/z = 41, attributable to acetonitrile, and although there is still evidence of dehydration reactions through the production of water, the production of m/z = 28 is diminished. Another known acid catalysed product of γ-Al2O3 supports is diethyl ether, which also has a significant fragment at m/z 31. We propose that the ammonia introduced as part of the catalyst preparation, binds strongly to the γ-Al2O3 support, and inhibits the acid catalysed dehydration of ethanol to ethene. Regarding the production of acetonitrile, other catalysts gave rise to minor amounts of m/z 41, which mirrored the production of acetaldehyde and is the consequence of a minor fragment of ethanol, acetaldehyde, and diethyl ether (Fig. 7). This is exemplified by the presence of m/z 41 for the virgin reduced catalyst, where exposure to ammonia has been avoided throughout (Fig. 7a and f). Again, there was a time delay between the acetaldehyde and acetonitrile production and that of the acid catalysed processes. Rooney and co-workers assessed different supports for the Pd mediated ammoxidation of ethanol.9 Their results showed that at 175 °C the selectivity of different systems to acetonitrile where in the following order ZrO2 > TiO2 > Al2O3 > SiO2. The Pd/ZrO2 catalyst achieved a maximum selectivity of ∼90% at a temperature of 170 °C, with the ethanol conversion <10%. The Pd/Al2O3 catalyst was found to have the highest support acidity. These findings are consistent with our results where we show that the acid catalysed side reactions can be minimised by working at high GHSV, where we are then able to achieve an acetonitrile selectivity of 99%.
To decouple the effects of the presence of PdNx and NH3 adsorbed on the catalyst, we decomposed the PdNx using a mild thermal treatment (Fig. 7d and l). When this catalyst was pulsed with ethanol, the presence of ammonia continued to restrict the production of ethene, however, although m/z 41 was observed it mirrored the trace associated with acetaldehyde and was much less intense than that observed when the nitride was present. When we assessed the ratio between m/z 41 and m/z 44 for all experiments, there was a clear distinction for the initial m/z 41 production for the PdNx samples (Fig. 7k–o). When we assessed the oxidised Pd surface, there was a large increase in m/z 44, prior to ethanol breakthrough, indicating rapid ethanol combustion to carbon dioxide as has been previously established (Fig. 7e and j).33–35 We also assessed the ratio of m/z 17 and m/z 18 to identify if ammonia was released during the pulsing experiments (Fig. 7k–o). For the PdNx samples, there is a strong correlation between increased acetonitrile production and ammonia production. Elsewhere, this is also found for the Pd/γ-Al2O3 with adsorbed oxygen; for this system we propose that the exothermic nature of ethanol combustion creates local hot spots, which result in the desorption of ammonia. When considering the rise in ammonia for the PdNx samples, this can originate from interstitial nitrogen reacting with liberated hydrogen (from ethanol dehydrogenation) or from local heat generation.
To probe the mechanism in more detail, we assessed the interaction of acetaldehyde with PdNx/γ-Al2O3 (Fig. 8), alongside the respective mass fragment ratios described previously. The acetaldehyde was readily converted to acetonitrile, with the highest m/z 41:m/z 44 ratio reported in this study. This indicates that acetaldehyde is the key intermediate in the formation of acetonitrile. Moreover, when we assessed the ratio of m/z 17 and m/z 18, there was limited variation. This confirmed that either there was minimal ammonia released, or ammonia released was quickly consumed in the process to make acetonitrile.
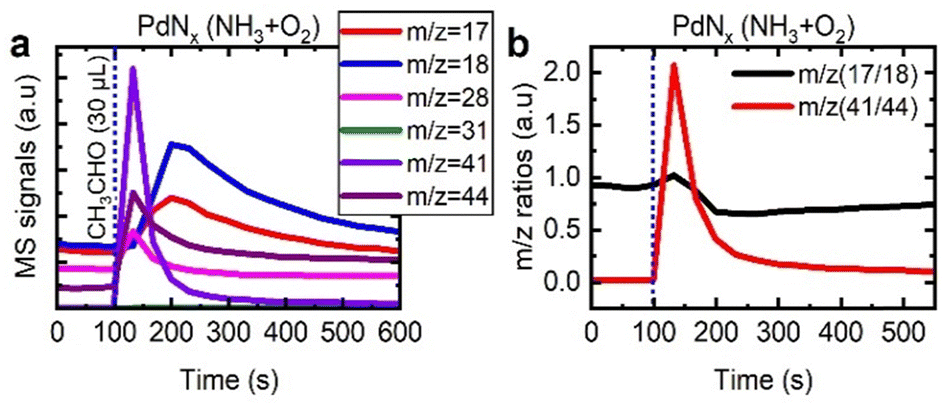 | ||
| Fig. 8 Traces of m/z = 31, 17, 18, 28, 31, 41, and 44 during the pulsing of 30 μl of acetaldehyde over the PdNx phase of a 1.5 wt% Pd/γ-Al2O3 catalyst (a) and the corresponding m/z (17/18) and m/z (41/44) ratios (b). | ||
From these experiments, we established that the role of PdNx goes beyond restricting oxygen adsorption; with no oxygen present and ammonia adsorbed on the γ-Al2O3 support, there is no indication of acetonitrile formation. Indeed, we propose that these results are consistent that the nitride structure plays a direct role in the reaction mechanism; whether this is through nitrogen abstraction or through alternative means remains unproven. Furthermore, the time delay between the ethanol pulse and the production of unwanted acid catalysed products is consistent with our observation that identified the selectivity could be further enhanced by operating at reduced contact times.
4. Conclusion
In this study, we demonstrate that Pd catalysed ammoxidation of ethanol is dependent on the dynamic structural evolution of the Pd nanoparticle during catalysis. Our operando and catalytic testing studies both demonstrate that the selectivity to acetonitrile has a strong temperature dependence. At temperatures below 240 °C, the operando EDE data is consistent with the presence of an interstitial structure type of Pd. The reaction conditions allow for the presence of both PdNx and PdCx, however, the increased spectral resolution of a traditional XAFS data acquisition confirmed the presence of PdNx post reaction. At the point where the first signs of surface oxidation is observed (240 °C), and by inference removal of the PdNx takes place, the selectivity towards acetonitrile drops markedly. As the catalyst becomes progressively more oxidised at higher temperatures, the selectivity to acetonitrile decreases further. Moreover, the catalytic testing data indicated the presence of multiple pathways that impact acetonitrile selectivity, that is not solely temperature dependent. Working at high GHSV and reducing the contact time reduced the impact of the parasitic side reactions and had a significant effect on acetonitrile selectivity, reaching a maximum of 99%. Our additional, studies looking at the reactivity of different surfaces to pulses of ethanol and acetaldehyde indicated that: (i) the nitride plays a more significant role in the reaction mechanism than limiting oxygen adsorption alone; (ii) acid catalysed side reactions have a clear time delay to other ethanol conversion products, indicating the benefit of working at high GHSV; and (iii) acetaldehyde readily converts to acetonitrile over PdNx surfaces. This work is an important first step in understanding how the production of acetonitrile from bioderived ethanol can be further optimised. Such processes are essential to achieve net zero as we seek to move towards a chemical industry not reliant on fossil-based carbon.Data access statement
The data that support the findings of this study are available from the University of Southampton repository with the identifier, https://doi.org/10.5258/SOTON/D3050.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to acknowledge EPSRC for funding (EP/V000691/1) and to Diamond Light Source for allocating beamtime (SP28666, and SP34632). We would like to acknowledge the support and assistance of other Diamond staff in maintaining the beamlines: Dr Diego Gianolio, Dr Veronica Celorrio, Dr Monica Amboage, and Professor Fred Mosselmans. Donato Decarolis and Peter Wells acknowledges funding received through the UK Catalysis Hub and the UK Catalysis Hub is kindly thanked for resources and support provided via our membership of the UK Catalysis Hub Consortium and funded by EPSRC grant: EP/R026815/1, EP/R026939/1, EP/R026645/1 or EP/R027129/1.References
- I. F. McConvey, D. Woods, M. Lewis, Q. Gan and P. Nancarrow, Org. Process Res. Dev., 2012, 16, 612–624 CrossRef CAS.
- F. Rezaie, V. Pirouzfar and A. Alihosseini, Thermal Sci. Eng. Progress, 2020, 16, 100463 CrossRef.
- A. Tripodi, E. Bahadori, D. Cespi, F. Passarini, F. Cavani, T. Tabanelli and I. Rossetti, ACS Sustainable Chem. Eng., 2018, 6, 5441–5451 CrossRef CAS.
- B. Reddy and B. Manohar, J. Chem. Soc., Chem. Commun., 1993, 234–235 RSC.
- S. Kulkarni, R. Rao, M. Subrahmanyam and A. Rao, J. Chem. Soc., Chem. Commun., 1994, 273 RSC.
- Y. Zhang, Y. Zhang, C. Feng, C. Qiu, Y. Wen and J. Zhao, Catal. Commun., 2009, 10, 1454–1458 CrossRef CAS.
- D. Zhang, Y. Zhang, Y. Wen, K. Hou and J. Zhao, Chem. Eng. Res. Des., 2011, 89, 2147–2152 CrossRef CAS.
- C. Feng, Y. Zhang, Y. Zhang, Y. Wen and J. Zhao, Catal. Lett., 2011, 141, 168–177 CrossRef CAS.
- C. Hamill, H. Driss, A. Goguet, R. Burch, L. Petrov, M. Daous and D. Rooney, Appl. Catal., A, 2015, 506, 261–267 CrossRef CAS.
- E. K. Dann, E. K. Gibson, R. H. Blackmore, C. R. A. Catlow, P. Collier, A. Chutia, T. E. Erden, C. Hardacre, A. Kroner, M. Nachtegaal, A. Raj, S. M. Rogers, S. F. R. Taylor, P. Thompson, G. F. Tierney, C. D. Zeinalipour-Yazdi, A. Goguet and P. P. Wells, Nat. Catal., 2019, 2, 157–163 CrossRef CAS.
- D. Decarolis, A. H. Clark, T. Pellegrinelli, M. Nachtegaal, E. W. Lynch, C. R. A. Catlow, E. K. Gibson, A. Goguet and P. P. Wells, ACS Catal., 2021, 11, 2141–2149 CrossRef CAS PubMed.
- L. G. Costley-Wood, K. Mohammed, M. Carravetta, D. Decarolis, A. Goguet, A. Kordatos, R. Vakili, H. Manyar, E. McPake, C. Skylaris, P. Thompson, E. K. Gibson and P. P. Wells, ChemCatChem, 2023, 15, e202300870 CrossRef CAS.
- A. L. Bugaev, O. A. Usoltsev, A. A. Guda, K. A. Lomachenko, I. A. Pankin, Y. V. Rusalev, H. Emerich, E. Groppo, R. Pellegrini, A. V. Soldatov, J. A. van Bokhoven and C. Lamberti, J. Phys. Chem. C, 2018, 122, 12029–12037 CrossRef CAS.
- M. W. Tew, M. Nachtegaal, M. Janousch, T. Huthwelker and J. A. van Bokhoven, Phys. Chem. Chem. Phys., 2012, 14, 5761–5768 RSC.
- M. W. Tew, M. Janousch, T. Huthwelker and J. A. van Bokhoven, J. Catal., 2011, 283, 45–54 CrossRef CAS.
- C. W. A. Chan, A. H. Mahadi, M. M.-J. Li, E. C. Corbos, C. Tang, G. Jones, W. C. H. Kuo, J. Cookson, C. M. Brown, P. T. Bishop and S. C. E. Tsang, Nat. Commun., 2014, 5, 5787 CrossRef CAS PubMed.
- J.-C. Labiche, O. Mathon, S. Pascarelli, M. A. Newton, G. G. Ferre, C. Curfs, G. Vaughan, A. Homs and D. F. Carreiras, Rev. Sci. Instrum., 2007, 78, 091301 CrossRef PubMed.
- P. J. Chupas, K. W. Chapman, C. Kurtz, J. C. Hanson, P. L. Lee and C. P. Grey, J. Appl. Crystallogr., 2008, 41, 822–824 CrossRef CAS.
- M. Basham, J. Filik, M. T. Wharmby, P. C. Y. Chang, B. El Kassaby, M. Gerring, J. Aishima, K. Levik, B. C. A. Pulford, I. Sikharulidze, D. Sneddon, M. Webber, S. S. Dhesi, F. Maccherozzi, O. Svensson, S. Brockhauser, G. Náray and A. W. Ashton, J. Synchrotron Rad., 2015, 22, 853–858 CrossRef PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- M. Newville, J. Synchrotron Rad., 2001, 8, 322–324 CrossRef CAS PubMed.
- T. Tabanelli, M. Mari, F. Folco, F. Tanganelli, F. Puzzo, L. Setti and F. Cavani, Appl. Catal., A, 2021, 619, 118139 CrossRef CAS.
- N. Iwasa, O. Yamamoto, R. Tamura, M. Nishikubo and N. Takezawa, Catal. Lett., 1999, 62, 179–184 CrossRef CAS.
- J. M. Davidson, C. M. McGregor (Née Shirrida) and L. K. Doraiswamy, Ind. Eng. Chem. Res., 2001, 40, 101–107 CrossRef CAS.
- A. Taherkhani, S. Z. Mortazavi, A. Reyhani, A. Tayal, W. A. Caliebe, M. A. Moradi and H. Noei, Int. J. Hydrogen Energy, 2023, 48, 9734–9747 CrossRef CAS.
- T. Fovanna, M. Nachtegaal, A. H. Clark, O. Kröcher and D. Ferri, ACS Catal., 2023, 13, 3323–3332 CrossRef CAS.
- E. K. Gibson, A. M. Beale, C. R. A. Catlow, A. Chutia, D. Gianolio, A. Gould, A. Kroner, K. M. H. Mohammed, M. Perdjon, S. M. Rogers and P. P. Wells, Chem. Mater., 2015, 27, 3714–3720 CrossRef CAS.
- S. Campisi, C. E. Chan-Thaw, L. E. Chinchilla, A. Chutia, G. A. Botton, K. M. H. Mohammed, N. Dimitratos, P. P. Wells and A. Villa, ACS Catal., 2020, 10, 5483–5492 CrossRef CAS.
- S. M. Rogers, C. R. A. Catlow, C. E. Chan-Thaw, A. Chutia, N. Jian, R. E. Palmer, M. Perdjon, A. Thetford, N. Dimitratos, A. Villa and P. P. Wells, ACS Catal., 2017, 7, 2266–2274 CrossRef CAS.
- M. Bowker, L. Cookson, J. Bhantoo, A. Carley, E. Hayden, L. Gilbert, C. Morgan, J. Counsell and P. Yaseneva, Appl. Catal., A, 2011, 391, 394–399 CrossRef CAS.
- M. A. Christiansen, G. Mpourmpakis and D. G. Vlachos, ACS Catal., 2013, 3, 1965–1975 CrossRef CAS.
- J. F. DeWilde, H. Chiang, D. A. Hickman, C. R. Ho and A. Bhan, ACS Catal., 2013, 3, 798–807 CrossRef CAS.
- F. Folco, J. V. Ochoa, F. Cavani, L. Ott and M. Janssen, Catal. Sci. Technol., 2017, 7, 200–212 RSC.
- A. F. Cunha, Y. J. Wu, J. C. Santos and A. E. Rodrigues, Ind. Eng. Chem. Res., 2012, 51, 13132–13143 CrossRef CAS.
- S. M. Sarathy, P. Oßwald, N. Hansen and K. Kohse-Höinghaus, Prog. Energy Combust. Sci., 2014, 44, 40–102 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ey00044g |
| This journal is © The Royal Society of Chemistry 2024 |