
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGelation phase diagrams of colloidal rod systems measured over a large composition space†
Shiqin Hea,
Marco Caggionib,
Seth Lindbergb and
Kelly M. Schultz *a
*a
aDepartment of Chemical and Biomolecular Engineering, Lehigh University, Bethlehem, PA, USA. E-mail: kes513@lehigh.edu; Fax: +1-610-758-5057; Tel: +1-610-758-2012
bProcess and Engineering Development, Procter & Gamble Co., West Chester, OH, USA
First published on 27th April 2022
Abstract
Rheological modifiers tune product rheology with a small amount of material. To effectively use rheological modifiers, characterizing the rheology of the system at different compositions is crucial. Two colloidal rod system, hydrogenated castor oil and polyamide, are characterized in a formulation that includes a surfactant (linear alkylbenzene sulfonate) and a depletant (polyethylene oxide). We characterize both rod systems using multiple particle tracking microrheology (MPT) and bulk rheology and build phase diagrams over a large component composition space. In MPT, fluorescent particles are embedded in the sample and their Brownian motion is measured and related to rheological properties. From MPT, we determine that in both systems: (1) microstructure is not changed with increasing colloid concentration, (2) materials undergo a sol–gel transition as depletant concentration increases and (3) the microstructure changes but does not undergo a phase transition as surfactant concentration increases in the absence of depletant. When comparing MPT and bulk rheology results different trends are measured. Using bulk rheology we observe: (1) elasticity of both systems increase as colloid concentration increases and (2) the storage modulus does not change when PEO or LAS concentration is increased. The differences measured with MPT and bulk rheology are likely due to differences in sensitivity and measurement method. This work shows the utility of using both techniques together to fully characterize rheological properties over a large composition space. These gelation phase diagrams will provide a guide to determine the composition needed for desired rheological properties and eliminate trial-and-error experiments during product formulation.
1 Introduction
Rheological modifiers are used in personal care and fabric and home care products to tune product properties to reach desired rheological behavior. To effectively design products, understanding how rheological properties and microstructure are changed by the addition of rheological modifiers at different component compositions is critical.1–7 Colloidal rods are commonly used materials for rheological modification because their anisotropic shape tunes rheology with the addition of a small amount of colloid.3,5 While colloidal rods are presently used as rheological modifiers, the strength of interactions between colloidal rods and other components in a formulation can affect the rheology of the final product and can be tuned by changing the amount of each component in the system.5,8–10 This tunability is advantageous but also complicates formulation because it presents a large parameter space to design materials. This large parameter space usually requires trial-and-error experiments to identify compositions that result in desired rheological properties. A better understanding of the effect of each component concentration within a formulation on rheological properties will enable product design while decreasing material and time used for trial-and-error experiments.Colloidal rods are currently used in products as rheological modifiers and their rheological properties have been previously characterized.1–6,11 In products, these rods can be used to simply change the viscosity or can induce sol–gel phase transitions during manufacturing processes and end-use.6,12 The state of a colloidal rod system depends on the colloid volume fraction, aspect ratio and interparticle interactions, which include depletion interactions, electrostatic forces, osmotic pressure gradients and steric forces.5,8–10,13,14 Most of these interactions are introduced by adding molecules to the colloidal rod system. The strength of these interactions can be tuned by simply changing the amount of each component. Therefore, it is crucial to characterize the role of each component in a given system and measure the combined effect on the final rheological properties and state of the material.
In this study, the rheological properties of two colloidal rod systems, hydrogenated castor oil (HCO) and polyamide (PA), are characterized over a large composition space. Both HCO and PA are long thin colloidal rods. Previous studies have determined that these systems have similar rheological properties during sol–gel transitions.6,8,12 Prior work focused on measuring material properties during phase transitions including the response of these colloidal rod systems to external driving forces and shear.6–8,15 In these studies, different compositions of materials are measured, but there are no systematic studies focusing on characterizing the state and rheology of these materials over a large composition space. In this work, we compare these two colloidal rod systems by mapping their rheological properties at different component compositions. The three components that are systematically varied are crystallized colloidal rods (HCO or PA), non-adsorbing polymer (polyethylene oxide, PEO) and surfactant (linear alkylbenzene sulfonate, LAS).
Each component in the system plays a separate role in the structure and rheology during phase transitions and at equilibrium. Non-adsorbing polymer, PEO, is a depletant which gels the system.8,12 By increasing PEO concentration, depletion interactions between the colloids increase and drive the rods to form a sample-spanning network. This sample-spanning network is the phase transition from a sol to a gel.8,9 LAS stabilizes the colloidal suspension by attaching its hydrophobic tail, a triglyceride, to the surface of the colloid. The hydrophilic end of LAS, a sulfonate group, introduces an electrostatic force between colloidal rods preventing aggregation and stabilizing the suspension.6,12,16 From our previous work, we determined that rheological evolution of these materials during a sol–gel transition is dependent on the amount of surfactant in the system. As the amount of surfactant changes the structure of the colloids change from stabilized single colloids to bundled colloids.12
Systematically changing the composition of the three components in our colloidal rod system will result in formulations with unique rheological properties. For each composition, we measure the rheological properties and create phase diagrams that will inform the design of products that use these materials for rheological modification. Both multiple particle tracking microrheology (MPT) and bulk rheology are used to characterize rheological properties. In MPT, florescent probe particles are embedded in the sample and their Brownian motion is recorded with video microscopy. The position of the probe particles in each frame is identified and tracked throughout a video using classic tracking algorithms to define particle trajectories.20,24 From these trajectories, the ensemble-averaged mean-squared displacement (MSD, 〈Δr2(τ)〉) and the logarithmic slope of the MSD, 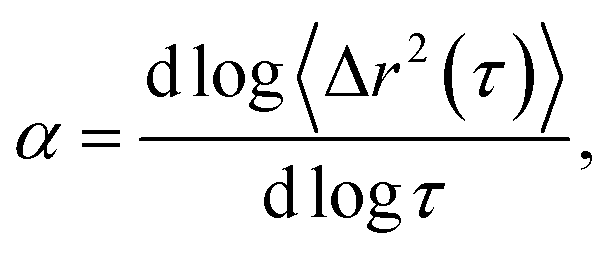 are calculated to determine the rheological properties and the state of the material. To quantitatively identify the state of the material, we compare the measured value of α to the critical relaxation exponent, n, which was previously measured for these materials.22,25–28,28–31 The MSD can also be related to other rheological properties using the Generalized Stokes–Einstein Relation.17–19,22,32,33
are calculated to determine the rheological properties and the state of the material. To quantitatively identify the state of the material, we compare the measured value of α to the critical relaxation exponent, n, which was previously measured for these materials.22,25–28,28–31 The MSD can also be related to other rheological properties using the Generalized Stokes–Einstein Relation.17–19,22,32,33
In this work, both MPT and bulk rheology are used to characterize these colloidal rod systems because they are complementary techniques that measure different moduli regimes and use different methods to measure rheological properties. In MPT, the Brownian motion of the particle applies a stress on the material enabling the rheological response to be measured with minimal shear applied.34–37 With bulk rheological measurements, we apply small angle oscillatory shear to the material and measure the bulk response of the material during a frequency sweep. This applies a much larger force on the material, but also enables measurement of materials with much higher elastic moduli than MPT.38,39 Because of these different measuring regimes and methods, using both MPT and bulk rheology to measure the same material provides more complete information about the system by broadening the frequency and measurable moduli range.
This work builds rheological phase diagrams for colloidal rod systems over a large parameter space. The current state-of-the-art generally does not fully map out parameter space. This provides a facile way to present colloidal material rheological properties under different component compositions and provides users with a lookup table for future compositions of rheological modification. In this study, we separately determine the effect of each component concentration on the rheology of HCO and PA colloidal rod systems. We use MPT and bulk rheology to characterize the rheology when the concentration of the colloid, non-adsorbing polymer and surfactant are varied independently. We first characterize the system using MPT. We measure that increasing colloid concentration does not change the rheology of either system and the material remains a sol. Next, we increase PEO concentration, which increases depletion interactions between the colloidal rods, and both systems transition from a sol to a gel. We then increase the LAS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :colloid ratio. For both colloidal rod systems, as LAS concentration increases the system remains in the sol state, but the structure changes from stable single colloidal rods to colloidal rod aggregates, agreeing with previous research.12 After separately determining the role of each component in HCO and PA, we build phase diagrams from MPT and bulk rheology measurements to provide a map of HCO and PA rheological modification. From the MPT phase diagram, when PEO concentration is near the critical depletant concentration (concentration where the system transitions from a sol to a gel, 0.34 ± 0.02 c/c* and 0.36 ± 0.04 c/c* for HCO and PA, respectively) the fraction of samples in the gel state is higher when LAS:HCO > 16 than LAS:HCO ≤ 16. From bulk rheological measurements we determine that the elasticity for both HCO and PA increases when more colloidal rods are added to the system. In addition, at higher colloid concentration HCO has higher elasticity than PA. We hypothesize that this is due to the polydispersity of HCO, which contains longer rods that are more likely to increase elasticity due to aggregation and entanglement. There is no measured change in elastic moduli when the concentration of PEO or LAS is changed. Comparing the phase diagrams between MPT and bulk rheology, different trends are measured when each component concentration is varied in our colloidal rod systems. These different trends are likely caused by the difference in the measurement methods. This work provides guidance for future product design by constructing phase diagrams for colloidal rod systems over a large composition space, which can minimize trial-and-error experiments by providing a lookup table for rheological properties.
:colloid ratio. For both colloidal rod systems, as LAS concentration increases the system remains in the sol state, but the structure changes from stable single colloidal rods to colloidal rod aggregates, agreeing with previous research.12 After separately determining the role of each component in HCO and PA, we build phase diagrams from MPT and bulk rheology measurements to provide a map of HCO and PA rheological modification. From the MPT phase diagram, when PEO concentration is near the critical depletant concentration (concentration where the system transitions from a sol to a gel, 0.34 ± 0.02 c/c* and 0.36 ± 0.04 c/c* for HCO and PA, respectively) the fraction of samples in the gel state is higher when LAS:HCO > 16 than LAS:HCO ≤ 16. From bulk rheological measurements we determine that the elasticity for both HCO and PA increases when more colloidal rods are added to the system. In addition, at higher colloid concentration HCO has higher elasticity than PA. We hypothesize that this is due to the polydispersity of HCO, which contains longer rods that are more likely to increase elasticity due to aggregation and entanglement. There is no measured change in elastic moduli when the concentration of PEO or LAS is changed. Comparing the phase diagrams between MPT and bulk rheology, different trends are measured when each component concentration is varied in our colloidal rod systems. These different trends are likely caused by the difference in the measurement methods. This work provides guidance for future product design by constructing phase diagrams for colloidal rod systems over a large composition space, which can minimize trial-and-error experiments by providing a lookup table for rheological properties.
2 Materials and methods
2.1 Hydrogenated castor oil colloidal rod synthesis
The HCO used in this work is provided by Procter & Gamble Co. and is synthesized using previously published protocols.3,4,6,16,40–42 Briefly, 4 wt% HCO flakes are dissolved in 16 wt% linear alkylbenzene sulfonate (LAS, Procter & Gamble Co., CAS# 68411-30-3). The mixture is heated to 92 °C for 5 min at 300 RPM to remove any existing crystal structure. To crystallize HCO into colloidal rods, the mixture is cooled at 1 °C min−1. This results in over 95% HCO colloidal rods having aspect ratios (AR = L/d where L is the length and d is the diameter) ranging from 50–2500. The exact size distribution of HCO is unknown. The length of these HCO rods range from 1–50 μm, with a width of approximately 20 nm.3,4,6,16,40,41 After synthesis, the final suspension is composed by 4 wt% HCO, 16 wt% LAS and 80 wt% water. Because HCO rods have a density close to the density of the solvent, 1.0 ± 0.01 g cm−3, the HCO rod suspension is stable without major rod sedimentation over time.2.2 Polyamide colloidal rod synthesis
Synthesis of PA rods is based on a previously published protocol.8 5 g batches are made by mixing 1 wt% PA powder (Disparlon 6650, King Industries), 16 wt% LAS and 83 wt% deionized (DI) water. This PA powder solution is rotated at 10 RPM overnight to fully mix all components. After mixing, a 1 cm magnetic stir bar is added to the PA solution and placed in an oil bath (silicon oil, Millipore Sigma) on a stirred hot plate (Fisher Scientific Co.). The solution is heated to 100 °C and agitated at 300 RPM for 30 min to melt the PA powder. After heating, the solution is removed from the oil bath and cooled to 25 °C to form PA colloidal rods. The PA solution is heated to 100 °C because this is the synthesis temperature that produces PA rods with the largest AR. This synthesis results in uniform PA rods with an AR = 279 ± 38 and a diameter of d = 84 ± 4 nm, which is characterized by differential interference contrast microscopy (DIC) and atomic force microscopy (AFM). Similar to HCO rods, PA rods have a density close to the solvent density, 0.99 ± 0.01 g cm−3, and the suspension is stable over time.2.3 Components of the colloidal rod system
The phase transition for both HCO and PA is driven by depletion interactions induced by the addition of polyethylene oxide (PEO, Alfa Aesar), a non-adsorbing polymer. We use PEO with a molecular weight of Mw = 100000 g mol−1 and a radius of gyration of Rg = 17.6 nm. The overlap concentration for PEO is c* = 0.07 mmol L−1, which is calculated using 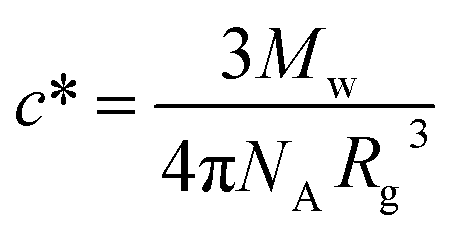 where NA is Avogadro's number.8,43,44 Both HCO and PA are stabilized by LAS with a molecular weight of Mw = 348.48 g mol−1.
where NA is Avogadro's number.8,43,44 Both HCO and PA are stabilized by LAS with a molecular weight of Mw = 348.48 g mol−1.
2.4 Multiple particle tracking microrheology
Multiple particle tracking microrheology (MPT) is used to characterize the rheological properties of both HCO and PA colloidal rod systems and to build phase diagrams as the colloid, depletant and surfactant concentration are varied. MPT is a passive microrheological technique that measures material properties in a low moduli range (G′ ≤ 4 Pa). MPT measures Brownian motion of probe particles embedded in the material.20–22,32,45 Poly (ethylene glycol) (PEG) grafted probe particles (2a = 0.53 ± 0.01 μm where a is the particle radius, Polysciences, Inc.) are used for MPT measurements. Particle PEGylation will be detailed in the next section. We use PEG grafted probes to prevent probe-colloid and probe–probe interactions in our colloidal rod systems.46 Prior to adding particles to our colloidal rod solutions, PEG grafted particles are washed 3× to remove any impurities by dilution and centrifugation (Eppendorf, Microcentrifuge 5424) at 7000 RPM for 5 min. After washing, probes are sonicated (Branson, M1800, 40 kHz) for 30 min to break apart any particle aggregates which might have formed during centrifugation. Probes are then added to the colloidal rod precursor solution at a final concentration of 0.15% solids volume−1. After the precursor solution is mixed, it is sealed in a sample chamber for MPT data collection.Sample chambers are made for all MPT data collection. The bottom of a sample chamber is constructed of a 25 × 75 × 1 mm glass slide (Fisher Scientific Co.). Two spacers are made by cutting a coverslip (22 × 22 × 0.13–0.17 mm, Fisher Scientific Co.) into two rectangular pieces. Spacers are glued onto the bottom glass slide parallel to each other to create the walls of the sample chamber. An additional coverslip is glued on top of the glass spacers, which creates the top of the chamber. A sample is pipetted into the opening of the chamber and capillary forces enable the solution to fill the entire chamber. The openings of the chamber are then sealed with UV adhesive (NOA 81 optical adhesive, Norland Products Inc.). This prevents the sample from being in contact with ambient air, which can prevent drift and sample evaporation.
All MPT data are collected with an inverted fluorescent microscope (Zeiss Observer Z1, Carl Zeiss AG) with a 63× water immersion objective (N.A. 1.3, 1× optovar, Carl Zeiss AG). A total of 800 frames are recorded at a frame rate of 30 frames per second and an exposure time of 1000 μs (Phantom Miro M120, 1024 × 1024 pixels, Vision Research Inc.). These video acquisition parameters are chosen to minimize static and dynamic particle tracking errors.22,23,47
Classical particle tracking algorithms are used to track probe particle motion.20,24 The brightness-weighted centroid of each probe is first determined in each frame of a video. A probability distribution function based on probe Brownian motion is then used to link the probe positions in successive frames into particle trajectories. The ensemble-averaged mean-squared displacement (MSD) is calculated from these particle trajectories. Additional rheological properties can be calculated from the MSD and will be discussed in detail in the Results and discussion. For all MPT measurements, the measurement is repeated three times with different solutions at the same composition.
2.5 PEG functionalization of probe particles
Bare probe particles can interact with HCO and PA colloidal rods which would make MPT measurements invalid.6,12 Therefore, the surface of probe particles are grafted with PEG before they are added to a precursor solution to prevent these interactions. This surface functionalization uses previously published protocols.46Briefly, probe particles are placed in dialysis tubing (10 kD cutoff, SpectraPor) during the surface modification reaction. This avoids the use of centrifugation to separate probe particles after the reaction, which can cause particle aggregation. Dialysis tubing is first submerged into 2-(N-morpholino)ethanesulfonic acid (MES) buffer (100 mmol, Millipore Sigma) at pH 6.0 for 2 hours under agitation. This allows the buffer with enough time to diffuse into the dialysis tubing and ensures the probes are at pH 6.0 for the formation of ester groups in the next step. The dialysis tubing is then moved to MES buffer with 5 mmol N-hydroxysuccinimide (NHS, MilliporeSigma), 15 mmol 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide (EDC, MilliporeSigma) and a 10-fold excess of amine-terminated methoxy poly (ethylene glycol) (mPEG-NH2, Mw = 750 g mol−1, Millipore Sigma) for 30 min. In this step, NHS and EDC react with carboxylic groups on the surface of the probe particles to form active ester groups. Excess mPEG-NH2 is added to the dialysis tubing in this step to ensure the immediate presence of mPEG-NH2 during the reaction in the next step. After 30 min, the dialysis tubing is submerged into borate buffer (50 mmol boric acid, 36 mmol sodium tetraborate, Millipore Sigma) at pH 8.5 with 5 mmol NHS, 15 mmol EDC and a 10-fold excess of mPEG-NH2 for at least 8 hours. In this step, active ester groups react with mPEG-NH2 to form amide bonds and graft mPEG-NH2 to the surface of the probe particles. This step is repeated three times with fresh borate buffer and reaction reagent. After the reaction is complete, the dialysis tubing is submerged in fresh borate buffer for at least 2 hours to wash out unreacted reagent.
PEG functionalized probes are stored at 4 °C and can be used for several months with no change in surface chemistry.46
2.6 Bulk rheology
Bulk rheology experiments measure frequency dependent material properties for both HCO and PA colloidal rod systems. An 86 μL sample is loaded onto the rheometer (Tmin = 0.05, where Tmin is the minimum torque, Ares G2, TA Instruments), and the modulus is measured using a sandblasted cone and plate geometry with a 2° 20 mm cone and 51 μm measurement gap. The measurement is performed in the linear viscoelastic (LVE) regime which is determined using a strain sweep. This measurement is provided in Fig. S1 in the ESI.† After the strain sweep, samples are measured at 25 °C using a frequency sweep from 0.08 to 10 rad s−1 at 0.1% strain within the LVE. At these conditions, three samples are measured of each colloidal rod composition.2.7 Microscopy
Differential interference contrast microscopy (DIC) is used to characterize the length of the PA rods. A representative image is provided in Fig. S2.† A Zeiss Axioplan 2 microscope (Carl Zeiss AG) equipped with DIC optics is used to collect images of PA rods. The rods are imaged with a Zeiss 20× Plan NeoFLUAR objective (N.A. 0.5) and captured with a Basler Ace 2.3 MP Mono.Atomic force microscopy (AFM, Bruker/VEECO DI-3000) is used to characterize the diameter of the PA rods. Samples are prepared by placing a drop of 0.02 wt% PA rod stock solution on a smooth mica surface (1 × 1 cm2, Novascan Technologies, Inc), and allowing it to dry at ambient conditions. The AFM scanner probes a 10 μm × 10 μm area on the mica sample with an AFM cantilever (TESPA-V2, Bruker) at 1.0 Hz scan rate. A representative image is provided in Fig. S3.† The diameter is determined from the height profile of the AFM measurements.
2.8 Experimental systems
The colloid, depletant and surfactant concentration is varied and the rheological properties and state of the material is characterized. This parameter space is traversed by increasing the concentration of one component and holding the other two component concentrations constant. Phase diagrams are built for both HCO and PA with various component compositions after the effect of a single component has been characterized. In the phase diagrams, the colloid concentration is varied from 0.2–0.8 wt%, the PEO concentration is varied from 0.14–0.75 c/c* and the LAS concentration is varied from 0.8–12.8 wt%.3 Results & discussion
This work characterizes the composition dependent structure, rheology and state of two colloidal rod systems: HCO and PA. We systematically vary the composition of these colloidal rod systems by changing the concentration of single components, namely the colloid, depletant and surfactant, while holding all other component concentrations constant. Multiple particle tracking microrheology (MPT) and bulk rheology measurements of this large composition space are used to build phase diagrams. These techniques are used because they measure rheology over different moduli ranges and use different measurement methods, which provide more complete characterization of these systems. Our results provide a road map for the design of products that use these colloidal rods as rheological modifiers. These phase diagrams tabulate composition dependent rheological modification over a large parameter space, eliminating the need for trial-and-error experiments to achieve desired material properties.In MPT, the Brownian motion of probe particles embedded in the sample material is recorded and this probe motion measures the state of the material and is related to material rheological properties by the mean-squared displacement (MSD) using the GSER.20,21,32,45 The MSD is calculated from probe particle trajectories and is defined as 〈Δr2(τ)〉 = 〈Δx2(τ)〉 + 〈Δy2(τ)〉, where τ is the lag time between frames of the captured video and Δx and Δy are the difference in particle positions in the x and y direction, respectively. The logarithmic slope of the MSD, 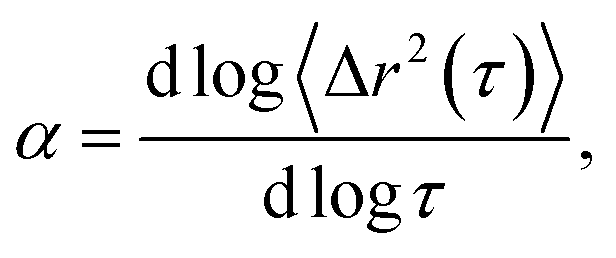 is also calculated and defines the state of the material. α = 1 indicates that probe particles are freely diffusing and the material is in a liquid state. α → 0 indicates that probe particle motion is completely restricted and the material is in a gel state. 0 < α < 1 indicates that the material is a viscoelastic sol or gel.17,22,23,48 To quantitatively determine whether the material is a viscoelastic sol or gel, α is compared to the critical relaxation exponent, n, which is calculated using time-cure superposition.6,12,22,28,31,49–53 When α > n, the material is in the viscoelastic sol state. α = n indicates that the material is at the sol–gel transition. α < n indicates that the material is a viscoelastic gel. In our previous work, we determined that gel evolution and fiber stability changes depending on the LAS:colloid ratio. Both measurements of rheology during gelation and the zeta potential of the starting colloidal rods showed that the material changes when LAS:colloid = 16 and LAS:colloid > 16. This is due to a difference in the starting solutions, we hypothesize that at LAS:colloid = 16 the solution is composed of stable single colloidal rods and at LAS:colloid > 16 these rods are bundled. n is determined for both HCO and PA and is dependent on LAS:colloid ratio. The values of n for these two materials are nLAS:HCO=16 = 0.37 ± 0.14 and nLAS:HCO>16 = 0.83 ± 0.10 for HCO and nLAS:PA=16 = 0.34 ± 0.07 and nLAS:PA>16 = 0.74 ± 0.13 for PA.12
is also calculated and defines the state of the material. α = 1 indicates that probe particles are freely diffusing and the material is in a liquid state. α → 0 indicates that probe particle motion is completely restricted and the material is in a gel state. 0 < α < 1 indicates that the material is a viscoelastic sol or gel.17,22,23,48 To quantitatively determine whether the material is a viscoelastic sol or gel, α is compared to the critical relaxation exponent, n, which is calculated using time-cure superposition.6,12,22,28,31,49–53 When α > n, the material is in the viscoelastic sol state. α = n indicates that the material is at the sol–gel transition. α < n indicates that the material is a viscoelastic gel. In our previous work, we determined that gel evolution and fiber stability changes depending on the LAS:colloid ratio. Both measurements of rheology during gelation and the zeta potential of the starting colloidal rods showed that the material changes when LAS:colloid = 16 and LAS:colloid > 16. This is due to a difference in the starting solutions, we hypothesize that at LAS:colloid = 16 the solution is composed of stable single colloidal rods and at LAS:colloid > 16 these rods are bundled. n is determined for both HCO and PA and is dependent on LAS:colloid ratio. The values of n for these two materials are nLAS:HCO=16 = 0.37 ± 0.14 and nLAS:HCO>16 = 0.83 ± 0.10 for HCO and nLAS:PA=16 = 0.34 ± 0.07 and nLAS:PA>16 = 0.74 ± 0.13 for PA.12
The effect of increasing colloid concentration on the rheology and state of each colloidal rod system is first characterized. The concentration of HCO and PA are increased from 0–0.8 wt% where the PEO and LAS concentration are held constant at 0 wt% and 12.8 wt%, respectively. Fig. 1 plots the logarithmic slope of the MSD, α, with increasing HCO or PA concentration. Plots of the corresponding MSDs are provided in Fig. S4 in the ESI.† The value of α ≈ 1 when there is no colloid in either system, 0 wt%. As the colloid concentration increases from 0.2–0.8 wt%, α is constant and remains at α = 0.95 ± 0.01. This means that probe particle motion is minimally restricted and the system remains in the liquid state regardless of colloid type or concentration.
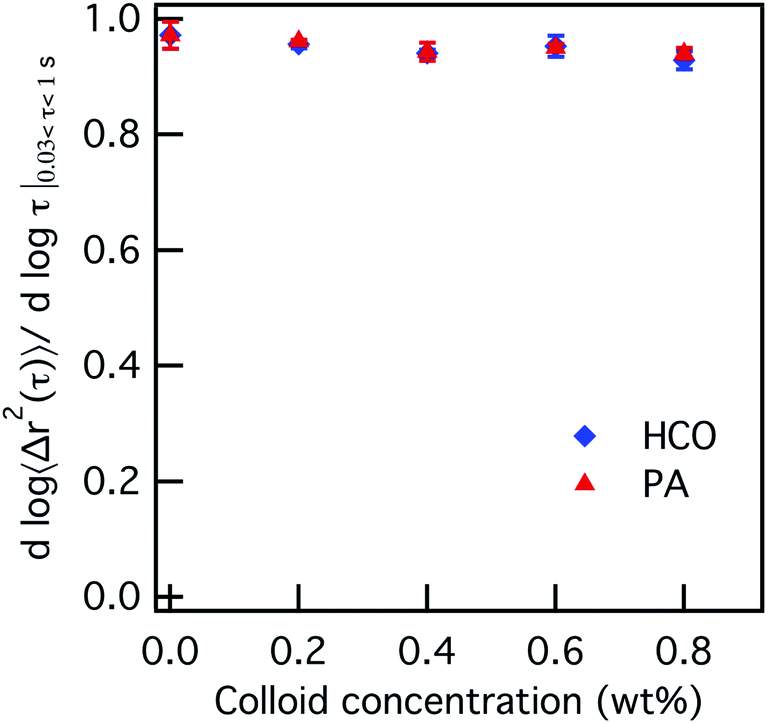 | ||
| Fig. 1 Microrheological measurements of HCO and PA with increasing colloid concentration. The logarithmic slope of the MSD, α, is constant with increasing colloid concentration from 0–0.8 wt%, indicating that the system remains in the sol phase (α ≈ 1). | ||
Next, we measure the change in each colloidal rod system structure and rheology when depletion interactions are increased. This is done by increasing PEO concentration. To measure these changes, LAS concentration is held constant and measurements taken at 12.8 wt% will be discussed here. These measurements are taken at other LAS concentrations and both plots of the MSD and α are provided in the ESI in Fig. S5–S13.† Colloid concentration is also held constant and PEO concentration is varied. This is done for sets of measurements at different colloid concentrations, 0.2, 0.4, 0.6 or 0.8 wt%. PEO concentration is increased in each system with constant colloid and LAS concentration from 0.14–0.75 c/c*, where c* is the overlap concentration and is 0.07 mmol L−1. From our previous study into the phase transitions of these colloidal rod systems, the phase transition point is different between LAS:colloid = 16 and LAS:colloid > 16 and is indicated in Fig. 2 by dashed lines.12
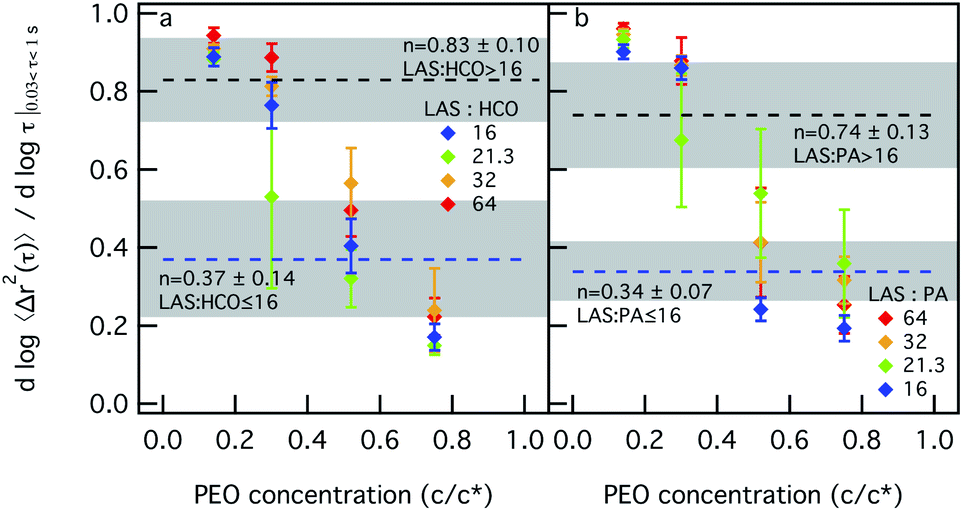 | ||
| Fig. 2 Microrheological measurements of (a) HCO and (b) PA with increasing PEO concentration, which increases depletion interactions. The blue dashed line indicates the critical relaxation exponent, n, for LAS:colloid ≤ 16. The black dashed line indicates the value of n for LAS:colloid > 16. The shaded area is the sol–gel transition region. In all measurements the LAS concentration is held constant at 12.8 wt%. The logarithmic slope of the MSD, α, decreases and passes through the sol–gel transition region for all colloidal gel systems as depletion interactions are increased, indicating that the system transitions from a sol to a gel. | ||
Fig. 2a and b show the values of α as a function of PEO concentration for different LAS:colloid ratios for both HCO and PA. For HCO at LAS:HCO = 16 in Fig. 2a, when PEO is 0.14 c/c*, α = 0.89 ± 0.02, which is greater than the critical relaxation exponent (nLAS:HCO=16 = 0.37 ± 0.14).12 This indicates that the HCO system is a sol at low PEO concentration. For this composition, α decreases with increasing PEO concentration entering the phase transition region when PEO is 0.52 c/c*. As PEO concentration is further increased, α decreases to 0.17 ± 0.03 and the material is in the gel state. A similar trend is measured for LAS:HCO > 16 in Fig. 2a, but at this LAS:HCO ratio the value of the critical relaxation exponent is different, nLAS:HCO>16 = 0.83 ± 0.10.12 Initially, α is greater than nLAS:HCO>16 when PEO is 0.14 c/c* and the system is a sol. As PEO increases to 0.3 c/c*, the LAS:HCO = 21.3 system is in the phase transition region while the LAS:HCO = 32 and LAS:HCO = 64 systems remain in the sol phase. When PEO is increased to 0.52 c/c*, all α values for all LAS:HCO ratios are less than nLAS:HCO>16, which indicates that the material is in the gel state. Fig. 2b shows the same characterization for PA with increasing depletion interactions. Similar trends are measured as PEO concentration and LAS:PA ratios are increased. For both colloidal rod systems, sol–gel transitions occur when depletion interactions are increased at any LAS:colloid ratio. These colloidal rod systems reach the gel state at different PEO concentrations, which is a function of the LAS:colloid ratio.
The effect of the surfactant (LAS) on phase transitions in our colloidal rod systems is characterized by changing the LAS concentration and holding the colloid concentration constant. These systems are characterized without depletion interactions (PEO concentration is 0 c/c*). LAS concentration is changed from 0.8–12.8 wt% while the colloid concentration is held constant. Sets of experiments are done for colloid concentrations of 0.2, 0.4, 0.6 or 0.8 wt%.
In our previous work, we determined that changing the LAS:colloid ratio can change the starting structure of the colloidal rods and, subsequently, the structure of the scaffold at the critical gel point and the gelation evolution.12 For LAS:colloid > 16, electrostatic forces between colloidal rods are weak, which enables aggregates or bundles of colloidal rods to form in the system. The attraction between the bundles are not strong enough to form sample-spanning networks. Therefore, the bundled colloids occupy less space in the sample enabling more space for probe particles to freely diffuse corresponding to values of α close to 1. For LAS:colloid ≤ 16, the electrostatic force between the colloidal rods are strong, stabilizing single colloidal rods in solution and preventing bundling. Probe particle motion is constrained by these overlapping single colloidal rods, corresponding to α values less than 1.12
Similar trends are measured in Fig. 3, which shows the change in the value of α as a function of LAS:colloid ratio. For 0.2 wt% HCO, α values first decrease and then increase as the LAS:HCO ratio increases. This indicates that for 0.2 wt% HCO, as the LAS:HCO ratio increases from 4–12, the system structure becomes less bundled and has more stable overlapping single colloids which restrict probe motion. As the LAS:HCO ratio further increases to 16, the colloids start to bundle. As the LAS:HCO ratio increases from 16–64, α values increase to 0.95, which indicates that the colloids are in the bundled phase where more space is available for probe particles to freely diffuse. For HCO concentrations greater than 0.2 wt%, as the LAS:HCO ratio increases from 1.6–16, α values increase. This indicates that probe motion is becoming less restricted as the LAS:HCO ratio increases, which indicates that bundles of colloidal rods form. Although changing the LAS:HCO ratio changes the microstructure of the colloidal rods, all changes in α are above the sol–gel transition, which indicates that the system is in the sol phase for all compositions measured.
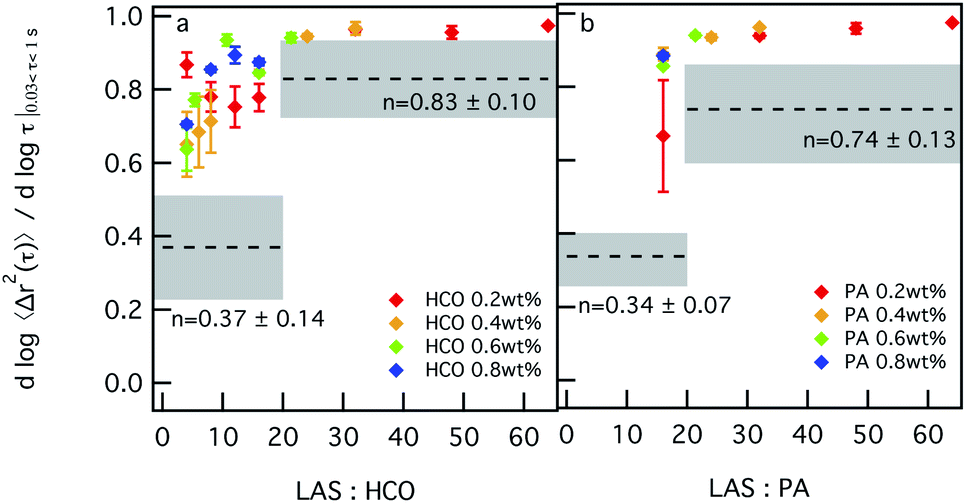 | ||
| Fig. 3 Microrheological measurements of (a) HCO and (b) PA with increasing LAS concentration. These samples do not contain depletant (PEO concentration is 0 c/c*). The logarithmic slope of the MSD, α, increases with increasing LAS concentration and remains above the sol–gel transition region for all concentrations measured. This indicates that the system microstructure does change with a change in LAS concentration but the material remains in the sol phase for all concentrations. | ||
PA is measured using the same composition range of colloid concentrations. LAS:PA < 16 cannot be measured because PA colloidal rods are stabilized with LAS during synthesis, which requires a minimum LAS:PA = 16. The effect of surfactant on the structure and rheology of the PA system is similar to that measured for HCO. As the LAS:PA ratio increases from 16–64, α values increase. Again, this indicates that the colloids are less stable and become bundled, corresponding to increasing probe particle diffusion. Similar to HCO, all values of α measured in the PA colloidal rod system are in the sol. Changing the concentration of LAS in both systems does not induce sol–gel transitions, but does change the structure of the colloidal rods in the system.
The effect of each component on the structure and rheological properties for both systems are measured and described in the previous paragraphs and in Fig. 1–3. To determine where rheology is modified in these systems, phase diagrams are constructed from MPT measurements for HCO and PA, Fig. 4a and b, respectively. 2D plots of some of the trends discussed here are provided in Fig. S12–S14 in the ESI.† In these phase diagrams, the concentration of HCO or PA is varied from 0.2–0.8 wt%, the concentration of PEO is varied from 0.14–0.75 c/c* and the concentration of LAS is varied from 0.8–12.8 wt%. The color of each data point indicates the value of α. Warm colors indicate α → 0 measuring that probe particle movement is restricted and the material is a gel. Cool colors indicate α → 1 where probe particles are diffusing and the material is a liquid. Phase transitions occur at the critical relaxation exponents, which are nLAS:HCO=16 = 0.37 ± 0.14 and nLAS:HCO>16 = 0.83 ± 0.10 for HCO and nLAS:PA=16 = 0.34 ± 0.07 and nLAS:PA>16 = 0.74 ± 0.13 for PA. These transitions correspond to orange data points for LAS:colloid = 16 and light blue data points for LAS:colloid > 16. With these phase diagrams, we can start to describe trends based on the composition of colloidal gel systems and lookup desired rheological properties to easily identify the corresponding colloidal gel composition.
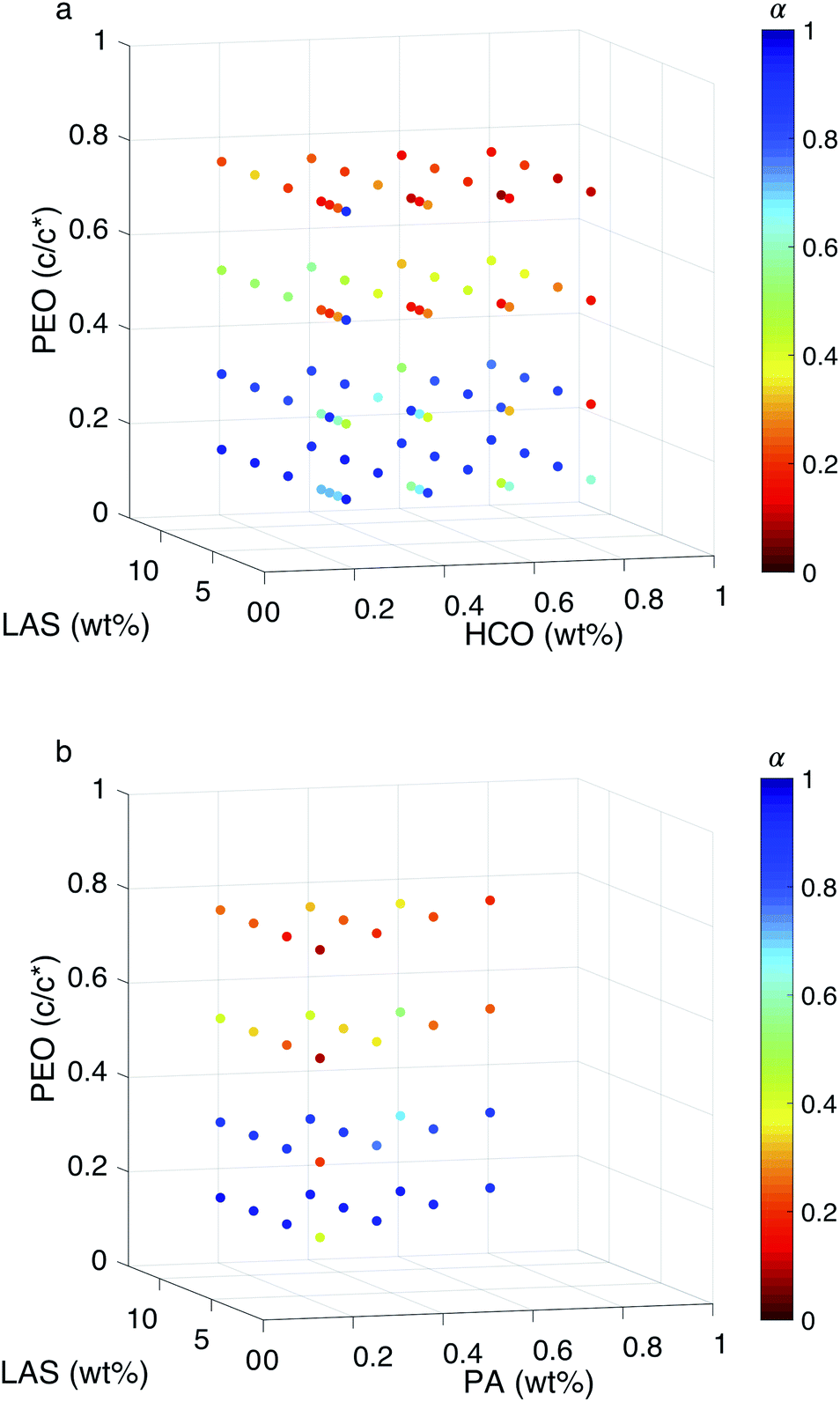 | ||
| Fig. 4 MPT phase diagrams for (a) HCO and (b) PA. The three axes are the concentration of each component and the color of the markers are the value of α. Warm colors are low α values where probe particle movement is restricted. Cool colors are high α values where probe particle movement is less restricted and, in some cases, freely diffusing. | ||
From Fig. 4a, the α value for HCO decreases with increasing PEO concentration at almost any combination of HCO and LAS. This indicates that probe movement is becoming restricted and the HCO system is gelling resulting in an arrested structure at all LAS:HCO ratio. The same trend is also measured in the PA system, Fig. 4b. This indicates that PEO induces gelation through depletion interaction at any LAS:colloid ratio.
When varying LAS concentration in the HCO system, α values only change significantly at moderate PEO concentrations, 0.3 and 0.52 c/c*, in Fig. 4a. Below 0.3 c/c* the HCO system is a sol and above 0.52 c/c* the material is a gel. At 0.3 c/c* and 0.52 c/c*, as LAS concentration decreases the material transitions from a sol to a gel. The same trend is measured in the PA system, Fig. 4b. It should be noted that in the PA colloidal rod system there is less data measured because of the high LAS concentration in the stock solution. This trend in both HCO and PA indicates that LAS can induce a phase change at moderate PEO concentrations. When there is a low concentration of PEO in the system, depletion interactions are too weak to drive gelation at any LAS concentration. At these low PEO concentrations, changing the LAS concentration in the system decreases the electrostatic forces between the colloids and drives colloidal rod bundling, but is not strong enough to induce a phase change. When there is a high amount of PEO in the system, depletion interactions gel the system regardless of LAS concentration. At moderate concentration of PEO, the effect of both depletion interactions and decreasing electrostatic forces, by changing LAS concentration, results in phase transitions of both systems.
According to our previous work, gelation evolution depends on the LAS:colloid ratio, and can be separated into two regimes: LAS:colloid > 16 and LAS:colloid ≤ 16.12 In these two regimes, the structure of the gel at the phase transition and critical phase transition point are different, as described above. In Fig. 5, a surface is constructed in both MPT phase diagrams to separate these two regimes. The compositions to the left of the surface is LAS:colloid > 16 and the compositions to the right of the surface is LAS:colloid ≤ 16. Data are presented using two colors to clearly indicate the state of each composition. Blue circles indicate that the system is in the sol state and red circles indicate that the system is in the gel state. The bi-color phase diagram for HCO, Fig. 5a, shows that when PEO concentration is low (0.14 c/c*), the HCO system is in the sol state for both LAS:HCO > 16 and LAS:HCO ≤ 16 regimes. Conversely, when PEO concentration is high (0.75 c/c*), the HCO system is in the gel state for both regimes. Comparing the state of the HCO system at moderate PEO concentrations (0.30 and 0.52 c/c*), the fraction of samples in the gel state for LAS:HCO > 16 is greater than samples with LAS:HCO ≤ 16. This indicates that PEO is more likely to induce gelation at moderate concentrations when LAS:HCO > 16. This is because at LAS:HCO > 16, the electrostatic forces between the colloidal rods are weak, and the starting material structure is bundles of colloidal rods. These larger bundles make it easier for PEO to drive gelation and create a sample-spanning network. At LAS:HCO ≤ 16 the electrostatic forces between the colloidal rods are strong and the rods remain stable single rods in solution, making the concentration of PEO required to drive gelation higher.
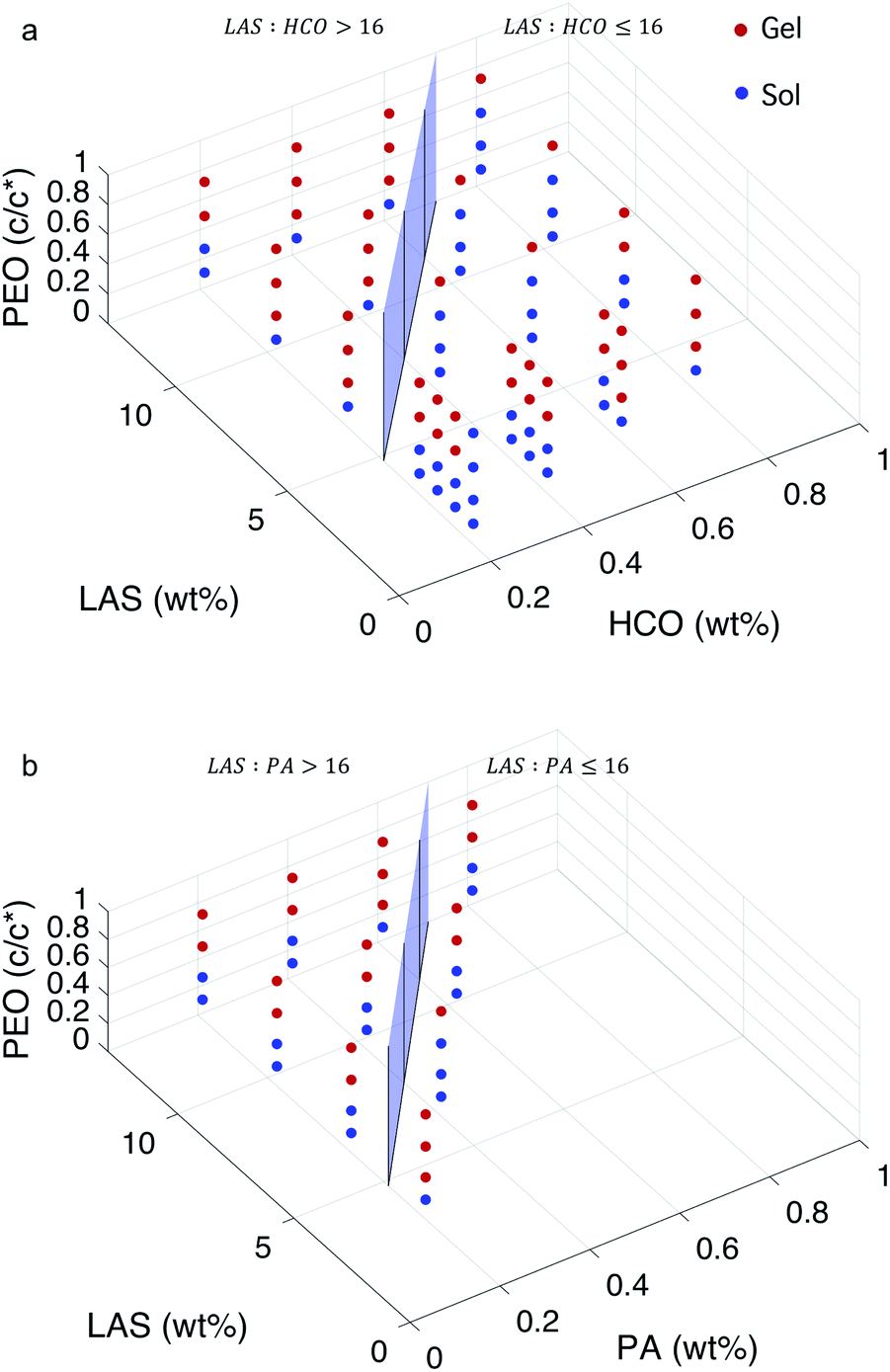 | ||
| Fig. 5 Bi-color MPT phase diagrams for (a) HCO and (b) PA. A surface is added to separate the phase diagrams into two regimes: LAS:colloid ≤ 16 and LAS:colloid > 16. The colors are the state of the material with red indicating the gel phase and blue indicating the sol phase. | ||
The bi-color phase diagram for PA, Fig. 5b, also shows that the PA system is in the sol state when PEO concentration is low (0.14 c/c*) and in the gel state when PEO concentration is high (0.75 c/c*) for both LAS:PA > 16 and LAS:PA ≤ 16 regimes. Due to the limited samples that can be made for PA at LAS:PA ≤ 16, we cannot compare the amount of samples at moderate PEO concentrations that are in each phase between LAS:PA > 16 and LAS:PA ≤ 16. Comparing the MPT phase diagrams between HCO and PA at LAS:colloid > 16, we measure that similar compositions are in the same state regardless of the type of colloid used. This indicates that HCO and PA have similar sol and gel phase distributions across the composition space measured.
To further investigate the ability of these colloidal rod systems to modify rheology, phase diagrams are constructed for both HCO and PA using bulk rheological measurements, Fig. 6a and b, respectively. 2D plots of some of the trends discussed here are provided in Fig. S15–S17 in the ESI.† Similar to the MPT phase diagrams (Fig. 4a and b), in Fig. 6 the three axes are the composition of the colloid (HCO or PA), PEO and LAS. Color indicates the value of the storage modulus, G′. Warm colors indicate higher storage moduli and cool colors indicate lower storage moduli. It should be noted that the maximum storage modulus measured is 25 Pa, which is a very low value and indicates that these materials are soft. From Fig. 6a, when HCO concentration is increased the storage modulus increases. The same trend is measured in the PA system. This indicates that increasing colloid concentration increases the elasticity in both systems. This is because when the amount of colloids in the system is increased the space between colloidal rods decreases. This increases colloid overlap, which would increase elasticity. Comparing the storage moduli between the HCO and PA systems at 0.6 and 0.8 wt%, HCO has higher storage moduli than PA. This means that at these colloid concentrations, HCO has more elasticity and can store more energy than PA. This is likely due to the polydispersity of HCO which includes longer colloids and allows more colloids to overlap compared with monodisperse PA. These measurements indicate that the elasticity of these colloidal systems can be tuned by changing the colloid concentration. Although this is a simple way to modify rheology this may not always be feasible in products since it requires adding more material to the formulation, therefore, we also measure the effect of LAS and PEO on the storage moduli.
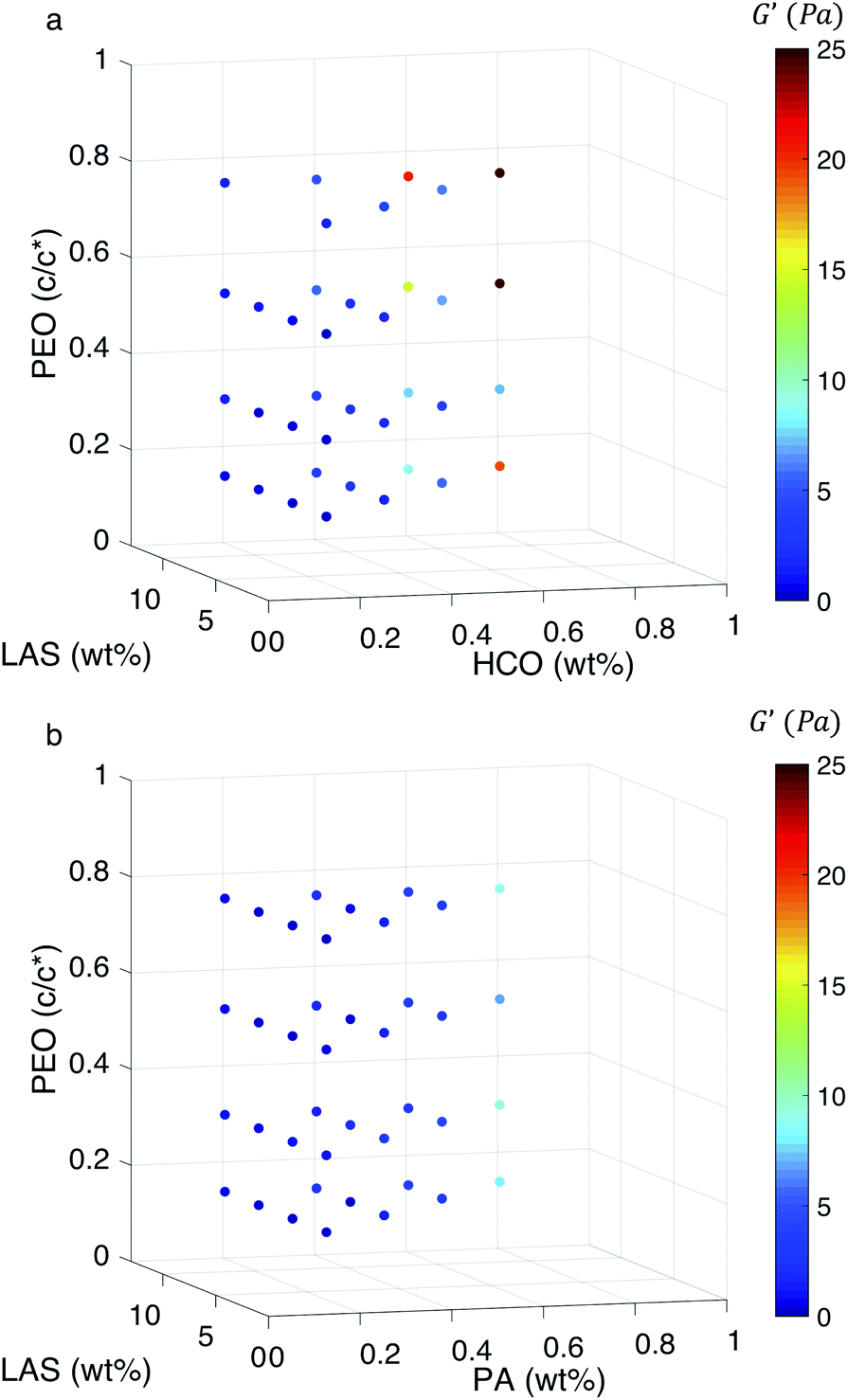 | ||
| Fig. 6 Bulk rheological measurements for varying concentrations of (a) HCO and (b) PA colloidal rod systems. The three axes are the concentration of each component and the color of the data indicates the value of the storage modulus, G′. Warm colors are the maximum measured storage moduli, which indicates higher elasticity of the material. Cool colors represent minimum storage moduli, which indicates low to no elasticity of the material. | ||
There is no measurable change in G′ when LAS and PEO concentration is varied using the procedure outlined in the Experimental section. From Fig. 6a, at low colloid concentration (0.2 and 0.4 wt%), the storage modulus is constant as PEO and LAS concentration is increased. We hypothesize that this is because the colloid concentration is too low to substantially overlap. At higher HCO concentration (0.6 and 0.8 wt%), we measure a small increase in storage modulus when PEO concentration is increased from 0.14–0.75 c/c*. From MPT measurements, we determine that increasing PEO concentration induces gelation and, therefore, we measure this as an increase in elasticity in bulk rheology measurements. For PA, the storage modulus is independent of PEO or LAS concentration, Fig. 6b. This is likely because PA is monodisperse, and the colloidal rods are shorter limiting their overlap. Additionally, both colloidal rod systems form delicate gel structures that may be broken by the shear applied during a bulk rheology measurement. This indicates that the elasticity does not change or the change is not measurable using the frequency sweep and the geometry outlined in the Experimental section. These moduli may be measurable with larger geometries, but due to the limited amount of PA synthesized, this is not possible for this work.
To directly compare the phase diagrams measured with MPT and bulk rheology, we normalize the storage modulus using 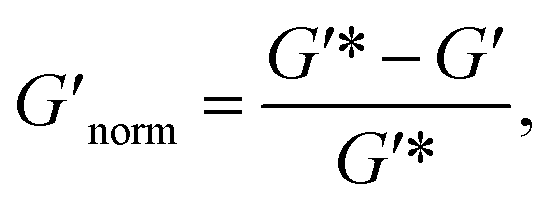 where G′* is the maximum storage modulus.
where G′* is the maximum storage modulus. 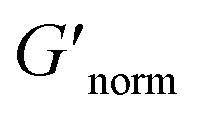 is used to compare bulk rheology measurements with MPT measurements on the same scale.
is used to compare bulk rheology measurements with MPT measurements on the same scale. 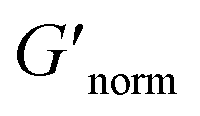 does not indicate the state of the material during gelation. Fig. 7a and b are the phase diagrams plotted with normalized storage modulus of HCO and PA, respectively. In these normalized phase diagrams the colors are similar to those in the MPT phase diagrams (Fig. 4a and b). Warm colors represent the maximum measured storage moduli and cool colors represent the minimum measured storage moduli. By comparing the normalized bulk rheology data with MPT data, different trends are measured for the same colloidal gel compositions with these two different techniques. As HCO concentration increases and LAS concentration is held constant MPT measures a constant α value and the system is in the sol state. When the same samples are measured with bulk rheology,
does not indicate the state of the material during gelation. Fig. 7a and b are the phase diagrams plotted with normalized storage modulus of HCO and PA, respectively. In these normalized phase diagrams the colors are similar to those in the MPT phase diagrams (Fig. 4a and b). Warm colors represent the maximum measured storage moduli and cool colors represent the minimum measured storage moduli. By comparing the normalized bulk rheology data with MPT data, different trends are measured for the same colloidal gel compositions with these two different techniques. As HCO concentration increases and LAS concentration is held constant MPT measures a constant α value and the system is in the sol state. When the same samples are measured with bulk rheology, 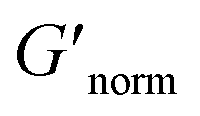 decreases indicating that elasticity is increasing. This means that MPT measures an unchanged microstructure and rheological properties as HCO concentration increases, but bulk rheology measures an increase in elasticity as HCO concentration increases. As PEO concentration increases and HCO and LAS concentration are held constant, MPT measures that α values decrease. Bulk rheology measures that
decreases indicating that elasticity is increasing. This means that MPT measures an unchanged microstructure and rheological properties as HCO concentration increases, but bulk rheology measures an increase in elasticity as HCO concentration increases. As PEO concentration increases and HCO and LAS concentration are held constant, MPT measures that α values decrease. Bulk rheology measures that 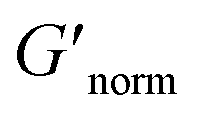 is nearly constant at 0.2 and 0.4 wt% HCO, and decreases slightly at 0.6 and 0.8 wt% HCO. Here, MPT measures that the system transitions to an arrested gel structure, but bulk rheology measures almost unchanged elasticity. At moderate PEO concentration (0.3 and 0.52 c/c*), as LAS concentration increases and PEO concentration is held constant, MPT measures that α increases but bulk rheology measures no change or a slight decrease in
is nearly constant at 0.2 and 0.4 wt% HCO, and decreases slightly at 0.6 and 0.8 wt% HCO. Here, MPT measures that the system transitions to an arrested gel structure, but bulk rheology measures almost unchanged elasticity. At moderate PEO concentration (0.3 and 0.52 c/c*), as LAS concentration increases and PEO concentration is held constant, MPT measures that α increases but bulk rheology measures no change or a slight decrease in 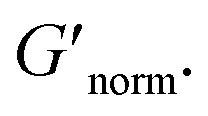 This indicates that MPT measures a less associated structure as LAS concentration increases, but bulk rheology measures unchanged elasticity. Similar differences in trends are also measured in PA.
This indicates that MPT measures a less associated structure as LAS concentration increases, but bulk rheology measures unchanged elasticity. Similar differences in trends are also measured in PA.
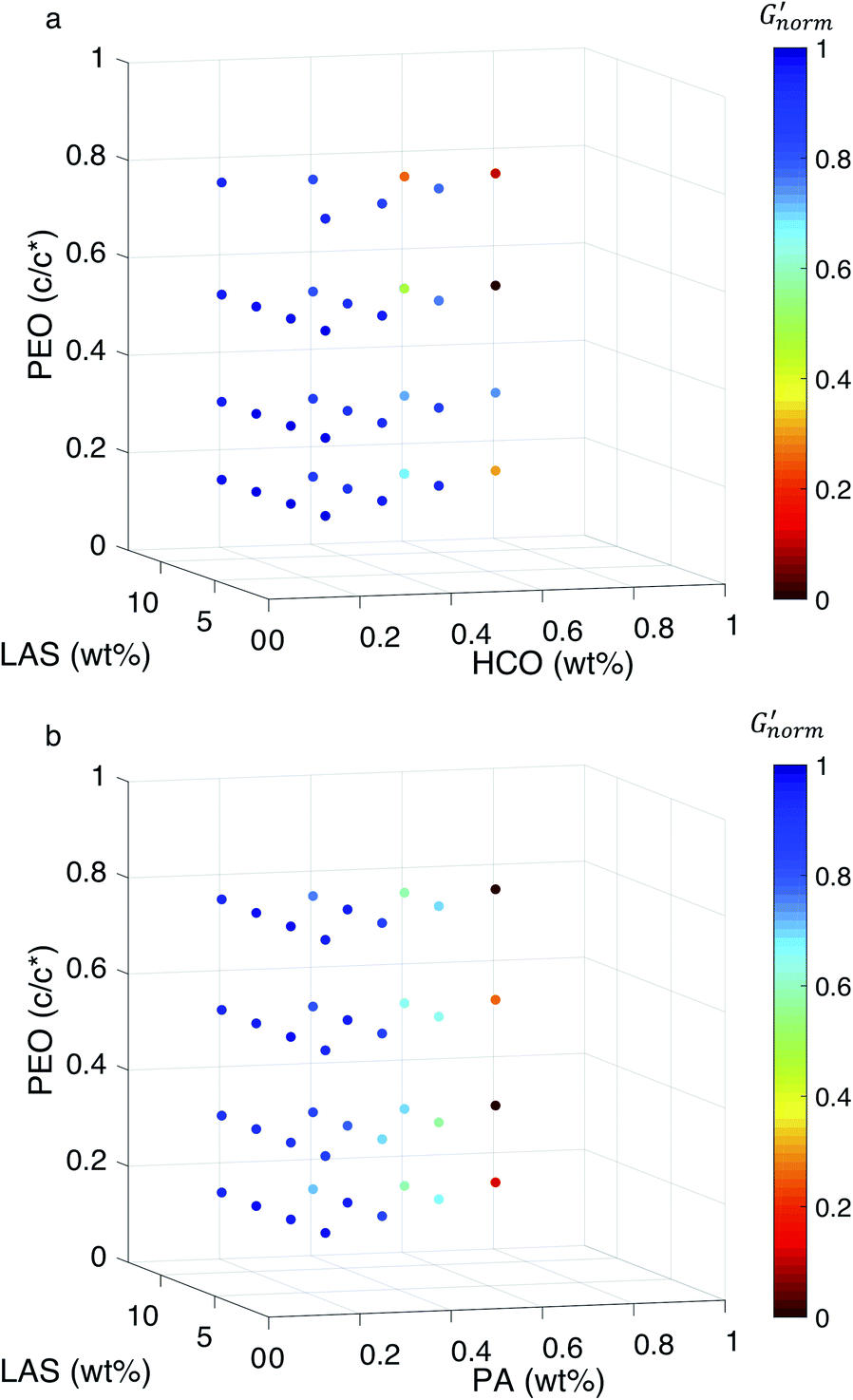 | ||
Fig. 7 Normalized bulk rheology phase diagrams for (a) HCO and (b) PA. The storage modulus is normalized using 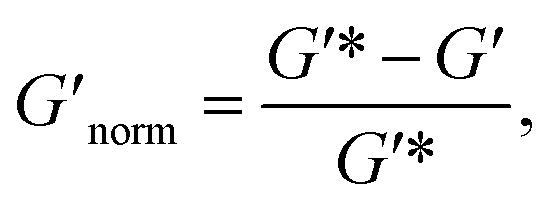 where G′* is the maximum storage modulus. Warm colors are higher where G′* is the maximum storage modulus. Warm colors are higher 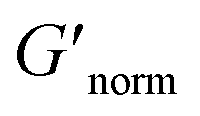 values and cool colors are lower values and cool colors are lower 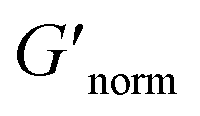 values. values. | ||
There are two reasons MPT and bulk rheology are measuring different trends in these colloidal gel systems. The applied force used to measure the rheological properties is different in MPT and bulk rheology. In MPT, the Brownian motion of the probe particles are used to characterize the rheological properties for the material. Brownian motion applies nearly no force on the system. In bulk rheology, shear is applied to the system and the response of the system to the applied force is measured. This could cause bulk rheological measurements to break delicate growing or established network structures that MPT can measure. This would result in measurements of unchanged elasticity once these structures have been broken by the bulk rheology measurements. The difference in the trends can also be the result of the difference in the range of moduli that MPT and bulk rheology can measure. MPT is sensitive to material with moduli G′ ≤ 4 Pa and bulk rheology measures material with higher moduli. This highlights the need to measure delicate colloidal gel materials with both techniques. This combination of complementary characterization techniques enable more complete characterization of the system and broadens the measurement range.
4 Conclusions
The phase behavior of two colloidal rod systems are characterized using multiple particle tracking microrheology and bulk rheology. In this work, the concentration of the colloid (HCO or PA), surfactant (LAS) and depletant (PEO) have been separately characterized to investigate the role of each component. For both HCO and PA, increasing colloid concentration from 0 to 0.8 wt% does not induce a phase change. As PEO concentration increases, both systems form sample-spanning networks and transition from a sol to a gel. With increasing LAS concentration, a change of the structure of both systems is measured. As LAS concentration increases, colloids aggregate and form bundles, resulting in a different material microstructure. Phase diagrams are constructed using MPT and bulk rheological measurements. From MPT phase diagrams, increasing PEO concentration induces phase transitions in both systems at any combination of LAS and colloid concentration. Changing LAS concentration only induces phase transitions at PEO concentrations near the critical depletant concentration. From bulk rheology phase diagrams, the elasticity of both systems increase as colloid concentration increases. This increase in elasticity is higher for HCO than PA. This is likely due to the polydispersity of HCO. HCO colloidal rods are a range of sizes, including longer colloidal rods, which help the colloids to overlap and increase G′. In addition, from the bulk rheology phase diagrams, no change in the storage modulus is measured when PEO or LAS concentration is increased for both systems using the frequency sweep and the geometry outlined in the Experimental section.Comparing phase diagrams from MPT and bulk rheology data, we measure differences when component concentrations are changed in both systems. As colloid concentration increases, MPT measures unchanged rheological properties, but bulk rheology measures an increase in elasticity. As PEO concentration increases, MPT measures a sol–gel transition at almost any colloid and LAS concentration. But the storage modulus remains constant in the bulk rheology phase diagram in the lower colloid concentration regime (0.2 and 0.4 wt%) and increases slightly in the higher colloid concentration regime (0.6 and 0.8 wt%). In addition, MPT measures sol–gel transitions as LAS concentration increases when PEO concentration is near the critical depletant concentration, but the storage modulus is constant for the same points. These different measured trends in MPT and bulk rheology are likely because the applied force is different during these measurements with bulk rheology potentially breaking delicate associated colloid structures. These techniques also measure different moduli ranges.
This study characterizes the role of three components in a colloidal rod suspension and constructs material rheology and phase diagrams over a large parameter space. These colloidal rod systems are used as rheological modifiers for consumer, fabric and home care products. Phase diagrams map the properties of the system at different component compositions, which can enable products to be designed with desired rheological properties and eliminate trial-and-error steps. In addition, supplementing microrheological measurements with bulk rheological measurements provides more information about these rheological modifiers and increases the measurement regime, which gives more complete information of the ability for these materials to modify the rheology of different products.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Funding for this work was provided by the National Science Foundation (NSF) under Grant No. GOALI-1933251. The authors would like to thank Dr Matthew Wehrman for the helpful discussions. The authors would like to thank Prof. Xiaohui Zhang and Dr Sajedehalsadat Yazdanparast Tafti for their help with AFM measurements.Notes and references
- M. H. Amaral, J. S. Lobo and D. Ferreira, AAPS PharmSciTech, 2001, 2, 14–21 CrossRef PubMed.
- A. van Blaaderen, Nature, 2006, 439, 545–546 CrossRef CAS PubMed.
- N. De Meirleir, L. Pellens, W. Broeckx and W. De Malsche, J. Cryst. Growth, 2013, 383, 51–56 CrossRef CAS.
- N. De Meirleir, L. Pellens, W. Broeckx, G. van Assche and W. De Malsche, Colloid Polym. Sci., 2014, 292, 2539–2547 CrossRef CAS.
- M. J. Solomon and P. T. Spicer, Soft Matter, 2010, 6, 1391–1400 RSC.
- M. D. Wehrman, S. Lindberg and K. M. Schultz, Soft Matter, 2016, 12, 6463–6472 RSC.
- M. D. Wehrman, S. Lindberg and K. M. Schultz, J. Rheol., 2018, 62, 437–446 CrossRef CAS.
- G. M. Wilkins, P. T. Spicer and M. J. Solomon, Langmuir, 2009, 25, 8951–8959 CrossRef CAS PubMed.
- H. N. Lekkerkerker and R. Tuinier, Colloids and the depletion interaction, Springer, 2011, pp. 57–108 Search PubMed.
- J. Mewis and N. J. Wagner, Colloidal suspension rheology, Cambridge University Press, 2012 Search PubMed.
- M. B. Gordon, C. J. Kloxin and N. J. Wagner, J. Rheol., 2017, 61, 23–34 CrossRef CAS.
- S. He, D. R. Pascucci, M. Caggioni, S. Lindberg and K. M. Schultz, AIChE J., 2021, 67, e17401 CAS.
- A. Yethiraj and A. van Blaaderen, Nature, 2003, 421, 513–517 CrossRef CAS PubMed.
- E. Zaccarelli, J. Phys.: Condens. Matter, 2007, 19, 323101 CrossRef.
- M. D. Wehrman, M. J. Milstrey, S. Lindberg and K. M. Schultz, Lab Chip, 2017, 17, 2085–2094 RSC.
- D. Yang, A. N. Hrymak and M. R. Kamal, Ind. Eng. Chem. Res., 2011, 50, 11594–11600 CrossRef CAS.
- T. G. Mason and D. A. Weitz, Phys. Rev. Lett., 1995, 74, 1250 CrossRef CAS PubMed.
- F. Gittes, B. Schnurr, P. Olmsted, F. C. MacKintosh and C. F. Schmidt, Phys. Rev. Lett., 1997, 79, 3286 CrossRef CAS.
- T. M. Squires and T. G. Mason, Annu. Rev. Fluid. Mech., 2010, 42, 413–438 CrossRef.
- J. C. Crocker and D. G. Grier, J. Colloid Interface Sci., 1996, 179, 298–310 CrossRef CAS.
- T. Mason, K. Ganesan, J. Van Zanten, D. Wirtz and S. C. Kuo, Phys. Rev. Lett., 1997, 79, 3282 CrossRef CAS.
- E. M. Furst and T. M. Squires, Microrheology, Oxford University Press, 2017 Search PubMed.
- J. A. McGlynn, N. Wu and K. M. Schultz, J. Appl. Phys., 2020, 127, 201101 CrossRef CAS.
- J. C. Crocker and E. R. Weeks, 2011, http://www.physics.emory.edu/Bweeks/idl/index.html.
- K. M. Schultz, A. D. Baldwin, K. L. Kiick and E. M. Furst, Soft Matter, 2009, 5, 740–742 RSC.
- K. M. Schultz, A. D. Baldwin, K. L. Kiick and E. M. Furst, Macromolecules, 2009, 42, 5310–5316 CrossRef CAS PubMed.
- M. Muthukumar and H. H. Winter, Macromolecules, 1986, 19, 1284–1285 CrossRef CAS.
- H. H. Winter and F. Chambon, J. Rheol., 1986, 30, 367–382 CrossRef CAS.
- A. Corrigan and A. Donald, Eur. Phys. J. E: Soft Matter Biol. Phys., 2009, 28, 457–462 CrossRef CAS PubMed.
- P. Martens, A. T. Metters, K. S. Anseth and C. N. Bowman, J. Phys. Chem. B, 2001, 105, 5131–5138 CrossRef CAS.
- F. Chambon and H. H. Winter, J. Rheol., 1987, 31, 683–697 CrossRef CAS.
- T. G. Mason, Rheol. Acta, 2000, 39, 371–378 CrossRef CAS.
- F. Escobar IV, K. S. Anseth and K. M. Schultz, Macromolecules, 2017, 50, 7351–7360 CrossRef.
- M. T. Valentine, P. D. Kaplan, D. Thota, J. C. Crocker, T. Gisler, R. K. Prud’homme, M. Beck and D. A. Weitz, Phys. Rev. E, 2001, 64, 061506 CrossRef CAS PubMed.
- L. Selvaggi, M. Salemme, C. Vaccaro, G. Pesce, G. Rusciano, A. Sasso, C. Campanella and R. Carotenuto, Methods, 2010, 51, 20–26 CrossRef CAS PubMed.
- A. V. Bayles, T. M. Squires and M. E. Helgeson, Rheol. Acta, 2017, 56, 863–869 CrossRef CAS.
- J. C. Crocker and B. D. Hoffman, Methods Cell Biol., 2007, 83, 141–178 CAS.
- M. Schenker, J. Schoelkopf, P. Gane and P. Mangin, Cellulose, 2018, 25, 961–976 CrossRef CAS.
- M. Caggioni, P. Spicer, D. Blair, S. Lindberg and D. Weitz, J. Rheol., 2007, 51, 851–865 CrossRef CAS.
- D. Yang and A. N. Hrymak, Ind. Eng. Chem. Res., 2011, 50, 11585–11593 CrossRef CAS.
- D. Yang and A. N. Hrymak, Appl. Rheol., 2013, 23, 1–9 Search PubMed.
- D. S. Ogunniyi, Bioresour. Technol., 2006, 97, 1086–1091 CrossRef CAS PubMed.
- W. Brown, Dynamic light scattering: the method and some applications, Clarendon Press Oxford, 1993, vol. 313 Search PubMed.
- M. Doi, S. F. Edwards and S. F. Edwards, The theory of polymer dynamics, Oxford University Press, 1988, vol. 73 Search PubMed.
- T. A. Waigh, Rep. Prog. Phys., 2005, 68, 685 CrossRef.
- M. Valentine, Z. Perlman, M. Gardel, J. H. Shin, P. Matsudaira, T. Mitchison and D. Weitz, Biophys. J., 2004, 86, 4004–4014 CrossRef CAS PubMed.
- T. Savin and P. S. Doyle, Biophys. J., 2005, 88, 623–638 CrossRef CAS PubMed.
- K. M. Schultz and K. S. Anseth, Soft Matter, 2013, 9, 1570–1579 RSC.
- T. H. Larsen and E. M. Furst, Phys. Rev. Lett., 2008, 100, 146001 CrossRef PubMed.
- A. M. Corrigan and A. M. Donald, Langmuir, 2009, 25, 8599–8605 CrossRef CAS PubMed.
- K. M. Schultz, A. D. Baldwin, K. L. Kiick and E. M. Furst, ACS Macro Lett., 2012, 1, 706–708 CrossRef CAS PubMed.
- D. Adolf and J. E. Martin, Macromolecules, 1990, 23, 3700–3704 CrossRef CAS.
- H. H. Winter, Polym. Eng. Sci., 1987, 27, 1698–1702 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra00609j |
| This journal is © The Royal Society of Chemistry 2022 |