
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConversion of diverse post-consumer PVC waste materials to PE via dual catalytic tandem dehydrochlorination–hydrogenation†
Galahad
O’Rourke
 a,
Alina
Skorynina
b,
Igor
Beckers
a,
Sam
Van Minnebruggen
a,
Christel
Colemonts
c,
Philippe
Gabriels
c,
Peter
Van der Veken
c and
Dirk
De Vos
*a
a,
Alina
Skorynina
b,
Igor
Beckers
a,
Sam
Van Minnebruggen
a,
Christel
Colemonts
c,
Philippe
Gabriels
c,
Peter
Van der Veken
c and
Dirk
De Vos
*a
aCentre for Membrane Separations, Adsorption, Catalysis and Spectroscopy for Sustainable Solutions (cMACS), KU Leuven, 3001 Leuven, Belgium. E-mail: dirk.devos@kuleuven.be
bALBA Synchrotron, Cerdanyola del Valles, E-08290 Barcelona, Spain
cVynova-group, 3980 Tessenderlo, Belgium
First published on 5th June 2024
Abstract
Chemical recycling of polyvinyl chloride (PVC) waste poses challenges due to its high chloride content and varied additive formulations. We present a dual catalytic system enabling full conversion of post-consumer PVC waste via tandem dehydrochlorination–hydrogenation. Using a ZnCl2 catalyst (0.1–0.2 eq.) for dehydrochlorination and a Ru catalyst (1.0 mol%) for hydrogenation, it directly converts PVC into a lower molecular weight polyethylene (PE)-like polymer. It prevents the problematic formation of polyenes and aromatic char during thermal processing. The system tolerates common additives (e.g. plasticisers and Pb-, Zn- and Ca/Zn-based stabilisers) and effectively dechlorinates materials with high inorganic filler content. The method can process PVC materials with a wide range of Mn values (29![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–120000 g mol−1). Methyl cyclohexanecarboxylate emerges as a suitable solvent for the tandem reaction, thereby producing 100% dechlorinated products with low molar mass averages (Mn ∼ 2400 g mol−1 and Mw ∼ 5000 g mol−1) and allows additive removal. X-ray absorption spectroscopy (XAS) and a study of the reactivity of a model compound elucidate the Ru-catalyst structure and the chain splitting mechanism. This tandem process yields soluble short-chained polymer fragments, facilitating industrial processing and additive removal from chlorinated plastic waste.
000–120000 g mol−1). Methyl cyclohexanecarboxylate emerges as a suitable solvent for the tandem reaction, thereby producing 100% dechlorinated products with low molar mass averages (Mn ∼ 2400 g mol−1 and Mw ∼ 5000 g mol−1) and allows additive removal. X-ray absorption spectroscopy (XAS) and a study of the reactivity of a model compound elucidate the Ru-catalyst structure and the chain splitting mechanism. This tandem process yields soluble short-chained polymer fragments, facilitating industrial processing and additive removal from chlorinated plastic waste.
Broader contextPost-consumer PVC waste originates from various commercial products ranging from construction materials to clothing and packaging. Additive leaching from PVC is environmentally concerning (e.g. toxic plasticizers and Pb-stabilizers) while no suitable methods for full additive removal exist in industry. Hence, mechanical PVC recycling is limited because of impure waste streams. Beside this, methods for chemical recycling of PVC by thermal processing involve polyene and aromatic char formation. These chars limit the industrial processability of PVC and are valuable losses as carbon fractions when considering a sustainable plastic resource management. This work focusses on a dual catalytic system using ZnCl2 as a benign Lewis acid catalyst for dehydrochlorination of PVC and allows full conversion to a PE-like material without residual chloride. A novel solvent with ester functionality provides a unique opportunity for full PVC conversion while also allowing separation of additives, such as stabilizers and plasticizers, from the waste materials. The dual catalytic tandem dehydrochlorination–hydrogenation mechanism is beneficial since it elicits C–C bond breakage and thereby reduces the molecular weight of the polymer product. |
Introduction
End-of-life management of post-consumer PVC poses many challenges due to the plastic's versatile applications and high chloride content. Environmental concerns from PVC usage arise from its tendency to leach toxic chemicals during its lifetime or after disposal in landfills. Thermal processing and the long-term durability of PVC are directly influenced by its additive formulation.1 As the third most produced polymer, PVC has primarily applications in the construction sector, in which the stability for outdoor applications is enhanced by the addition of heavy metal stabilizers.2 Besides this, other additives like lubricants, chalk or plasticizers can account for more than 50% of the total mass. Therefore, the primary technology of mechanical recycling is only possible for a limited fraction of post-consumer waste, as additives used in PVC can contaminate recycling streams.3–6Chemical recycling is a promising emerging technology for sustainable plastics end-of-life handling and waste management. Industrial plants have difficulty in processing PVC and mixed plastic waste streams containing chlorinated plastics. Pyrolysis processes are readily perturbed by the presence of organochlorine bonds. Typically, there is a tolerance of maximally 2% PVC and chlorine containing pyrolysis oils are of little value.7 The HCl that is released from PVC readily reacts with olefins, while concomitant formation of polyacetylene and aromatic tar leads to undesired losses of carbon.8,9
So far, few investigations have succeeded in preventing this undesirable PVC conversion (Fig. 1). Firstly, two reductive dechlorination methods by Fieser et al. applied trialkylsilanes as the reducing agent. Combining this reduction with silylium catalyzed Friedel–Crafts alkylation in aromatic solvents resulted in conversion of the PVC to a polyethylene-co-styrene type polymer, with stoichiometric formation of trialkylchlorosilane as the by-product.10 Complementarily, a (Xantphos)RhCl reduction catalyst was applied for controlled hydrodechlorination of PVC using a range of reducing agents, including trialkylsilanes and NaH.10,11 However, the selectivity for hydrodechlorination versus the competing dehydrochlorination is not always easily controlled. Although full dechlorination is possible for virgin PVC, several limitations persist, such as the use of costly silanes or organic H-sources as reagents with poor atom economy, the use of aromatic solvents, and the necessity of specialized ligands to enhance the activity of Ru. Another strategy by Goulas et al. used an excess of amine base (2–5 eq.) for dehydrochlorination of virgin PVC to polyene.12 While this product was converted by hydrogenation to a PE-like wax using a Pt/C catalyst, with 90% saturation, only a limited wax yield of 40% was obtained. With unrecovered products and char being amongst other major product fractions, the incompatibility of a heterogeneous hydrogenation catalyst with the insoluble polyenes and char seems to inherently limit the efficiency of this process.9,12 Therefore, we recently explored the activity of ionic liquids for a tandem dehydrochlorination–hydrogenation to promote HCl release from PVC, while preventing accumulation of unwanted polyenes.13–15 This approach enables the conversion of the double bonds formed in degraded PVC to PE-like segments, using H2 as a green reagent, and a spatially homogeneous product transformation of the polymer without char formation, as demonstrated for virgin PVC.16 In an industrial setting however, ionic liquids can be disadvantageous because of their high viscosity and potential cost for large scale operations.17
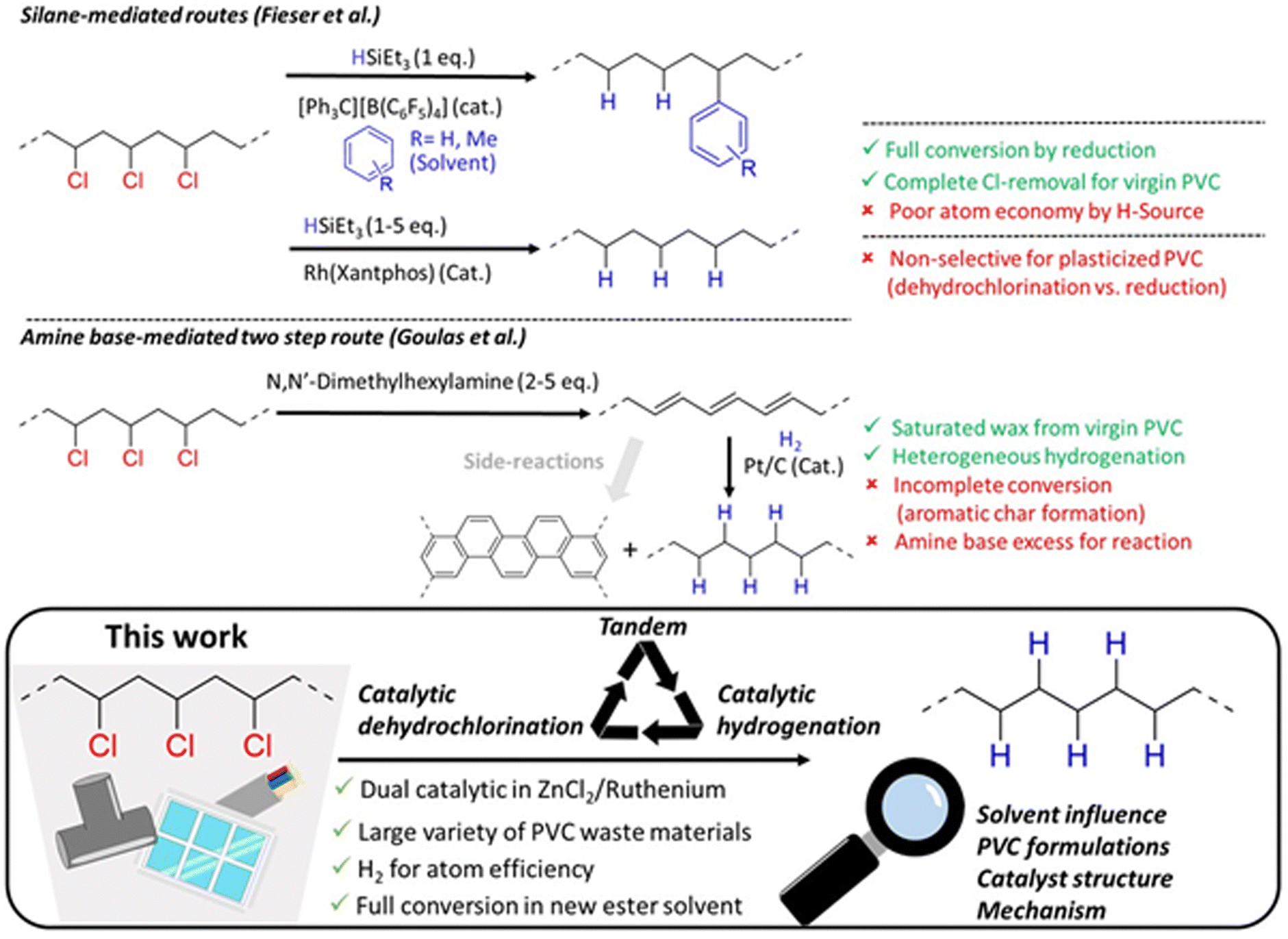 | ||
| Fig. 1 Concept of recycling post-consumer polyvinylchloride by solvent based dual catalytic tandem dehydrochlorination–hydrogenation, with comparison to previous literature reports.10–12 | ||
In this work, we report on the development of a dual catalytic system for tandem dehydrochlorination–hydrogenation, which enables full dechlorination of a wide range of waste PVC materials. A benign ZnCl2 catalyst is shown to exhibit unprecedented activity for dehydrochlorination. The consecutive hydrogenation by a homogeneous Ru catalyst avoids polyene side product formation. With particular focus on the influence of solvents and the composition of PVC waste formulations, parameters that result in complete conversion of PVC to PE are identified. The chemical structure of the polymer products is characterized in detail, and the reactivity of a model compound for PVC is studied, leading to mechanistic considerations.
Results and discussion
Solvent screening for Lewis acid promoted dehydrochlorination–hydrogenation
As an attractive alternative to ionic liquids, we hypothesized that a Lewis acid catalyst could promote the dehydrochlorination of PVC.16 Although bases such as hydroxides or alkoxide salts could be viable reagents to promote dehydrochlorination, we opted for metal salts as substoichiometric promotors of HCl release.18–20 After initial screening, none of the investigated Fe, Cu or Mg salts demonstrated significant PVC dehydrochlorination activity (ESI,† Section S1). In contrast, employing ZnCl2 as a Lewis acid did show good potential.Solvent based processes for mixed plastic separation or reactive polymer conversion are decisively influenced by the affinity of the solvent for each plastic fraction or polymer product.11,21–23 For PVC, solvents containing a carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) moiety have previously been reported as good solvents.21 Hence, initial reactions were done in 2-butanone, in which 62 mg PVC was dissolved per mL (1 mmol mL−1 based on monomer unit, 62.5 g per mol per unit), together with 0.2 mmol of ZnCl2 and 1.0 mol% of H(CO)Ru(PPh3)3 as the homogeneous hydrogenation catalyst. Char formation is an undesirable consequence of polyacetylene formation from PVC in sub-optimal hydrogenation conditions;12 it was observed that under 20 bar of H2, minimally 0.5 mol% but ideally 1 mol% of HCl(CO)Ru(PPh3)3 catalyst was necessary to keep the transformed polymers soluble, without precipitation of black residue. When using 1 mol% Ru, the obtained solution of the reaction product was entirely transparent and clear of any polymer precipitate (indicative for optimal hydrogenation activity), while the polymer dispersion obtained with 0.5 mol% Ru had the appearance of a suspension of white particles (limited polyene formation but sub-optimal hydrogenation). Parallelly, increasing the ZnCl2 catalyst loading from 0.2 to 0.5 mmol gave a white suspension of the polymer, while higher loadings of 0.6–0.8 mmol led to black residue formation, because of too rapid dechlorination.
O) moiety have previously been reported as good solvents.21 Hence, initial reactions were done in 2-butanone, in which 62 mg PVC was dissolved per mL (1 mmol mL−1 based on monomer unit, 62.5 g per mol per unit), together with 0.2 mmol of ZnCl2 and 1.0 mol% of H(CO)Ru(PPh3)3 as the homogeneous hydrogenation catalyst. Char formation is an undesirable consequence of polyacetylene formation from PVC in sub-optimal hydrogenation conditions;12 it was observed that under 20 bar of H2, minimally 0.5 mol% but ideally 1 mol% of HCl(CO)Ru(PPh3)3 catalyst was necessary to keep the transformed polymers soluble, without precipitation of black residue. When using 1 mol% Ru, the obtained solution of the reaction product was entirely transparent and clear of any polymer precipitate (indicative for optimal hydrogenation activity), while the polymer dispersion obtained with 0.5 mol% Ru had the appearance of a suspension of white particles (limited polyene formation but sub-optimal hydrogenation). Parallelly, increasing the ZnCl2 catalyst loading from 0.2 to 0.5 mmol gave a white suspension of the polymer, while higher loadings of 0.6–0.8 mmol led to black residue formation, because of too rapid dechlorination.
Under the optimal conditions (0.2 mmol ZnCl2, 1 mol% HCl(CO)Ru(PPh3)3, 16 h under 20 bar H2 at 180 °C), a higher concentration of 1.5 mmol mL−1 (S/L mass–volume ratio of 94 mg mL−1) still led to good results for low molar weight (LMW)-PVC. In general, the Lewis acidity of ZnCl2 led to some adverse side reactions of the butanone solvent. As confirmed by GC-MS, the butanone is prone to aldol condensation under the applied reaction conditions. After purification of the polymer product, the 1H-NMR was indicative for complete disappearance of CHCl-groups in the polymer backbone. Additionally, converted PVC product seemed to have a large CH3- to –CH2-ratio after conversion in 2-butanone.
In order to identify a solvent that could be used repeatedly in the tandem dehydrochlorination–hydrogenation reaction, a solvent screening was performed to find good solvents for PVC and ideally, also for the PE-like reaction product. Since solubility is heavily influenced by the molar mass of the polymer, high molecular weight (HMW)-PVC was used to visually observe polymer dissolution at 100 °C (0.75 mmol PVC per mL solvent; solid to liquid (S/L), mass–volume ratio of 47 mg mL−1; Mn and Mw in ESI,† Fig. S9). Tandem dehydrochlorination reactions of PVC were performed in all investigated solvents. The degree of dechlorination of PVC was determined by 1H-NMR in order to quantify the extent of dechlorination, according to our previously developed procedure (Experimental, ESI,† Section S2).16 Additionally, Fourier Transform Infrared (FTIR) spectra were recorded of the isolated polymer products, to evaluate qualitative changes of the isolated polymer products (ESI,† Section S3).
Although known ketone solvents showed excellent dissolution capacity for PVC (Fig. 2, left), these were unstable due to the aforementioned aldol side reaction. For cyclopentanone and cyclohexanone, this led to high boiling side-products which are hard to separate from the polymeric products. Alcohols are typically reported as anti-solvents for PVC, and cyclohexanol was indeed found unsuitable for dissolution of PVC; additionally no dechlorination of the polymer happened in this solvent (ESI,† Fig. S6 and S7). In ethers, like tetrahydrofuran (THF), a common polymer solvent, or the biomass-derived ether tetrahydropyran (THP), PVC showed excellent solubility; these solvents have been described as highly acid-stable solvents for plastic dissolution, even at room temperature.24 On the contrary, as the only ether, cyclopentyl methyl ether (CPME) showed no solubility for PVC. Neither the reactions in THF, nor in THP led to complete dechlorination of PVC, reaching maximally 50% and 64% dechlorination, respectively. This suggests that the ZnCl2 catalyst is less active for dehydrochlorination when used in ether solvents, even if these solvents are stable under the reaction conditions. Use of tetrahydrofurfuryl alcohol (THFA), which has both an ether and alcohol group, still allowed good polymer dissolution but showed similar low dechlorination. Alternatively, ester solvents displayed partial to good solubility of PVC, depending on solvent structure. While reactions in butyl acetate led to charring due to incomplete dissolution of PVC, methyl cyclohexanecarboxylate (MCC) showed to be an excellent solvent for PVC, in which complete dechlorination was accomplished. The choice of MCC relies on its structural properties, with a sterically poorly accessible α-H atom, which prevent it from undergoing Claisen condensation as an undesirable solvent side reaction. Additionally, the PE-like reaction product seems to have high affinity for a solvent containing a cyclohexyl ring; note that cyclohexane itself is a known apolar solvent for PE dissolution.25,26 FTIR spectra of the products (Fig. 2, right) after treatment in THP (Fig. 2A) and MCC (Fig. 2B) showed significantly more intense vibrational peaks of C–H stretching (2913, 2846 cm−1) and diminished intensity of C–Cl stretching (691 cm−1) and CH bending (δCHCl, 1250 cm−1) vibrations, indicating extensive dechlorination with subsequent hydrogenation. In agreement with 1H-NMR results, the product obtained after the reaction in MCC had virtually no C–Cl stretching vibrational peaks, with its FTIR corresponding to virgin PE. However, some residual peaks of the ester solvent, or CO stretching peaks possibly resulting from double bond oxidation, were observed as impurities (1730 cm−1).
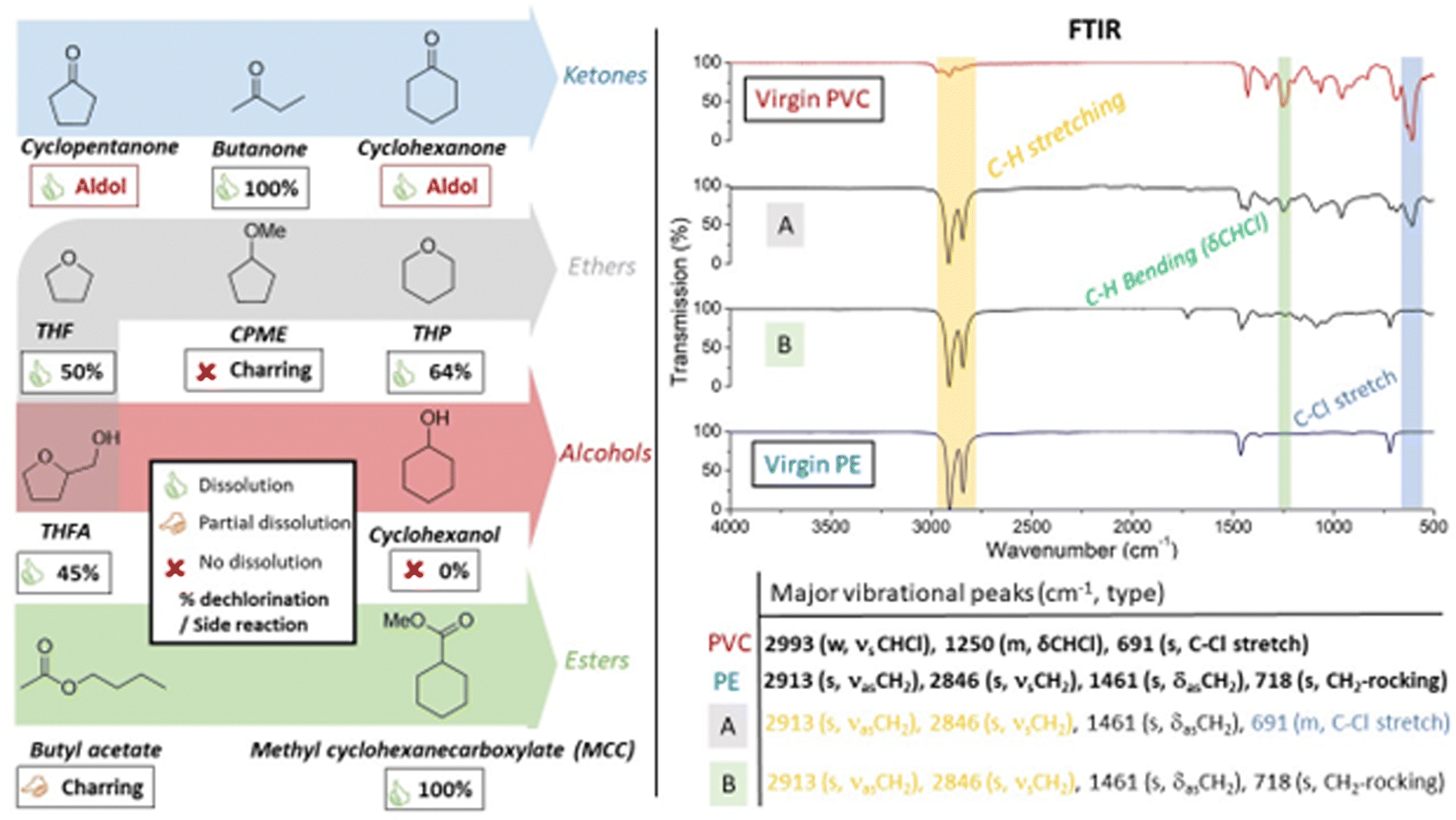 | ||
| Fig. 2 (left) Solvents screened for the tandem dehydrochlorination–hydrogenation of HMW-PVC. Conditions: 1.5 mmol PVC, 0.2 mmol ZnCl2, 0.65 mol% HCl(CO)Ru(PPh3)3, 2 mL solvent, 180 °C under 20 bar H2 for 16 h. Solubility of PVC in the solvent was observed visually at 100 °C. The degree of dechlorination of isolated polymer products was determined by liquid 1H-NMR (CDCl3, 400 MHz). (right) ATR FTIR spectra and vibrational assignments of virgin PVC and PE, and reaction products obtained after treatment in (A) THP and (B) MCC. | ||
Hence, methyl cyclohexanecarboxylate was found to be a stable solvent in which the tandem dehydrochlorination–hydrogenation of high molecular weight (HMW)-PVC can be pursued to completion. In this solvent-based dechlorination process, a noticeable influence of the viscosity was observed for the virgin PVC samples. As a rheological quantity, the Fikentscher K-value represents an estimation of the molecular weight distribution of PVC samples, based on dilute solution viscosity measurements (Experimental). Typically, higher K-values are correlated with higher molecular weights of the PVC, which is in agreement with GPC results of our investigated PVC samples, with Fikentscher values ranging from K57 to K70. Solution viscosity of PVC dissolved in MCC allowed a concentration of 1.5 mmol mL−1 (mass–volume S/L of 93 mg mL−1) for complete dechlorination of LMW-PVC samples with K-values up to 57. Reacting virgin PVC samples with K-values of 68 or higher, as for HMW-PVC, led to charring when the same concentration (S/L ratio of 93 mg mL−1) was used. This can be attributed to insufficient hydrogenation activity of the homogeneous Ru-catalyst due to the high viscosity of the polymer solution. Under these conditions, the charred reaction products were insoluble in all investigated solvents, and had a black appearance (ESI,† Fig. S8). Fortunately, consistent use of a lower concentration of 0.75 mmol mL−1 (mass–volume S/L ratio of 47 mg mL−1) enabled complete dechlorination-hydrogenation for the entire range of investigated molecular weights of virgin PVC samples (ESI,† Fig. S9). While concentrated solutions of PVC in the solvent displayed viscidness before reaction, after treatment, markedly runny solutions were obtained. Since this reduced viscosity of the fluid may be related to the molar mass of the polymer solute, this aspect will be scrutinized further on. Additionally, in contrast to charred products, polymer products with complete dechlorination had the appearance of white-yellow powders after the work-up procedure.
Conversion of waste PVC materials
PVC is found in many applications, with its primary use in the construction sector. Therefore, the feasibility of solvent-based dehydrochlorination–hydrogenation using ZnCl2 and homogeneous Ru was explored on ground PVC waste samples that were recovered from construction materials (ESI,† Fig. S10). The investigated samples contained various types of metal carboxylate salts, including Pb, Sn-stabilisers and CaZn-costabilisers which are typically added to PVC for thermal stability during extrusion. To investigate the effect of the formulation of each waste material, thermogravimetric analysis (TGA) was used to quantify the inorganic filler contents (ESI,† Section S5). Additionally, the organic additive content was characterized by liquid 1H-NMR (d8-THF, 400 MHz) of the dissolved powders. Finally, the molecular weight distribution of the polymer component itself was investigated by GPC (Experimental, ESI,† Section S6).Because of its particular effectiveness for PVC conversion, MCC was used as the solvent for waste conversion reactions. In general, maximally 2–3% of metal salt stabilizer was found in all waste materials, whilst the content of inorganic CaCO3 fillers was often higher. The Pb-stabilised materials contained the highest fraction of these fillers (entries 1–3, Fig. 3). Tandem reactions of the pipe and profile, stabilised by Pb (entries 1 and 2), led to charring when a S/L ratio of 93 mg mL−1 was used but could be accomplished by using a lower S/L ratio of 47 mg mL−1 and pre-dissolving the ground sample in the solvent before reaction. Hence, even with inclusion of Pb-stabilizer and 16% filler material, near complete-conversion to PE was viable. However, when the inorganic filler content was too high, as for the Pb-stabilised cable (entry 3), a solvent dissolution–precipitation pretreatment was necessary to suppress char formation. For this pre-treatment, the crude PVC sample was firstly dissolved in THF, after which a centrifugation of the solvent mixture led to the possibility to remove the filler materials as solid residue. After precipitation of the PVC from the solvent by anti-solvent addition, this PVC was dissolved in the reaction solvent, enabling successful conversion. The example of the CaZn-stabilised pipe (entry 5) illustrates the necessity of a pretreatment: without such pretreatment, the dechlorination was incomplete, which seems due to partial charring or insufficient hydrogenation. In contrast, the lower additive content of the investigated Sn-stabilised rigid film (entry 4) and CaZn-stabilised pipe/film (entries 6 and 7) allowed full conversion of the ground powders without the necessity for a pre-dissolution in the solvent. Additionally shoe soles (entry 8) were investigated, which were heterogeneous in nature for sampling, but all contained approximately 90% PVC, and lacked residual metals or inorganic fillers, as was confirmed by TGA. Because this material also had a relatively low molecular weight, nor its viscosity nor the inorganic filler content required a pretreatment of the polymer and the reaction could be done in a more concentrated solution, using a higher S/L ratio of 93 mg mL−1.
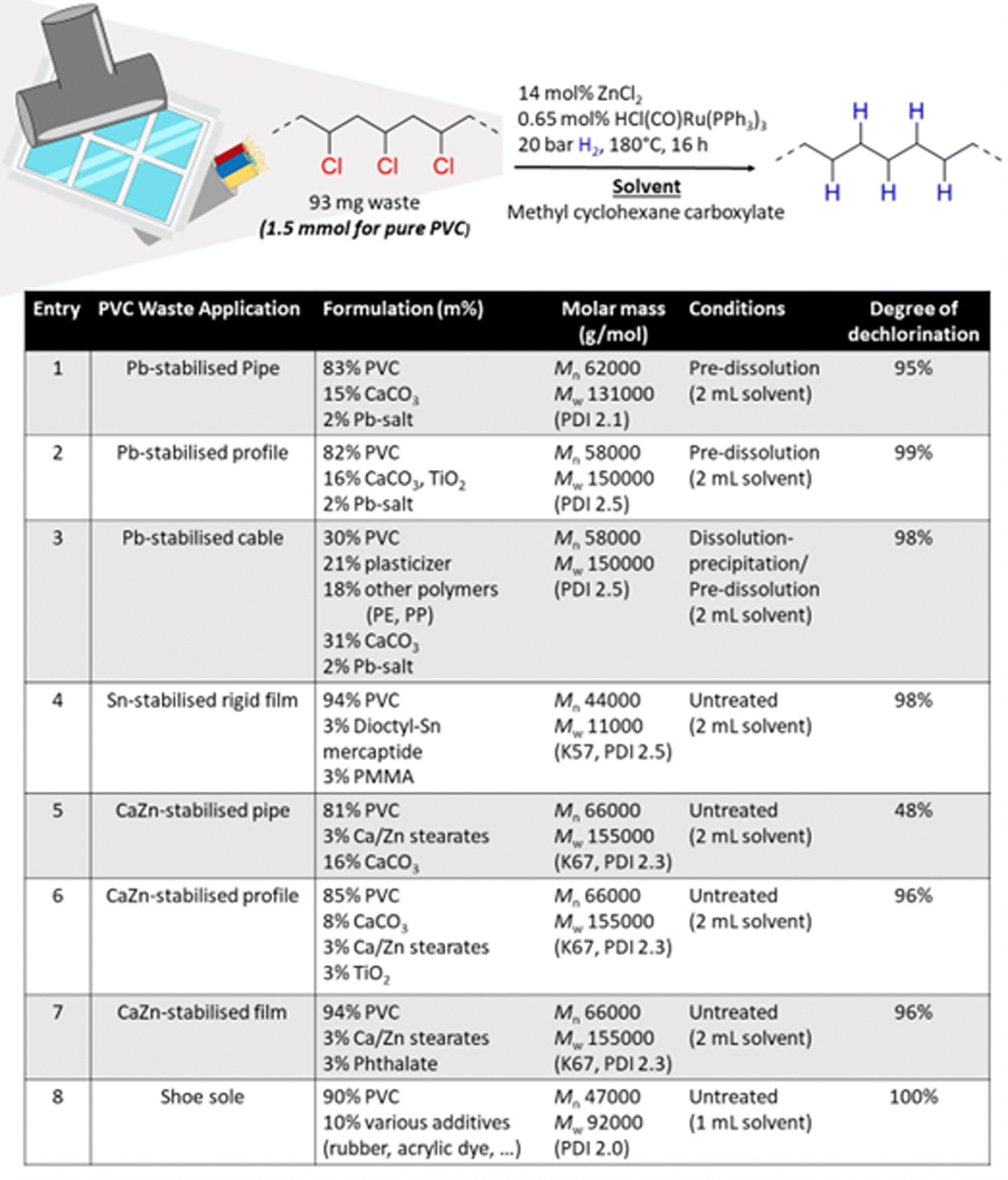 | ||
| Fig. 3 (scheme) Reaction conditions for conversion of PVC waste to PE. (table) Composition of investigated PVC waste materials and determined molar mass averages by GPC (Mn, Mw) and viscometry (K-values). The conditions describe the solvent pretreatment steps that were applied to get efficient dechlorination. Degree of dechlorination of products was determined by liquid 1H-NMR (400 MHz, CDCl3). | ||
Liquid 1H-NMR spectra of the dissolved waste materials (Fig. 4A) all contain predominant peaks of PVC at δ 4.5 ppm (CHCl) and δ 2.4–2.5 ppm (CH2–CHCl, i.e. α-CH2-groups), but they may additionally show signals related to organic additives. Indicative for the presence of these additives are for instance peaks of phthalate plasticizers, with multiplets at δ 7.7 and 7.6 ppm from the aromatic rings, and peaks at 4.1 ppm which originate from –O–CH2-groups of the ester branches that are connected to these rings. Other polymers such as PE and polypropylene (PP), as present in the Pb-stabilised cable, have also characteristic peaks for –CH2-groups and CH3-groups at δ 1.49 and 0.9 ppm, respectively. In contrast to the starting material samples, the reaction products were readily soluble in both THF and chloroform at room temperature. After reactions, the spectra of polymer products (CDCl3, 400 MHz) showed significant conversion as illustrated for the CaZn stabilised film and pipe (Fig. 4B). As determined by integration of 1H-NMR signals (Experimental, ESI,† Sections S2 and S3), the fraction of CH2 groups (δ 2.00–1.28 ppm) relatively to CHCl-groups (δ 4.5–4.0 ppm) is indicative for the completeness of the reaction. A very low quantity of double bonds was found in the products under these conditions, with a corresponding CH-group δ of 5.4 ppm. Additionally, peaks of relatively low intensity at 0.9 ppm can be attributed to formed CH3-groups at the chain ends of the PE-like polymer products. Ostensibly, the polymer products with near-complete dechlorination, e.g. of ≥95%, had no apparent residual CHCl-groups, as these were only detected in the noise of the spectrum. However, the reacted CaZn stabilised film, for which insufficient pretreatment was done beforehand, still contained residual CHCl-groups and the α-CH2-groups were detected in considerable quantity. Because of the solvent treatment after tandem reactions, the products were free from any organic additives such as plasticizers originating from the waste material.
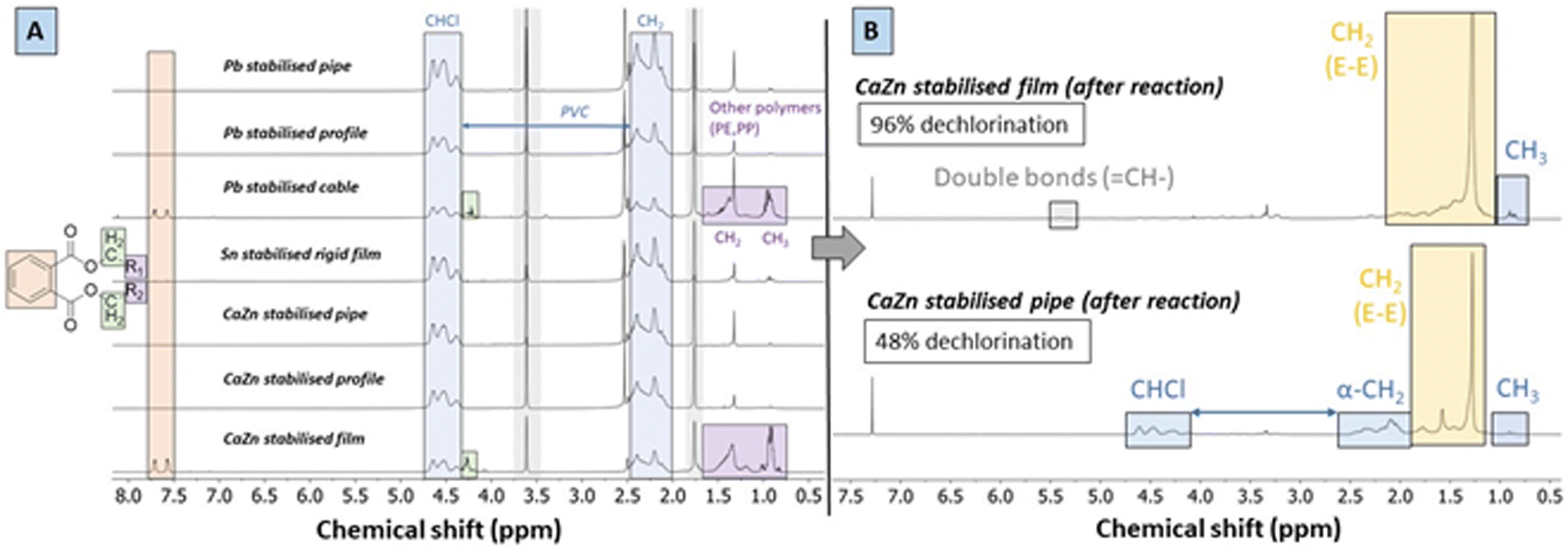 | ||
| Fig. 4 Changes of polymer structure and additive composition of PVC after catalytic tandem-dehydrochlorination–hydrogenation. (A) 1H-NMR spectra (d8-THF, 400 MHz) of dissolved waste PVC samples; peaks in grey boxes at 3.57 ppm and 1.77 ppm originate from THF. (B) 1H-NMR spectra (CDCl3, 400 MHz) of converted polymer from CaZn stabilised film and CaZn stabilised pipe, with predominantly peaks of CH2 groups in the hydrogenated product backbone at 1.29 ppm. | ||
Solvent influence on polymer product
To further understand the tandem conversion process using the most suitable solvents for PVC, reactions were also performed with addition of HCl prior to the reaction, and the resulting products were analyzed. (Experimental) While using only the Ru-catalyst without ZnCl2 leads to 42% dechlorination in MCC, adding HCl reduces the degree of dechlorination of the polymer product, suggesting that the dehydrochlorination is reversible (Fig. 5A). Upon addition of ZnCl2 a complete dechlorination is achieved, leading to a polymer product with mainly –CH2-groups that are in ethylene–ethylene sequences (E–E) as in polyethylene, with a CH3-end group/CH2 ratio of 1/100. This relatively high CH3/CH2 group ratio compared to the starting material (i.e. <0.2% CH3-groups in PVC) is indicative for some chain branching or splitting during the tandem reactions. This means that the formed product has a polymer backbone with approximately one CH3-group for every 50 CH2–CH2 (i.e. ethylene) repeating units, and a theoretical number average molar mass (Mn) of only 5600 g mol−1 (according to NMR integration). In 2-butanone (Fig. 5B), using only the Ru-catalyst yields 8% dechlorination and added HCl hardly changes the dechlorination degree. Notably, products from reactions with ZnCl2/Ru in 2-butanone are highly branched with methyl groups, since the CH3/CH2-group ratio is 30/70 when using ZnCl2–Ru. In the third investigated solvent, THP, using the ZnCl2/Ru combination only allows incomplete dechlorination (64% dechlorination) (Fig. 5C). Additionally, using only Ru or Ru with added HCl in THP, leads to similar low dechlorination as under the same conditions as in 2-butanone, but this does not involve quick formation of CH3-branches. Hence, the ZnCl2-catalyst leads to a higher activity in 2-butanone and MCC than in THP. HCl addition in THP leads to some suppression of the forward dechlorination, as is observed for reactions in MCC. Products of the reaction in MCC had markedly less double bonds when HCl was added; this confirms that the double bonds are consumed not only by a hydrogenation on the Ru catalyst, but also by reverse addition of HCl. While there is little branching in MCC and THP, there is extensive formation of methyl branches for reaction in 2-butanone. This shows that (i) the Ru catalyst has poor access to the chains, resulting in slow hydrogenation of the CC bonds, and (ii) that even the reaction of carbocationic species with chloride proceeds insufficiently. Consequently, there is ample time for the (linear) carbocations to rearrange to methylbranched motifs, likely via protonated cyclopropanes.
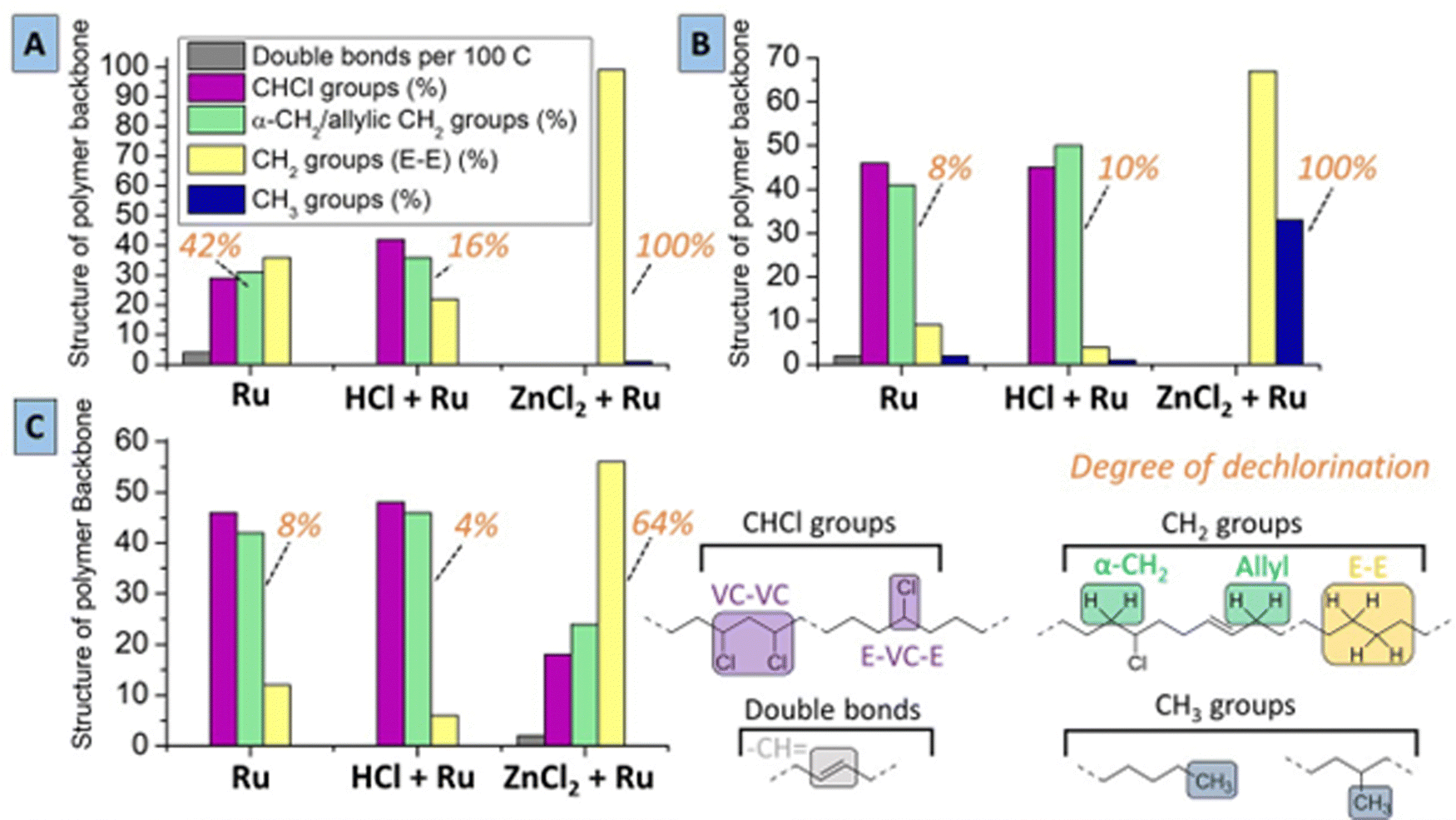 | ||
| Fig. 5 Structural composition of formed polymer products after reactions, upon using (A) MCC, (B) 2-butanone, (C) THP (conditions: 1.5 mmol LMW-PVC, 0.2 mmol ZnCl2, 1.0 mol% HCl(CO)Ru(PPh3)3, 2 mL solvent, 20 bar H2, 180 °C for 16 h). | ||
Molecular weight changes by chain splitting and branching
In order to further elucidate mechanistic aspects of the dual catalytic system, conditions using only one of the catalysts were applied on virgin LMW-PVC samples (Mn ∼ 29800 g mol−1 and Mw ∼ 66000 g mol−1). Without Ru-catalyst, when applying only 0.2 mmol ZnCl2 per 1.5 mmol PVC in MCC at 180 °C, black char formation was observed almost immediately after the start of the reaction (ESI,† Section S7). When monitoring this process by UV-Vis spectroscopy, a rapidly increasing intensity of the absorbance peak at 254 nm suggests fast formation of double bonds and highly conjugated systems, possibly even aromatics in the first minutes of reaction (Fig. S13, ESI†). Using both ZnCl2 and Ru-catalysts led to conversion of PVC to a fully saturated PE-like polymer after 16 h (Fig. 6A). Employing only the Ru-catalyst, also gradually increased the degree of dechlorination of the polymer products with reaction time, resulting in 42% dechlorination after 16 h. A control reaction without any catalyst showed no dechlorination activity. Hence, besides its activity in hydrogenation, the presence of Ru-catalyst can also elicit a steady but slow dehydrochlorination in absence of the ZnCl2. When applying GPC on the polymer products after 16 h reaction (Fig. 6B), the products with full dechlorination by Zn–Ru had a remarkable reduction in molar weight. The number average molar mass (Mn) and weight average molar mass (Mw) of these products are in the range of ∼2000 and ∼4000 g mol−1, respectively. Considering the Mn of the polymer with full dechlorination, one would expect a theoretical Mn of 14100 g mol−1 for this 53% mass reduction in the reaction (net Cl-loss and H-gain). Hence, this large decrease in molar mass of the polymer may be attributed to chain splitting reactions of the polymer, beside dehydrochlorination–hydrogenation, which is most apparent when high degrees of dechlorination are obtained by the ZnCl2/Ru-catalyst combination. Chain splitting may be caused by carbocation intermediates of the polymer or possibly by hydrogenolysis activity of the Ru catalyst. The thermal properties of the completely dechlorinated products were found to be similar to those of virgin low molecular weight-PE (ESI,† Section S10).
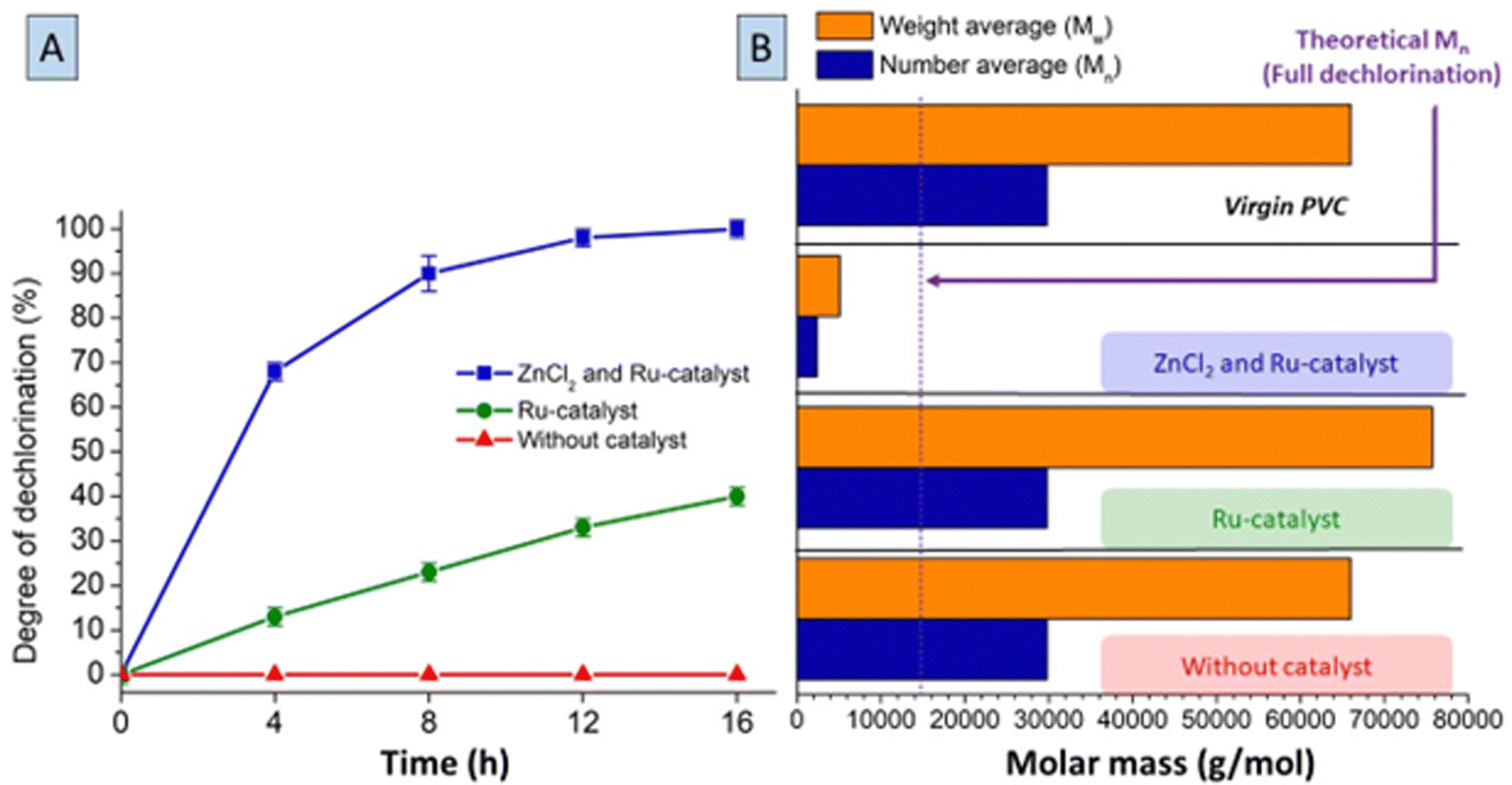 | ||
| Fig. 6 (A) Conversion of LMW-PVC under tandem dehydrochlorination–hydrogenation conditions in time (general conditions with 1.5 mmol LMW-PVC, 0.2 mmol ZnCl2, 0.01 mmol HCl(CO)Ru(PPh3)3, 1 mL methyl cyclohexanecarboxylate at 180 °C under 20 bar H2). (B) Change in molar mass averages (number average (Mn) and weight average (Mw)) of the polymer, after reactions for 16 h. | ||
Control reactions, without catalyst, led to no change of the molar mass of the products. On the contrary, applying solely the Ru-catalyst gave a slight increase in molar mass, which can be explained by cross-linking reactions under influence of HCl during the slow dechlorination process. 1H-NMR and 13C-NMR spectra of the polymer products (ESI,† Fig. S14 and S15), showed residual double bonds under these conditions.
In order to better understand the molar mass reduction of PVC by the ZnCl2/Ru-catalytic system, the conversion of 2-chloroheptane was monitored by gas chromatography (GC), as a model reaction for PVC, under similar conditions. To avoid bimolecular reaction pathways, these reactions were performed at a slightly lower temperature of 140 °C, and using tetradecane as an inert, diluting solvent. A control reaction without catalyst gave no conversion of the 2-chloroheptane starting material (Fig. 7). When using only ZnCl2, slightly higher conversion of the starting material than for the ZnCl2/Ru system was obtained, leading to mostly heptene formation by HCl elimination. While further hydrogenation to heptane happens extensively when applying the Ru-catalyst, also isomerization reactions to 3-chloroheptane were observed under these conditions. This proves the addition of HCl to formed double bonds after initial elimination. Additionally, various side products were detected, which were identified as branched (chloro-)alkanes, with 7% and 10% yield of the branched products in the presence of ZnCl2 and ZnCl2/Ru-catalyst, respectively. Methylhexane and chlorinated methylhexanes were found amongst these products. These can be formed via protonated cyclopropane intermediates, which shift internal carbons of the alkane, to external (branched) methyl positions on the chain.27 Carbocation intermediates that undergo β-scission reactions may also explain the detection of fragmentation products like isobutene and chloropropane. Additionally, the mass balance of these reactions was not complete, as an unknown fraction of, most probably, volatile short-chained isomers (i.e. alkenes and alkanes) from these rearrangements eluded detection. Hence, the loss in molar mass of the polymer products of PVC, when using the ZnCl2-catalyst, originates from its Lewis acidity which accelerates HCl release. Subsequently, the Brønsted acidity of HCl leads to carbocation rearrangements that split the initially linear polymer backbone into shorter chains.
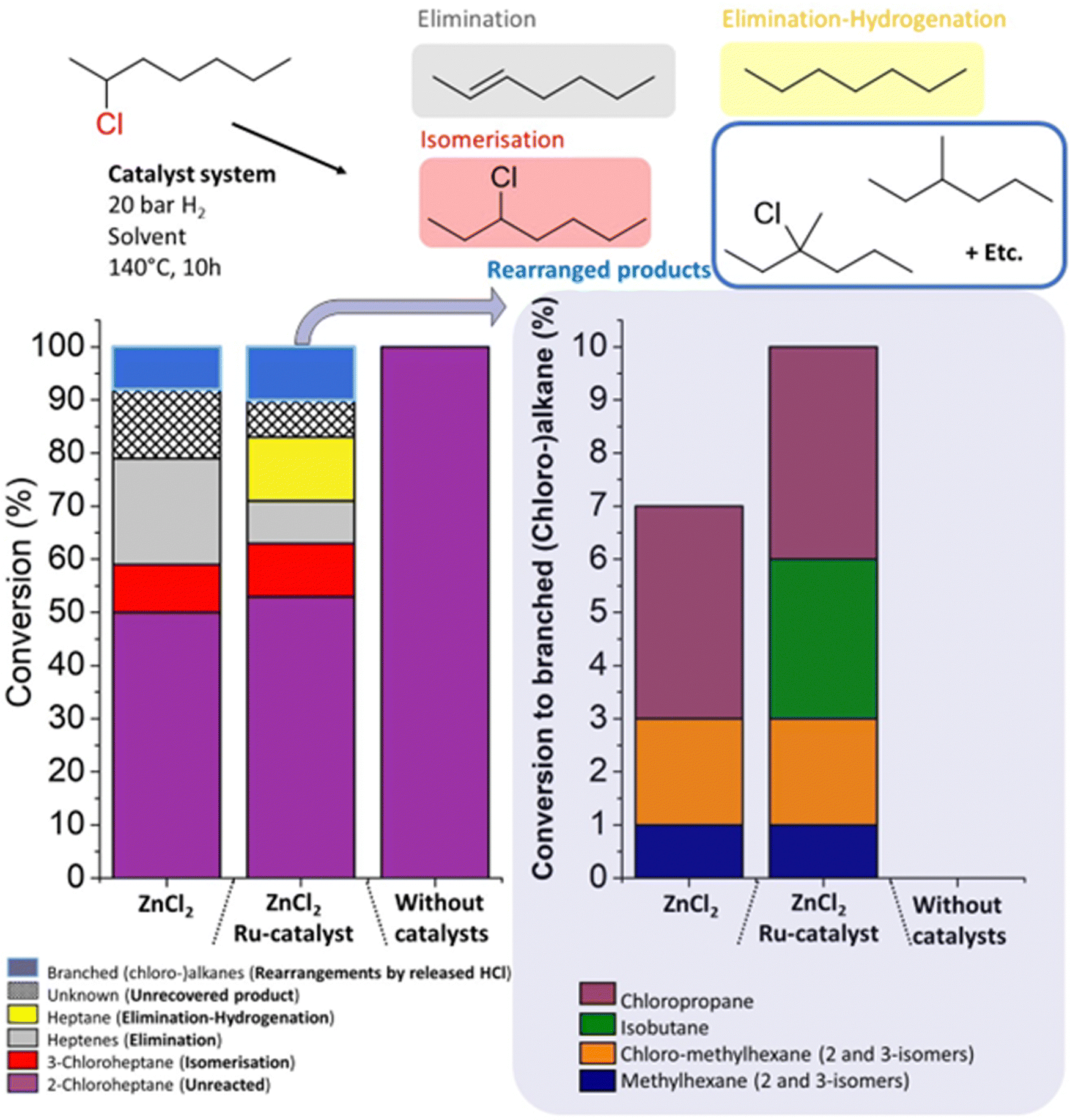 | ||
| Fig. 7 Influence of the investigated catalytic system on the conversion of 2-chloroheptane, a model compound for PVC. Various side reactions occur beside elimination and hydrogenation. Using ZnCl2, rearrangements by released HCl lead to branching and lower molecular weight products. Reactions were performed with 0.2 mmol ZnCl2, 0.01 mmol HCl(CO)Ru(PPh3)3 and 0.5 mmol 2-chloroheptane in tetradecane as the solvent (1 mL), at 140 °C under 20 bar H2 for 10 h. | ||
Characterization of active ruthenium catalyst
To identify the hydrogenation catalyst as formed during reactions, X-ray absorption spectroscopy (XAS) was performed (Experimental) for the characterization of the Ru-catalyst at its K-edge (22.1 keV) in methyl cyclohexanecarboxylate, as the solvent. Firstly, X-ray absorption near edge structure (XANES) spectra (Fig. 8A) demonstrated no shift in K-edge position of the Ru-catalyst, without ZnCl2 (after 16 h of reaction), compared to fresh HCl(CO)Ru(PPh3)3 pellets. Even after applying the actual reaction conditions, employing the Ru-catalyst with ZnCl2, the oxidation state of +2 of the initial precursor is retained. However, the extended X-ray absorption fine structure (EXAFS) spectra (Fig. 8B) do show a change in the ligand coordination of the Ru-catalyst, when using ZnCl2. Under these conditions, additional Ru–Cl bonding is observed, while also Ru–Ru bonding is detected. This suggests that bimetallic or oligonuclear Ru catalyst clusters form during the reaction, although the catalyst remains homogeneous throughout the entire tandem dehydrochlorination–hydrogenation conversion of PVC. While the ZnCl2-catalyst was found to be stable for at least three reaction cycles of PVC conversion (ESI,† Section S11), the Ru-complex was found to be deactivated after a 2nd reaction cycle. This is most likely caused by Ru-clustering due to the HCl-concentration buildup when reusing the spent solvent (which still contains ZnCl2 and accumulated the formed HCl). Thereby, solid Ru-particle formation was observed in the solvent after the 2nd cycle, which was also accompanied by charring of the PVC products. Since the proposed procedure uses a dual homogeneous catalytic system, a reuse of the methyl cyclohexanecarboxylate solvent with dissolved ZnCl2 from a first reaction was possible when adding fresh HCl(CO)Ru(PPh3)3 catalyst. This approach also led to complete conversion of the PVC to PE-like material for at least three reaction cycles.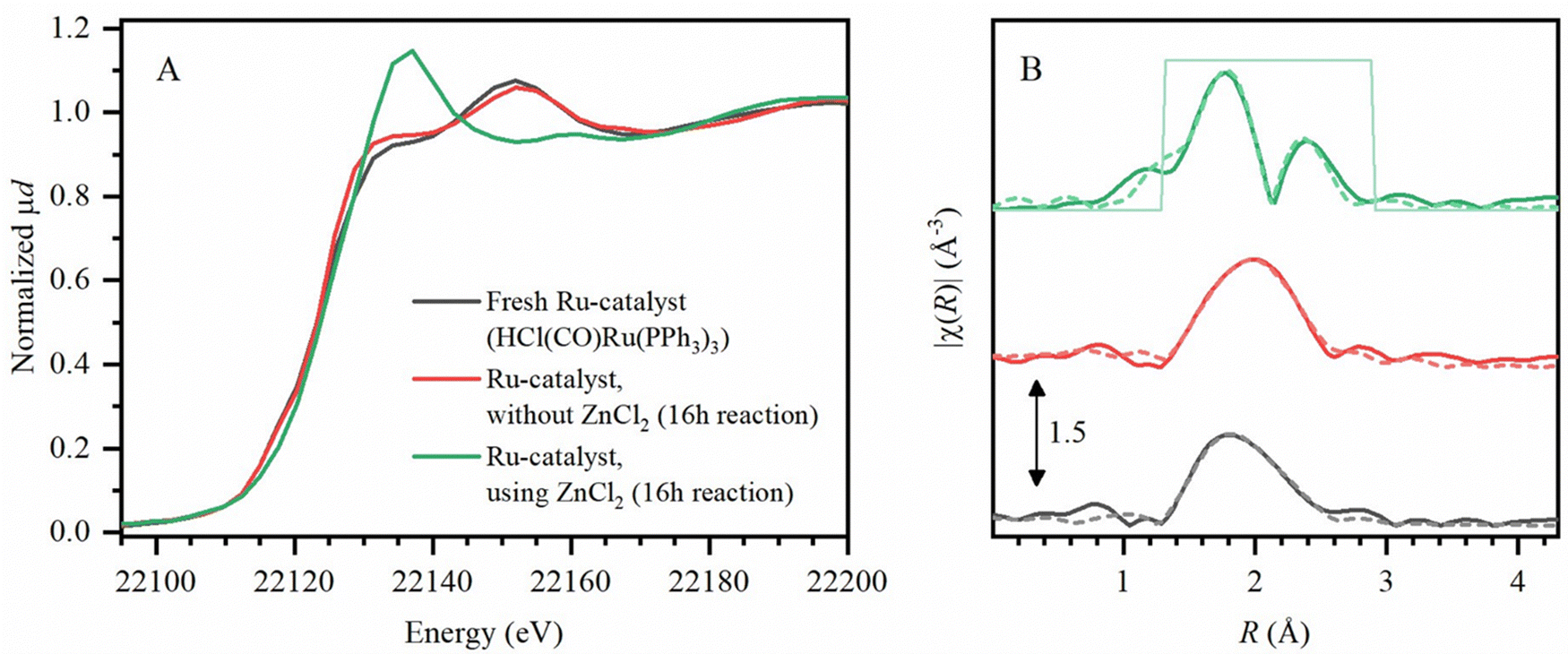 | ||
| Fig. 8 Ru K-edge (A) normalized XANES spectra, (B) k2-weighted Fourier transform EXAFS spectra (solid lines) and fitted models (dashed lines) for fresh Ru-catalyst (gray) and measurements of Ru-catalyst after 16 h reaction: without and with added ZnCl2 (conditions: 1.5 mmol PVC, 0.01 mmol Ru-catalyst, 0.2 mmol for added ZnCl2, 20 bar H2 at 180 °C). | ||
Mechanism of PVC conversion
Based on these insights, a mechanism can be proposed for the tandem reaction. In comparison with earlier approaches, e.g. based on ionic liquids, the use of ZnCl2 enables a mild and complete conversion to a saturated polymer chain.16 Previously a zipper effect has been reported for release of HCl from PVC, which implies that labile allylic chlorides on the polymer backbone react faster by dehydrochlorination, thereby accelerating successive release of HCl molecules from PVC.13 In the case of the ZnCl2–Ru catalyzed reaction, there is no clear evidence for a rate acceleration via labile allylic chlorides; it seems that the Lewis acid ZnCl2 elicits fast double bond formation, and that the following Ru-catalysed hydrogenation occurs fast enough to avoid formation of extensive conjugated systems (Fig. 9A). Several elements indicate that the dehydrochlorination is reversible, such as the effect of added HCl on the PVC reaction in methyl cyclohexanecarboxylate, or the formation of the 3-Cl-heptane isomerization product in the reaction of 2-Cl-heptane. Side reactions can be rationalized based on the reactions of carbocations that are formed from olefins and the increasingly large amount of HCl released. Methyl branching of the originally linear polymer chain can proceed via protonated cyclopropane intermediates (PCP+) when olefins in the chain are protonated (Fig. 9B).27,28 The carbocationic intermediates can either undergo various rearrangements, or even β-scission reactions when the chain is increasingly branched, with preferentially formation of stable olefins and stable cations, i.e. secondary or tertiary carbocations. This explains for instance the formation of isobutane and 2-Cl-propane as side-products in the 2-chloroheptane model reactions. For the reaction of the PVC, such splitting reactions explain the decrease of the molecular weight of the polymer product. Finally, because of the tandem hydrogenation that is catalyzed by Ru, the split olefins are converted to a saturated alkane product.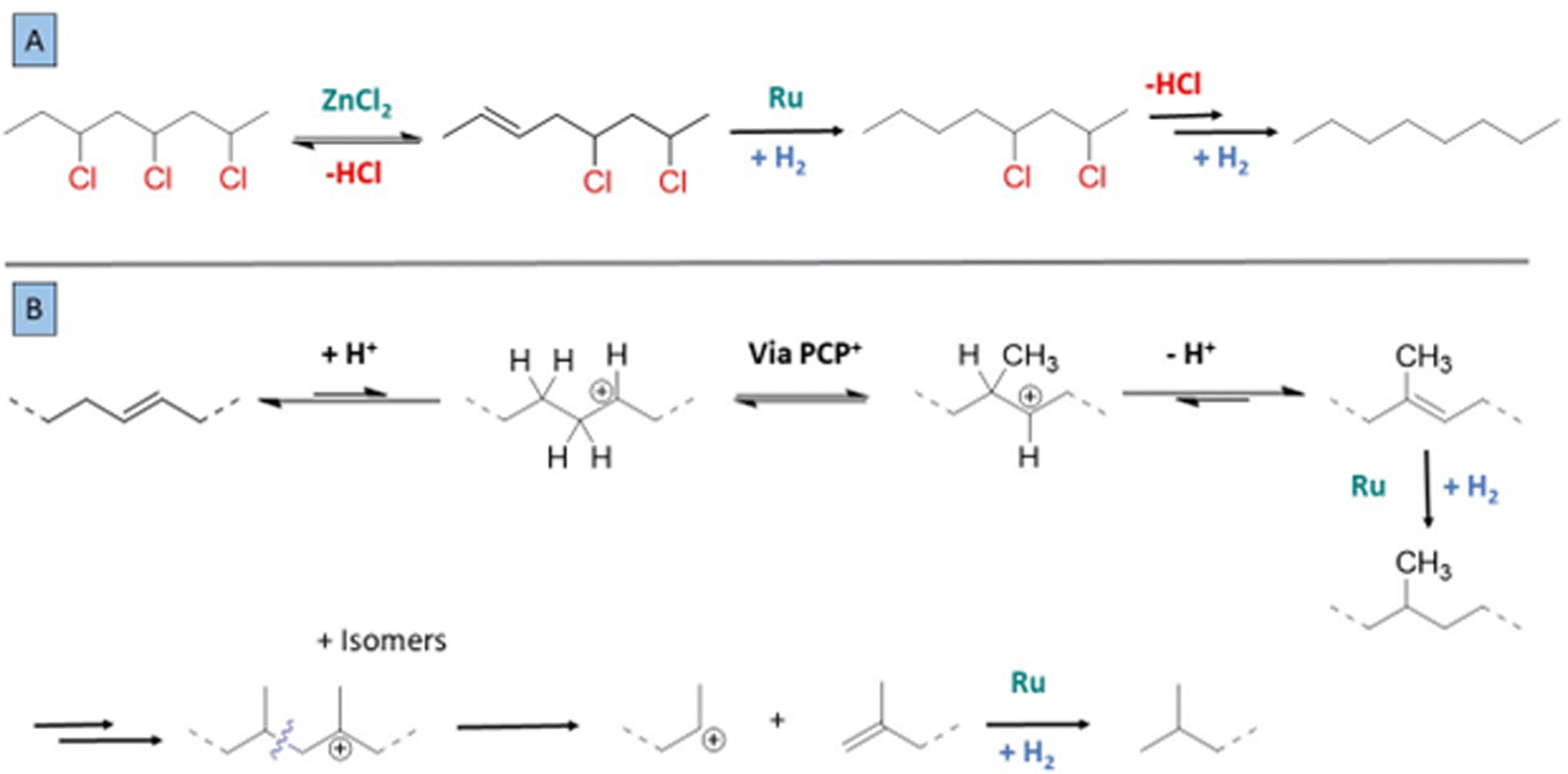 | ||
| Fig. 9 Mechanisms involved in the chemical conversion of PVC with (A) the dual catalyst for the tandem dehydrochlorination–hydrogenation, with HCl release by ZnCl2 and hydrogenation by the active Ru complex. (B) HCl addition to formed double bonds leads to isomerization or ZnCl2/H+-promoted branching of the saturated polymer product. Protonated cyclopropane intermediates form branched carbocations. | ||
Conclusion
In spite of the large differences in formulation and the presence of diverse additives, the developed tandem dehydrochlorination–hydrogenation procedure demonstrated an unprecedented versatility in the conversion of a wide array of post-consumer PVC waste materials. So far, most literature procedures rely on a stoichiometric reducing agent or base to enable Cl-release from PVC, with unfavourable atom efficiency.9–12 Our findings show that ZnCl2 can be used in low catalytic loadings as an effective dehydrochlorination catalyst to achieve complete HCl removal, with homogeneous Ru as a complementary hydrogenation catalyst. The chemical recycling of typical waste materials such as piping, window profiles and cables was successfully demonstrated, in spite of the typical contaminants that are otherwise problematic in mechanical recycling. This chemical recycling method is highly solvent dependent. Not the most common solvents for PVC, such as ketones or ethers, were suitable media to achieve full conversion but a novel ester solvent (i.e. methyl cyclohexanecarboxylate) did show excellent capacity. It was found that combining the methyl cyclohexanecarboxylate with an alcohol solvent (e.g. methanol or ethanol) after reaction, is a useful method for product isolation and solvent recovery. The alcohol anti-solvent is low-boiling and can be evaporated to purify the ester solvent. Methyl cyclohexanecarboxylate is a high boiling solvent (boiling point of 183 °C) compared to typical ether solvents for PVC (e.g. boiling points of THF and THP are 66 °C and 88 °C, respectively). Therefore, in regard to sustainable solvent selection,29 it has less issues related to VOC emission. Additionally, the solvent shares structural similarities with cyclohexane and ester containing-moieties, which are generally known to have few known health hazards. Finally, mechanistic elucidation of the tandem dechlorination reaction shows the occurrence of carbocation mediated side reactions during the process, which leads to branching in some solvents, and also involves chain scissions resulting in lower molecular weight polymer products. This process, yielding soluble short-chained polymer fragments with a strongly reduced Cl content is potentially promising for industrial application, easing additive removal and thermal processing of chlorinated plastic waste.Experimental
Materials
Virgin LMW and HMW PVC powders were obtained from Sigma. Virgin PVC with K-values ranging between 57 and 70 and waste PVC samples were acquired from Vynova. All solvents were supplied by Sigma and were used without drying or additional purification. Employed metal salts (e.g. ZnCl2) and HCl(CO)Ru(PPh3)3 were purchased in anhydrous form, but were handled under air without considering atmospheric humidity.Solubility tests of PVC
To a crimp neck vial (7 mL), PVC (1.5 mmol, 93.5 mg) and the solvent (2 mL) were added, after which the vial was sealed. Upon stirring, the mixture was heated at a temperature of 100 °C whereby the dissolution of the polymer was monitored visually. The dissolution temperature is noted to be the temperature at which the liquid phase became transparent with complete dissolution of the solid polymer particles after maximally 1 h.Molecular weight of polymer samples
The molecular weight of virgin PVC samples was measured using viscometry (determination of Fikentscher K-value) and Gel Permeation Chromatography (GPC). For viscometry, the elution time of solutions of PVC in cyclohexanone (0.5 m% PVC) was measured using a Ubelhode viscometer. When comparing the elution time of the solution (t1) with that of pure cyclohexanone (t0), a viscosity index (VI) was calculated as the difference in elution time. According to this, the K-value is a viscometry determined value that describes the molecular weight of the polymer (ESI,† Fig. S9). GPC calibration was done according to polystyrene standards, on an Agilent 1260 Infinity II GPC device, with THF as the eluent (1 mL min−1) with a column temperature of 35 °C. The molar weight averages of polymer products, after tandem reaction, were measured using GPC under the same conditions.Pretreatment of waste powders
Before reaction of waste materials, aliquots of the ground waste powders (93 mg) were subjected to one of following procedures: (1) when pre-dissolved, the powder was dissolved in methyl cyclohexanecarboxylate (2 mL) by magnetically stirring at 100 °C for 1 h; (2) when applying dissolution–precipitation/pre-dissolution, the powder was firstly dissolved by stirring it for 30 min at 50 °C in THF (1 mL). After cooling to room temperature, ethanol (2 mL) was added to the solution to precipitate the polymer. After solvent decantation, the polymer product was pre-dissolved in methyl cyclohexanecarboxylate (2 mL) by stirring at 100 °C for 1 h.Tandem dehydrochlorination–hydrogenation reactions
Virgin PVC (1.5 mmol based on the monomer unit with molar mass of 62.6 g mol−1, 93 mg), the pre-treated waste sample (obtained from 93 mg waste powder), or varying quantities as described otherwise, were brought into a glass liner. To the glass liner, ZnCl2 (0.2 mmol, 27.3 mg, 14 mol% per 1.5 mmol PVC) and HCl(CO)Ru(PPh3)3 (0.01 mmol, 0.65 mol% per 1.5 mmol PVC, 9.4 mg) were added. For some mechanistic reactions, HCl in solution (1.5 mmol, 0.38 mL, 4 M solution in dioxane) was added at this stage. The reactions were performed in sealable home-made autoclave reactors. Hence, the glass liner was brought into the reactor, after which this was sealed and purged with N2-gas before bringing it under 20 bar of H2. The reaction mixture was then stirred (750 rpm) at 180 °C, under the applied H2-pressure, for the designated time. After heating, the reactor was cooled in an ice bath. The polymer products were isolated from the solvent by the addition of an excess of ethanol, and subsequent centrifugation (4700 rpm, 2 min) for precipitation. After solvent removal, the product was redissolved in THF (0.3 mL) at 50 °C for purification. The polymer product was then recovered by ethanol addition to the solution, followed by centrifugation (4700 rpm, 2 min) and solvent decantation. Then, the polymer product was dried overnight on a Schlenk line, under reduced pressure. Products were characterised by Attenuated Total Reflection-Fourier Transform Infrared Spectroscopy (ATR-FTIR) and liquid NMR. ATR-FTIR spectra were recorded on a benchtop Cary 630 FTIR Spectrometer.Determination of degree of dechlorination
The polymer products (samples of ±10 mg) were dissolved in CDCl3 at 50 °C. Subsequently, 1H-NMR spectra were recorded on a 400 MHz Bruker Avance III HD spectrometer at room temperature. Integration of the 1H-NMR spectrum was used to determine the degree of dechlorination of products, according to our previously described procedure.16 Each integral value is divided by the theoretical number of protons per corresponding carbon group. The theoretical number of protons is 1 for CHCl groups (δ 4.7–4.3 ppm), 2 for α-CH2-groups (δ 2.4–2.0 ppm) and 2 for ethylene–ethylene type units of CH2 (δ 2.0–1.28 ppm). The peak of –CH groups (δ 5.40 ppm) was indicative for the presence of residual double bonds. CH3-groups at chain ends of the polymer backbone had a peak with a shift at 0.9 ppm, accounting for 3 protons. All integrations enabled a calculation of the relative fractions of each type of carbon in the polymer chain. For this, the sum of the corrected integral values was normalised to 100%. Half of the fraction of CH-groups in products represents the number of double bonds per 100 carbon atoms. Additionally, the fraction of CHCl groups indicated the extent of completion for reactions. For this quantification, the degree of dechlorination was calculated as 100% (1 − (fraction of CHCl groups in products)/(fraction of CHCl in PVC)). In Section S2 of the ESI,† examples of the calculation are illustrated for polymer products with varying degree of dechlorination.
2-Chloroheptane model reaction
A stock solution of 2-chloroheptane (3.5 mmol, 0.5 M) in tetradecane (7.0 mL) was prepared with decane (3.5 mmol, 0.5 M) as internal standard. For reactions, the stock solution (0.5 mmol 2-chloroheptane, 1.0 mL) was added to a glass liner, loaded with ZnCl2 (0.2 mmol) and in some cases HCl(CO)Ru(PPh3)3 (0.01 mmol). The reaction mixture was then heated and magnetically stirred at 140 °C under 20 bar H2, in a sealed autoclave reactor. After 16 h, the reactor was cooled in an ice bath and the reaction mixture was analyzed by gas chromatography (GC) and GC-mass spectrometry (MS). GC-MS was used for identification of product peaks, consistent with data from the NIST database, while GC was used for quantification of the product peaks. The conversion of 2-chloroheptane was calculated relative to the internal standard, using effective carbon numbers (ECN) to correct for Flame Ionisation Detector (FID) response factors (ECN-values: 6.88 for 2-chloroheptane, 7 for heptane, 6.9 for chloro-methylhexane, 7 for methylhexanes, 4 for isobutane and 2.88 for chloropropane).XAS data collection
Ru K-edge XAS spectra were collected in transmission mode, by continuous energy scans using a Si(220) monochromator on the SAMBA beamline at the SOLEIL Synchrotron (Saint-Aubin, France). Simultaneous measurement of a metallic ruthenium foil, was used as a reference for the energy alignment. For measurements, the HCl(CO)Ru(PPh3)3 was pressed into a pellet (fresh Ru-catalyst), while the structure of Ru-catalyst after 16 h of PVC dehydrochlorination–hydrogenation was measured in solution (using methyl cyclohexanecarboxylate as solvent) in 7 mL crimp cap vials. The applied reaction conditions and data processing are described in ESI,† Section S9.Data availability
Original data are available within KULeuven's OneDrive system. The ESI† contains additional characterisation data and conditions of experiments.Author contributions
Galahad O’Rourke: conceptualization, methodology, validation, formal analysis, investigation, resources, data curation, writing – original draft, writing – review & editing, visualization. Alina Skorynina: investigation, formal analysis, visualization. Igor Beckers: writing – review & editing. Sam Van Minnebruggen: resources, methodology, writing – review & editing. Christel Colemonts: resources, investigation. Philippe Gabriels: project administration, resources. Peter Van der Veken: project administration, resources. Dirk De Vos: conceptualization, methodology, writing – review & editing, visualization, supervision, project administration, funding acquisition.Conflicts of interest
The authors declare that there are no competing interests.Acknowledgements
This work was financially supported by Vynova. The authors acknowledge SOLEIL for provision of synchrotron radiation facilities and would like to thank Andrea Zitolo for assistance in using beamline SAMBA. The authors thank Robin Lemmens for support with the GPC.References
- L. J. González-Ortiz, M. Arellano, M. J. Sánchez-Peña, E. Mendizábal and C. F. Jasso-Gastinel, Polym. Degrad. Stab., 2006, 91, 2715–2722 CrossRef.
- G. Akovali, Plastic materials: polyvinyl chloride (PVC), Woodhead Publishing Limited, 2012 Search PubMed.
- K. Lewandowski and K. Skórczewska, Polymers, 2022, 14, 3035 CrossRef CAS PubMed.
- I. Janajreh, M. Alshrah and S. Zamzam, Sustainable Cities Soc., 2015, 18, 13–20 CrossRef.
- E. Eckert, F. Münch, T. Göen, A. Purbojo, J. Müller and R. Cesnjevar, Chemosphere, 2016, 145, 10–16 CrossRef CAS PubMed.
- A. Marcilla, S. García and J. C. García-Quesada, J. Anal. Appl. Pyrolysis, 2004, 71, 457–463 CrossRef CAS.
- G. Jiang, D. A. Sanchez Monsalve, P. Clough, Y. Jiang and G. A. Leeke, ACS Sustainable Chem. Eng., 2021, 9, 1576–1589 CrossRef CAS.
- H. Li, H. A. Aguirre-Villegas, R. D. Allen, X. Bai, C. H. Benson, G. T. Beckham, S. L. Bradshaw, J. L. Brown, R. C. Brown, V. S. Cecon, J. B. Curley, G. W. Curtzwiler, S. Dong, S. Gaddameedi, J. E. García, I. Hermans, M. S. Kim, J. Ma, L. O. Mark, M. Mavrikakis, O. O. Olafasakin, T. A. Osswald, K. G. Papanikolaou, H. Radhakrishnan, M. A. Sanchez Castillo, K. L. Sánchez-Rivera, K. N. Tumu, R. C. Van Lehn, K. L. Vorst, M. M. Wright, J. Wu, V. M. Zavala, P. Zhou and G. W. Huber, Green Chem., 2022, 24, 8899–9002 RSC.
- P. A. Kots, B. C. Vance, C. M. Quinn, C. Wang and D. G. Vlachos, Nat. Sustainability, 2023, 6, 1258–1267 CrossRef.
- M. K. Assefa and M. E. Fieser, J. Mater. Chem. A, 2023, 11, 2128–2132 RSC.
- N. G. Bush, M. K. Assefa, S. Bac, S. Mallikarjun Sharada and M. E. Fieser, Mater. Horiz., 2023, 10, 2047–2052 RSC.
- S. Svadlenak, S. Wojcik, O. Ogunlalu, M. Vu, M. Dor, B. W. Boudouris, D. Wildenschild and K. A. Goulas, Appl. Catal., B, 2023, 338, 123065 CrossRef CAS.
- D. Glas, J. Hulsbosch, P. Dubois, K. Binnemans and D. E. Devos, ChemSusChem, 2014, 7, 610–617 CrossRef CAS PubMed.
- T. Zhao, Q. Zhou, X. L. He, S. Di Wei, L. Wang, J. M. N. van Kasteren and Y. Z. Wang, Green Chem., 2010, 12, 1062–1065 RSC.
- K. Oster, A. Tedstone, A. J. Greer, N. Budgen, A. Garforth and C. Hardacre, Green Chem., 2020, 22, 5132–5142 RSC.
- G. O’Rourke, T. Hennebel, M. Stalpaert, A. Skorynina, A. Bugaev, K. Janssens, L. Van Emelen, V. Lemmens, R. D. O. Silva, C. Colemonts, P. Gabriels, D. Sakellariou and D. De Vos, Chem. Sci., 2023, 14, 4401–4412 RSC.
- K. Binnemans and P. T. Jones, J. Sustainable Metall., 2023, 9, 423–438 CrossRef.
- T. Kameda, M. Ono, G. Grause, T. Mizoguchi and T. Yoshioka, Ind. Eng. Chem. Res., 2008, 47, 8619–8624 CrossRef CAS.
- T. Yoshioka, T. Kameda, S. Imai and A. Okuwaki, Polym. Degrad. Stab., 2008, 93, 1138–1141 CrossRef CAS.
- T. Kameda, M. Ono, G. Grause, T. Mizoguchi and T. Yoshioka, Polym. Degrad. Stab., 2009, 94, 107–112 CrossRef CAS.
- G. Grause, S. Hirahashi, H. Toyoda, T. Kameda and T. Yoshioka, J. Mater. Cycles Waste Manage., 2017, 19, 612–622 CrossRef CAS.
- G. Pappa, C. Boukouvalas, C. Giannaris, N. Ntaras, V. Zografos, K. Magoulas, A. Lygeros and D. Tassios, Resour., Conserv. Recycl., 2001, 34, 33–44 CrossRef.
- G. O’Rourke, M. Houbrechts, M. Nees, M. Roosen, S. De Meester and D. De Vos, Green Chem., 2022, 24, 6867–6878 RSC.
- R. G. Dastidar, M. S. Kim, P. Zhou, Z. Luo, C. Shi, K. J. Barnett, D. J. McClelland, E. Y. X. Chen, R. C. Van Lehn and G. W. Huber, Green Chem., 2022, 24, 9101–9113 RSC.
- H. Matsuda, K. Araki, H. Fujimatsu and S. Kuroiwa, Polym. J., 1981, 13, 587–590 CrossRef CAS.
- N. Vidyarthi and S. R. Palit, J. Polym. Sci., Part A: Polym. Chem., 1980, 18, 3315–3317 Search PubMed.
- D. J. S. Sandbeck, D. J. Markewich and A. L. L. East, J. Org. Chem., 2016, 81, 1410–1415 CrossRef PubMed.
- K. Y. Cheung, S. Schouterden, S. Van Minnebruggen, P. Tomkins, T. De Baerdemaeker, A. N. Parvulescu, T. Yokoi and D. De Vos, ChemCatChem, 2023, 15(6), e202300070 CrossRef CAS.
- C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, E. Shuster and H. F. Sneddon, Green Chem., 2016, 18, 3879–3890 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ey00082j |
| This journal is © The Royal Society of Chemistry 2024 |