
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis, in silico docking, ADMET and anticancer evaluations of thiazolidine-2,4-diones bearing heterocyclic rings as dual VEGFR-2/EGFRT790M tyrosine kinase inhibitors†
Nada A. A. M. Aziz*a,
Riham F. George b,
Khaled El-Adl*ac and
Walaa R. Mahmoud*b
b,
Khaled El-Adl*ac and
Walaa R. Mahmoud*b
aPharmaceutical Chemistry Department, Faculty of Pharmacy, Heliopolis University for Sustainable Development, Cairo, Egypt. E-mail: eladlkhaled74@azhar.edu.eg; khaled.eladl@hu.edu.eg; eladlkhaled74@yahoo.com; nada.aziz@hu.edu.eg
bPharmaceutical Chemistry Department, Faculty of Pharmacy, Cairo University, Cairo 11562, Egypt. E-mail: walaa.abozaid@pharma.cu.edu.eg
cPharmaceutical Chemistry Department, Faculty of Pharmacy, Al-Azhar University, Cairo, Egypt
First published on 27th April 2022
Abstract
Fourteen recent thiazolidine-2,4-diones bearing furan and/or thiophene heterocyclic rings have been designed, synthesized and assessed for their anticancer activities against four human tumor cell lines HepG2, A549, MCF-7 and HCT-116 targeting both VEGFR-2 and EGFR tyrosine kinases. Molecular design was carried out to investigate the binding mode of the proposed compounds with VEGFR-2 and EGFR receptors. HepG2 was the most susceptible cell line to the influence of our derivatives. Compounds 5g and 4g revealed the highest activities against HepG2 (IC50 = 3.86 and 6.22 μM), A549 (IC50 = 7.55 and 12.92 μM), MCF-7 (IC50 = 10.65 and 10.66 μM) and HCT116 (IC50 = 9.04 and 11.17 μM) tumor cell lines. Sorafenib (IC50 = 4.00, 4.04, 5.58 and 5.05 μM) and elotinib (IC50 = 7.73, 5.49, 8.20 and 13.91 μM) were used as reference standards. Furthermore, the most active cytotoxic compounds 4d, 4e, 4f, 4g, 5d, 5e, 5f and 5g were selected to assess their VEGFR-2 inhibitory effects. Derivatives 5g, 4g and 4f were observed to be the highest effective derivatives that inhibited VEGFR-2 at the submicromolar level (IC50 = 0.080, 0.083 and 0.095 μM respectively) in comparison to sorafenib (IC50 = 0.084 μM). As well, compounds 4d, 4e, 4f, 4g, 5d, 5e, 5f and 5g were additionally assessed for their inhibitory activities against mutant EGFRT790M. Compounds 5g and 4g could interfere with the EGFRT790M activity exhibiting stronger activities than elotinib with IC50 = 0.14 and 0.23 μM respectively. Finally, our derivatives 4g, 5f and 5g showed a good in silico calculated ADMET profile. The obtained results showed that our compounds could be useful as a template for future design, optimization, adaptation and investigation to produce more potent and selective dual VEGFR-2/EGFRT790M inhibitors with higher anticancer activity.
1. Introduction
The thiazolidine-2,4-diones (TZDs) as heterocyclic compounds have been disclosed to be a potential scaffold that play an important role in cancer therapy. TZDs were proved to exhibit anticancer activity in a wide variety of experimental cancer models by affecting the cell cycle, induction of cell differentiation and apoptosis as well as by inhibiting tumor angiogenesis.1–3 Tumor angiogenesis is a fundamental marker for the growth and metastasis of malignant tumors.4Lung cancer is the chief reason of cancer mortality, with nearly 85% of lung cancers being NSCLC. Since 2004, EGFR-sensitizing mutations have been established as driver oncogenes for NSCLC that predict response to EGFR tyrosine kinase inhibitors (TKIs).5 EGFR mutations occur in 7% to 37% of NSCLC cases in white patients and 40% to 64% in Asian patients.6 Regardless of ethnicity, these mutations more often present in women, nonsmokers, and those who have adenocarcinoma histologic diagnosis.6 In approximately 90% of cases, EGFR-mutant NSCLC tumors harbor an exon 19 deletion or exon 21 L858R mutations, both of which render tumors sensitive to EGFR TKIs patients.7 EGFRTKIs have been established as first-line therapy, in the metastatic setting, because of its progression-free survival (PFS) benefit and excellent tolerability.5 Resistance to EGFR TKI therapy inevitably arises, even though initially effective and patients' progress. The 5 year survival rate for patients with EGFR-mutant metastatic lung cancer is around 15%.8 Thus, the foundation of novel therapeutic strategies is necessary to improve patient outcomes. One aspiring complementary target of EGFR suppression in NSCLC is the VEGF passageway. VEGF signaling plays an important role in neoangiogenesis, and its blocking is a key curative strategy in cancer treatment.5 During oncogenesis and acquired therapeutic resistance VEGF and EGF may function exclusively of one another where they share common downstream signaling pathways. It has been suggested that EGFR-mutant tumors are more VEGF-dependent than EGFR wild-type tumors.9 Therefore, mutual VEGF and EGFR inhibition represent a rational combination strategy for EGFR-mutant NSCLC treatment.
VEGFR-2 is the fundamental regulator of VEGF-driven responses in endothelial cells and can control proliferation, differentiation, and microvascular permeability. Over and above, it has shown to be a prerequisite signal transducer in both physiologic and pathologic angiogenesis.10 VEGFR-2 is overexpressed in several malignancies, including hepatocellular carcinoma, breast, colorectal, ovarian and thyroid cancer, melanoma and medulloblastoma.11 Therefore, VEGFR-2 has been identified as an excellent therapeutic target for the production of novel anticancer agents.12 Numerous TZD derivatives, were reported to be potent anticancer agents and inhibitors of angiogenesis targeting VEGFR-2 via decreasing the VEGF production in an in vitro model e.g. ciglitazone I13 compound (II).9 Due to the important role of VEGFR-2 in angiogenesis, this receptor is the most vital target in anti-angiogenic therapy against cancer. A number of effective VEGFR-2 inhibitors have been established and permitted as antiangiogenic drugs for treatment of numerous cancers, e.g. sunitinib (III)14 and sorafenib (Nexavar)® (IV)15 (Fig. 1).
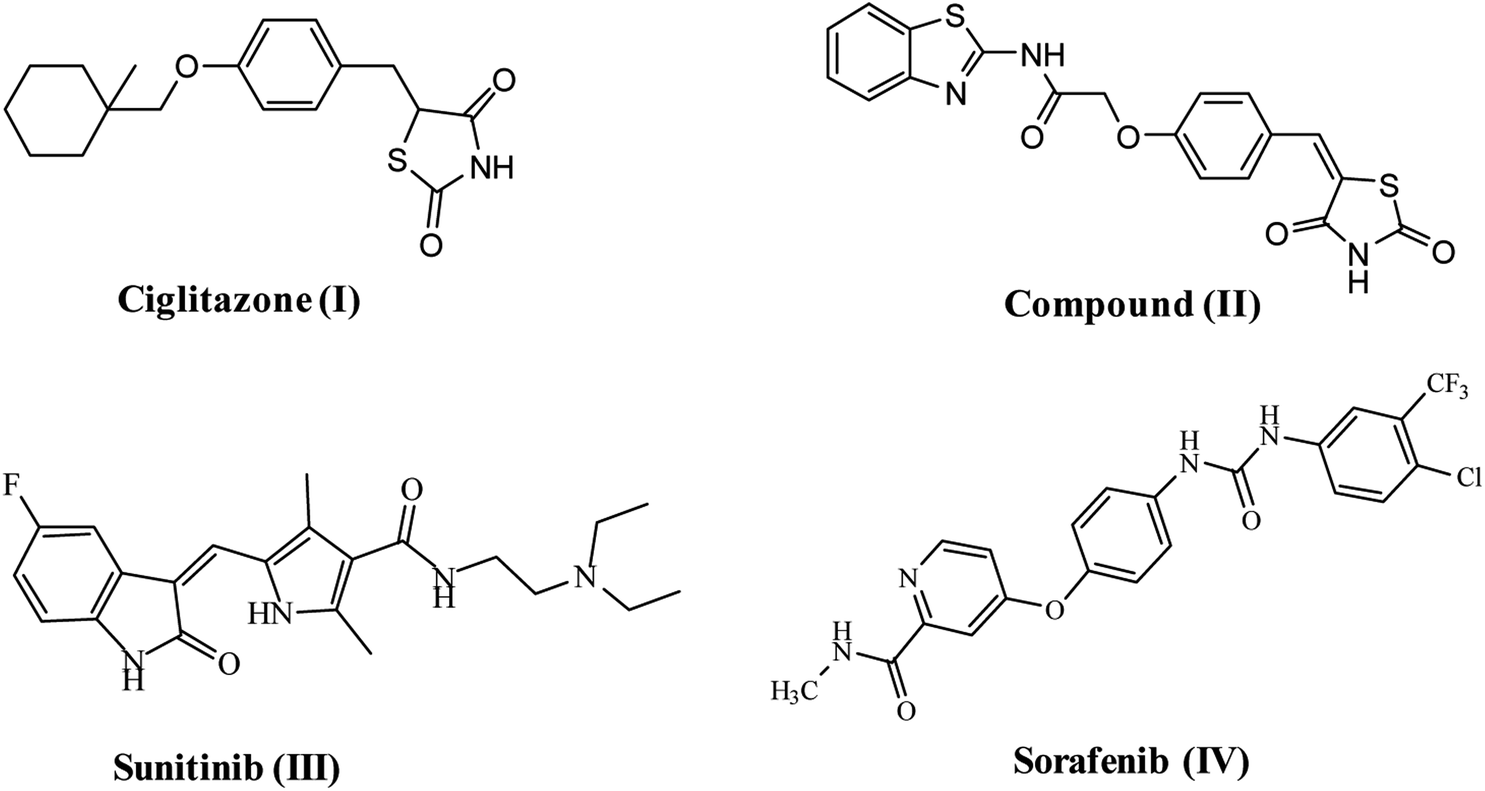 | ||
| Fig. 1 Reported VEGFR-2 inhibitors. | ||
Moreover, numerous TZD derivatives were reported to be potent EGFR inhibitors.16 Overexpression of EGFR-tyrosine kinaseis common in many human solid tumors17 as breast cancer18 and hepatocellular carcinoma (HCC).19 Therefore, EGFR-TK is considered as a rational target for the design of new anticancer agents.20
VEGFR-2 and EGFR are involved in the progression of different kinds of tumors and pathological disorders. They are closely related and sharing partial common downstream signaling pathways. The functional relationship between VEGFR-2 and EGFR has been well-recognized: antitumor effect of EGFR inhibitors was increased by VEGFR-2 signaling pathway inhibition, while activation of VEGFR-2 independent of EGFR signaling may give rise to the resistance of EGFR inhibitors.21 Thus, the simultaneous blockade of both EGFR and VEGFR signaling pathways appears to be an attractive methodology to cancer therapy.22
VEGFR-2 inhibitors can be categorized into three types. Type I inhibitors that occupy the ATP binding region forming a hydrogen bond with the hinge region amino acid Cys919. Type II inhibitors are occupying the ATP binding site and extend over the gate area into the adjacent allosteric hydrophobic back pocket. Type III inhibitors that block the receptor through hydrophobic interactions and they accommodate the allosteric hydrophobic back pocket of VEGFR-2. The affinity and selectivity of the type II inhibitors are preferred over type I inhibitors. Moreover, type II inhibitors increase their drug-target residence time so prolong TK suppression.23 Therefore, diverse strategies have been employed to develop novel type II VEGFR-2 inhibitors (Fig. 2A).
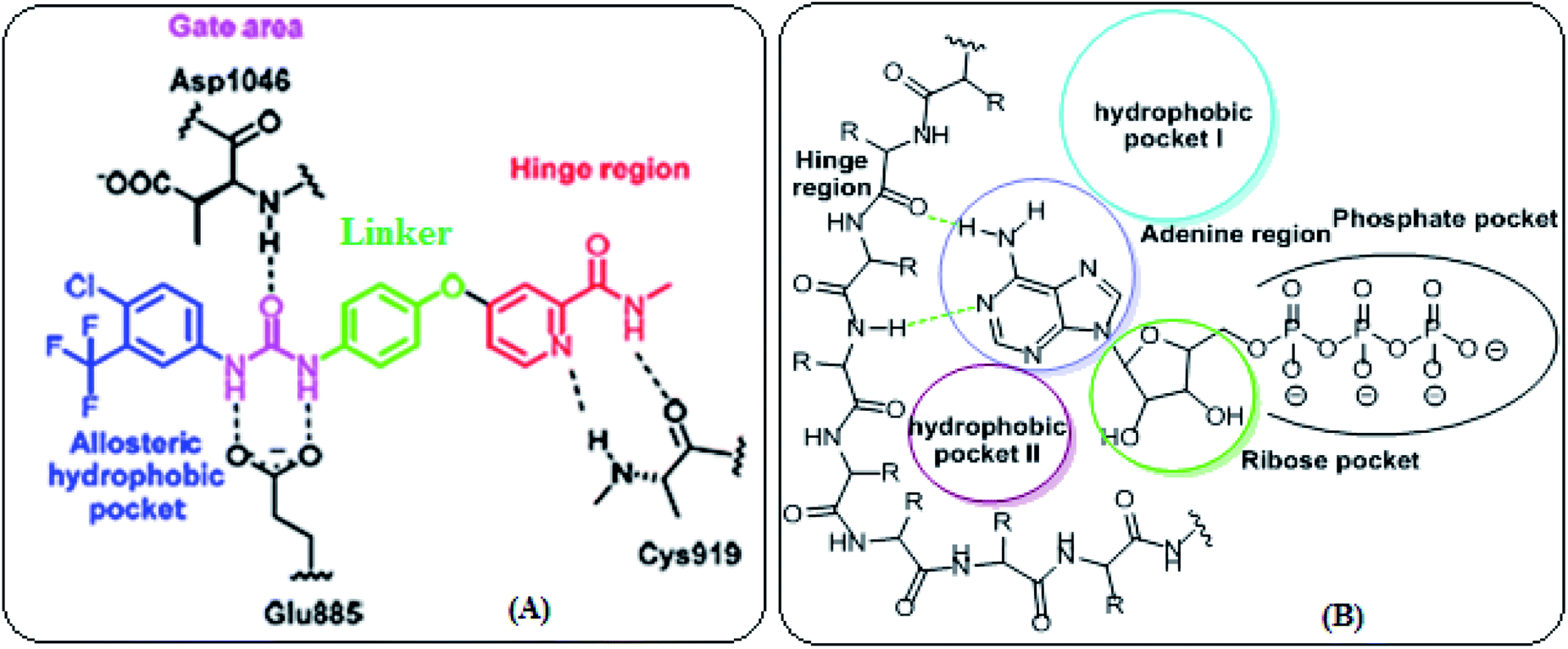 | ||
| Fig. 2 (A) VEGFR-2 active site (B) EGFR-TK active site. | ||
Moreover, the ATP binding pocket of EGFR-TK consists of five main regions; (a) adenine binding pocket contains key amino acids which can form hydrogen bonds with the adenine ring, (b) sugar region (hydrophilic ribose pocket), (c) hydrophobic region I, plays an important role in inhibitor selectivity, (d) hydrophobic region II, may be exploited for inhibitor specificity, (e) phosphate binding region that can be used for improving inhibitor pharmacokinetics (Fig. 2B).24
In continuation of our preceding efforts toplot and synthesis new anticancer agents25–32 we designed and synthesized novel thiazolidine-2,4-diones bearing heterocyclic rings as dual VEGFR-2/EGFR-mutant tyrosine kinase inhibitors for treatment of NSCLC lung (A549), hepatocellular carcinoma (HCC) type (HepG2), breast cancer (michigan cancer foundation-7 (MCF-7)) and human colorectal carcinoma-116 (HCT-116).
1.1. Rationale and structure-based design
Our idea in the rational of our derivatives comes from that our derivatives showed the required pharmacophoric features of both EGFR and VEGFR-2 inhibitors (Fig. 3). Sorafenib and multiple VEGFR-2 inhibitors had four main features:33,34 (i) a flat hetero aromatic ring which occupied the adenosine triphosphate (ATP)-binding domain.34 (ii) A central hydrophobic spacer.35 (iii) A linker containing a functional group (e.g. amino or urea) that possesses both H-bond acceptor (HBA) and donor (HBD) in order to bind along with two crucial residues (Glu885 and Asp1046). The NH motifs of the urea or amide moiety commonly make one hydrogen bond with Glu885, whereas the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O motif forms a further hydrogen bond with Asp1046.36 (iv) The terminal hydrophobic moiety that occupies the allosteric hydrophobic pocket37 (Fig. 3A).
O motif forms a further hydrogen bond with Asp1046.36 (iv) The terminal hydrophobic moiety that occupies the allosteric hydrophobic pocket37 (Fig. 3A).
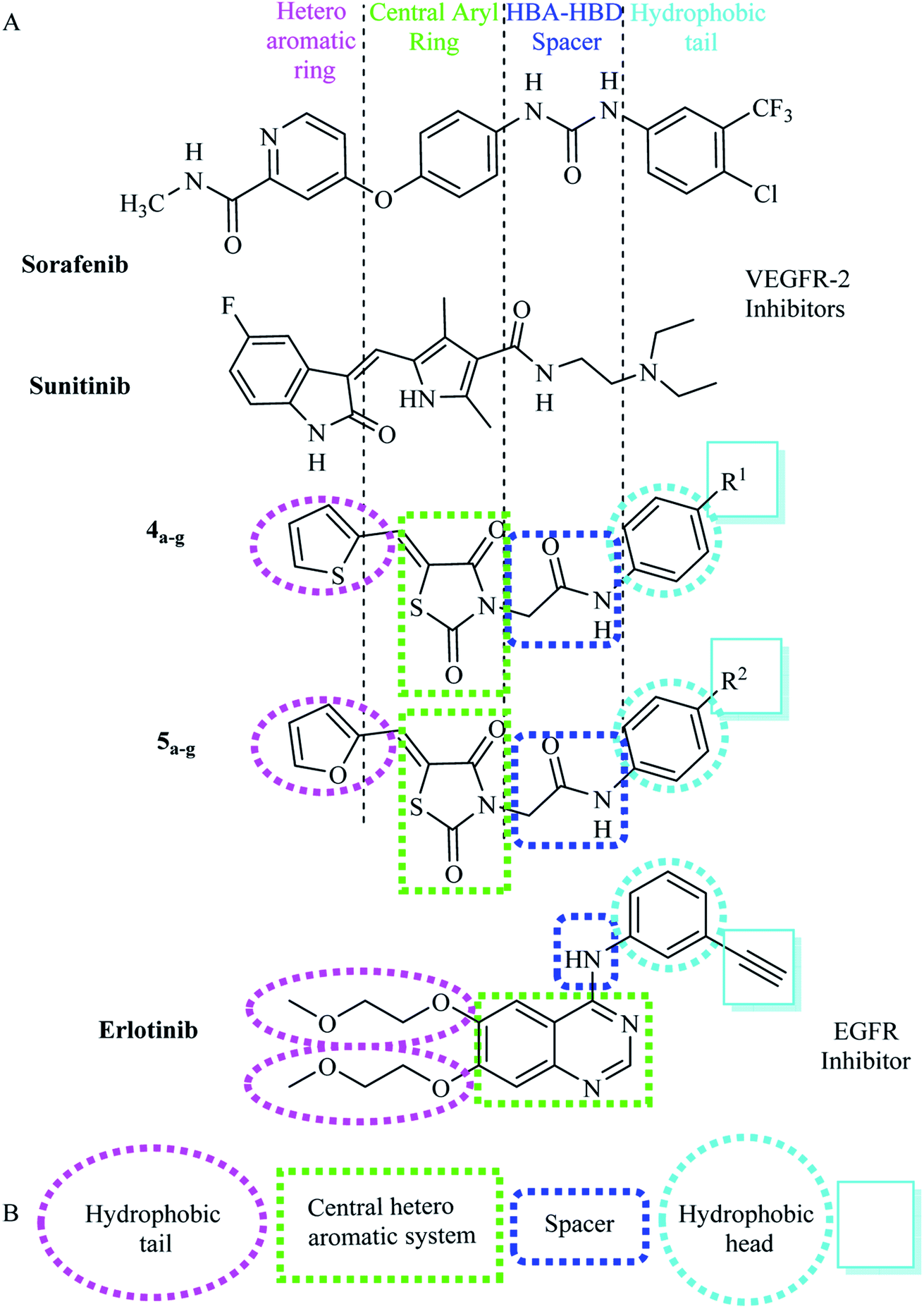 | ||
| Fig. 3 (A) Basic structural features of EGFR inhibitors. (B) Basic structural features of VEGFR-2 inhibitors. | ||
The main core of our molecular design rationale was carried out by bioisosteric modification strategies of VEGFR-2 inhibitors (sorafenib & sunitinib) at four different positions (Fig. 3A).
In addition, the common pharmacophoric features of EGFR-TKIs are: (i) terminal hydrophobic head to be inserted in the hydrophobic region I. (ii) A flat hetero aromatic system, occupying the adenine binding pocket. This hetero aromatic system can participate in hydrogen bonding interactions with Met793, Thr854,and Thr790 residues38 (iii) NH spacer which can form important hydrogen bond interaction to amino acid residues in the linker region. (iv) Hydrophobic tail which occupies the hydrophobic region II.39
In this work, a series of thiazolidine-2,4-diones derivations having the fundamental pharmacophoric characters of EGFR-TKIs have been framed and synthesized. Such compounds comprised structural modification of elotinib at four different positions (Fig. 3B).
As VEGFR-2 inhibitors the first bioisosteric modification was adopted in the target furan and/or thiophene rings to replace the pyridine and 5-fluoro-2-oxoindolin-3-ylidene moieties of the reference ligands sorafenib and sunitinib, respectively. The second strategy is to use thiazolidine-2,4-dione to replace the central aryl and the five membered pyrrole rings of the reference ligands sorafenib and sunitinib, respectively aiming to increase VEGFR-2 binding affinity. The third strategy is using acetamide linkers containing HBA-HBD functional groups that possess H-bond acceptors and/or donors. In addition, the forth strategy where the hydrophobic diethylamino and 4-chloro-3-(trifluoromethyl)phenyl tail of sorafenib and sunitinib correspondingly, was replaced by phenyl one 4-substituted with different electronic and lipophilic environments (Fig. 4A).
 | ||
| Fig. 4 (A) Superimposition of 5g and sorafenib. (B) Superimposition of 5g and elotinib. | ||
As EGFR inhibitors the first position was the quinazoline moiety (hetero aromatic system), which was replaced by thiazolidine-2,4-dione ring as a biological isostere to occupy the adenine binding region. The second position was the terminal phenyl ring (hydrophobic head), where phenyl group with various substituents was used. The third position was the NH linker, where acetamide linker was utilized. The fourth position was the two 2-methoxyethoxy groups (hydrophobic tail), where thiophen-2-yl/furan-2-ylmethylene were incorporated at position-5 of thiazolidin-2,4-dione nucleus to be fitted in the hydrophobic region II of ATP binding site (Fig. 4B).
All modifications encouraged us to study the structure–activity relationship of the hypothesized compounds as anti-cancer agents. The most active structures were examined for their anti-proliferative activities against a number of human cancer cell lines. Promising compounds were examined for their activities against VEGFR-2 and EGFRT790M. Furthermore, a molecular docking was performed to rationalize and emphasize the mechanism of action of the produced compounds as VEGFR-2 and EGFR-TKIs.
2. Results and discussion
2.1. Chemistry
The synthetic strategy for preparation of the target compounds 4a–g and 5a–g is depicted in Scheme 1. Synthesis was initiated by cyclocondensation of thiourea with chloroacetic acid to afford thiazolidine-2,4-dione (1)27 which underwent further condensation reaction with thiophene-2-carbaldehyde and/or furan-2-carbaldehyde via Knoevenagel condensation27 to afford 5-(thiophen-2-ylmethylene)thiazolidine-2,4-dione 2 and 5-(furan-2-ylmethylene)thiazolidine-2,4-dione 3 respectively. Subsequent heating of 2 and 3 under reflux with the appropriate chloroacetamide derivative in acetone in the presence of potassium carbonate afforded the corresponding amide derivatives 4a–g and 5a–g correspondingly. | ||
| Scheme 1 Synthetic route for preparation of the target compounds 4a–g and 5a–g. | ||
It is advantageous to state that there is no risk for a nitrosamine formation in our derivatives. The nitrosamine formation on the amino NH group depends on the presence of nitrosating agent, basicity of NH group and the steric accessibility to the nitrogen atom.40 As our derivatives containing electron withdrawing group attached to NH group (forming amide linkers) and also NH attached to electron with drawing benzene ring directly so the electrons on the NH groups is not available to be donated to attack or form nitrosamine. Moreover, there is highly steric hindrance to reach to the NH atom as it lies between two bulky thiazolidine-2,4-dione and phenyl groups.
2.2. Docking studies
Molsoft software was applied for molecular docking studies. All experiments utilized VEGFR-2 and EGFRT790M (PDB ID 4ASD)40 and (PDB ID 3W2O)39 respectively.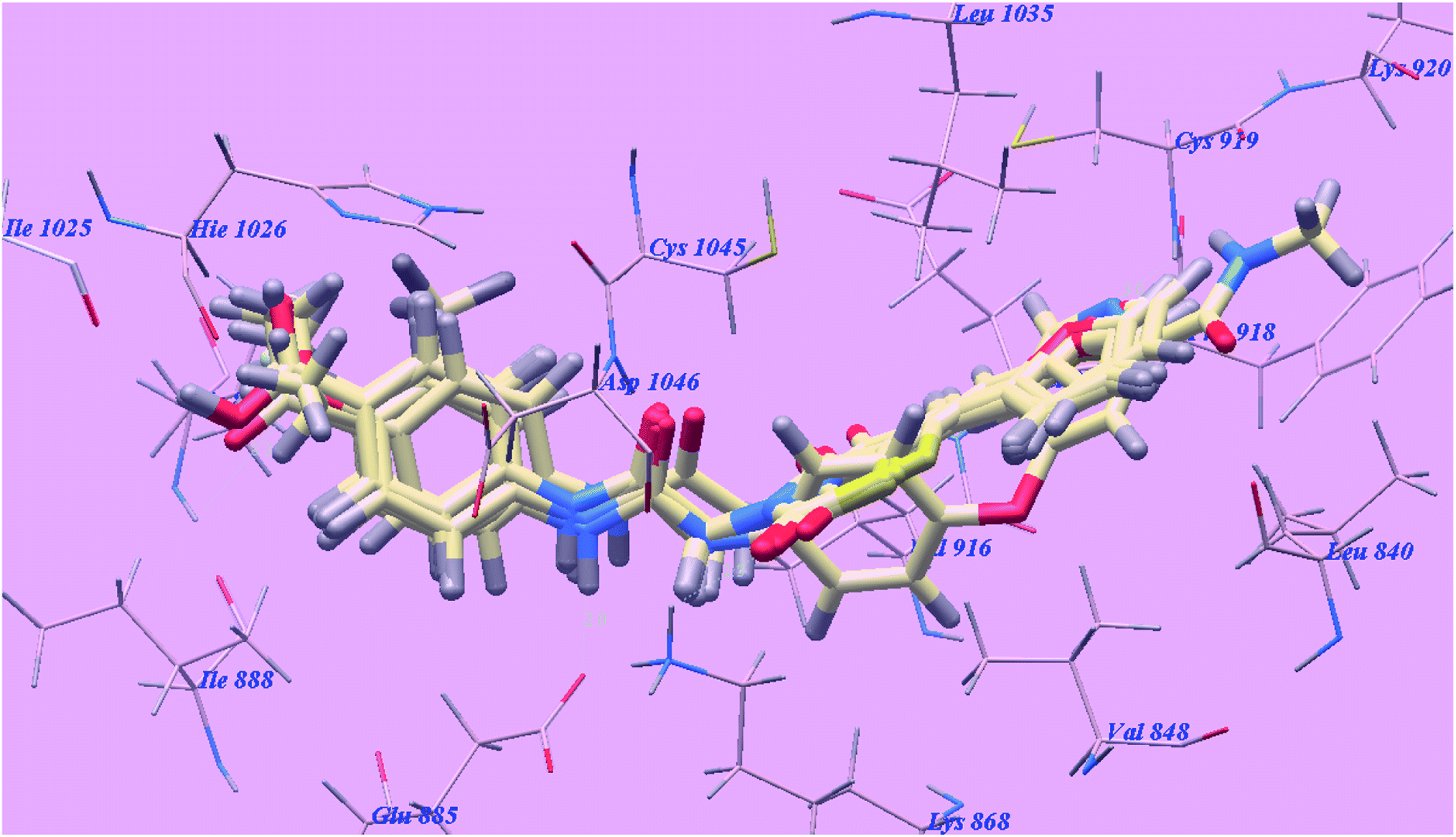 | ||
| Fig. 5 Superimposition of some docked compounds inside the binding pocket of 4ASD. | ||
| Compound | ΔG [kcal mol−1] | Compound | ΔG [kcal mol−1] |
|---|---|---|---|
| 4a | −84.77 | 5b | −88.80 |
| 4b | −87.36 | 5c | −89.65 |
| 4c | −88.18 | 5d | −91.06 |
| 4d | −91.36 | 5e | −90.52 |
| 4e | −89.77 | 5f | −99.85 |
| 4f | −91.24 | 5g | −102.80 |
| 4g | −100.12 | Sorafenib | −99.50 |
| 5a | −85.04 |
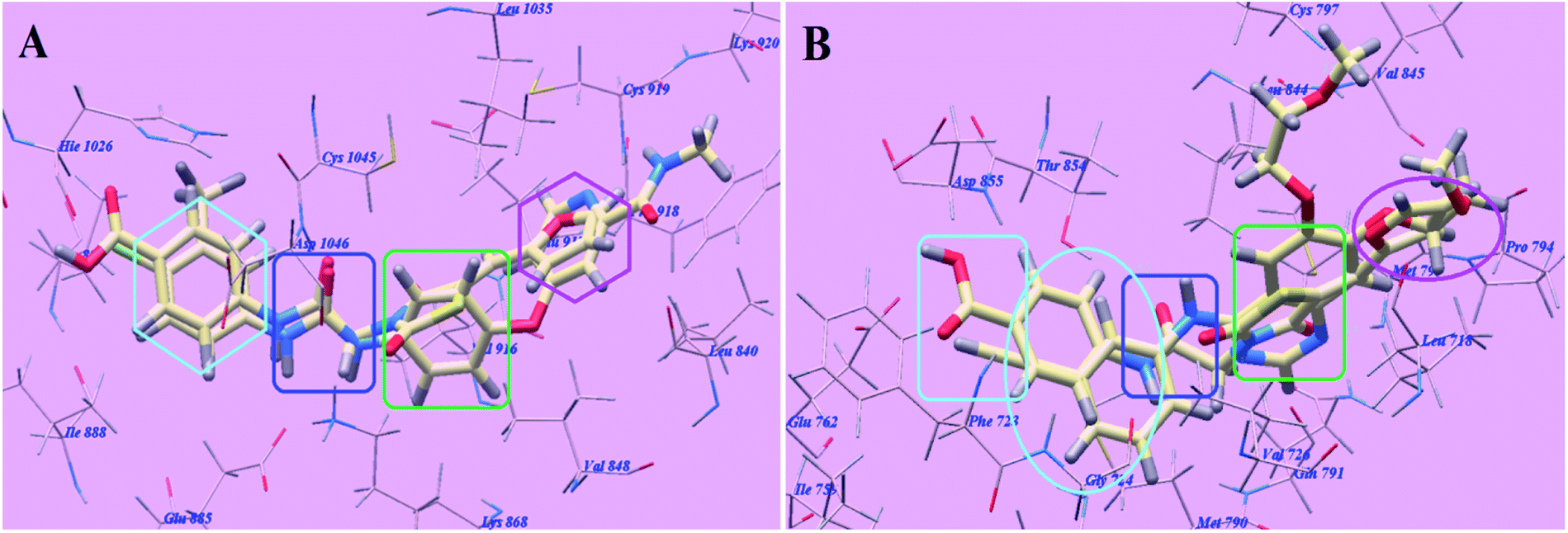
Sorafenib suggested binding mode showed affinity value −99.50 kcal mol−1 and formed 5 H-bonds. It formed 2 H-bonding interactions with Cysteine919 (2.51 Å and 2.10 Å), 2 H-bonds with Glutamate885 (1.77 Å and 2.75 Å) and one H-bonding interaction with Aspartate1046 (1.50 Å). The N-methylpicolinamide group occupied the pocket made via Leucine1035, Lysine920, Cysteine919, Phenylalanine918, Glutamate917, Valine848 and Leucine840. Furthermore, the central phenyl linker occupied the hydrophobic groove made by Cysteine1045, Leucine1035, Threonine916, Lysine868 and Valine848. Additionally, the terminal 3-trifluromethyl-4-chlorophenyl group occupied the hydrophobic channel formed by Aspartate1046, Cysteine1045, Histidine1026, Isoleucine892, Isoleucine888 and Glutamate885 (Fig. 6). The urea linker had a significant function in the binding with VEGFR-2 enzyme; however, the linker had important role responsible for high binding affinity of sorafenib. These conclusions promote us to use acetamide linker wishing to get effective VEGFR-2 inhibitors.
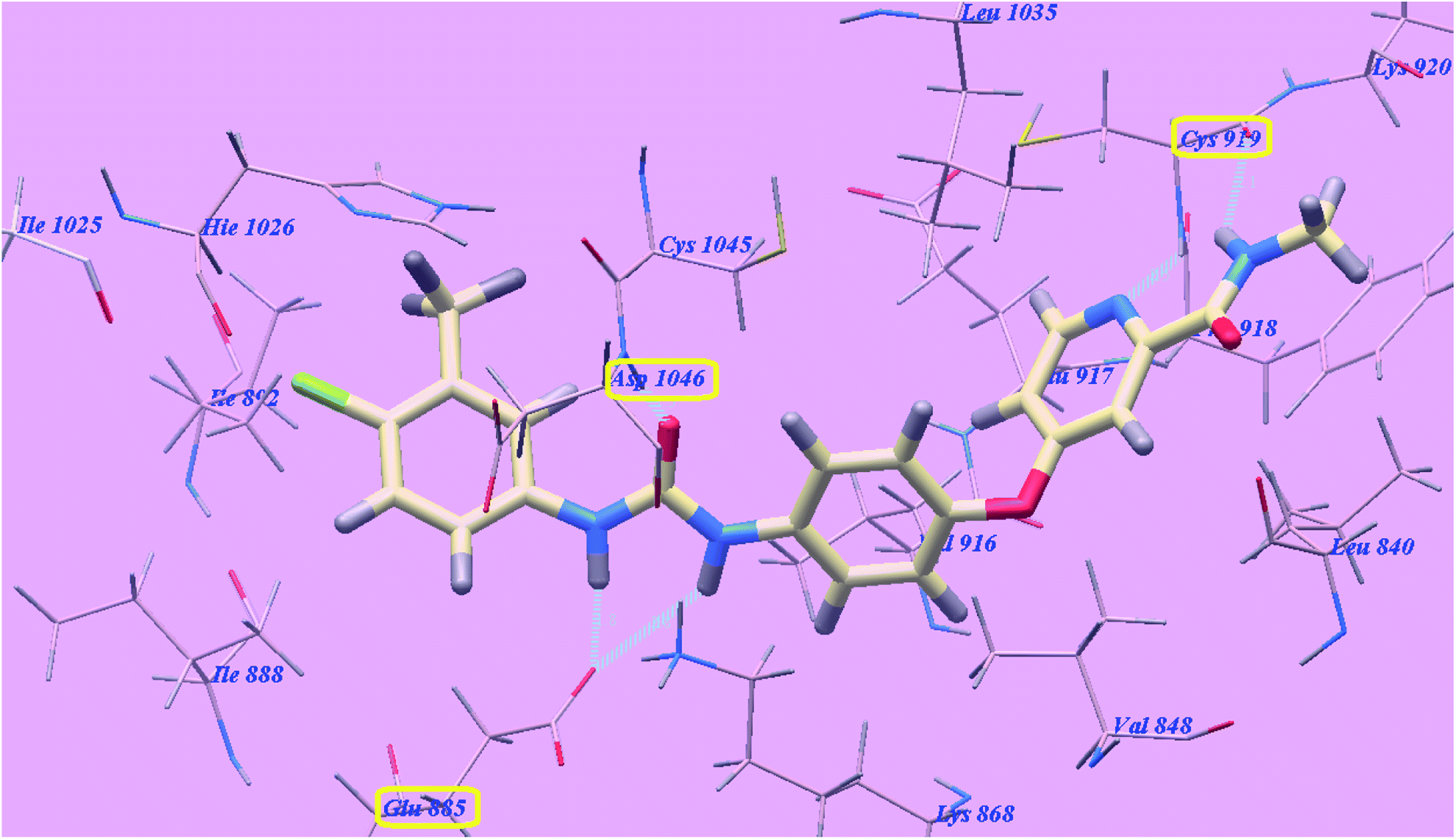 | ||
| Fig. 6 Predicted binding mode for sorafenib with 4ASD. H-bonded atoms are indicated by dotted lines. | ||
The proposed binding mode of compound 5g is virtually the same as that of sorafenib which revealed affinity value of −102.80 kcal mol−1 and 5 H-bonds. The furan ring formed one H-bond with Cysteine919 (2.98 Å). The carbonyl group of the acetamide linker formed one H-bond with Aspartate1046 (1.41 Å) while its NH group formed another H bond with Glutamate885 (2.22 Å). The carbonyl group at position-2 of thiazolidine-2,4-dione formed one H-bond with Lysine868 (2.13 Å). Furthermore, the OH group of the distal carboxylic acid moiety formed one H-bond with Arginine1025 (2.23 Å). The heterocyclic furan ring occupied the pocket made by Leucine1035, Cysteine919, Phenylalanine918, Glutamate917, Valine848 and Leucine840. Furthermore, the thiazolidine-2,4-dione linker occupied the hydrophobic groove made by Cysteine1045, Leucine1035, Threonine916, Lysine868 and Valine848. Additionally, the terminal phenyl tall occupied the hydrophobic channel formed by means ofAspartate1046, Cysteine1045, Histidine1026, Isoleucine892, Isoleucine888 and Glutamate885 (Fig. 7). These interactions of compound 5g may explain the highest anticancer activity.
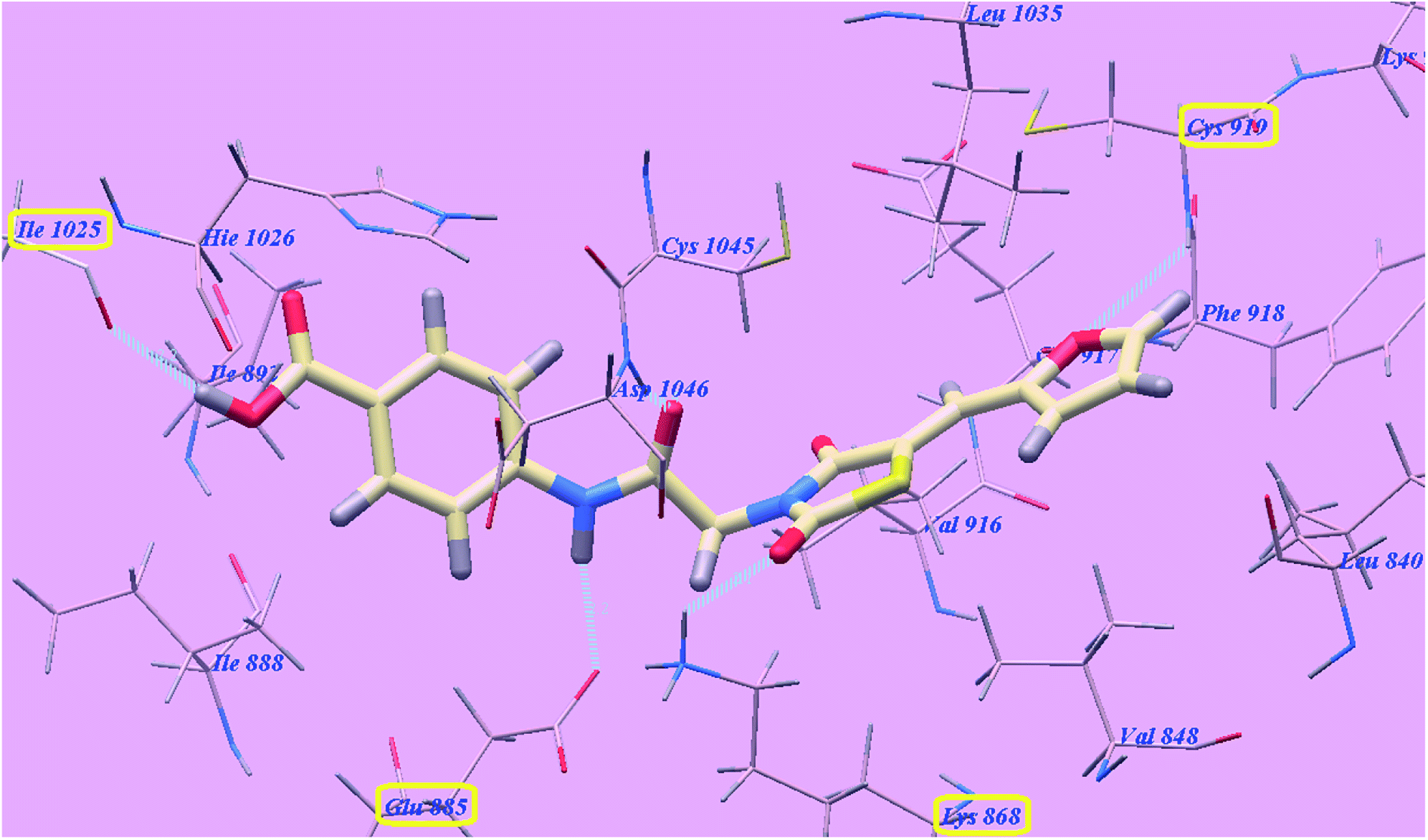 | ||
| Fig. 7 Predicted binding mode for 5g with 4ASD. | ||
The proposed binding mode of compound 4g is virtually the same as that of sorafenib and 5g which revealed affinity value of −100.12 kcal mol−1 and 4 H-bonds. The carbonyl group of the acetamide linker formed 1 H-bond with Aspartate1046 (1.37 Å) while its NH group formed another H bond with Glutamate885 (2.27 Å). The carbonyl group at position-2 of thiazolidine-2,4-dione formed one H-bond with Lysine868 (2.11 Å). Furthermore, the OH group of the distal carboxylic acid moiety formed 1 H-bond with Arginine1025 (2.63 Å). The heterocyclic thiophene ring occupied the pocket made by Leucine1035, Cysteine919, Phenylalanine918, Glutamate917, Valine848 and Leucine840. Furthermore, the thiazolidine-2,4-dione linker occupied the hydrophobic groove made by Cysteine1045, Leucine1035, Threonine916, Lysine868 and Valine848. Additionally, the terminal phenyl tall occupied the hydrophobic channel formed by Aspartate1046, Cysteine1045, Histidine1026, Isoleucine892, Isoleucine888 and Glutamate885 (Fig. 8). These interactions of compound 4g may explain its high anticancer activity.
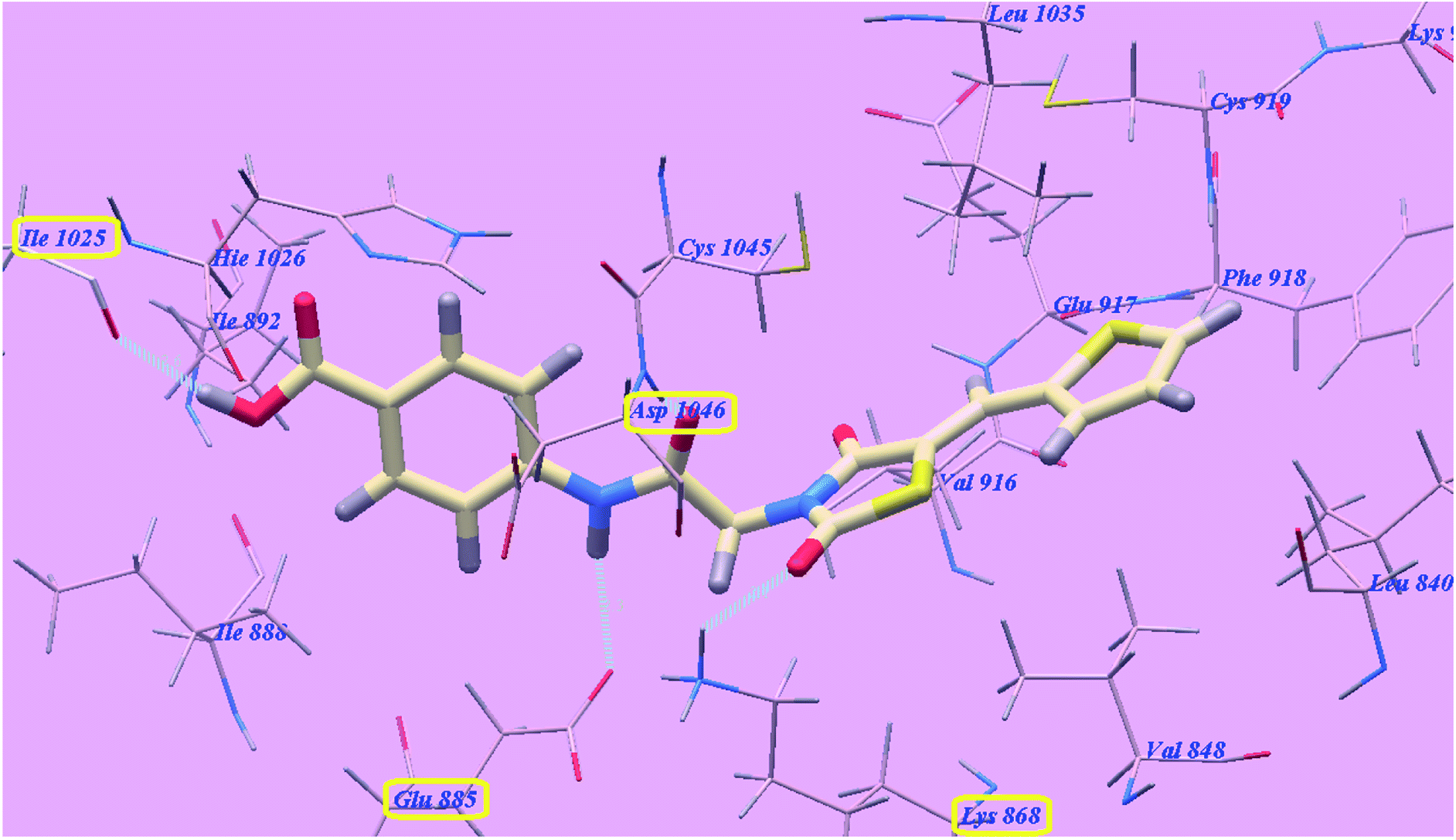 | ||
| Fig. 8 Predicted binding mode for 4g with 4ASD. | ||
The proposed binding mode of compound 5f is virtually the same as that of 5g with affinity value of −99.85 kcal mol−1 and 4 H-bonds (Fig. 9).
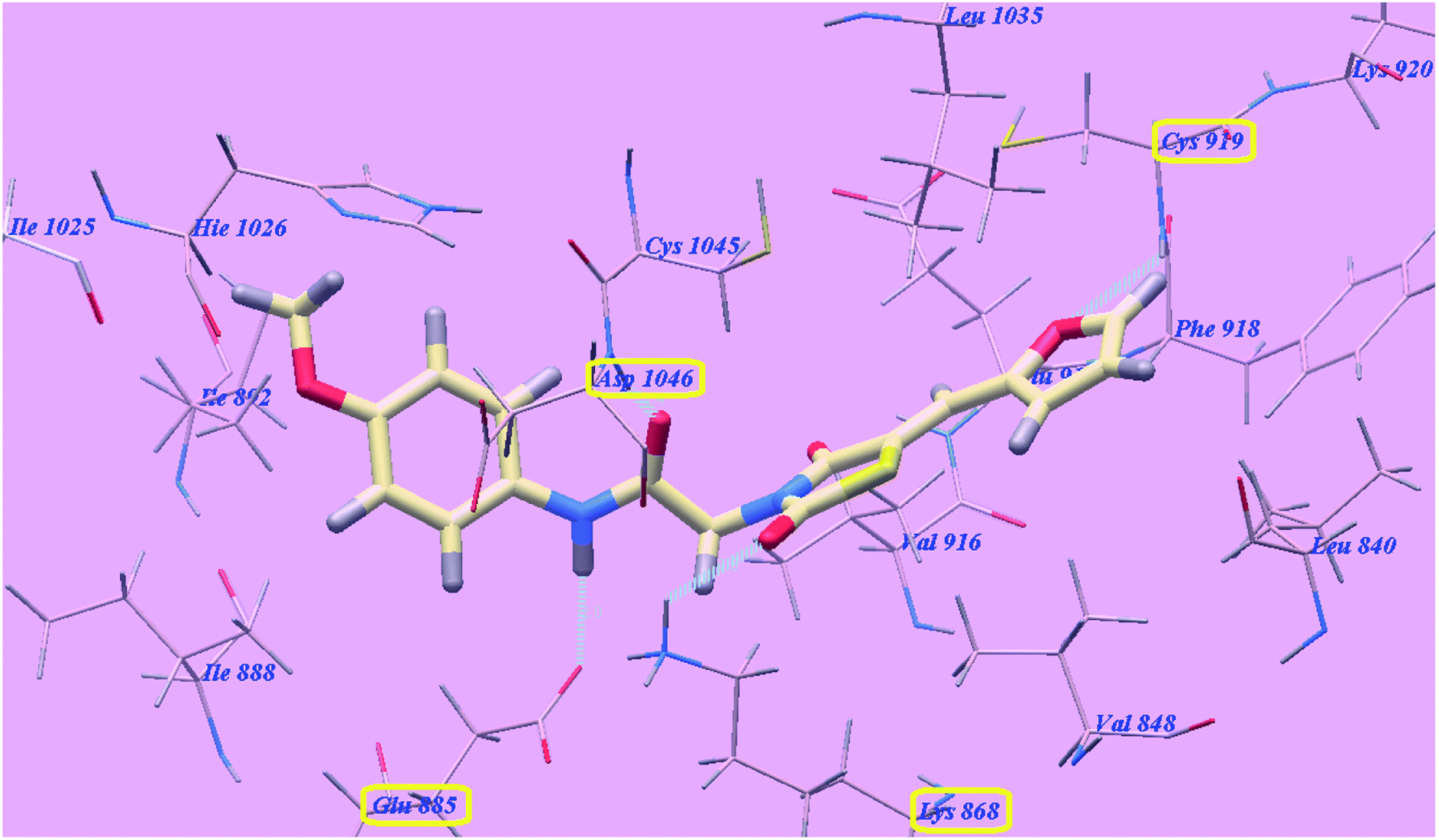 | ||
| Fig. 9 Predicted binding mode for 5f with 4ASD. | ||
From the accomplished docking results (Table 1), we concluded that, the acetamide linker occupied the same groove occupied by urea linker of sorafenib and played the same role which is essential for higher affinity towards VEGFR-2 enzyme. The heterocyclic furan ring increased affinities towards VEGFR-2 enzyme than the heterocyclic thiophene one. This may due to formation of H-bond with the oxygen of the furan ring with Cysteine919. The carbonyl group at position-2 of thiazolidine-2,4-dione derivatives form new H-bond with the amino acid Lysine866.
 | ||
| Fig. 10 Superimposition of some docked compounds inside the binding pocket of 3W2O. | ||
| Compound | ΔG [kcal mol−1] | Compound | ΔG [kcal mol−1] |
|---|---|---|---|
| 4a | −65.49 | 5b | −74.50 |
| 4b | −73.10 | 5c | −74.49 |
| 4c | −68.41 | 5d | −72.15 |
| 4d | −74.45 | 5e | −77.02 |
| 4e | −68.13 | 5f | −73.60 |
| 4f | −71.89 | 5g | −81.06 |
| 4g | −77.08 | Erlotinib | −82.77 |
| 5a | −66.09 |
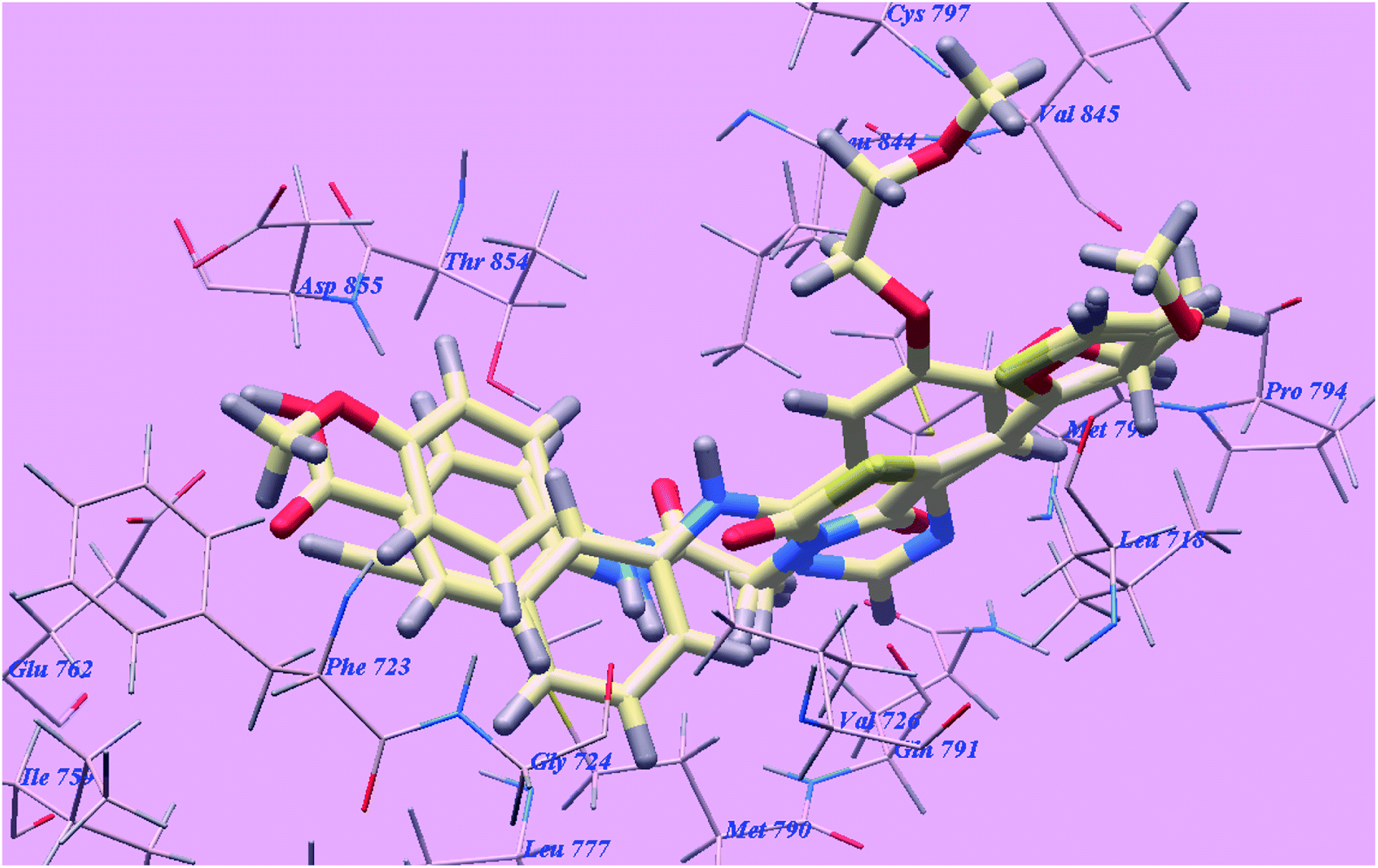
The proposed binding mode of elotinib unveiled affinity value of −82.77 kcal mol−1 and 4 H-bonds. One of the two 2-methoxyethoxy groups formed one H-bond with the inhabitant amino acid Cysteine797 (2.05 Å). The quinazoline moiety was stabilized by formation of two H bonds with Methionine793 (1.82 Å) and Valine726 (2.97 Å). The NH spacer formed one H-bond with the essential amino acid Threonine854 (2.99 Å). The 3-ethynylphenyl head occupied the hydrophobic region I formed by Aspartate855, Threonine854, Glutamine791, Methionine790, Leucine777, Glutamate762, Isoleucine759, Valine726, Glycine724 and Phenylalanine723. Moreover, the 2-methoxyethoxy tail occupied the hydrophobic region II formed through Valine845, Leucine844, Proline794, Methionine793 and Leucine718 (Fig. 11).
 | ||
| Fig. 11 Predicted binding mode for elotinib with 3W2O. H-bonded atoms are indicated by dotted lines. | ||
The proposed binding mode of compound 5g is virtually the same as that of elotinib that showed affinity value of −81.06 kcal mol−1 and 4 H-bonds. The carbonyl group of the acetamide linker formed 1 H-bond with the chief amino acid Threonine854 (2.78 Å). The carbonyl group at position-4 of thiazolidine-2,4-dione formed 1 H-bond with Methionine793 (2.02 Å). Moreover, the carboxylate group at position-4 of the phenyl tail was stabilized by formation of two H-bonds with Aspartate855 (2.61 Å) and Glutamate762 (2.61 Å). The terminal phenyl group occupied the hydrophobic region I formed by Aspartate855, Threonine854, Glutamine791, Methionine790, Leucine777, Glutamate762, Isoleucine759, Valine726, Glycine724 and Phenylalanine723. Moreover, the heterocyclic furan ring occupied the hydrophobic region II formed by Valine845, Leucine844, Proline794, Methionine793 and Leucine718 (Fig. 12). These interactions of compound 5g may explain the highest anticancer activity.
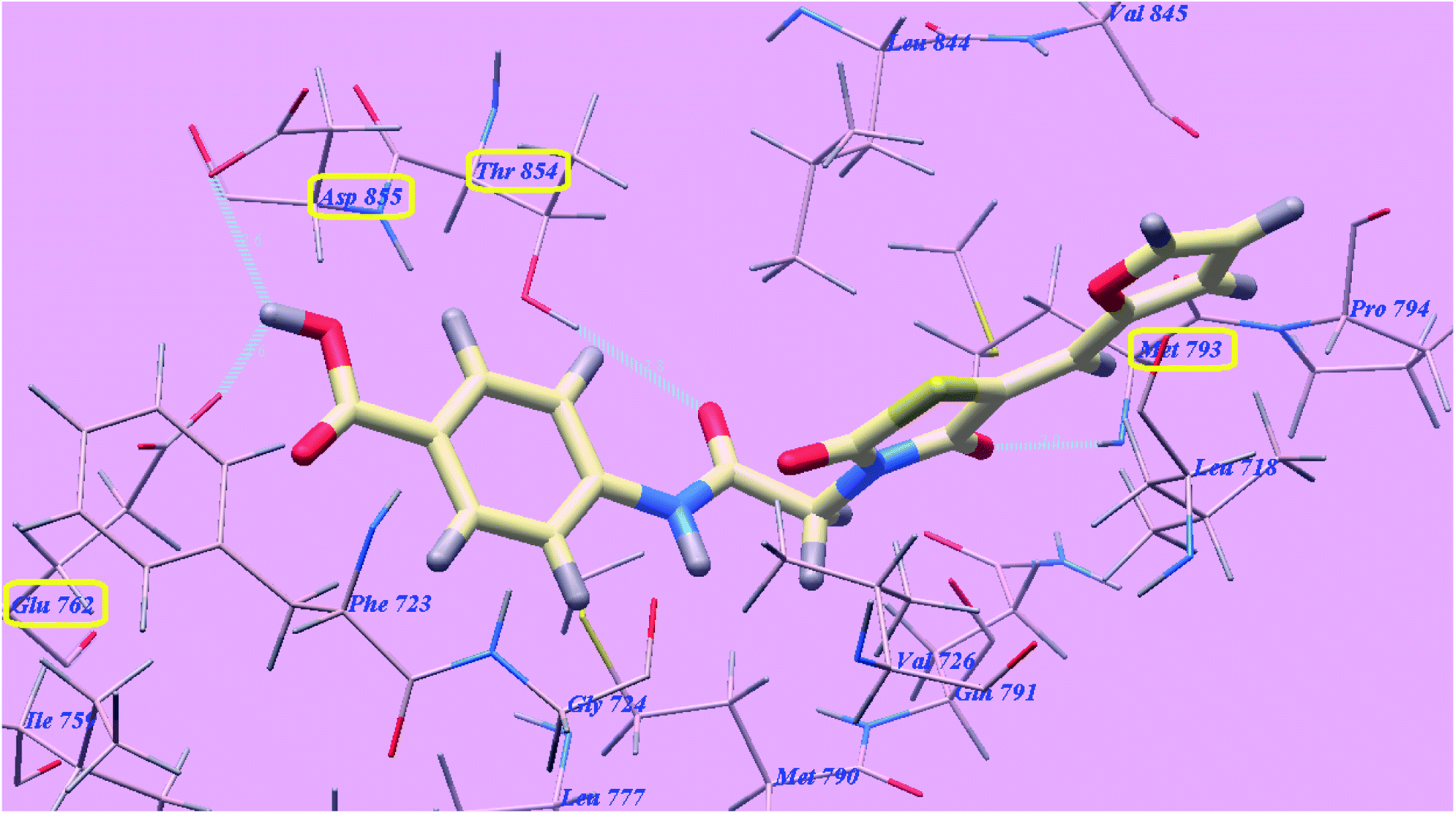 | ||
| Fig. 12 Predicted binding mode for 5g with 3W2O. | ||
The proposed binding mode of compound 4g is virtually the same as that of elotinib and 5g revealed affinity value of −77.08 kcal mol−1 and 4 H-bonds. The carbonyl group of the acetamide linker formed one H-bond with the key amino acid Threonine854 (2.77 Å). The carbonyl group at position-4 of thiazolidine-2,4-dione formed one H-bond with Methionine793 (2.04 Å). Moreover, the carboxylate group at position-4 of the phenyl tail was stabilized by formation of two H-bonds with Aspartate855 (2.60 Å) and Glutamate762 (2.62 Å). The terminal phenyl group occupied the hydrophobic region I formed by Aspartate855, Threonine854, Glutamine791, Methionine790, Leucine777, Glutamate762, Isoleucine759, Valine726, Glycine724 and Phenylalanine723. Moreover, the heterocyclic thiophene ring occupied the hydrophobic region II formed by Valine845, Leucine844, Proline794, Methionine793 and Leucine718 (Fig. 13).
 | ||
| Fig. 13 Predicted binding mode for 4g with 3W2O. | ||
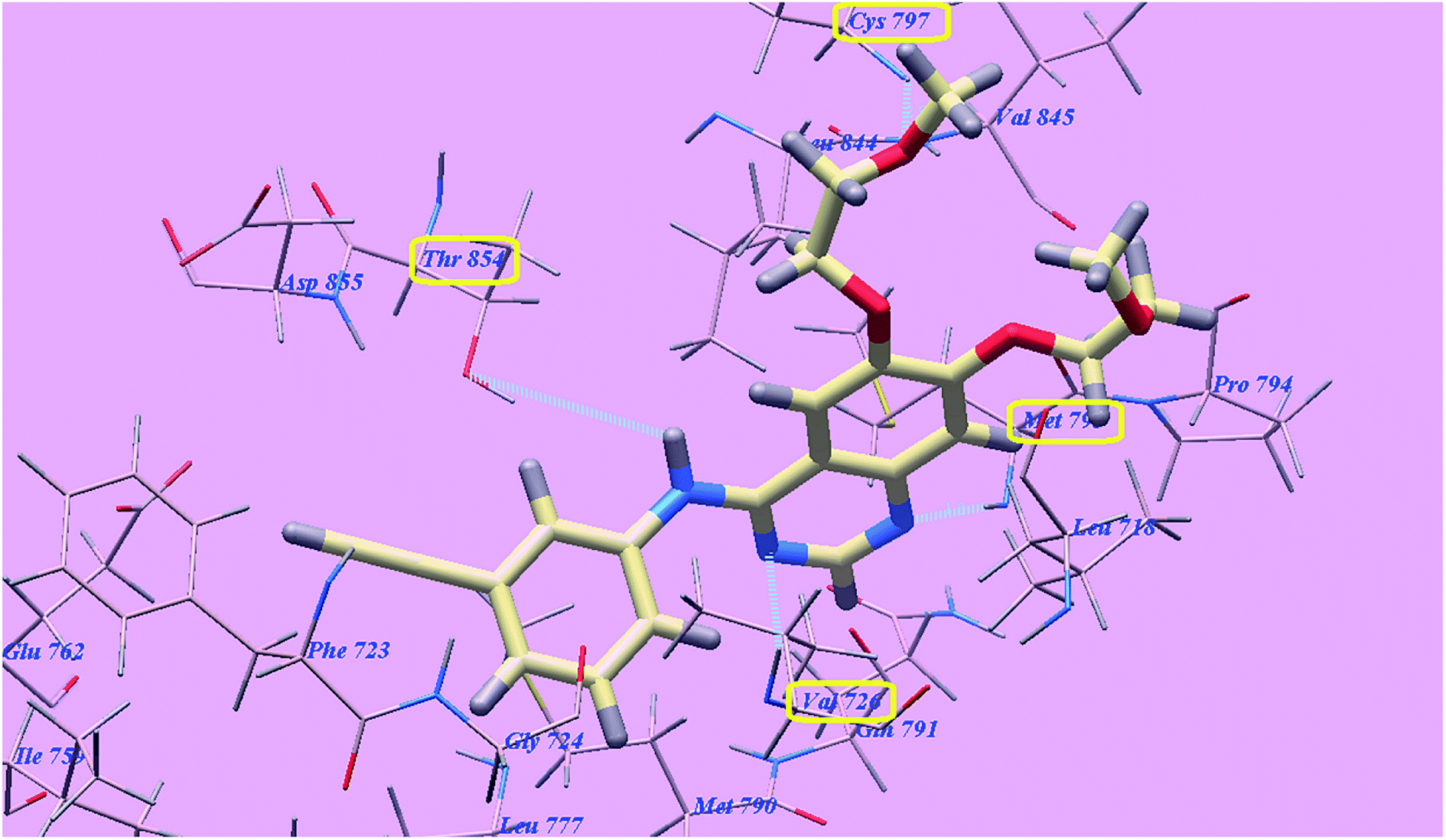
The proposed binding mode of compound 4f is virtually the same as that of 4g and revealed affinity value of −71.89 kcal mol−1 and 3 H-bonds with Threonine854 (2.71 Å), Methionine793 (2.20 Å) and Aspartate855 (2.96 Å). The terminal 4-methoxyphenyl group occupied the hydrophobic region I formed by Aspartate855, Threonine854, Glutamine791, Methionine790, Leucine777, Glutamate762, Isoleucine759, Valine726, Glycine724 and Phenylalanine723. Moreover, the heterocyclic thiophene ring occupied the hydrophobic region II formed by Valine845, Leucine844, Proline794, Methionine793 and Leucine718 (Fig. 14).
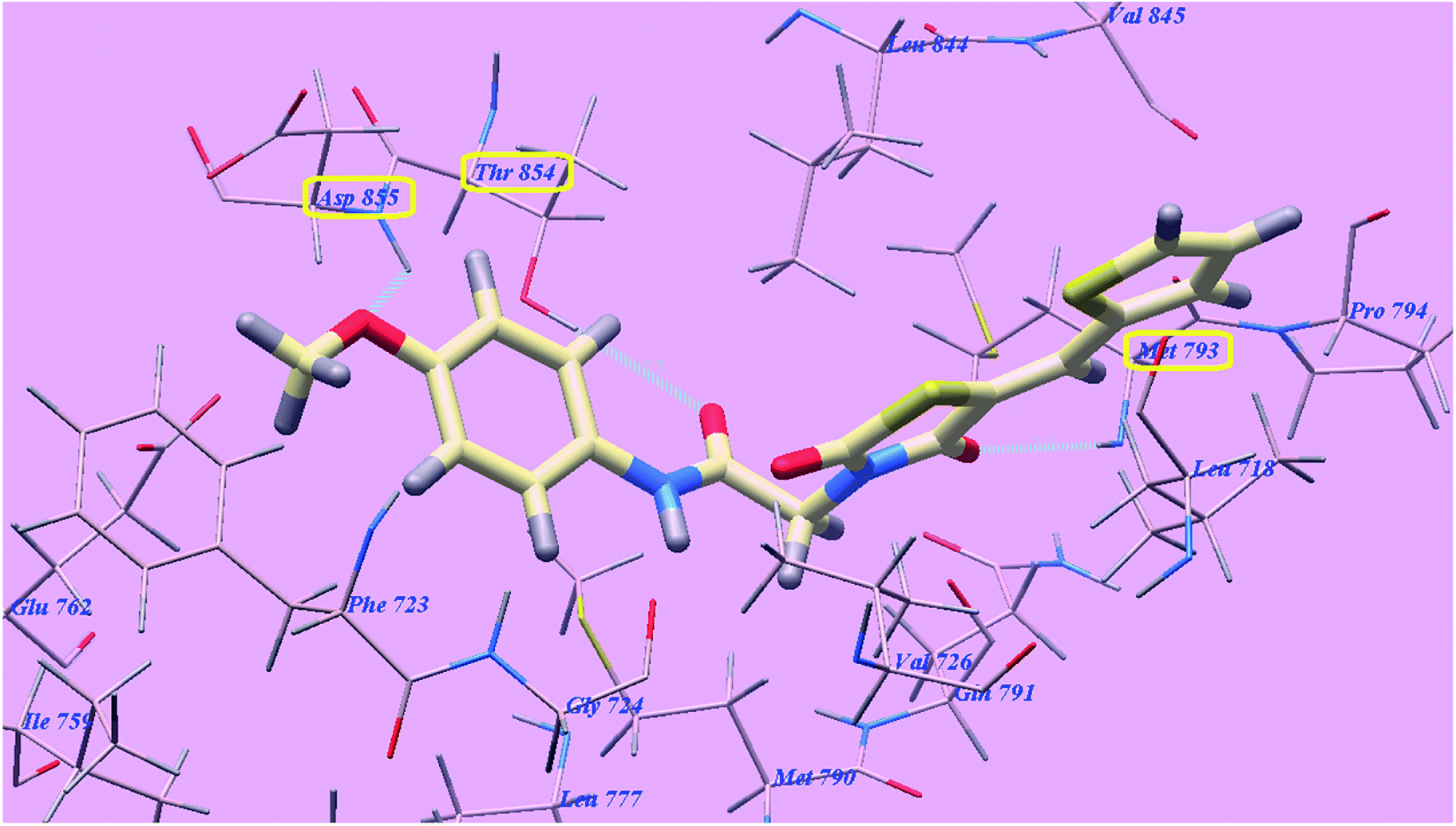 | ||
| Fig. 14 Predicted binding mode for 4f with 3W2O. | ||
2.3. In vitro cytotoxic activity
Anti-proliferative action of the newly prepared 5-benzylidenethiazolidine-2,4-dione derivatives 4a–g–5a–g was examined versus four human tumor cell lines namely, hepatocellular carcinoma (HepG2), lung cancer (A549), breast cancer (MCF-7) and colorectal carcinoma (HCT-116) using 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) colorimetric assay as described by Mosmann.41,42 Sorafenib and elotinib were included in the experiments as reference cytotoxic drugs. The results were expressed as growth inhibitory concentration (IC50) values and summarized in Table 3. From the obtained results, it was explicated that most of the prepared compounds showed excellent to modest growth inhibitory activity toward the tested cancer cell lines. In general, investigations of the cytotoxic activity indicated that HepG2 was the most liable cell line to the effect of the new derivatives. Especially, compounds 5g and 4g were observed to be the best effective compounds against HepG2 (IC50 = 3.86 and 6.22 μM), A549 (IC50 = 7.55 and 12.92 μM), MCF-7 (IC50 = 10.65 and 10.66 μM) and HCT116 (IC50 = 9.04 and 11.17 μM) tumor cell lines. Compound 5g showed higher activities than sorafenib (IC50 = 4.00, 4.04, 5.58 and 5.05 μM) against HepG2 but lower activities versus A549, MCF-7 and HCT116 correspondingly. However, this structure demonstrated higher actions than elotinib (IC50 = 7.73, 5.49, 8.20 and 13.91 μM), toward HepG2 and HCT116 but lower activities versus both A549 and MCF-7 correspondingly. However compound 4g presented lower activities than sorafenib against the four cell lines, it also demonstrated higher actions than elotinib against HepG2 and HCT116 but lower activities versus both A549 and MCF-7 correspondingly.| Compound | IC50a (μM) | |||||
|---|---|---|---|---|---|---|
| HepG2 | A549 | MCF-7 | HCT116 | VEGFR-2 | EGFRT790M | |
| a IC50 values are the mean ± S.D. of three separate experiments.b NT: compounds not tested. | ||||||
| 4a | 52.11 ± 5.2 | 51.00 ± 2.5 | 63.63 ± 3.8 | 58.22 ± 0.7 | NTb | NTb |
| 4b | 35.40 ± 5.1 | 36.11 ± 1.9 | 36.89 ± 1.3 | 38.35 ± 3.9 | NTb | NTb |
| 4c | 34.23 ± 5.1 | 38.32 ± 1.9 | 38.96 ± 1.3 | 42.52 ± 3.9 | NTb | NTb |
| 4d | 13.47 ± 3.9 | 18.97 ± 3.9 | 22.73 ± 3.9 | 16.59 ± 3.9 | 0.255 ± 3.9 | 0.42 ± 3.9 |
| 4e | 24.54 ± 3.9 | 33.97 ± 3.9 | 25.22 ± 3.9 | 21.32 ± 3.9 | 0.288 ± 3.9 | 0.52 ± 3.9 |
| 4f | 10.24 ± 3.9 | 22.73 ± 3.9 | 22.29 ± 3.9 | 17.71 ± 3.9 | 0.095 ± 0.08 | 0.36 ± 3.9 |
| 4g | 6.22 ± 3.9 | 12.92 ± 3.9 | 10.66 ± 3.9 | 11.17 ± 3.9 | 0.083 ± 3.9 | 0.23 ± 3.9 |
| 5a | 52.11 ± 5.2 | 53.22 ± 2.5 | 59.63 ± 3.8 | 48.34 ± 0.7 | NTb | NTb |
| 5b | 23.78 ± 3.9 | 36.22 ± 3.9 | 25.69 ± 3.9 | 28.22 ± 3.9 | NTb | NTb |
| 5c | 24.12 ± 2.5 | 35.38 ± 1.9 | 28.61 ± 1.3 | 35.67 ± 3.9 | NTb | NTb |
| 5d | 23.59 ± 3.9 | 29.66 ± 4.1 | 25.28 ± 4.5 | 26.41 ± 1.4 | 0.276 ± 3.9 | 0.52 ± 3.9 |
| 5e | 23.76 ± 3.9 | 32.90 ± 3.9 | 14.54 ± 3.9 | 10.66 ± 3.9 | 0.263 ± 3.9 | 0.41 ± 3.9 |
| 5f | 11.01 ± 3.9 | 13.02 ± 3.9 | 18.53 ± 3.9 | 15.68 ± 3.9 | 0.247 ± 3.9 | 0.35 ± 3.9 |
| 5g | 3.86 ± 3.9 | 7.55 ± 3.9 | 10.65 ± 3.9 | 9.04 ± 3.9 | 0.080 ± 0.05 | 0.14 ± 3.9 |
| Sorafenib | 4.00 ± 0.33 | 4.04 ± 0.33 | 5.58 ± 0.55 | 5.05 ± 0.50 | 0.084 ± 0.04 | NTb |
| Erlotinib | 7.73 ± 0.67 | 5.49 ± 0.45 | 8.20 ± 0.34 | 13.91 ± 1.3 | NTb | 0.24 ± 0.22 |
Regarding HepG2, compounds 4d, 4e, 4f, 5b, 5c, 5d, 5e and 5f exhibited the greatest anticancer effects with IC50 ranging from 10.24 to 24.54 μM. Derivatives 4b and 4c with IC50 = 35.40 and 34.23 μM consequently, showed good cytotoxicity. Derivatives 4a and 5a with the same IC50 = 52.11 μM exhibited moderate cytotoxicity.
Concerning A549, compounds 4d, 4f and 5f exhibited the greatest anticancer effects with IC50 = 18.97, 22.73 and 13.02 μM consequently. Derivatives 4b, 4c, 4e, 5b, 5c, 5d and 5e with IC50 ranging from 29.66 to 38.32 μM, showed good cytotoxicity. Derivatives 4a, and 5a with IC50 = 51.00 and 53.22 μM exhibited moderate cytotoxicity.
Derivatives 4d, 4f, 5e and 5f exhibited the ultimate anticancer effect with IC50 = 22.73, 22.29, 14.54 and 18.53 μM correspondingly upon assessment against MCF-7. Derivatives 4b, 4c, 4e, 5b, 5c and 5d with IC50 ranging from 25.22 to 38.96 μM correspondingly, showed great cytotoxic effect. Derivative 4a and 5a with IC50 = 63.63 and 59.63 μM respectively showed mild cytotoxicity.
Derivatives 4d, 4e, 4f, 5e and 5f exhibited the greatest anticancer effect with IC50 = 16.59, 21.32, 17.71, 10.66 and 15.68 μM in that order against HCT-116. Moreover, derivatives 4b, 4c, 5a, 5b, 5c and 5d with IC50 ranging from 26.41 to 48.34 μM, showed potent cytotoxic effect. On the other hand derivative 3 with IC50 = 58.22 μM showed moderate cytotoxic activity.
2.4. In vitro VEGFR-2 kinase inhibitory assay
Additionally, as VEGFR-2 is mostly overexpressed in several malignancies such as HepG2, MCF-7 and HCT116, our most active cytotoxic compounds 4d, 4e, 4f, 4g, 5d, 5e, 5f and 5g were selected to assess their VEGFR-2 inhibitory effects as a result of applying an anti-phosphotyrosine antibody with the Alpha Screen system (PerkinElmer, USA).43 The results were described as IC50 (50% inhibition concentration value) in Table 3. In this assessment, sorafenib was applied as a positive standard. The assessed derivatives demonstrated high to low inhibitory effect with IC50vary between 0.080 to 0.288 μM. Derivatives 5g, 4g and 4f were observed to be the highest effective derivatives that inhibited VEGFR-2 at IC50 = 0.080, 0.083 and 0.095 μM respectively. 5g and 4g showed more potent activities than that of sorafenib (IC50 = 0.084 μM). Moreover, compounds 4d, 4e, 5d, 5e and 5f possessed moderate VEGFR-2 inhibitory effect with IC50 ranging from 0.247 to 0.288 μM.2.5. EGFRT790M kinase inhibitory assay
Overexpression of EGFR is related to the increased poor prognosis, metastases and aggressiveness in various cancers such as A549 and HepG2 cell lines.44 As a result, derivatives with effective cytotoxic activities 4d, 4e, 4f, 4g, 5d, 5e, 5f and 5g were additionally assessed to evaluate their inhibitory activities against mutant EGFRT790M kinase inhibitory activities. Homogeneous time resolved fluorescence (HTRF) assay was applied in this test.45 Erlotinib was used as standard with IC50 = 0.24. Table 3 indicates a comparison of IC50 of the experienced derivatives.Derivatives 5g and 4g could interfere with the EGFRT790M activity exhibiting stronger activities than elotinib with IC50 = 0.14 and 0.23 μM sequentially. Candidates 4d, 4f, 5e and 5f substantially inhibited EGFRT790M at IC50 ranging from 0.35 to 0.42 μM. Alternatively compounds 4e and 5d moderately inhibited EGFRT790M at the same IC50 = 0.52 μM.
2.6. Structure activity relationship (SAR)
The preliminary SAR study has focused on the effect of replacement of pyridine and the two 2-methoxyethoxy groups moieties of sorafenib and elotinib respectively by the heterocyclic furan and thiophene moieties. Also, it focused on the effect of replacement of the urea and amino linkers of sorafenib and elotinib respectively with acetamide linkers which interacting as H-bond acceptors through its carbonyl group and as H-bond donor through its NH atom. Moreover, the thiazolidine-2,4-dione was designed to replace the central aryl and quinazoline rings of the reference ligands sorafenib and elotinib respectively. On the other hand different substituents were introduced to the phenyl tail of our derivatives with different lipophilicity and electronic nature in order to study their effects on the anticancer activity. The data obtained revealed that, the tested structures displayed different levels of antitumor activity and possessed a distinctive pattern of selectivity toward the HepG2 cell lines. Generally, heterocyclic rings, the spacer, linker (HBA-HBD), lipophilicity and electronic nature of substituents exhibited an important role in anticancer activity. The furan ring as in compounds 5a–g exhibited higher anticancer activities than the thiophene ones. Generally, the substituted phenyl tail as in compounds 4a–f and 5a–f showed higher anticancer activities than the unsubstituted 4g and 5g ones against the four HepG2, A549, MCF-7 and HCT116 cell lines.From the structure of the synthesized derivatives and the data shown in Table 3 we can divide these tested compounds into two groups. The first group is compounds 4a–g which containing thiophene heterocyclic ring; where derivative 4g with electron withdrawing hydrophilic carboxylic group showed higher activities than 4f with electron donating hydrophobic methoxy one against the four HepG2, A549, MCF-7 and HCT116 cell lines. Compound 4f showed higher activities than 4d with electron withdrawing hydrophobic bromo group against both HepG2 and MCF-7 but lower activities against both A549 and HCT116 cell lines. Furthermore, compound 4e with hydrophobic electron donating methyl group showed higher activities than 4c with electron withdrawing hydrophobic chloro group against the four cancer cell lines. In addition, derivative 4b with chloro group exhibited higher activities than derivative 4c with flouro one against A549, MCF-7 and HCT116 but lower activities against HepG2 cell lines. Finally, the unsubstituted derivative 4g showed the least activities regarding the four cell lines.
The second group is compounds 5a–g which containing furan heterocyclic ring; where derivative 5g with electron withdrawing hydrophilic carboxylic group showed higher activities than 5f with electron donating hydrophobic methoxy and 5d with electron donating hydrophobic methyl ones against the four HepG2, A549, MCF-7 and HCT116 cell lines. Compound 5e with electron donating hydrophobic isopropyl group showed higher activities than 5f and 5d against both MCF-7 and HCT116 but lower activities against both HepG2 and A549 cell lines respectively. Furthermore, compound 5b with electron withdrawing hydrophobic chloro group exhibited higher activities than derivative 4c with flouro one against HepG2, MCF-7 and HCT116 but lower activities against A549 cell lines. Finally, the unsubstituted derivative 5g showed the least activities against the four cell lines.
2.7. ADMET; in silico studies profile
In silico report of the highly active derivatives 4g, 5f and 5g was conducted for their physicochemical characters evaluation and the proposed ADMET profile. It was predicted using pkCSM descriptor algorithm procedures46 and matched to rule of five described by Lipinski.47 The good absorption properties were expected for the molecules that accomplish at least three rules: (i) hydrogen bond donors are not more than five; (ii) partition coefficient (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P) is not more than 5, (iii) molecular weight less than 500, (iv) hydrogen bond acceptors are not more than 10. In the current work, the standard anticancer agent sorafenib violates one rule while our new compounds 4g, 5f and 5g and elotinib don't violate any one.
P) is not more than 5, (iii) molecular weight less than 500, (iv) hydrogen bond acceptors are not more than 10. In the current work, the standard anticancer agent sorafenib violates one rule while our new compounds 4g, 5f and 5g and elotinib don't violate any one.
As a result of obtaining data (Table 4), we can assume that compound 5f have very good GIT absorption in human (91.952) while compounds 4g and 5g have good absorption (56.765 and 55.558 respectively) which indicates easier to cross different biological membranes.48 So, they may show a significant high bioavailability through GIT. Concerning CNS penetrability, our prepared compounds have the capability to reach CNS (CNS permeability values −2.413 to −3.009), lower than sorafenib (CNS permeability −2.007) but higher than elotinib (CNS permeability −3.216).
| Parameter | 4g | 5f | 5g | Sorafenib | Erlotinib |
|---|---|---|---|---|---|
| Physicochemical properties | |||||
| Molecular weight | 388.426 | 358.375 | 372.358 | 464.831 | 393.443 |
| LogP | 3.1213 | 2.9632 | 2.6528 | 5.5497 | 3.4051 |
| Rotatable bonds | 5 | 5 | 5 | 5 | 10 |
| Acceptors | 6 | 6 | 6 | 4 | 7 |
| Donors | 2 | 1 | 2 | 3 | 1 |
| Surface area | 155.861 | 147.173 | 151.015 | 185.111 | 169.532 |
|
|||||
| Absorption | |||||
| Water solubility | −3.663 | −4.262 | −3.674 | −4.822 | −4.736 |
| Caco2 permeability | 1.012 | 1.088 | 0.839 | 0.689 | 1.431 |
| Human intest. Absorption | 56.765 | 91.952 | 55.558 | 89.043 | 94.58 |
| Skin permeability | −2.735 | −3.11 | −2.735 | −2.767 | −2.741 |
| Substrate for P-glycoprotein | + | + | + | + | — |
| Inhibitor of P-glycoprotein I | — | — | — | + | + |
| Inhibitor of P-glycoprotein II | — | — | — | + | + |
|
|||||
| Distribution | |||||
| VDss (human) | −1.173 | −0.389 | −1.294 | −0.29 | 0.199 |
| Human unbound fraction | 0.213 | 0.224 | 0.352 | 0.065 | 0.059 |
| Permeability throughout BBB | −1.447 | −0.657 | −1.496 | −1.684 | −0.745 |
| Permeability to CNS | −2.413 | −2.918 | −3.009 | −2.007 | −3.216 |
|
|||||
| Metabolism | |||||
| CYP2D6 substrate | — | — | — | — | — |
| CYP3A4 substrate | — | — | — | + | + |
| Inhibition of CYP3A4 | — | — | — | + | + |
| Inhibition of CYP2D6 | — | — | — | + | + |
| Inhibition of CYP2C9 | — | — | — | + | + |
| Inhibition of CYP2C19 | — | — | — | — | — |
| Inhibition of CYP1A2 | — | + | — | + | + |
|
|||||
| Excretion | |||||
| Clearance | −0.199 | 0.054 | −0.052 | −0.219 | 0.702 |
| Renal OCT2 substrate | — | — | — | — | — |
|
|||||
| Toxicity | |||||
| AMES toxicity | — | — | — | — | — |
| Human max. tolerated dose | 0.199 | −0.198 | 0.894 | 0.549 | 0.839 |
| Inhibitor of hERG I | — | — | — | — | — |
| hERG II inhibitor | — | + | — | + | + |
| Acute toxic activity (LD50) | 2.131 | 2.972 | 2.208 | 2.538 | 2.393 |
| Chronic toxic activity (LOAEL) | 0.914 | 0.627 | 0.3 | 1.198 | 1.37 |
| Hepatotoxic effect | + | — | + | + | + |
| Skin sensitization | — | — | — | — | — |
| T. Pyriformis toxicity | 0.287 | 0.951 | 0.286 | 0.383 | 0.309 |
| Minnow toxic activity | 0.395 | 0.794 | 1.524 | 0.189 | −0.1 |
It well known that CYP3A4, the major drug metabolizing enzyme, could be inhibited by sorafenib and elotinib but 4g, 5f and 5g could not. This is as well possibly for the superior lipophilicity of sorafenib and elotinib. Elimination was expected depending on the total clearance which is a considerable factor in deciding dose intervals. The data showed that elotinib confirmed higher clearance rates compared with sorafenib and our new compounds which demonstrated very low clearance values. Thus, elotinib, could be eliminated faster, and as a result supposed to have shorter dosing intervals. Unlike elotinib, the prepared compounds exhibited slowly clearance rate, which signifies longer duration of action and extended dosing intervals. Toxicityis the final ADMET profile studied factor. Like presented in Table 4, sorafenib, elotinib, and the novel compounds 4g and 5g shared the drawback of unwanted hepatotoxic actions but compound 5f did not. Elotinib and 5g demonstrated the highest maximum tolerated dose. In contrast, sorafenib, 4g and 5f demonstrated the lowest maximum tolerated doses which involve the advantage of the broad therapeutic index of 5g and elotinib respectively. Lastly, the oral chronic toxic doses of the novel compounds are lower than sorafenib and elotinib.
Finally, ADMET is in a good agreement with the experimental data obtained in many studies for thiazolidine-2,4-dions.49
3. Conclusion
In summary, fourteen new thiazolidine-2,4-dione-based derivatives have been designed, synthesized and evaluated for their anticancer activities against four human tumor cell lines HepG2, A549, MCF-7 and HCT-116 targeting both and VEGFR-2 and EGFR tyrosine kinases. The molecular design was performed to investigate the binding mode of the proposed compounds with VEGFR-2 and EGFR receptors. The data obtained from the docking studies were highly correlated with that obtained from the biological screening. All the tested compounds showed variable anticancer activities. HepG2 was the most sensitive cell line to the influence of the new derivatives. In particular, compounds 5g and 4g were observed to be the best effective compounds against HepG2 (IC50 = 3.86 and 6.22 μM), A549 (IC50 = 7.55 and 12.92 μM), MCF-7 (IC50 = 10.65 and 10.66 μM) and HCT116 (IC50 = 9.04 and 11.17 μM) tumor cell lines. Compound 5g exhibited higher activities than sorafenib (IC50 = 4.00, 4.04, 5.58 and 5.05 μM) against HepG2 but lower activities versus A549, MCF-7 and HCT116 correspondingly. However, this compound demonstrated higher actions than Erlotinib (IC50 = 7.73, 5.49, 8.20 and 13.91 μM), against HepG2 and HCT116 but lower activities versus both A549 and MCF-7 correspondingly. However compound 4g exhibited lower activities than sorafenib against the four cell lines, it also demonstrated higher actions than Erlotinib against HepG2 and HCT116 but lower activities versus both A549 and MCF-7 correspondingly. Derivatives 5g, 4g and 4f were observed to be the highest effective derivatives that inhibited VEGFR-2 at IC50 = 0.080, 0.083 and 0.095 μM respectively. 5g and 4g showed more potent activities than that of sorafenib (IC50 = 0.084 μM). As well, compounds 5g and 4g could interfere with the EGFRT790M activity exhibiting stronger activities than Erlotinib with IC50 = 0.14 and 0.23 μM respectively. Moreover, ADMET profile was in silico calculated for the three most active compounds 4g, 5f and 5g in comparing to sorafenib and Erlotinib as reference drugs. Our derivative 5f have very good GIT absorption in human (91.952) while compounds 4g and 5g have good absorption (56.765 and 55.558 respectively) which indicates easier to cross different biological membranes. Unlike Erlotinib, the prepared compounds exhibited slowly clearance rate, which signifies longer duration of action and extended dosing intervals. Compound 5f don't show the drawback of unwanted hepatotoxic actions. The obtained results showed that, our compounds specially 4g and 5g could be useful as a template for future design, optimization and investigation to produce more potent and selective dual VEGFR-2/EGFRT790M inhibitors with higher anticancer analogs.4. Experimental
4.1. Chemistry
All compounds were crystallized from ethanol and their NMR spectra were made in DMSO-d6 solvent at 400 MHz for 1HNMR and 100 MHz for 13CNMR.
4.1.2.1. 2-[2,4-Dioxo-5-(thiophen-2-ylmethylene)thiazolidin-3-yl]-N-phenylacetamide (4a). Yield, 87%; mp 230–2 °C; IRνmax (cm−1): 3197 (NH), 3055 (CH aromatic), 2960 (CH aliphatic), 1714, 1667 (3C
O amide); 1HNMR: 4.27 (s, 2H, CH2), 6.72–6.79 (m, 1H, Ar–H, H-4 of phenyl), 7.02–7.07 (m, 1H, Ar–H, H-4 of thiophene), 7.19 (d, 1H, Ar–H, H-3 of thiophene), 7.54–7.58 (m, 2H, Ar–H, H-3 & H-5 of phenyl), 7.60 (s, 1H, CCH), 7.61–7.82 (m, 2H, Ar–H, H-2 & H-6 of phenyl), 8.00–8.01 (d, 1H, Ar–H, H-5 of thiophene), 10.50 (s, 1H, NH, D2O exchangeable); anal. calcd for C16H12N2O3S2 (344.4): C, 55.80; H, 3.51; N, 8.13. Found: C, 55.90; H, 3.58; N, 8.09.
4.1.2.2. N-(4-Chlorophenyl)-2-[2,4-dioxo-5-(thiophen-2-ylmethylene)thiazolidin-3-yl]acetamide (4b). Yield, 84%; mp 240–2 °C; IRνmax (cm−1): 3250 (NH), 3100 (CH aromatic), 2917 (CH aliphatic), 1704, 1649 (3C
O amide); 1HNMR: 4.10 (s, 1H, CH2), 4.52 (s, 1H, CH2), 7.26–7.34 (m, 2H, Ar–H, H-2 & H-6 of phenyl), 7.37–7.40 (m, 1H, Ar–H, H-4 of thiophene), 7.53–7.60 (m, 2H, Ar–H, H-3 & H-5 of phenyl), 7.77 (s, 1H, CCH), 7.99 (d, 1H, Ar–H, H-3 of thiophene), 8.09–8.28 (d, 1H, Ar–H, H-5 of thiophene), 10.58 (s, 1H, NH, D2O exchangeable); LC-MS ESI spectrometry in the negative ion mode: the mass spectrum of compound 4b showed M + 2 molecular ion peak at 379.0852 and base peak at 377.1044; anal. calcd for C16H11ClN2O3S2 (378.8): C, 50.73; H, 2.93; N, 7.39. Found: C, 50.76; H, 2.95; N, 7.44.
4.1.2.3. 2-[2,4-Dioxo-5-(thiophen-2-ylmethylene)thiazolidin-3-yl]-N-(4-fluorophenyl)acetamide (4c). Yield, 73%; mp 245–7 °C; IRνmax (cm−1): 3288 (NH), 3050 (CH aromatic), 2950 (CH aliphatic), 1782, 1712, 1650 (3C
O amide); 1HNMR: 4.50 (s, 2H, CH2), 7.15–7.31 (m, 2H, Ar–H, H-2 & H-6 of phenyl), 7.38–7.42 (m, 1H, Ar–H, H-4 of thiophene), 7.56–7.59 (m, 2H, Ar–H, H-3 & H-5 of phenyl), 7.76 (s, 1H, CCH), 7.77 (d, 1H, Ar–H, H-3 of thiophene), 8.27 (d, 1H, Ar–H, H-5 of thiophene), 10.44 (s, 1H, NH, D2O exchangeable); 13CNMR: 44.53, 115.84, 116.06, 118.61, 121.47, 121.55, 125.42, 127.44, 129.29, 134.31, 135.19, 137.41, 164.25, 165.52, 167.07, 171.49; LC-MS ESI spectrometry in the positive ion mode: the mass spectrum of compound 4c showed M molecular ion peak at 362.4517 and base peak at 318.4247; LC-MS ESI spectrometry in the negative ion mode: M − 1 molecular ion peak at 361.1305 which also was the base peak. It showed M molecular ion peak at 362.1415 and M + 1 molecular ion peak at 363.1041; anal. calcd forC16H11FN2O3S2 (362.4): C, 53.03; H, 3.06; N, 7.73; found: C, 52.98; H, 3.04; N, 7.66.
4.1.2.4. N-(4-Bromophenyl)-2-[2,4-dioxo-5-(thiophen-2-ylmethylene)thiazolidin-3-yl]acetamide (4d). Yield, 70%; mp 238–9 °C; IRνmax (cm−1): 3150 (NH), 3050 (CH aromatic), 2950 (CH aliphatic), 1747, 1669 (3C
O amide); 1HNMR: 4.53 (s, 2H, CH2), 7.14–7.32 (m, 1H, Ar–H, H-4 of thiophene), 7.34–7.44 (m, 3H, Ar–H, H-2 & H-6 of phenyl & H-3 of thiophene), 7.76–7.77 (m, 2H, Ar–H, H-3 & H-5 of phenyl), 8.07 (s, 1H, CCH), 8.28 (d, 1H, Ar–H, H-5 of thiophene), 10.60 (s, 1H, NH, D2O exchangeable); 13CNMR: 44.61, 118.09, 118.54, 119.19, 124.00, 127.51, 129.62, 131.11, 133.67, 134.33, 135.89, 137.38, 140.18, 164.76, 165.49, 166.95; anal. calcd for C16H11BrN2O3S2 (423.3): C, 45.40; H, 2.62; N, 6.62; found: C, 45.40; H, 2.55; N, 6.60.
4.1.2.5. 2-[2,4-Dioxo-5-(thiophen-2-ylmethylene)thiazolidin-3-yl]-N-(p-tolyl)acetamide (4e). Yield, 70%; mp 278–9 °C; IRνmax (cm−1): 3368 (NH), 3050 (CH aromatic), 2950 (CH aliphatic), 1697 (3C
O amide); 1HNMR: 2.25 (s, 3H, CH3), 4.49 (s, 2H, CH2), 7.12–7.14 (m, 2H,Ar–H, H-3 & H-5 of phenyl), 7.32–7.34 (m, 1H, Ar–H, H-4 of thiophene), 7.42–7.44 (m, 2H, Ar–H, H-2 & H-6 of phenyl), 7.76 (d, 1H, Ar–H, H-3 of thiophene), 8.07 (d, 1H, Ar–H, H-5 of thiophene), 8.27 (s, 1H, CCH), 10.28 (s, 1H, NH, D2O exchangeable); 13CNMR: 20.66, 56.67, 83.08, 100.64, 106.69, 113.92, 119.97 (2), 130.15 (3), 133.69, 135.91, 167.49, 183.13, 203.35, 208.81; anal. calcd for C17H14N2O3S2 (358.4): C, 56.97; H, 3.94; N, 7.82; found: C, 57.06; H, 4.05; N, 7.77.
4.1.2.6. 2-[2,4-Dioxo-5-(thiophen-2-ylmethylene)thiazolidin-3-yl]-N-(4-methoxyphenyl)acetamide (4f). Yield, 73%; mp 284–6 °C; IRνmax (cm−1): 3250 (NH), 3068 (CH aromatic), 2933 (CH aliphatic), 1779, 1714 (4C
O amide); 1HNMR: 3.80 (s, 3H, CH3), 4.08 (s, 2H, CH2), 6.99–7.02 (m, 2H,Ar–H, H-3 & H-5 of phenyl), 7.11–7.13 (m, 2H, Ar–H, H-2 & H-6 of phenyl), 7.23–7.27 (m, 1H, Ar–H, H-4 of thiophene), 7.74 (s, 1H, CCH), 7.75–7.97 (d, 1H, Ar–H, H-3 of thiophene), 7.98–8.27 (d, 1H, Ar–H, H-5 of thiophene), 10.25 (s, 1H, NH, D2O exchangeable); 13CNMR: 31.09, 55.78, 114.50, 114.65, 118.56, 121.36, 128.41, 128.66 (2), 130.09, 132.26, 133.16, 136.18, 138.09, 159.27, 164.18, 168.60; MS (m/z): 375 (M+ + 1, 6.54%), 374 (M+, 47.45%), 331 (100%, base beak), 274 (7.33%), 95 (13.49%); anal. calcd for C17H14N2O4S2 (374.4): C, 54.53; H, 3.77; N, 7.48; found: C, 54.44; H, 3.75; N, 7.55.
4.1.2.7. 4-{2-[2,4-Dioxo-5-(thiophen-2-ylmethylene)thiazolidin-3-yl]acetamido}benzoic acid (4g). Yield, 80%; mp 260–2 °C; IRνmax (cm−1): 3450 (OH), 3114 (NH), 3025 (CH aromatic), 2965 (CH aliphatic), 1729, 1672 (3C
O amide); 1HNMR: 4.26 (s, 1H, CH2), 4.54 (s, 1H, CH2), 7.14–7.42 (m, 4H, Ar–H of phenyl), 7.52–7.61 (m, 2H, Ar–H, H-3 & H-4 of thiophene), 7.62 (d, 1H, Ar–H, H-5 of thiophene), 8.30 (s, 1H, CCH), 10.50 (s, 1H, NH, D2O exchangeable), 10.65 (s, 1H, OH, D2O exchangeable); MS (m/z): 388 (M+, 59.76%), 326 (94.26%), 302 (92.19%), 225 (91.25%), 84 (98.76%), 43 (100%, base beak); LC-MS ESI spectrometry in the positive ion mode: M molecular ion peak at 388.2247 which also was the base peak. It showed M + 2 and M + 3 molecular ion peaks at 390.2077 and 391.2493 respectively; anal. calcd for C17H12N2O5S2 (388.4): C, 52.57; H, 3.11; N, 7.21; found: C, 52.51; H, 3.15; N, 7.15.
4.1.3.1. 2-[5-(Furan-2-ylmethylene)-2,4-dioxothiazolidin-3-yl]-N-phenylacetamide (5a). Yield, 77%; mp 240–2 °C; IRνmax (cm−1): 3212 (NH), 3050 (CH aromatic), 2950 (CH aliphatic), 1713, 1667 (3C
O amide); 1HNMR: 4.47 (s, 2H, CH2), 7.04–7.10 (m, 2H, Ar–H, H-4 of furan & H-4 of phenyl), 7.27–7.31 (m, 2H, Ar–H, H-3 & H-5 of phenyl), 7.35–7.41 (m, 2H, Ar–H, H-2 & H-6 of phenyl), 7.44–7.54 (m, 2H, Ar–H, H-3 of furan & CCH), 7.56–7.71 (m, 1H, Ar–H, H-5 of furan), 10.38 (s, 1H, NH, D2O exchangeable); anal. calcd for C16H12N2O4S (328.3): C, 58.53; H, 3.68; N, 8.53. Found: C, 58.46; H, 3.73; N, 8.55.
4.1.3.2. N-(4-Chlorophenyl)-2-[5-(furan-2-ylmethylene)-2,4-dioxothiazolidin-3-yl]acetamide (5b). Yield, 75%; mp 247–9 °C; IRνmax (cm−1): 3114 (NH), 3025 (CH aromatic), 2965 (CH aliphatic), 1729, 1672 (3C
O amide); 1HNMR: 4.56 (s, 1H, CH2), 4.65 (s, 1H, CH2), 7.25–7.28 (m, 2H, Ar–H, H-3 & H-5 of phenyl), 7.62–7.63 (m, 2H, Ar–H, H-2 & H-6 of phenyl), 7.71–7.73 (m, 1H, Ar–H, H-4 of furan), 7.90 (s, 1H, CCH), 7.92–8.00 (m, 2H, Ar–H, H-3 & H-5 of furan), 10.73 (s, 1H, NH, D2O exchangeable); anal. calcd for C16H11ClN2O4S (362.8): C, 52.97; H, 3.06; N, 7.72. Found: C, 53.07; H, 3.12; N, 7.74.
4.1.3.3. N-(4-Fluorophenyl)-2-[5-(furan-2-ylmethylene)-2,4-dioxothiazolidin-3-yl]acetamide (5c). Yield, 72%; mp 242–4 °C; IRνmax (cm−1): 3160 (NH), 3041 (CH aromatic), 2950 (CH aliphatic), 1665 (3C
O amide); 1HNMR: 4.49 (s, 2H, CH2), 6.76–6.78 (m, 1H, Ar–H, H-4 of furan), 7.13–7.18 (m, 3H, Ar–H, H-3 & H-5 of phenyl & H-3 of furan), 7.57–7.60 (m, 2H, Ar–H, H-2 & H-6 of phenyl), 7.81 (s, 1H, CCH), 8.08 (d, 1H, Ar–H, H-5 of furan), 10.50 (s, 1H, NH, D2O exchangeable); LC-MS ESI spectrometry in the negative ion mode: M − 1 molecular ion peak at 345.1374 which also was the base peak; anal. calcd for C16H11FN2O4S (346.3): C, 55.49; H, 3.20; N, 8.09. Found: C, 55.55; H, 3.24; N, 8.25.
4.1.3.4. 2-[5-(Furan-2-ylmethylene)-2,4-dioxothiazolidin-3-yl]-N-(p-tolyl)acetamide (5d). Yield, 77%; mp 247–9 °C; IRνmax (cm−1): 3138 (NH), 3092 (CH aromatic), 2956 (CH aliphatic), 1668 (3C
O amide); 1HNMR: 2.24 (s, 3H, CH3), 4.23 (s, 2H, CH2), 6.88–6.95 (m, 1H, Ar–H, H-4 of furan), 7.00–7.14 (m, 2H, Ar–H, H-3 & H-5 of phenyl), 7.25–7.41 (m, 2H, Ar–H, H-2 & H-6 of phenyl), 7.43–7.69 (m, 2H, H-3 of furan & CCH), 7.70 (d, 1H, Ar–H, H-5 of furan), 10.23 (s, 1H, NH, D2O exchangeable); 13CNMR: 20.80, 61.89, 120.02, 120.12, 123.52, 124.94, 128.17, 129.42, 129.57, 130.30, 132.95, 133.85, 135.85, 144.11, 165.14, 167.19, 168.24; MS (m/z): 344 (M+ + 2, 3.97%), 342 (M+, 73.12%), 273 (81.03%), 271 (82.51%), 115 (100%, base beak); anal. calcd for C17H14N2O4S (342.4): C, 59.64; H, 4.12; N, 8.18. Found: C, 59.75; H, 4.15; N, 8.20.
4.1.3.5. 2-[5-(Furan-2-ylmethylene)-2,4-dioxothiazolidin-3-yl]-N-(4-isopropylphenyl)acetamide (5e). Yield, 80%; mp 287–9 °C; IRνmax (cm−1): 3320 (NH), 3100 (CH aromatic), 2958 (CH aliphatic), 1669 (3C
O amide); 1HNMR: 1.17 (d, 6H, 2CH3), 2.83–2.87 (m, 1H, CH), 4.44 (s, 2H, CH2), 7.01–7.08 (m, 1H, Ar–H, H-4 of furan), 7.18–7.20 (m, 2H, Ar–H, H-3 & H-5 of phenyl), 7.36–7.47 (m, 3H, Ar–H, H-2 & H-6 of phenyl & CCH), 7.67–7.71 (m, 2H, Ar–H, H-3 & H-5 of furan), 10.30 (s, 1H, NH, D2O exchangeable); 13CNMR: 24.30 (2), 33.27, 44.23, 119.78 (2), 122.41, 123.44, 127.09 (2), 128.40, 129.48, 130.60, 134.50, 135.79, 144.77, 145.08, 165.34, 167.64; anal. calcd for C19H18N2O4S (370.4): C, 61.61; H, 4.90; N, 7.56. Found: C, 61.55; H, 4.95; N, 7.50.
4.1.3.6. 2-[5-(Furan-2-ylmethylene)-2,4-dioxothiazolidin-3-yl]-N-(4-methoxyphenyl)acetamide (5f). Yield, 76%; mp 283–5 °C; IRνmax (cm−1): 3300 (NH), 3050 (CH aromatic), 2950 (CH aliphatic), 1670 (3C
O amide); 1HNMR: 3.72 (s, 3H, OCH3), 4.46 (s, 2H, CH2), 6.78–6.80 (m, 1H, Ar–H, H-4 of furan), 6.88 (d, 2H, Ar–H, H-3 & H-5 of phenyl), 7.18–7.25 (m, 1H, Ar–H, H-3 of furan), 7.25 (d, 2H, Ar–H, H-2 & H-6 of phenyl), 7.82 (s. 1H, CCH), 8.10 (d, 1H, Ar–H, H-5 of furan), 10.30 (s, 1H, NH, D2O exchangeable); 13CNMR: 44.21, 55.63, 114.19, 114.45, 118.05, 120.07, 120.42, 121.24, 128.60, 131.92, 148.43, 149.60, 155.97, 163.82, 165.60, 168.39, 183.79; MS (m/z): 359 (M+ + 1, 5.97%), 358 (M+, 25.42%), 236 (12.33%), 208 (46.57%), 124 (90.17%), 123 (100%, base beak); anal. calcd for C17H14N2O5S (358.4): C, 56.98; H, 3.94; N, 7.82. Found: C, 57.11; H, 4.06; N, 7.76.
4.1.3.7. 4-{2-[5-(Furan-2-ylmethylene)-2,4-dioxothiazolidin-3-yl]acetamido}benzoic acid (5g). Yield, 83%; mp 275–7 °C; IRνmax (cm−1): 3375 (OH), 3200 (NH), 3050 (CH aromatic), 2950 (CH aliphatic), 1730, 1655 (4C
O amide); 1HNMR: 4.74 (s, 2H, CH2), 7.31–7.42 (m, 2H, Ar–H, H-4 of furan & CCH), 7.60–7.62 (m, 2H, Ar–H, H-3 & H-5 of furan), 7.64–7.74 (m, 2H, Ar–H, H-2 & H-6 of phenyl), 7.75–7.92 (m, 2H, Ar–H, H-3 & H-5 of phenyl), 10.02 (s, 1H, OH, D2O exchangeable), 10.92 (s, 1H, NH, D2O exchangeable); anal. calcd for C17H12N2O6S (372.4): C, 54.84; H, 3.25; N, 7.52. Found: C, 55.06; H, 3.35; N, 7.60.
Author contributions
K. El-Adl, Riham F. George and Walaa R. Mahmoud were responsible for the conception and rational design of the work. K. El-Adl and Nada A. A. M. Aziz were responsible for the data collection and synthesis of the new compounds. K. El-Adl performed the molecular docking study. Riham F. George, Walaa R. Mahmoud and K. El-Adl were responsible for spectral data analysis. Walaa R. Mahmoud and Nada A. A. M. Aziz conducted the cytotoxicity assay. Nada A. A. M. Aziz and K. El-Adl conducted the in silico pharmacokinetic study. All authors discussed the results and contributed to the writing and revision of the original manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors extend their appreciation and thanking to Dr Mohamed Ragab, Pharmacology & Toxicology Department, Faculty of Pharmacy, Al-Azhar University, Cairo, Egypt for helping in the pharmacological part.References
- D. Panigrahy, et al., PPARγ ligands inhibit primary tumor growth and metastasis by inhibiting angiogenesis, The Journal of clinical investigation, 2002, 110(7), 923–932 CrossRef CAS PubMed.
- H. Joshi, T. Pal and C. Ramaa, A new dawn for the use of thiazolidinediones in cancer therapy, Expert Opin. Invest. Drugs, 2014, 23(4), 501–510 CrossRef CAS PubMed.
- U. Bhanushali, et al., 5-Benzylidene-2, 4-thiazolidenedione derivatives: Design, synthesis and evaluation as inhibitors of angiogenesis targeting VEGR-2, Bioorg. Chem., 2016, 67, 139–147 CrossRef CAS PubMed.
- E. S. Ch'ng, H. Jaafar and S. E. Tuan Sharif, Breast tumor angiogenesis and tumor-associated macrophages: histopathologist's perspective, Pathol. Res. Int., 2011, 2011, 1–15 CrossRef PubMed.
- X. Le, M. Nilsson, J. Goldman, M. Reck, K. Nakagawa, T. Kato, L. P. Ares, B. Frimodt-Moller, K. Wolff, C. Visseren-Grul, J. V. Heymach and E. B. Garon, Dual EGFR-VEGF Pathway Inhibition: A Promising Strategy for Patients With EGFR-Mutant NSCLC, J. Thorac. Oncol., 2021, 16(2), 205–215, DOI:10.1016/j.jtho.2020.10.006.
- A. Midha, S. Dearden and R. McCormack, EGFR mutation incidencein non-small-cell lung cancer of adenocarcinoma histology: a systematic review and global map by ethnicity (mutMapII), Am. J. Cancer Res., 2015, 5, 2892–2911 Search PubMed.
- P. A. Jänne, J. C. Yang and D. W. Kim, et al., AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer, N. Engl. J. Med., 2015, 372, 1689–1699 CrossRef PubMed.
- J. J. Lin, S. Cardarella and C. A. Lydon, et al., Five-year survival in EGFR-mutant metastatic lung adenocarcinoma treatedwith EGFR-TKIs, J. Thorac. Oncol., 2016, 11, 556–565 CrossRef PubMed.
- A. K. Larsen, D. Ouaret, K. El Ouadrani and A. Petitprez, Targeting EGFR and VEGF(R) pathway cross-talk in tumor survivaland angiogenesis, Pharmacol. Ther., 2011, 131, 80–90 CrossRef CAS PubMed.
- P. M. Hoff and K. K. Machado, Role of angiogenesis in the pathogenesis of cancer, Cancer Treat. Rev., 2012, 38(7), 825–833 CrossRef CAS PubMed.
- Z. K. Otrock, J. A. Makarem and A. I. Shamseddine, Vascular endothelial growth factor family of ligands and receptors, Blood Cells, Mol., Dis., 2007, 38(3), 258–268 CrossRef CAS PubMed.
- E. Gershtein, et al., Vascular endothelial growth factor and its type 2 receptor in hepatocellular carcinoma, Bull. Exp. Biol. Med., 2010, 149(6), 749 CrossRef CAS PubMed.
- D. K. Shah, et al., Thiazolidinediones decrease vascular endothelial growth factor (VEGF) production by human luteinized granulosa cells in vitro, Fertil. Steril., 2010, 93(6), 2042–2047 CrossRef CAS PubMed.
- S. Takahashi, Vascular endothelial growth factor (VEGF), VEGF receptors and their inhibitors for antiangiogenic tumor therapy, Biol. Pharm. Bull., 2011, 34(12), 1785–1788 CrossRef CAS PubMed.
- P. Wu, T. E. Nielsen and M. H. Clausen, FDA-approved small-molecule kinase inhibitors, Trends Pharmacol. Sci., 2015, 36(7), 422–439 CrossRef CAS PubMed.
- T. Francis, S. R. Dixit and B. R. P. Kumar, Discovery of Rhodanine and Thiazolidinediones as Novel Scaffolds for EGFR Inhibition: Design, Synthesis, Analysis and CoMSIA Studies, Polycyclic Aromat. Compd., 2020, 1–17, DOI:10.1080/10406638.2020.1836004.
- Y. Chen, J. Wu, A. Wang, Z. Qi, T. Jiang, C. Chen, F. Zou, C. Hu, W. Wang and H. Wu, Discovery of N-(5-((5-chloro-4-((2-(isopropylsulfonyl) phenyl) amino) pyrimidin-2-yl) amino)-4-methoxy-2-(4-methyl-1, 4-diazepan-1-yl) phenyl) acrylamide (CHMFL-ALK/EGFR-050) as a potent ALK/EGFR dual kinase inhibitor capable of overcoming a variety of ALK/EGFR associated drug resistant mutants in NSCLC, Eur. J. Med. Chem., 2017, 139, 674–697 CrossRef CAS PubMed.
- J. Chang, H. Ren, M. Zhao, Y. Chong, W. Zhao, Y. He, Y. Zhao, H. Zhang and C. Qi, Development of a series of novel 4-anlinoquinazoline derivatives possessing quinazoline skeleton: Design, synthesis, EGFR kinase inhibitory efficacy, and evaluation of anticancer activities in vitro, Eur. J. Med. Chem., 2017, 138, 669–688 CrossRef CAS PubMed.
- Y. A. Elshaier, M. A. Shaaban, M. K. A. El Hamid, M. H. Abdelrahman, M. A. Abou-Salim, S. M. Elgazwi and F. Halaweish, Design and synthesis of pyrazolo [3, 4-d] pyrimidines: Nitric oxide releasing compounds targeting hepatocellular carcinoma, Biorg. Med. Chem., 2017, 25(12), 2956–2970 CrossRef CAS PubMed.
- H.-Q. Zhang, F.-H. Gong, J.-Q. Ye, C. Zhang, X.-H. Yue, C.-G. Li, Y.-G. Xu and L.-P. Sun, Design and discovery of 4-anilinoquinazoline-urea derivatives as dual TK inhibitors of EGFR and VEGFR-2, Eur. J. Med. Chem., 2017, 125, 245–254 CrossRef CAS PubMed.
- M. L. D. C. Barbosa, L. M. Lima, R. Tesch, C. M. R. SantAnna, F. Totzke, M. H. G. Kubbutat, C. Schachtele, S. A. Laufer and E. J. Barreiro, Novel 2-chloro-4-anilino-quinazoline derivatives as EGFR and VEGFR-2 dual inhibitors, Eur. J. Med. Chem., 2014, 71, 1–14 CrossRef PubMed.
- A. Garofalo, L. Goossens, A. Lemoine, S. Ravez, P. Six, M. Howsam, A. Farce and P. Depreux, [4-(6,7-Disubstituted quinazolin-4-ylamino)phenyl] carbamic acid esters: a novel series of dual EGFR/VEGFR-2 tyrosine kinase inhibitors, Med. Chem. Commun, 2011, 2, 65–72 RSC.
- K. El-Adl, A.-H. Abdel-Rahman, A. M. Omar, M. Alswah and N. M. Saleh, Arch. Pharm., 2021, e2100237, DOI:10.1002/ardp.202100237.
- V. Gandin, A. Ferrarese, M. Dalla Via, C. Marzano, A. Chilin and G. Marzaro, Targeting kinases with anilinopyrimidines: discovery of N-phenyl-N’-[4-(pyrimidin-4-ylamino) phenyl] urea derivatives as selective inhibitors of class III receptor tyrosine kinase subfamily, Sci. Rep., 2015, 5, 16750 CrossRef CAS PubMed.
- K. El-Adl, H. Sakr, M. Nasser and F. M. A. Shoman, Arch. Pharm., 2020, e2000079, DOI:10.1002/ardp.202000079.
- K. El-Adl, A. A. El-Helby, H. Sakr and S. S. A. El-Hddad, Arch. Pharm., 2020, e2000068, DOI:10.1002/ardp.202000068.
- K. El-Adl, A. A. El-Helby, H. Sakr, I. H. Eissa, S. S. A. El-Hddad and F. M. A. Shoman, Bioorg. Chem., 2020, 102, 104059, DOI:10.1016/j.bioorg.2020.104059.
- A. M. Sayed, F. A. Taher, M. R. K. Abdel-Samad, M. S. A. El-Gaby, K. El-Adl and N. M. Saleh, Bioorg. Chem., 2021, 108, 104669, DOI:10.1016/j.bioorg.2021.104669 , Epub 2021 Jan 21. PMID: 33515863, .
- N. M. Saleh, M. S. A. El-Gaby, K. El-Adl and N. E. A. Abd El-Sattar, Bioorg. Chem., 2020, 104, 104350, DOI:10.1016/j.bioorg.2020.104350.
- K. El-Adl, A. A. El-Helby, H. Sakr and A. Elwan, Bioorg. Chem., 2020, 105, 104399, DOI:10.1016/j.bioorg.2020.104399.
- K. El-Adl, A. A. El-Helby, H. Sakr and A. Elwan, New J. Chem., 2021, 45, 881, 10.1039/D0NJ02990D.
- N. E. A. Abd El-Sattar, K. El-Adl, M. A. El-Hashash, S. A. Salama and M. M. Elhady, Bioorg. Chem., 2021, 22(115), 105186, DOI:10.1016/j.bioorg.2021.105186 . Epub ahead of print. PMID: 34314914, .
- Q.-Q. Xie, et al., Pharmacophore modeling studies of type I and type II kinase inhibitors of Tie2, J. Mol. Graphics Modell., 2009, 27(6), 751–758 CrossRef CAS PubMed.
- K. Lee, et al., Pharmacophore modeling and virtual screening studies for new VEGFR-2 kinase inhibitors, Eur. J. Med. Chem., 2010, 45(11), 5420–5427 CrossRef CAS PubMed.
- V. A. Machado, et al., Synthesis, antiangiogenesis evaluation and molecular docking studies of 1-aryl-3-[(thieno [3, 2-b] pyridin-7-ylthio) phenyl] ureas: Discovery of a new substitution pattern for type II VEGFR-2 Tyr kinase inhibitors, Bioorg. Med. Chem., 2015, 23(19), 6497–6509 CrossRef CAS PubMed.
- Z. Wang, et al., Dietary compound isoliquiritigenin inhibits breast cancer neoangiogenesis via VEGF/VEGFR-2 signaling pathway, PLoS One, 2013, 8(7), e68566 CrossRef CAS PubMed.
- J. Dietrich, C. Hulme and L. H. Hurley, The design, synthesis, and evaluation of 8 hybrid DFG-out allosteric kinase inhibitors: A structural analysis of the binding interactions of Gleevec®, Nexavar®, and BIRB-796, Bioorg. Med. Chem., 2010, 18(15), 5738–5748 CrossRef CAS PubMed.
- Z. Zhao, H. Wu, L. Wang, Y. Liu, S. Knapp, Q. Liu and N. S. Gray, Exploration of type II binding mode: a privileged approach for kinase inhibitor focused drug discovery?, ACS Chem. Biol., 2014, 9(6), 1230–1241 CrossRef CAS PubMed.
- S. A. Elmetwally, K. F. Saied, I. H. Eissa and E. B. Elkaeed, Design, synthesis and anticancer evaluation of thieno[2,3-d]pyrimidine derivatives as dual EGFR/HER2 inhibitors and apoptosis inducers, Bioorg. Chem., 2019, 88, 102944, DOI:10.1016/j.bioorg.2019.102944.
- F. Khedr, M.-K. Ibrahim, I. H. Eissa, H. S. Abulkhair and K. El-Adl, Arch. Pharm., 2021, e2100201, DOI:10.1002/ardp.202100201.
- T. Mosmann, Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays, J. Immunol. Methods, 1983, 65(1–2), 55–63 CrossRef CAS PubMed.
- F. M. Freimoser, et al., The MTT [3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide] assay is a fast and reliable method for colorimetric determination of fungal cell densities, Appl. Environ. Microbiol., 1999, 65(8), 3727–3729 CrossRef CAS PubMed.
- S. M. Abou-Seri, et al., 1-Piperazinylphthalazines as potential VEGFR-2 inhibitors and anticancer agents: synthesis and in vitro biological evaluation, Eur. J. Med. Chem., 2016, 107, 165–179 CrossRef CAS PubMed.
- C. Deng, J. Xiong, X. Gu, X. Chen, S. Wu, Z. Wang, D. Wang, J. Tu and J. Xie, Novel recombinant immunotoxin of EGFR specific nanobody fused with cucurmosin, construction and antitumor efficiency in vitro, Oncotarget, 2017, 8(24), 38568–38580 CrossRef PubMed.
- Y. Jia, C. M. Quinn, A. I. Gagnon and R. Talanian, Homogeneous time-resolved fluorescence and its applications for kinase assays in drug discovery, Anal. Biochem., 2006, 356(2), 273–281 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Deliv. Rev., 1997, 23, 3, DOI:10.1016/S0169-409X(96)00423-1.
- D. E. V. Pires, T. L. Blundell and D. B. Ascher, J. Med. Chem., 2015, 58, 4066, DOI:10.1021/acs.jmedchem.5b00104.
- A. Beig, R. Agbaria and A. Dahan, PLoS One, 2013, 8, e68237, DOI:10.1371/journal.pone.0068237.
- G. Marc, A. Stana, A. Pîrnău, L. Vlase, D. C. Vodnar, M. Duma, B. Tiperciuc and O. Oniga, 3,5-Disubstituted Thiazolidine-2,4-Diones: Design, Microwave-Assisted Synthesis, Antifungal Activity, and ADMET Screening, SLAS Discovery, 2018, 23(8), 807–814, DOI:10.1177/2472555218759035.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra01119k |
| This journal is © The Royal Society of Chemistry 2022 |